

Abstract
Objective:
Glucocorticoids (GCs) are used as standard treatment for acute attacks of multiple sclerosis (MS). However, GCs eventually lose efficacy and do not prevent disease progression. Macrophage migration inhibitory factor (MIF) is the only known proinflammatory cytokine induced by GCs that inhibits their anti-inflammatory effects. Therefore, we investigated whether MIF plays a role in resistance to GC treatment in experimental autoimmune encephalomyelitis (EAE), an animal model of MS.
Methods:
EAE was induced in wild-type (Wt) and MIF knockout (MIF−/−) mice followed by treatment with dexamethasone (Dex) before or upon disease onset. Splenocytes and brain mononuclear cells were harvested for cytokine ELISPOT assay and flow cytometry analysis.
Results:
Treatment of EAE with Dex was substantially more efficacious in MIF−/− mice than Wt mice. Dex treatment decreased MOG35-55–induced cytokine production by Wt or MIF−/− CD4+ T cells only at the onset of EAE but inhibited upregulation of T-bet during acute and chronic phases of disease, particularly in MIF−/− mice. Furthermore, passive EAE induced by adoptive transfer of T cells showed that Dex was highly effective in ameliorating disease induced by MIF−/− CD4+ T cells but not by Wt CD4+ T cells. The expression of T-bet and VLA-4 was decreased in CD4+ T cells in MIF−/− mice compared with Wt mice.
Conclusions:
Our data establish MIF as a key molecule in resistance of pathogenic CD4+ T cells to GC treatment in EAE and as a potential target to enhance the effectiveness of steroid treatment in neuroinflammatory disorders.
Multiple sclerosis (MS) is a chronic inflammatory disease of the CNS characterized by demyelination, axonal loss, and varying degrees of immune infiltrates, including T cells.1,2 Studies in experimental autoimmune encephalomyelitis (EAE) have demonstrated a critical role for T cells in the disease process via the release of proinflammatory cytokines.3,4
Previously, macrophage migration inhibitory factor (MIF) was shown to promote EAE by enhancing T cell effector function5–7; however, the underlying mechanisms have not been fully resolved. MIF inhibits random migration of macrophages, activates T cells, and enhances macrophage tumor necrosis factor α and nitric oxide production.8,9 MIF has the unique property of being induced by glucocorticoids (GCs) and counterregulating their anti-inflammatory effects.10–13 Along these lines, it was recently suggested that MIF may promote progression of EAE via antagonizing endogenous GCs.6
GCs are the standard treatment for acute attacks of MS and effectively induce clinical remission.14,15 However, GC treatment does not cure MS and does not prevent long-term disease progression.15 GC treatment has numerous side effects and some patients develop resistance.15,16 Thus, optimizing GC treatment and dosage may decrease side effects and prevent development of GC resistance.
Here, we show that treatment of EAE with dexamethasone (Dex) was substantially more efficacious in MIF−/− mice. The data suggest that MIF promotes disease in EAE via inhibition of immunosuppressive GC effects on pathogenic T cells, conceivably via T-bet and VLA-4 expression.
METHODS
Mice.
C57BL/6 mice (6–8 weeks old) were purchased from The Jackson Laboratory (Bar Harbor, ME). C57BL/6 MIF−/− mice were generated as described.11 All animals were maintained under pathogen-free conditions in the American Association for Laboratory Animal Science facility at the University of Texas at San Antonio.
Standard protocol approvals.
All experiments were approved by the Institutional Animal Care and Use Committee and performed in accordance with the relevant guidelines and regulations.
EAE induction.
Active EAE was induced in wild-type (Wt) and MIF−/− mice by subcutaneous (SC) injection of 300 μg MOG35-55 peptide (United Biochemical Research, Seattle, WA) in 50 µL of complete Freund's adjuvant (CFA). Mice also received intraperitoneal (IP) injections of 400 ng pertussis toxin on days 0 and 2. For induction of passive EAE by adoptive transfer, donor mice were immunized SC with 300 µg of MOG35-55 in CFA followed by IP injections of 10 mg/kg Dex or phosphate-buffered saline (PBS) for 3 continuous days starting on day 7. Splenocytes and draining lymph nodes were collected from donor mice on day 10 and restimulated for 3 days at 37°C with 20 µg/mL of MOG35-55 peptide in complete Dulbecco modified Eagle medium containing 10% fetal bovine serum, 20 ng/mL of recombinant mouse interleukin (IL)-23 (R&D Systems, Minneapolis, MN), and 10 µg/mL of anti-interferon (IFN)-γ monoclonal antibody (mAb) (Bio X Cell, West Lebanon, NH; R4-6A2), with or without 10−8 M of Dex.17,18 Recipient mice were injected with 2 × 107 restimulated donor cells. For recipients in pretreatment groups, 10 mg/kg Dex or PBS control was given by IP injections for 3 continuous days prior to adoptive transfer.
EAE evaluation.
Mice were monitored and graded daily for clinical signs of EAE using the following scoring system19: 0, no abnormality; 1, limp tail; 2, moderate hind limb weakness; 3, complete hind limb paralysis; 4, quadriplegia or premoribund state; 5, death.
Cytokine ELISPOT assay.
Cytokine ELISPOT assay was performed and spots analyzed as described previously.20 In brief, ELISPOT plates (Millipore, Billerica, MA) were precoated with anti-mouse-IFN-γ mAb (eBioscience, San Diego, CA; AN-18) and anti-mouse-IL-17 mAb (Bio X Cell; 17F3). Splenocytes (1 × 106 cells per well) and brain-infiltrating mononuclear cells (MNCs; 2–5 × 104 cells per well) were restimulated with MOG35-55 peptide in HL-1 medium (Lonza, Basel, Switzerland) at 37°C for 24 hours. Biotinylated anti-mouse-IFN-γ mAb (eBioscience; R4-6A2) and anti-mouse-IL-17 mAb (BioLegend, San Diego, CA; TC11-8H4) were then added overnight at 4°C, followed by incubation with streptavidin alkaline phosphatase (Invitrogen, Waltham, MA) for 2 hours at room temperature and developing with BCIP/NBT Phosphatase Substrate (KPL, Gaithersburg, MD). After plate developing, image analysis of spots was performed on a Series 2 Immunospot analyzer (Cellular Technology Limited, Cleveland, OH).
Flow cytometry analysis.
EAE brains were collected and depleted of myelin using myelin removal beads (Miltenyi, Bergisch Gladbach, Germany). Brain MNCs were quantified by hemocytometer. For cell surface staining, cell suspensions were stained with fluorochrome-conjugated anti-mouse-CD4 (eBioscience; GK1.5), anti-mouse-CD8b (eBioscience; eBioH35-17.2), anti-mouse-CD11c (eBioscience; N418), anti-mouse-CD11b (eBioscience; M1/70), anti-mouse-VLA-4 (ATCC, Manassas, VA; PS/2), and anti-mouse-IL-23R (R&D Systems; 258010) mAbs, followed by red blood cell lysis (Beckman Coulter, Brea, CA). For intracellular staining (ICS), cells were restimulated at 5–10 × 105 cells/mL with 5 μg/mL of MOG35-55 peptide in HL-1 medium for 24 hours, followed by cell surface staining and then ICS with anti-T-bet mAb (eBioscience; 4B10) using Transcription Factor Staining Buffer (eBioscience). For tetramer staining, preadoptive transfer donor cells were stained with fluorochrome-conjugated anti-mouse-CD4 mAb (BD, Franklin Lakes, NJ; RM4-5) and IAb-MOG38-49 or IAb-hCLIP tetramers (NIH Tetramer Core Facility, Atlanta, GA) as controls by incubation at 4°C for 1.5 hours. Samples were costained with fluorochrome-conjugated anti-mouse-VLA-4 mAb or subsequently with anti-T-bet mAb. For Annexin V staining, cells were incubated for 15 minutes in the dark with Annexin V in binding buffer (BD) after cell surface staining of anti-mouse-CD4 mAb and tetramers. All samples were analyzed by BD FACSAria II.
Statistical analysis.
Analysis of variance (ANOVA) was used for statistical analysis involving multiple groups (SigmaPlot 12.5). Nonparametric comparison (Mann-Whitney test) was used as indicated if ANOVA did not show significance. A t test was used for pairwise comparison of groups as indicated.
RESULTS
Treatment of EAE with Dex is more efficacious in MIF−/− mice.
To test whether MIF inhibited the disease-ameliorating effects of GCs in EAE, C57BL/6 Wt and MIF−/− mice were immunized with MOG35-55 peptide and observed and scored for clinical disease as described in the Methods. The mice were treated daily with Dex or PBS starting on day 6 postimmunization until the peak of disease.
As shown in figure 1A, Wt and MIF−/− mice treated with PBS developed EAE with a similar onset of disease. EAE scores were somewhat lower in MIF−/− mice, but the differences did not reach statistical significance (figure 1A, closed circles and triangles). Consistent with previous reports, treatment of C57BL/6 Wt mice with Dex delayed EAE onset and significantly reduced disease severity (figure 1A, open circles).21 However, once the Dex treatment was stopped, EAE rebounded in Wt mice and the animals developed disease with severity similar to PBS-treated control mice. In strong contrast, EAE was inhibited by Dex treatment in MIF−/− mice and remained significantly suppressed after the treatment was ended (figure 1A, open triangles).
Figure 1. EAE severity and CNS pathology in Dex-treated MIF−/− and Wt mice.
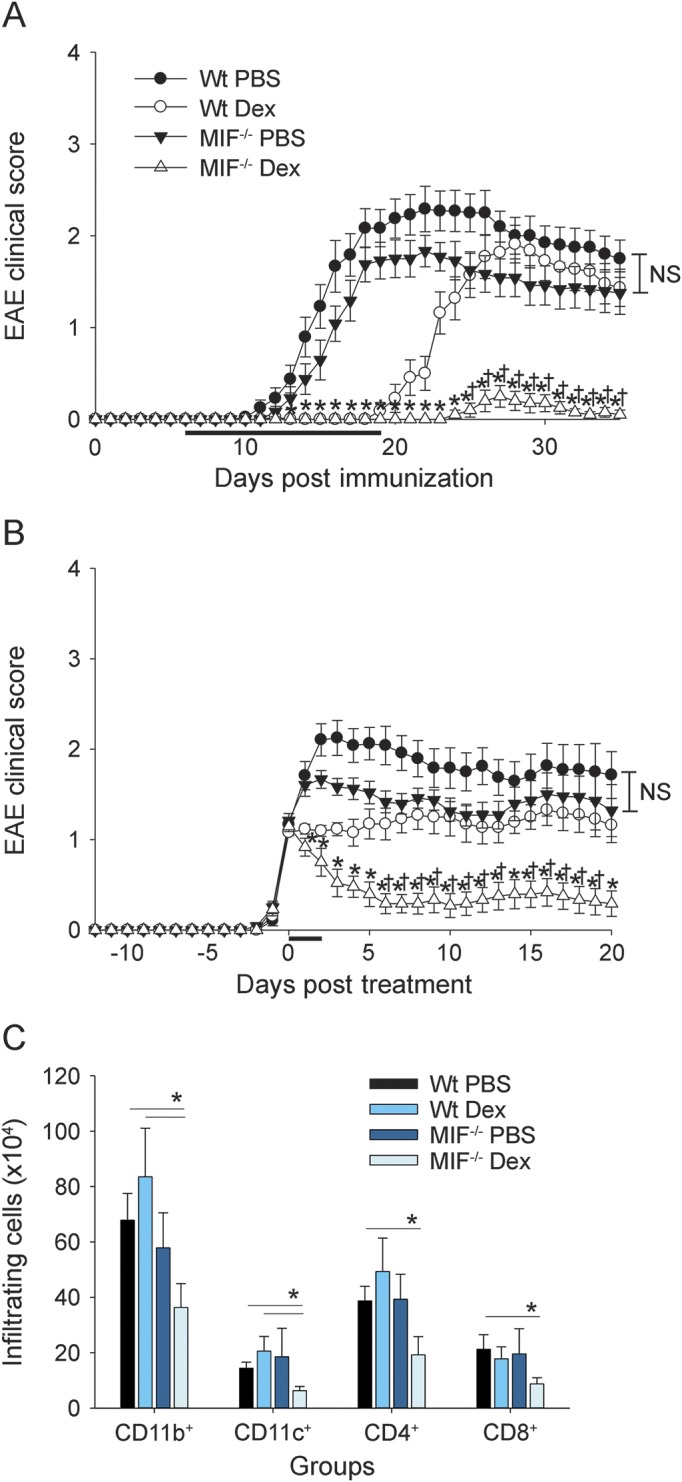
Wild-type (Wt) and macrophage migration inhibitory factor (MIF)−/− mice were immunized to induce experimental autoimmune encephalomyelitis (EAE) as described in the Methods. Mice were injected intraperitoneally with (A) 5 mg/kg dexamethasone (Dex) vs phosphate-buffered saline (PBS) control every day starting on day 6 and then every other day after onset until the peak of disease (Wt PBS, n = 12; Wt Dex, n = 11; MIF−/− PBS, n = 12; MIF−/− Dex, n = 10) or (B) 10 mg/kg Dex vs PBS control for 3 continuous days upon onset of disease (Wt PBS, n = 12; Wt Dex, n = 13; MIF−/− PBS, n = 12; MIF−/− Dex, n = 12). Mice were scored daily for EAE as described in the Methods. Shown are pooled data from 2 to 3 independent experiments (mean ± SEM). *indicates significant difference between MIF−/− Dex group and Wt PBS group or MIF−/− PBS group; †indicates significant difference between MIF−/− Dex group and Wt Dex group (analysis of variance [ANOVA], p < 0.05). EAE mice from (B) were sacrificed for analysis (C) at the acute stage (day 16–24 postimmunization, whenever PBS groups reached clinical score of 2 or higher) of disease and then quantified for CD11b+, CD11c+, CD4+, and CD8+ cellular infiltration in the brain by flow cytometry. Shown are pooled data from 3 independent experiments (mean ± SEM; n = 7–8 mice per group; ANOVA, no significant difference; post hoc Mann-Whitney test for indicated groups; *p < 0.05). NS = not significant.
To further investigate the effectiveness of Dex treatment in MIF−/− vs Wt mice, the treatment period with Dex was shortened to 3 days starting at the onset of clinical symptoms of EAE (score 1–1.5, as described in the Methods).
The shorter treatment regimen only modestly decreased the clinical EAE severity in Wt mice (figure 1B, open circles). In strong contrast, the shorter course of Dex treatment was still highly effective at suppressing clinical EAE in MIF−/− mice and no significant disease rebound was observed (figure 1B, open triangles).
Histopathologic evaluation of the CNS of PBS-treated Wt and MIF−/− mice at the peak of EAE by immunofluorescence staining (figure e-1 at Neurology.org/nn) and quantification of inflammatory cells by flow cytometry corroborated the clinical EAE results and showed large numbers of infiltrating inflammatory cells consisting of CD11c+ dendritic cells, CD11b+ microglia/infiltrating macrophages, CD4+ T cells, and CD8+ T cells (figure 1C). In contrast, Dex-treated MIF−/− mice showed significantly reduced numbers of inflammatory cells in the CNS during EAE compared with Wt mice.
Taken together, the results showed that GC treatment was significantly more efficacious in MIF−/− mice, even under conditions that were suboptimal clinically in Wt animals.
Temporary inhibition of IFN-γ and IL-17 by Dex in Wt and MIF−/− CD4+ T cells.
Immunosuppressive effects of GCs on cytokine production by inflammatory cells have been reported.22 Therefore, we tested whether Dex treatment was more effective at inhibiting the production of the pathogenic cytokines IFN-γ or IL-17 by Wt or MIF−/− CD4+ T cells.
Spleen- and brain-infiltrating MNCs were procured from Wt and MIF−/− mice at different time points after induction of EAE and treatment for 3 days with Dex or PBS and tested by cytokine ELISPOT assay for MOG35-55–induced production of IFN-γ and IL-17.
The results showed that Dex treatment substantially decreased the production of IFN-γ by MOG35-55–reactive spleen CD4+ T cells at the onset of EAE in both Wt and MIF−/− mice compared with PBS-injected animals (figure 2A). In contrast, Dex treatment significantly decreased IL-17 production only in MIF−/− mice treated with Dex at disease onset (figure 2B). The inhibitory effects of Dex on cytokine production by spleen CD4+ T cells were no longer observed at later time points, irrespective of whether the mice were Wt or MIF−/− (figure 2, A and B). No shift toward the production of Th2 cytokines was observed (data not shown).
Figure 2. Effect of Dex on cytokine production by MIF−/− or Wt T cells in EAE.

Wild-type (Wt) and macrophage migration inhibitory factor (MIF)−/− mice were immunized to induce experimental autoimmune encephalomyelitis (EAE) and injected intraperitoneally with 10 mg/kg dexamethasone (Dex) vs phosphate-buffered saline (PBS) control for 3 continuous days upon onset of disease, as shown in figure 1B. Spleens (A, B) and brain mononuclear cells (MNCs) (C, D) of EAE mice were harvested at different time points and restimulated with MOG35-55 peptide. The frequencies of Th1 and Th17 cytokine-producing T cells were measured by ELISPOT assay as described in the Methods. Shown are pooled data from 2 to 4 independent experiments (mean ± SEM; onset, n = 7–14 mice per group from day 10 postimmunization and after 3 injections of Dex or PBS; acute, n = 5 mice per group from day 16–24 postimmunization when PBS groups reached peak score of 2 or higher; chronic, n = 6–8 mice per group from day 26–36, usually 10 days after PBS groups remitted from peak of disease; analysis of variance; *p < 0.05; **p < 0.01). IFN = interferon; IL = interleukin; NS = not significant; SFC = spot-forming cells.
In the CNS, no significant effect of Dex on IFN-γ or IL-17 production was observed during the acute phase of EAE in Wt or MIF−/− mice (figure 2, C and D).
Collectively, the results showed an inhibitory effect of Dex on IFN-γ production by Wt and MIF−/− T cells that was most pronounced at disease onset. Dex suppressed IL-17 only in MIF−/− mice.
T-bet expression is decreased in MIF−/− CD4+ T cells during EAE.
Expression of T-bet has been suggested to be critical for the function of encephalitogenic T cells and can be downregulated by GCs.23–25 Therefore, we investigated the effect of MIF on the regulation of T-bet expression by GCs in EAE.
EAE was induced in Wt and MIF−/− mice and the animals were treated once daily for 3 days with Dex or PBS starting at the onset of EAE, as described earlier. CD4+ T cells were recovered from the CNS at the acute or chronic stage of EAE and reactivated with MOG35-55 peptide in culture, and the expression of T-bet was determined by flow cytometry analysis.
The results show that the expression of T-bet in CD4+ T cells was comparable between naive Wt and MIF−/− mice upon activation with anti-CD3 mAb (figure 3F). Moreover, T-bet was highly expressed in CD4+ T cells recovered from the CNS of PBS-treated Wt mice during the acute stage of EAE (figure 3A upper left; figure 3B). Treatment of Wt mice with Dex resulted in an approximately 30% reduction of T-bet expression by CD4+ T cells, which did not reach statistical significance (figure 3A upper right; figure 3B). It is interesting that T-bet expression was significantly lower in CD4+ T cells from both PBS- and Dex-treated MIF−/− mice compared with Wt animals (figure 3A, lower panels; figure 3B, p < 0.05). Dex treatment of MIF−/− mice showed a decrease in the expression of T-bet in CD4+ T cells and the total numbers of CD4+T-bet+ T cells compared with PBS treatment of MIF−/− mice, particularly during the chronic phase, but statistical significance was not achieved (figure 3, C–E). However, T-bet expression by CD4+ T cells remained significantly decreased in Dex-treated MIF−/− mice during the chronic phase of EAE compared with PBS-treated Wt mice (figure 3C, p < 0.05).
Figure 3. Downregulation of T-bet expression in brain-infiltrating MIF−/− CD4+ T cells.

Macrophage migration inhibitory factor (MIF)−/− and wild-type (Wt) mice were immunized to induce experimental autoimmune encephalomyelitis (EAE) and injected intraperitoneally with 10 mg/kg dexamethasone (Dex) vs phosphate-buffered saline (PBS) control for 3 continuous days upon onset of disease, as shown in figure 1B. Brain mononuclear cells (MNCs) of EAE mice from acute and chronic stages were harvested and cultured for 24 hours with MOG35-55 peptide as described in the Methods. Ag-recalled brain MNCs were stained for CD4 and T-bet for flow cytometry analysis. Shown are representative intracellular staining results of T-bet expression gated on CD4+ T cells (A), pooled results from 2 independent experiments for T-bet expression (B, C), and absolute number of brain-infiltrating T-bet+CD4+ T cells (D, E) from acute stage and chronic stage of EAE (mean ± SEM; acute stage, n = 6–8 mice per group from day 16–24 postimmunization when PBS groups reached peak score of 2 or higher; chronic stage, n = 5–9 mice per group from day 26–36, usually 10 days after PBS groups remitted from peak of disease; analysis of variance; *p < 0.05). (F) T-bet expression in activated CD4+ T cells under non-EAE conditions. Splenocytes from naive Wt and MIF−/− mice were cultured with plate-bound anti-CD3 monoclonal antibody for 48 hours prior to staining for CD4 and T-bet for flow cytometry analysis (Wt, n = 11; MIF−/−, n = 12; shown are pooled results from 4 independent experiments; mean ± SEM; t test). NS = not significant.
Since T-bet can regulate the expression of IL-23R and VLA-4, which are critical molecules for the effector function and CNS migration of encephalitogenic T cells,26–28 we investigated whether MIF promotes EAE by antagonizing GC effects on these molecules.
EAE was induced in Wt or MIF−/− mice followed by treatment with Dex or PBS as indicated. Brain-infiltrating MNCs were analyzed during the chronic stage of EAE by flow cytometry for IL-23R and VLA-4 expression on CD4+ T cells. As shown in figure e-2A, the expression of IL-23R was decreased by CNS-infiltrating CD4+ T cells from PBS-treated MIF−/− mice, but the results did not achieve statistical significance. Dex treatment did not significantly affect IL-23R expression by Wt or MIF−/− CD4+ T cells. Similarly, VLA-4 expression by CD4+ T cells was lower in MIF−/− mice than in Wt mice (figure e-2B). Furthermore, Dex had a moderate, but not statistically significant, inhibitory effect on VLA-4 expression in MIF−/− but not Wt mice.
Overall, the data show that the expression of T-bet was significantly decreased in MIF−/− mice with EAE, but its expression was not further suppressed by exogenous GCs. Furthermore, the expression of IL-23R and VLA-4 by CD4+ T cells was decreased in MIF−/− mice compared with Wt mice, but Dex treatment did not provide a significant additional inhibitory effect, irrespective of whether the mice were MIF deficient.
MIF promotes resistance of autoreactive T cells to Dex treatment.
Recently, it has been suggested that MIF has an important role in the activation of microglia and the recruitment of macrophages to the CNS, which are critical in promoting EAE, whereas other studies showed a role for this cytokine in supporting the function of pathogenic T cells.5–7
Thus, we investigated whether MIF promotes EAE by inhibiting GC effects on infiltrating inflammatory cells or on CNS-resident tissue cells in our model.
To begin to address this question, passive EAE was induced by adoptive transfer of activated MOG35-55–reactive T cells from Dex- or PBS-treated Wt or MIF−/− mice into Wt recipient mice as described in the Methods. Dex-conditioned MOG35-55–specific MIF−/− donor T cells were significantly less potent at inducing EAE in Wt recipient mice compared with Dex-treated Wt T cells or PBS-treated MIF−/− or Wt T cells (figure 4A). In contrast, pretreatment of Wt or MIF−/− recipient mice with Dex did not significantly attenuate EAE induced by adoptive transfer of MOG35-55–specific Wt donor cells (figure 4B).
Figure 4. Increased susceptibility of MIF−/− CD4+ T cells to immunosuppression by Dex in EAE.

(A) MOG35-55 peptide-activated donor cells from wild-type (Wt) or macrophage migration inhibitory factor (MIF)−/− mice pretreated with phosphate-buffered saline (PBS) or dexamethasone (Dex) were adoptively transferred into Wt recipient mice as described in the Methods. (B) Transfer of Wt donor cells into Wt or MIF−/− recipient mice pretreated with PBS or Dex. Shown is 1 representative experiment of 2–3 independent experiments (n = 5 mice per group; mean ± SEM). *indicates significant difference between recipient groups that received MIF−/− Dex donor cells vs Wt PBS donor cells; †indicates significant difference between recipient groups that received MIF−/− Dex donor cells vs Wt Dex or MIF−/− PBS donor cells (analysis of variance [ANOVA], p < 0.05; NS = not significant). Spleen (C) and brain mononuclear cells (MNCs) (D) of recipient mice on day 23 (A) were harvested and restimulated with MOG35-55 peptide. The frequencies of Th1 and Th17 cytokine-producing T cells from recipients were measured by ELISPOT assay as described in the Methods (n = 5 mice per group; mean ± SEM; ANOVA, *p < 0.05). (E) Preadoptive transfer donor cells were stained after 3-day Ag recall and analyzed by flow cytometry for MOG35-55–specific CD4+ T cells using IAb-MOG38-49 tetramer as described in the Methods. Shown are absolute numbers of Ag-specific CD4+ T cells of different donor groups from pooled data of 5 independent experiments (n = 9–16 mice per group; mean ± SEM; ANOVA, NS). (F, G) Cytokine ELISPOT assays were performed using preadoptive transfer donor cells after 3-day Ag recall as described in the Methods. Shown are pooled data from 3 to 5 independent experiments (F: interleukin (IL)-17, n = 11–15 mice per group; G: interferon (IFN)-γ, n = 9–12 mice per group; mean ± SEM; ANOVA, *p < 0.05, NS). In parallel, preadoptive transfer donor cells were stained and analyzed by flow cytometry for (H) VLA-4 and (I) T-bet expression gated on MOG35-55–specific CD4+ T cells, as described in the Methods. Shown are pooled data from 3 to 5 independent experiments (n = 10–17 mice per group; mean ± SEM; ANOVA, *p < 0.05, **p < 0.01, NS). SFC = spot-forming cells.
To determine whether Dex treatment of the MIF−/− donor cells altered T cell effector function, IFN-γ and IL-17 production by spleen- and CNS-infiltrating T cells was measured by cytokine ELISPOT assay at the end of the adoptive transfer studies.
As shown in figure 4, a significant decrease in the frequencies of IFN-γ– and IL-17–producing MOG35-55–specific CD4+ T cells was observed in the spleen of mice that had received Dex-treated MIF−/− donor T cells compared with recipients of Dex- or PBS-treated Wt cells (figure 4C, p < 0.05). Moreover, adoptive transfer of Dex-treated MIF−/− cells substantially decreased the number of IL-17–producing T cells in the CNS by day 23 posttransfer compared with PBS-treated MIF−/− cells (figure 4D).
Next, we investigated whether the decrease in EAE severity and numbers of MOG35-55–reactive T cells observed after adoptive transfer of Dex-treated MIF−/− T cells was due to deletion or silencing of pathogenic T cells in the recipient mice or whether these cells were already decreased during restimulation in vitro.
To directly determine the frequencies of Ag-specific CD4+ T cells, we used IAb-MOG-38-49 tetramer staining to quantify MOG35-55–specific CD4+ T cells, as described in the Methods. The results showed no significant difference in the numbers of MOG35-55–specific CD4+ T cells in Dex-treated Wt and MIF−/− CD4+ T cells compared with PBS-treated Wt and MIF−/− T cells (figure 4E; figure e-3A). Moreover, no significant difference was observed in the frequencies of IL-17–producing MOG35-55–specific T cells between PBS- or Dex-treated Wt and MIF−/− mice (figure 4F). Consistent with our earlier results, Dex treatment decreased the frequencies of IFN-γ–producing MOG-specific T cells, but no significant difference was observed between Wt and MIF−/− T cells (figure 4G).
Next, we investigated whether the reduced ability of Dex-conditioned MIF−/− T cells to induce EAE was due to increased apoptosis or altered VLA-4 or T-bet expression. As shown in figure e-3B, a modest, but not statistically significant, increase in apoptosis was observed in Dex-treated MIF−/− T cells compared with the other groups. However, we noted a significant decrease in the expression of VLA-4 by MOG35-55–specific CD4+ T cells from Dex-treated MIF−/− donors compared with PBS-treated Wt and MIF−/− donor cells (figure 4H). Furthermore, T-bet expression was significantly downregulated in Ag-specific CD4+ T cells from both Dex-treated MIF−/− and Wt donors compared with PBS-treated Wt donors, and its expression in Ag-specific CD4+ T cells was the lowest in the Dex-treated MIF−/− group (figure 4I). Taken together, the results suggest that MIF promotes EAE by antagonizing the suppressive effects of Dex on T-bet and VLA-4 expression by pathogenic T cells.
DISCUSSION
In this study, we show that MIF is an important mediator of resistance to Dex treatment in EAE by antagonizing its suppressive effects on T-bet and VLA-4 expression by encephalitogenic T cells.
MIF is an important mediator of autoimmune pathology.29,30 Studies in the EAE model in mice showed that MIF has an important role in promoting disease by enhancing T cell effector function and survival and migration of pathogenic T cells into the CNS.5–7 MIF is highly expressed in human leukocytes and it has been implicated in several human disease conditions, including MS.31–33 Treatment with anti-MIF mAb improved disease in mouse models of EAE and other autoimmune diseases.5,34,35
An intriguing property of MIF is that it can be induced by GCs and subsequently counterregulates anti-inflammatory GC effects. Therefore, MIF may also promote autoimmune pathology by antagonizing the immunosuppressive effects of endogenous and/or exogenous GCs.
Here, we tested this question in the MOG35-55–induced EAE model in C57BL/6 Wt and MIF−/− mice in combination with Dex treatment. Consistent with previous studies, we found that Dex treatment of Wt mice substantially attenuated, but did not completely prevent, EAE.36 Moreover, EAE was decreased in MIF−/− mice, although the effect in our studies was not as pronounced as in previous studies, possibly due to differences in animal backgrounds, housing conditions, and scoring system.6,7 In contrast, EAE was profoundly suppressed in Dex-treated MIF−/− mice, even at a suboptimal dosage that failed to suppress disease in Wt mice.
When investigating the underlying mechanisms, we found that Dex similarly suppressed the production of IFN-γ in Wt and MIF−/− mice, but the effect was temporary and waned over the course of disease. In contrast, Dex was more effective at decreasing IL-17 production in MIF−/− mice than in Wt mice. Thus, the results suggest that MIF interfered with additional immunosuppressive mechanisms of Dex in EAE beyond suppression of T cell cytokine production.
Recent work suggested that regulation of T-bet is a critical factor for the pathogenicity of autoreactive T cells, irrespective of their Th1/Th17 cytokine profiles.25 GCs have been reported to interfere with the transcription of T-bet in animal models and in immune cells from patients with MS.23,24 Therefore, we examined whether MIF interfered with the downregulation of T-bet by GCs. Expression of T-bet was significantly decreased in CD4+ T cells of Dex-treated MIF−/− mice compared with Wt T cells and remained downregulated during the chronic stage of EAE.
It is known that T-bet, in combination with other transcription factors such as Runx, is important for the development of Th1-like Th17 cells, which are thought to be the most pathogenic T cell population, and can regulate the expression of IL-23R and VLA-4.37,38 We observed that VLA-4 expression was significantly suppressed by Dex in MIF−/− CD4+ T cells compared with Wt T cells, but the effects on IL-23R expression were negligible. Thus, by antagonizing Dex effects on T-bet and VLA-4, MIF may promote the migration and effector function of pathogenic T cells in the CNS. Our studies did not test whether MIF could antagonize suppression of CXCR4 by GCs in light of recent reports that MIF can modulate chemokine signaling via CXCR4.39,40
Using the adoptive transfer EAE model, we found that treatment with Dex completely abrogated EAE mediated by MIF−/− T cells but not by Wt T cells. In contrast, Dex treatment of Wt or MIF−/− recipient mice did not show differences in onset or severity of EAE, suggesting that MIF was important for antagonizing Dex effects on encephalitogenic T cells and to a lesser degree on CNS tissue–resident cells. Nevertheless, while our results show that MIF antagonizes GC effects primarily on encephalitogenic T cells independently, it may promote EAE via additional effects on CNS tissue–resident cells, as has been suggested.7
There remain unresolved questions regarding our understanding of the signaling pathways targeted by MIF to interfere with immunosuppressive GC effects in pathogenic T cells, which could potentially be explored as a mechanism to optimize GC treatment and reduce side effects by allowing for lower doses.
Overall, our studies identified MIF as a central molecule in promoting autoimmune tissue damage by undoing the immunoregulatory effects of GCs on encephalitogenic T cells. We posit that T-bet is one of the key molecules modulated by MIF to promote pathogenicity of autoreactive T cells. Moreover, measuring the expression of MIF may serve as a biomarker for treatment efficacy of GCs in MS. MIF has been targeted by mAbs as well as small molecule inhibitors. Therefore, targeting MIF in combination with immunosuppressive agents is feasible and could prove superior to current therapies.
Supplementary Material
GLOSSARY
- ANOVA
analysis of variance
- CFA
complete Freund's adjuvant
- Dex
dexamethasone
- EAE
experimental autoimmune encephalomyelitis
- GC
glucocorticoid
- ICS
intracellular staining
- IFN
interferon
- IL
interleukin
- IP
intraperitoneal
- mAb
monoclonal antibody
- MIF
macrophage migration inhibitory factor
- MNC
mononuclear cell
- MS
multiple sclerosis
- PBS
phosphate-buffered saline
- SC
subcutaneous
- Wt
wild-type
Footnotes
Supplemental data at Neurology.org/nn
AUTHOR CONTRIBUTIONS
N.J. and T.G.F. designed the study, analyzed data, and drafted the manuscript. N.J. and A.K. performed the experiments. G.F.-R. and M.N.G. gave conceptual advice and discussed the results. G.F.-R. generated and contributed the MIF−/− C57BL/6 mice. N.J. performed the statistical analysis with advice and assistance from M.N.G. T.G.F. supervised the study.
STUDY FUNDING
This work was supported by grants NS-52177 and 2G12RR013646-11 from the NIH, grants RG3499 and RG3701 from the National Multiple Sclerosis Society (T.G.F.), and grant DFG 712/2 (G.F.-R.).
DISCLOSURE
N. Ji and A. Kovalovsky report no disclosures. G. Fingerle-Rowson has been employed by Janssen Pharmaceuticals and F. Hoffman-LaRoche Ltd. M.N. Guentzel holds patents for Biomarkers of Francisella Infection, Methods and composition to prevent or treat bacterial infections and received research support from SWRI-UTSA. T.G. Forsthuber provides expert review for Clinical Immunology, is an associate editor for Journal of Immunology, is an associate editor for PLos One, is an editor for Frontiers in Multiple Sclerosis and Neuroimmunology, is on the editorial board for Immunotherapy, and receives research support from National MS Society. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Lucchinetti C, Brück W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol 2000;47:707–717. [DOI] [PubMed] [Google Scholar]
- 2.Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med 1998;338:278–285. [DOI] [PubMed] [Google Scholar]
- 3.McFarland HF, Martin R. Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol 2007;8:913–919. [DOI] [PubMed] [Google Scholar]
- 4.Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol 2005;23:683–747. [DOI] [PubMed] [Google Scholar]
- 5.Denkinger CM, Denkinger M, Kort JJ, Metz C, Forsthuber TG. In vivo blockade of macrophage migration inhibitory factor ameliorates acute experimental autoimmune encephalomyelitis by impairing the homing of encephalitogenic T cells to the central nervous system. J Immunol 2003;170:1274–1282. [DOI] [PubMed] [Google Scholar]
- 6.Powell ND, Papenfuss TL, McClain MA, et al. Macrophage migration inhibitory factor is necessary for progression of experimental autoimmune encephalomyelitis. J Immunol 2005;175:5611–5614. [DOI] [PubMed] [Google Scholar]
- 7.Cox GM, Kithcart AP, Pitt D, et al. Macrophage migration inhibitory factor potentiates autoimmune-mediated neuroinflammation. J Immunol 2013;191:1043–1054. [DOI] [PubMed] [Google Scholar]
- 8.Bacher M, Metz CN, Calandra T, et al. An essential regulatory role for macrophage migration inhibitory factor in T-cell activation. Proc Natl Acad Sci USA 1996;93:7849–7854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jüttner S, Bernhagen J, Metz CN, Röllinghoff M, Bucala R, Gessner A. Migration inhibitory factor induces killing of Leishmania major by macrophages: dependence on reactive nitrogen intermediates and endogenous TNF-alpha. J Immunol 1998;161:2383–2390. [PubMed] [Google Scholar]
- 10.Calandra T, Bernhagen J, Metz CN, et al. MIF as a glucocorticoid-induced modulator of cytokine production. Nature 1995;377:68–71. [DOI] [PubMed] [Google Scholar]
- 11.Fingerle-Rowson G, Petrenko O, Metz CN, et al. The p53-dependent effects of macrophage migration inhibitory factor revealed by gene targeting. Proc Natl Acad Sci USA 2003;100:9354–9359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mitchell RA, Liao H, Chesney J, et al. Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: regulatory role in the innate immune response. Proc Natl Acad Sci USA 2002;99:345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fingerle-Rowson G, Koch P, Bikoff R, et al. Regulation of macrophage migration inhibitory factor expression by glucocorticoids in vivo. Am J Pathol 2003;162:47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Milligan NM, Newcombe R, Compston DA. A double-blind controlled trial of high dose methylprednisolone in patients with multiple-sclerosis: 1. Clinical effects. J Neurol Neurosurg Psychiatry 1987;50:511–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tischner D, Reichardt HM. Glucocorticoids in the control of neuroinflammation. Mol Cell Endocrinol 2007;275:62–70. [DOI] [PubMed] [Google Scholar]
- 16.Webster JI, Tonelli L, Sternberg EM. Neuroendocrine regulation of immunity. Annu Rev Immunol 2002;20:125–163. [DOI] [PubMed] [Google Scholar]
- 17.Ito K, Barnes PJ, Adcock IM. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1beta-induced histone H4 acetylation on lysines 8 and 12. Mol Cell Biol 2000;20:6891–6903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ito K, Jazrawi E, Cosio B, Barnes PJ, Adcock IM. p65-activated histone acetyltransferase activity is repressed by glucocorticoids: mifepristone fails to recruit HDAC2 to the p65-HAT complex. J Biol Chem 2001;276:30208–30215. [DOI] [PubMed] [Google Scholar]
- 19.Hofstetter HH, Shive CL, Forsthuber TG. Pertussis toxin modulates the immune response to neuroantigens injected in incomplete Freund's adjuvant: induction of Th1 cells and experimental autoimmune encephalomyelitis in the presence of high frequencies of Th2 cells. J Immunol 2002;169:117–125. [DOI] [PubMed] [Google Scholar]
- 20.Kawamura K, McLaughlin KA, Weissert R, Forsthuber TG. Myelin-reactive type B T cells and T cells specific for low-affinity MHC-binding myelin peptides escape tolerance in HLA-DR transgenic mice. J Immunol 2008;181:3202–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wust S, van den Brandt J, Tischner D, et al. Peripheral T cells are the therapeutic targets of glucocorticoids in experimental autoimmune encephalomyelitis. J Immunol 2008;180:8434–8443. [DOI] [PubMed] [Google Scholar]
- 22.Almawi WY, Beyhum HN, Rahme AA, Rieder MJ. Regulation of cytokine and cytokine receptor expression by glucocorticoids. J Leukoc Biol 1996;60:563–572. [DOI] [PubMed] [Google Scholar]
- 23.Frisullo G, Nociti V, Lorio R, et al. Glucocorticoid treatment reduces T-bet and pSTAT1 expression in mononuclear cells from relapsing remitting multiple sclerosis patients. Clin Immunol 2007;124:284–293. [DOI] [PubMed] [Google Scholar]
- 24.Liberman AC, Refojo D, Druker J, et al. The activated glucocorticoid receptor inhibits the transcription factor T-bet by direct protein-protein interaction. FASEB J 2007;21:1177–1188. [DOI] [PubMed] [Google Scholar]
- 25.Yang YH, Weiner J, Liu Y, et al. T-bet is essential for encephalitogenicity of both Th1 and Th17 cells. J Exp Med 2009;206:1549–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baron JL, Madri JA, Ruddle NH, Hashim G, Janeway CA. Surface expression of alpha 4 integrin by CD4 T cells is required for their entry into brain parenchyma. J Exp Med 1993;177:57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gocke AR, Cravens PD, Ben LH, et al. T-bet regulates the fate of Th1 and Th17 lymphocytes in autoimmunity. J Immunol 2007;178:1341–1348. [DOI] [PubMed] [Google Scholar]
- 28.Lord GM, Rao RM, Choe H, et al. T-bet is required for optimal proinflammatory CD4(+) T-cell trafficking. Blood 2005;106:3432–3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bucala R, Lolis E. Macrophage migration inhibitory factor: a critical component of autoimmune inflammatory diseases. Drug News Perspect 2005;18:417–426. [DOI] [PubMed] [Google Scholar]
- 30.Denkinger CM, Metz C, Fingerle-Rowson G, Denkinger MD, Forsthuber T. Macrophage migration inhibitory factor and its role in autoimmune diseases. Arch Immunol Ther Exp (Warsz) 2004;52:389–400. [PubMed] [Google Scholar]
- 31.Leng L, Wang W, Roger T, et al. Glucocorticoid-induced MIF expression by human CEM T cells. Cytokine 2009;48:177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mizue Y, Nishihira J, Miyazaki T, et al. Quantitation of macrophage migration inhibitory factor (MIF) using the one-step sandwich enzyme immunosorbent assay: elevated serum MIF concentrations in patients with autoimmune diseases and identification of MIF in erythrocytes. Int J Mol Med 2000;5:397–403. [DOI] [PubMed] [Google Scholar]
- 33.Niino M, Ogata A, Kikuchi S, Tashiro K, Nishihira J. Macrophage migration inhibitory factor in the cerebrospinal fluid of patients with conventional and optic-spinal forms of multiple sclerosis and neuro-Behçet's disease. J Neurol Sci 2000;179:127–131. [DOI] [PubMed] [Google Scholar]
- 34.Matsui Y, Okamoto H, Jia N, et al. Blockade of macrophage migration inhibitory factor ameliorates experimental autoimmune myocarditis. J Mol Cell Cardiol 2004;37:557–566. [DOI] [PubMed] [Google Scholar]
- 35.Cvetkovic I, Al-Abed Y, Miljkovic D, et al. Critical role of macrophage migration inhibitory factor activity in experimental autoimmune diabetes. Endocrinology 2005;146:2942–2951. [DOI] [PubMed] [Google Scholar]
- 36.Reder AT, Thapar M, Jensen MA. A reduction in serum glucocorticoids provokes experimental allergic encephalomyelitis: implications for treatment of inflammatory brain disease. Neurology 1994;44:2289–2294. [DOI] [PubMed] [Google Scholar]
- 37.Chen Y, Langrish CL, Mckenzie B, et al. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J Clin Invest 2006;116:1317–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y, Godec J, Ben-Aissa K, et al. The transcription factors T-bet and Runx are required for the ontogeny of pathogenic interferon-γ-producing T helper 17 cells. Immunity 2014;40:355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lue HQ, Dewor M, Leng L, Bucala R, Bernhagen J. Activation of the JNK signalling pathway by macrophage migration inhibitory factor (MIF) and dependence on CXCR4 and CD74. Cell Signal 2011;23:135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwartz V, Lue HQ, Kraemer S, et al. A functional heteromeric MIF receptor formed by CD74 and CXCR4. FEBS Lett 2009;583:2749–2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.