

Abstract
Tissue fibrosis is associated with structural and functional changes that limit blood flow and oxygen availability. In the kidney, tubulointerstitial fibrosis, which leads to progressive destruction of renal tissue and irreversible loss of kidney function, is associated with reduced tissue oxygen levels and activation of hypoxia-inducible factor (HIF) signaling. Although cytoprotective in acute injury models, HIF-1 was found to promote fibrosis in an experimental model of chronic renal injury following unilateral ureteral obstruction. Pharmacological inhibition of lysyl oxidases phenocopied the effects of genetic HIF-1 ablation on cell motility in vitro and on fibrogenesis in vivo, suggesting that lysyl oxidases are important mediators of profibrotic HIF signaling. These findings support the notion that HIF-mediated cellular responses differ under conditions of acute and chronic oxygen deprivation. Under certain conditions, these responses may lead to further tissue destruction by promoting fibrogenesis.
Keywords: hypoxia-inducible factor (HIF), fibrosis, chronic kidney disease (CKD), epithelial to mesenchymal transition (EMT), transcriptional repressors, lysyl oxidases, extracellular matrix
Evidence for Hypoxia in Chronic Renal Diseases
Despite the very large blood volume they receive (~20% of total cardiac output), kidneys have to carry out complex and energy-consuming cellular transport functions under markedly reduced oxygen tension. Regional tissue oxygen levels vary and under physiological conditions can be as low as 10 mm Hg in the medulla. This is partly due to the unique anatomy of the renal vasculature, which generates an oxygen shunt that shifts molecular oxygen away from the medulla, and makes the kidney particularly sensitive to both acute and chronic ischemic injuries.1 Under pathophysiological conditions, reduced renal tissue oxygen levels have been demonstrated in a variety of chronic kidney diseases (CKD), both in human patients and in experimental animal models, and result from a combination of structural and functional changes that are commonly found in fibrotic kidneys. These include a reduction in peritubular blood flow and supply as a consequence of capillary rarefaction, glomerular injury, luminal narrowing of atherosclerotic vessels, as well as vascular constriction due to altered expression of vasoactive factors and signaling molecules in the injured kidney (e.g., angiotensin II, endothelin, nitric oxide) (Fig. 1). The ensuing discrepancy between oxygen supply and demand is furthermore exacerbated by extracellular matrix (ECM) expansion, limiting oxygen diffusion, by increased oxygen demand from hyperfiltration, and tubular hypertrophy, and by renal anemia.2,3 Blood oxygen level-dependent (BOLD) MRI, various molecular and histological techniques, and direct measurement of renal tissue oxygen levels with microelectrodes are methods that have been used to assess oxygenation in CKD, such as diabetic and IgA nephropathy, unilateral ureteral obstruction (UUO), 5/6 nephrectomy, and anti-Thy1 glomerulonephritis (for an overview of these studies see Refs. 1–4). Consistent with decreased renal oxygenation in experimental models5,6 is the increased expression of the oxygen-sensitive α-subunit of hypoxia-inducible factor (HIF)-1 in renal biopsy material from patients with diabetic nephropathy7 (Fig. 2). Genome-wide gene expression analysis of microdissected renal biopsy tissues (glomeruli were excluded) detected that approximately 45 confirmed or putative HIF-regulated genes were differentially expressed between control and diabetic patients.7 Moreover, the degree of HIF-1α expression (tubular epithelial cells and glomerular cells) correlated with the degree of tissue injury and fibrosis,7 supporting the notion that CKD progression is associated with further impairment of renal oxygenation. HIF transcription factors, which are activated under hypoxic conditions and in response to certain cytokines and other non-hypoxic stimuli,1 play key roles in cellular adaptation to hypoxia by regulating various biological processes that are usually aimed at improving tissue oxygenation and cell survival in a low oxygen environment, such as vascular remodeling, erythropoiesis, and anaerobic glycolysis. Recent experimental evidence, however, suggests that prolonged activation of HIF signaling in certain renal cells may enhance maladaptive responses, such as tissue fibrosis, which will ultimately result in further tissue destruction.
Figure 1.

HIF signaling in CKD. Potential contributions of activated HIF signaling in resident kidney cells to the pathogenesis of renal fibrosis. As a result of hypoxia, HIF activation in renal epithelial cells is capable of promoting fibrosis as shown experimentally in proximal tubule-specific HIF-1α knockout mice (UUO model) and in mice with increased tubular HIF-1α expression.7,10 Epithelial HIF acts as a profibrotic transcription factor by modulating ECM production and processing through regulation of matrix-modifying factors and enzymes, such as CTGF, PAI1, TIMP1, and MMPs, and the modulation of EMT-triggering pathways. The role of non-epithelial HIF in the pathogenesis of renal fibrogenesis is not known. Abbreviations: CTGF, connective tissue growth factor; ECM, extracellular matrix; EMT, epithelial-to-mesenchymal transition; NO, nitric oxide; PAI1, plasminogen activator inhibitor 1; ROS, reactive oxygen species; TIMP1, tissue-inhibitor of metalloproteinase 1.
Figure 2.
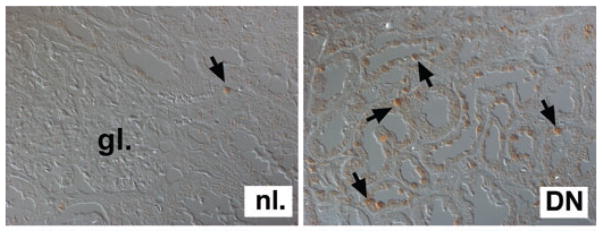
HIF expression in CKD. Shown is HIF-1α immunostaining of normal kidney tissue (nl.) and renal biopsy tissue from a patient with advanced diabetic nephropathy (DN) analyzed by differential interference contrast microscopy. Shown is a representative photograph from a diabetic kidney with >50% of epithelial cells stained positive. Arrows depict cells with nuclear HIF-1α staining; (gl.) indicates glomerulus. Magnification ×400.
Activated HIF Signaling in Ischemic Renal Diseases
The biological outcome of HIF activation appears to be different under conditions of acute and chronic renal hypoxia. As a consequence of acute renal ischemia, HIF-α proteolysis is inhibited, and HIF-1α can be detected in the nucleus of renal tubular epithelial cells, where it dimerizes with HIF-1β to form transcriptionally active HIF-1. Whereas HIF-1 is active in renal epithelial and in endothelial cells, HIF-2α is expressed in renal interstitial fibroblasts and in endothelial and glomerular cells (Fig. 1). As global regulator of cellular adaptation to hypoxia, HIF controls biological processes that are important for cell survival, such as anaerobic glycolysis (Pasteur effect), cellular proliferation, apoptosis, and reactive oxygen species (ROS) scavenging. Genetic studies in mice have demonstrated that reduced expression of HIF-1α or HIF-2α worsens the clinical outcome of acute ischemic renal injury.8,9 Although the relative contributions of different renal cells to HIF-mediated cytoprotection remains to be determined, endothelial HIF-2 appears to ameliorate renal ischemia reperfusion injury through increased expression of ROS scavenging enzymes, such as superoxide dismutase (SOD).8
In the context of chronic renal injury the biological consequences of HIF activation are different and appear to negatively impact clinical outcome. Our laboratory has recently identified tubular epithelial HIF-1α as a promoter of renal fibrosis7 and demonstrated that genetic inactivation of HIF-1α in the proximal renal tubule led to a reduction in collagen accumulation, inflammatory cell infiltration, and the number of FSP1 (S100A4)-expressing interstitial cells following UUO.7 The notion of epithelial HIF-1 acting as a profibrotic transcription factor is supported by overexpression studies in vivo.10 In addition, we and others have shown that hypoxia and HIF activation promotes cell motility and epithelial to mesenchymal transition (EMT) of renal epithelial cells in vitro,7,11 which most likely contributes to HIF’s fibrogenic role in vivo. EMT is a developmentally vital, molecularly complex cellular process by which epithelial cells lose apico–basal polarity and cell–cell contact, become motile, and acquire mesenchymal characteristics. In the context of CKD, renal tubular epithelial cells acquire a mesenchymal phenotype and then migrate into the interstitium, where together with resident cells they produce ECM as myofibroblasts.12 The accumulation of myofibroblasts is closely correlated with the degree of interstitial damage and the risk of disease progression. In vivo studies with genetically tagged renal epithelial cells have suggested that up to 36% of interstitial myofibroblasts are EMT-derived and approximately 15% are bone marrow-derived, while the rest are derived from resident fibroblast proliferation.13 However, the degree to which EMT contributes to the renal myofibroblast pool is still under investigation, and more recent studies have emphasized that pericytes14 and/or endothelial cells15 can become major sources of ECM-producing renal interstitial myofibroblasts.
HIF as a Promoter of Fibrosis
The cellular responses to HIF activation in the context of renal fibrogenesis are complex and most likely highly cell type-dependent. While HIF signaling in non-epithelial renal cells is still being investigated, more experimental information exists about the role of tubular epithelial HIF in CKD progression. Renal epithelial cells contribute to the development of renal fibrosis, as they increase and remodel ECM when stimulated with TGF-β1, angiotensin II, and other cytokines,16,17 or when they transition into myofibroblasts as a result of EMT.12,18 There are several potential mechanisms by which epithelial HIF-1 may promote renal fibrosis. Work by our laboratory and by others suggests that activation of HIF signaling in resident kidney cells promotes renal fibrosis by at least three mechanisms: (a) by direct regulation of gene products that control ECM turnover; (b) by functional co-operation with TGF-β1, which is a potent profibrotic factor; and (c) by promoting EMT. In addition, HIF-1 is important for the normal function of immune cells under hypoxic conditions, and thus will most likely impact the outcome of renal injury through its ability to modulate the activity of infiltrating inflammatory cells, which are usually present in CKD tissues. Experimental studies, which examine the functional role of HIF in these cells as it pertains to CKD progression, are still pending.
HIF Regulation of ECM-Modifying Factors and Functional Interaction with TGF-β Signaling
Hypoxia has the potential to drive fibrogenesis through direct transcriptional increase in the expression of collagen genes or gene products that are directly involved in the regulation of ECM turnover. Hypoxia induces collagen I, decreases matrix-metallopeptidase 2 (MMP-2) in renal epithelial cells,19 and increases plasminogen activator inhibitor-1 (PAI-1),20 tissue-inhibitor of metalloproteinase-1 (TIMP-1),19 as well as connective tissue growth factor (CTGF),21 through HIF-mediated transcriptional responses. Synergistic co-operation between HIF and non-HIF pathways, such as the TGF-β1/SMAD3 signaling pathway, may further enhance the expression of oxygen-regulated genes in the setting of CKD. HIF/TGF-β1 have been shown to co-regulate vascular endothelial growth factor (VEGF ),22 endoglin23 and erythropoietin (EPO),24 and further support for this notion comes from the observation that hypoxia and TGF-β1 synergize with regard to the production of certain collagens in fibroblasts.25,26 TGF-β1 is a member of the TGF-β superfamily, which encompasses other multifunctional cytokines such as bone morphogenetic proteins, activins, and inhibins. TGF-β1 expression is strongly correlated with tissue fibrosis and is largely responsible for the observed increase in ECM deposition in fibrotic diseases through stimulation of profibrogenic gene expression in a wide variety of different cells. It has been identified as a main contributing factor to the development of renal fibrosis in a variety of renal disease settings, most notably diabetic nephropathy,27 and regulates gene expression through the activation of SMAD transcription factors. Although the molecular basis of functional interactions between hypoxia, HIF, and TGF-β signaling is not well-understood, close proximity of HIF and SMAD-binding DNA sequences in regulatory regions of certain hypoxia-sensitive genes suggests that both pathways cross-talk at a transcriptional level, as proposed for the regulation of VEGF expression.22 Even though a direct role for HIF has yet to be demonstrated, hypoxia increases TGF-β1 protein19 and SMAD3 mRNA levels, and it promotes the thrombospondin-dependent release of latent TGF-β2, thereby activating TGF-β signaling.28 This would suggest that hypoxia and HIF modulate TGF-β signaling at multiple levels.
HIF Regulation of EMT and Kidney Fibrosis
There is growing experimental evidence that hypoxia and HIF promote EMT by (a) regulating the expression levels of EMT inducers or their receptors (e.g., TGF-β1; c-met proto-oncogene (receptor for hepatocyte growth factor)), (b) affecting the expression levels and activity of EMT-associated signaling molecules and downstream effectors (e.g., modulation of Notch and β-catenin-mediated transcriptional activity29,30), or (c) by regulating the expression and activity levels of EMT-inducing transcriptional repressors (Fig. 3). Epithelial repressors are transcription factors, which recruit specific chromatin-remodeling complexes and downregulate the expression of gene products important in the maintenance of the epithelial phenotype. These include cadherins, claudins, cytokeratins, mucins, plakophilin, occludin, and ZO proteins (for a recent review, see Ref. 31). The loss of E-cadherin expression is consistently observed during EMT and represents an important molecular event during this process. E-cadherin as a key component of cell–cell adhesion junctions is essential for the formation of epithelia during embryonic development and maintenance of adult epithelial homeostasis. Some epithelial repressors can also directly or indirectly induce mesenchymal gene products that are important in matrix remodeling as demonstrated by overexpression studies. An example of this is the activation of zinc finger-containing transcriptional repressor SNAI1, which indirectly increases MMP2 and MMP9 expression, as well as the expression of TIMP1 and PAI1.32–34 Imai et al. observed that SNAI1 was increased by hypoxia in ovarian cancer cells, which correlated with increased HIF-α levels and decreased E-cadherin expression.35 HIF control of EMT-inducing epithelial repressors was subsequently demonstrated in pVHL defective renal cancer cells,36,37 which lack the ability to target hydroxylated HIF-α for ubiquitination and subsequent proteasomal degradation, resulting in constitutive activation of HIF signaling. HIF-1 activation was associated with decreased E-cadherin expression, loss of cell–cell adhesion, and induction of EMT through increased expression of epithelial repressors TCF3, ZEB1/δEF1, and ZEB2/SIP1.36 Similarly, Evans et al. proposed that inactivation of pVHL resulted in E-cadherin suppression via HIF-dependent induction of SNAI1 and ZEB2/SIP1,37 while Esteban et al. suggested that HIF-2 may be more potent in suppressing E-cadherin levels than HIF-1.38 In the kidney, SNAI1 seems to be particularly important for the maintenance of epithelial homeostasis, as forced expression of SNAI1 induces renal fibrosis in transgenic mice.39
Figure 3.

HIF as a modulator of EMT. Simplified overview of hypoxia/HIF-regulated EMT-triggering signaling pathways and effector molecules. The composition and integrity of the basement membrane is crucial for the maintenance of epithelial homeostasis, and its disruption is a critical event during the EMT process. Under pathological conditions, transitioned epithelial cells migrate into the interstitium, where they produce ECM as myofibroblasts. Migration is likely to be modulated by HIF via increased expression of proteins that regulate and balance ECM degradation and turnover, such as PAI1 and TIMP1. Abbreviations: CTGF, connective tissue growth factor; PAI1, plasminogen activator inhibitor 1; TIMP1, tissue-inhibitor of metalloproteinase 1; RTyrKinases, receptor tyrosine kinases; SNAI1, snail homologue 1; TCF3, transcription factor 3 (E2A immunoglobulin enhancer-binding factors E12/E47); ZEB, zinc finger E-box-binding homeobox.
Another HIF-regulated transcriptional repressor that may be relevant for renal fibrogenis is the basic helix-loop-helix transcriptiona factor TWIST, which represses E-cadherin via E-boxes that are also targeted by SNAI1 and ZEB2/SIP1. TWIST is directly HIF-regulated in Caenorhabditis elegans40 and in mammalian cells,40,41 and it mediates hypoxia/HIF-induced EMT in renal epithelial cell lines.42 However, increased transcription of TWIST, ZEB2/SIP1, or SNAI1 was not observed following exposure of primary renal epithelial cells to short term hypoxia; this may be a reflection of cell type and context-dependent differences in hypoxic gene regulation or the duration of hypoxia exposure.7
HIF-1 induction of lysyl oxidase (LOX) and lysyl oxidase-like (LOXL)-2 has been shown to promote migration of primary renal epithelial cells and human breast and cervical cancer cells, and to downregulate E-cadherin expression.7,43 Although initially identified by their ability to cross-link collagen and elastin fibers, LOX and LOXL proteins carry out intracellular functions and display a range of biological activities that extend beyond ECM cross-linkage. These include the regulation of ras- and NF-κB-signaling, possibly through the modification of DNA–histone and histone–histone interactions.44 A recent report by Peinado et al. indicated that LOXL2 and LOXL3 have the potential to regulate EMT by stabilizing and promoting the activity of transcriptional repressor SNAI1.45 Because LOX and LOXL2 are direct transcriptional targets of HIF, LOXL-mediated control of SNAI1 activity may represent an important molecular mechanism in the oxygen-dependent regulation of renal EMT and in the pathogenesis of kidney fibrosis.7,39
HIF as Therapeutic Target in Renal Fibrosis
Normoxic stabilization of HIF-α can be achieved by pharmacological inhibition of prolyl-4-hydroyxlase domain (PHD) proteins with 2-oxoglutarate analogues. PHDs are 2-oxoglutarate-dependent dioxygenases, which act as molecular oxygen sensors and hydroxylate specific proline residues within the oxygen-dependent degradation domain of HIF-α. This is a prerequisite for rapid HIF inactivation under conditions of normal oxygen tension. Notwithstanding HIF’s profibrogenic actions in vivo and its role in the regulation of EMT-triggering pathways, systemic inhibition of PHDs (e.g., with cobalt chloride or 2-oxoglurate analogues, which both inhibit HIF prolyl-4-hydroxylases) has been shown to be cy-toprotective not only in acute ischemia, but also in certain chronic renal disease models (for an overview of studies, see Refs. 1,3,4). It is unclear, however, which cell type, signaling pathway, or HIF homologue confers cytoprotection or aids in the preservation of renal function. Cell type-specific genetic studies in mice—studies that target individual HIF homologues—are needed to better understand the contributions of individual HIF transcription factors to chronic renal injury. Because HIF is an upstream regulator of multiple biological processes, which include the regulation of erythropoiesis and iron metabolism, systemic pharmacological inhibition of HIF for the treatment of fibrotic disorders, in particular CKD, is not desirable. Certain HIF-regulated proteins, however, may represent better drug targets to retard CKD progression. Pharmacological inhibition of HIF-regulated lysyl oxidases phenocopied the effects of genetic HIF-1α inactivation on cell motility in vitro and on fibrogenesis in vivo, suggesting that lysyl oxidases are important contributors to the pathogenesis of renal fibrosis. In keeping with this notion, increased LOXL2 expression was found in renal biopsy tissues from patients with CKD.7 Further investigation is warranted to determine if pharmacological inhibition of lysyl oxidases is feasible in clinical practice to slow the progression of CKD.
Summary
Recent studies have identified epithelial HIF-1 as an oxygen-regulated transcription factor capable of promoting renal fibro-sis through increased expression of ECM-modifying factors, lysyl oxidase genes, and the facilitation of EMT. These findings have immediate clinical implications as they encourage therapies that aim at improving tissue oxygenation to halt the progression of fibrosis. The relevance of non-epithelial HIF-1 and HIF-2 signaling to the pathogenesis of renal fibrosis, however, remains to be determined. Moreover, HIF-mediated cellular responses appear to differ under conditions of acute and chronic oxygen deprivation, resulting in different clinical outcomes. This notion of cell type- and context-dependent HIF biological functions, including those in immune and inflammatory cells, will stimulate further studies into the role of HIF signaling in acute and chronic ischemic injuries.
Acknowledgments
VHH is supported by the Krick–Brooks Chair in Nephrology and grants from the National Cancer Institute (NCI), and the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK).
Footnotes
Conflicts of Interest
The author declares no conflicts of interest.
References
- 1.Haase VH. Hypoxia-inducible factors in the kidney. Am J Physiol Renal Physiol. 2006;291:F271–F281. doi: 10.1152/ajprenal.00071.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eckardt KU, et al. Role of hypoxia in the pathogenesis of renal disease. Kidney Int Suppl. 2005:S46–S51. doi: 10.1111/j.1523-1755.2005.09909.x. [DOI] [PubMed] [Google Scholar]
- 3.Heyman SN, et al. Renal parenchymal hypoxia, hypoxia response and the progression of chronic kidney disease. Am J Nephrol. 2008;28:998–1006. doi: 10.1159/000146075. [DOI] [PubMed] [Google Scholar]
- 4.Nangaku M, et al. Hypoxia and hypoxia-inducible factor in renal disease. Nephron Exp Nephrol. 2008;110:e1–e7. doi: 10.1159/000148256. [DOI] [PubMed] [Google Scholar]
- 5.Edlund J, et al. Reduced oxygenation in diabetic rat kidneys measured by T2* weighted magnetic resonance micro-imaging. Adv Exp Med Biol. 2009;645:199–204. doi: 10.1007/978-0-387-85998-9_31. [DOI] [PubMed] [Google Scholar]
- 6.Ries M, et al. Renal diffusion and BOLD MRI in experimental diabetic nephropathy. Blood oxygen level-dependent. J Magn Reson Imaging. 2003;17:104–113. doi: 10.1002/jmri.10224. [DOI] [PubMed] [Google Scholar]
- 7.Higgins DF, et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. 2007;117:3810–3820. doi: 10.1172/JCI30487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kojima I, et al. Protective role of hypoxia-inducible factor-2alpha against ischemic damage and oxidative stress in the kidney. J Am Soc Nephrol. 2007;18:1218–1226. doi: 10.1681/ASN.2006060639. [DOI] [PubMed] [Google Scholar]
- 9.Hill P, et al. Inhibition of hypoxia inducible factor hydroxylases protects against renal ischemia-reperfusion injury. J Am Soc Nephrol. 2008;19:39–46. doi: 10.1681/ASN.2006090998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kimura K, et al. Stable expression of HIF-1alpha in tubular epithelial cells promotes interstitial fibrosis. Am J Physiol Renal Physiol. 2008;295:F1023–F1029. doi: 10.1152/ajprenal.90209.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manotham K, et al. Transdifferentiation of cultured tubular cells induced by hypoxia. Kidney Int. 2004;65:871–880. doi: 10.1111/j.1523-1755.2004.00461.x. [DOI] [PubMed] [Google Scholar]
- 12.Liu Y. Epithelial to mesenchymal transition in renal fibrogenesis: pathologic significance, molecular mechanism, and therapeutic intervention. J Am Soc Nephrol. 2004;15:1–12. doi: 10.1097/01.asn.0000106015.29070.e7. [DOI] [PubMed] [Google Scholar]
- 13.Iwano M, et al. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110:341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin SL, et al. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol. 2008;173:1617–1627. doi: 10.2353/ajpath.2008.080433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zeisberg EM, et al. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol. 2008;19:2282–2287. doi: 10.1681/ASN.2008050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen X, et al. STAT proteins mediate angiotensin II-induced production of TIMP-1 in human proximal tubular epithelial cells. Kidney Int. 2003;64:459–467. doi: 10.1046/j.1523-1755.2003.00133.x. [DOI] [PubMed] [Google Scholar]
- 17.Yu L, et al. TGF-beta isoforms in renal fibrogenesis. Kidney Int. 2003;64:844–856. doi: 10.1046/j.1523-1755.2003.00162.x. [DOI] [PubMed] [Google Scholar]
- 18.Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112:1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orphanides C, et al. Hypoxia stimulates proximal tubular cell matrix production via a TGF-beta1-independent mechanism. Kidney Int. 1997;52:637–647. doi: 10.1038/ki.1997.377. [DOI] [PubMed] [Google Scholar]
- 20.Kietzmann T, et al. Induction of the plasminogen activator inhibitor-1 gene expression by mild hypoxia via a hypoxia response element binding the hypoxia-inducible factor-1 in rat hepatocytes. Blood. 1999;94:4177–4185. [PubMed] [Google Scholar]
- 21.Higgins DF, et al. Hypoxic induction of Ctgf is directly mediated by Hif-1. Am J Physiol Renal Physiol. 2004;287:F1223–F1232. doi: 10.1152/ajprenal.00245.2004. [DOI] [PubMed] [Google Scholar]
- 22.Sanchez-Elsner T, et al. Synergistic cooperation between hypoxia and transforming growth factor-beta pathways on human vascular endothelial growth factor gene expression. J Biol Chem. 2001;276:38527–38535. doi: 10.1074/jbc.M104536200. [DOI] [PubMed] [Google Scholar]
- 23.Sanchez-Elsner T, et al. Endoglin expression is regulated by transcriptional cooperation between the hypoxia and transforming growth factor-beta pathways. J Biol Chem. 2002;277:43799–43808. doi: 10.1074/jbc.M207160200. [DOI] [PubMed] [Google Scholar]
- 24.Sanchez-Elsner T, et al. A cross-talk between hypoxia and TGF-beta orchestrates erythropoietin gene regulation through SP1 and SMADs. J Mol Biol. 2004;336:9–24. doi: 10.1016/j.jmb.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 25.Falanga V, et al. Low oxygen tension stimulates collagen synthesis and COL1A1 transcription through the action of TGF-beta1. J Cell Physiol. 2002;191:42–50. doi: 10.1002/jcp.10065. [DOI] [PubMed] [Google Scholar]
- 26.Saed GM, et al. Alteration of type I and III collagen expression in human peritoneal mesothelial cells in response to hypoxia and transforming growth factor-beta1. Wound Repair Regen. 1999;7:504–510. doi: 10.1046/j.1524-475x.1999.00504.x. [DOI] [PubMed] [Google Scholar]
- 27.Bottinger EP, Bitzer M. TGF-beta signaling in renal disease. J Am Soc Nephrol. 2002;13:2600–2610. doi: 10.1097/01.asn.0000033611.79556.ae. [DOI] [PubMed] [Google Scholar]
- 28.Zhang H, et al. Cellular response to hypoxia involves signaling via SMAD proteins. Blood. 2003;101:2253–2260. doi: 10.1182/blood-2002-02-0629. [DOI] [PubMed] [Google Scholar]
- 29.Sahlgren C, et al. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc Natl Acad Sci USA. 2008;105:6392–6397. doi: 10.1073/pnas.0802047105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaidi A, et al. Interaction between beta-catenin and HIF-1 promotes cellular adaptation to hypoxia. Nat Cell Biol. 2007;9:210–217. doi: 10.1038/ncb1534. [DOI] [PubMed] [Google Scholar]
- 31.Peinado H, et al. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–428. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 32.Moreno-Bueno G, et al. Genetic profiling of epithelial cells expressing E-cadherin repressors reveals a distinct role for Snail, Slug, and E47 factors in epithelial-mesenchymal transition. Cancer Res. 2006;66:9543–9556. doi: 10.1158/0008-5472.CAN-06-0479. [DOI] [PubMed] [Google Scholar]
- 33.Jorda M, et al. Upregulation of MMP-9 in MDCK epithelial cell line in response to expression of the Snail transcription factor. J Cell Sci. 2005;118:3371–3385. doi: 10.1242/jcs.02465. [DOI] [PubMed] [Google Scholar]
- 34.Taki M, et al. Involvement of Ets-1 transcription factor in inducing matrix metalloproteinase-2 expression by epithelial-mesenchymal transition in human squamous carcinoma cells. Int J Oncol. 2006;28:487–496. [PubMed] [Google Scholar]
- 35.Imai T, et al. Hypoxia attenuates the expression of E-cadherin via up-regulation of SNAIL in ovarian carcinoma cells. Am J Pathol. 2003;163:1437–1447. doi: 10.1016/S0002-9440(10)63501-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krishnamachary B, et al. Hypoxia-inducible factor-1-dependent repression of E-cadherin in von Hippel-Lindau tumor suppressor-null renal cell carcinoma mediated by TCF3, ZFHX1A, and ZFHX1B. Cancer Res. 2006;66:2725–2731. doi: 10.1158/0008-5472.CAN-05-3719. [DOI] [PubMed] [Google Scholar]
- 37.Evans AJ, et al. VHL promotes E2 box-dependent E-cadherin transcription by HIF-mediated regulation of SIP1 and snail. Mol Cell Biol. 2007;27:157–169. doi: 10.1128/MCB.00892-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Esteban MA, et al. Regulation of E-cadherin expression by VHL and hypoxia-inducible factor. Cancer Res. 2006;66:3567–3575. doi: 10.1158/0008-5472.CAN-05-2670. [DOI] [PubMed] [Google Scholar]
- 39.Boutet A, et al. Snail activation disrupts tissue homeostasis and induces fibrosis in the adult kidney. Embo J. 2006;25:5603–5613. doi: 10.1038/sj.emboj.7601421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gort EH, et al. The TWIST1 oncogene is a direct target of hypoxia-inducible factor-2alpha. Oncogene. 2008;27:1501–1510. doi: 10.1038/sj.onc.1210795. [DOI] [PubMed] [Google Scholar]
- 41.Yang MH, et al. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol. 2008;10:295–305. doi: 10.1038/ncb1691. [DOI] [PubMed] [Google Scholar]
- 42.Sun S, et al. Hypoxia-inducible factor-1alpha induces Twist expression in tubular epithelial cells subjected to hypoxia, leading to epithelial-to-mesenchymal transition. Kidney Int. 2009;75:1278–1287. doi: 10.1038/ki.2009.62. [DOI] [PubMed] [Google Scholar]
- 43.Erler JT, et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature. 2006;440:1222–1226. doi: 10.1038/nature04695. [DOI] [PubMed] [Google Scholar]
- 44.Lucero HA, Kagan HM. Lysyl oxidase: an oxidative enzyme and effector of cell function. Cell Mol Life Sci. 2006;63:2304–2316. doi: 10.1007/s00018-006-6149-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peinado H, et al. A molecular role for lysyl oxidase-like 2 enzyme in snail regulation and tumor progression. Embo J. 2005;24:3446–3458. doi: 10.1038/sj.emboj.7600781. [DOI] [PMC free article] [PubMed] [Google Scholar]