

Abstract
Despite recent therapeutic advances, multiple myeloma (MM) remains largely incurable. Herein we report results of a phase I/II trial to evaluate the safety and activity of autologous T-cells engineered to express an affinity-enhanced T-cell receptor (TCR) recognizing a naturally processed peptide shared by the cancer-testis antigens NY-ESO-1 and LAGE-1. Twenty patients with antigen-positive MM received an average 2.4×109 engineered T cells two days after autologous stem cell transplant (ASCT). Infusions were well-tolerated without clinically apparent cytokine release syndrome, despite high IL-6 levels. Engineered T-cells expanded, persisted, trafficked to marrow and exhibited a cytotoxic phenotype. Persistence of engineered T cells in blood was inversely associated with NY-ESO-1 levels in the marrow. Disease progression was associated with loss of T cell persistence or antigen escape, consistent with the expected mechanism of action of the transferred T cells. Encouraging clinical responses were observed in 16 of 20 patients (80%) with advanced disease, with a median progression free survival of 19.1 months. NY-ESO-1/LAGE-1 TCR-engineered T-cells were safe, trafficked to marrow and showed extended persistence that correlated with clinical activity against antigen-positive myeloma.
Allogeneic stem cell transplants can eradicate myeloma through the T-cell mediated “graft-vs-myeloma” (GVM) effect but success is limited by morbidity and mortality from infections and organ toxicity. Autologous stem cell transplantation (ASCT) is less toxic but rarely curative due in part to the lack of GVM effect 1-6. Better clinical outcomes following ASCT for myeloma are associated with rapid post-transplant lymphocyte recovery 7,8. Tumor-reactive T-cells present at low frequencies in the marrow and blood of myeloma patients have the potential to target myeloma cells upon activation 9,10. Thus autologous immune-mediated control of myeloma may be possible.
We and others have studied whether cancer vaccines and autologous T-cell transfer administered post-ASCT could enhance immune reconstitution and improve post-transplant clinical outcomes in myeloma 11-16. A key problem with these approaches however, is that post-transplant tumor responses remain inadequate. A likely reason for this is that tumor antigens are typically self-antigens which would result in deletion of high affinity T-cells capable of recognizing effective tumor antigens during the process of thymic maturation17,18. Moreover, advanced cancers are often immune edited resulting in reduced antigen presentation, thus rendering low affinity T cells incapable of tumor interaction 19,20. Synthetic biology may help to overcome these problems by enabling the genetic engineering of autologous T cells to express either chimeric antigen receptors (CARs) or affinity-enhanced T-cell receptors (TCRs) that recognize known tumor target antigens. Early clinical results using CAR-modified T-cells have been encouraging but also highlight the risks from cytokine release syndrome (CRS) 21-23.
TCR engineered T cells have been employed in a number of early-stage clinical trials for melanoma 24,25, although very short-term expression of these transgenic TCRs (usually < 1 month) likely compromised their clinical impact 26. We generated a human-derived affinity-enhanced TCR that recognizes the NY-ESO-1/LAGE-1-derived SLLMWITQC peptide in complex with HLA-A*0201 (NY-ESOc259) as previously described 27,28 and clinically tested in patients with metastatic synovial cell sarcoma and melanoma 29,30. NY-ESO-1 (also known as CTAG-1B) is an immunogenic cancer testis antigen (CTA) associated with spontaneous and vaccine-induced immunity that can lead to clinical cancer responses 31,32. Up to 60% of advanced myelomas have been reported to express NY-ESO-1, a feature correlated to tumor proliferation and high risk features 33-37. We hypothesized that adoptive transfer of NY-ESOc259 TCR-engineered T-cells would improve the duration and depth of post-ASCT clinical responses in HLA-A201 –positive patients with advanced NY-ESO-1/LAGE-1-expressing MM.
Our results Indicate that engineered cells engrafted long term, trafficked to sites of tumor, and retained polyfunctionality and cytotoxic potential over time, despite the lack of systemic IL-2 administration used in prior studies with this TCR 29,30. The temporal pattern of tumor regression, the relationship between disease relapse and loss of T cell persistence or loss of target antigen, and robust IL-6 production at the peak of T cell expansion, all provide evidence to support bioactivity of the NY-ESOc259 T-cells in vivo.
Results
Patients
A flow diagram depicting the trial design is shown in Figure 1 and a consort diagram is provided in Supplementary Figure 1. We screened HLA-A201 positive patients for expression of NY-ESO-1 and/or the related cancer testis antigen LAGE-1 in their myeloma cells.
Figure 1. Overview of clinical study.
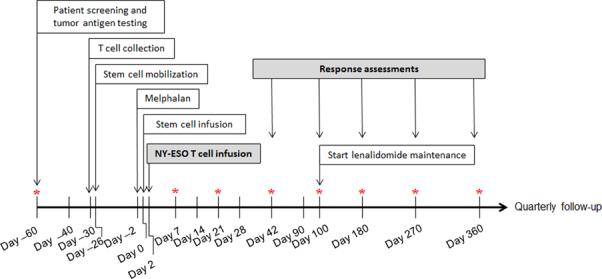
Patient screening, including HLA testing and tumor antigen testing, and apheresis scheduling requires 2-4 weeks. Manufacture of gene-modified cells takes 3-4 weeks. Patients received high dose melphalan two days prior to stem cell infusion, and four days prior to T-cell infusion. Response assessments were performed at day 42, 100, 180 and quarterly thereafter. Optional bone marrow biopsies are indicated by asterisk. For eligible patients, maintenance lenalidomide was given starting at day 100. Once off study, patients are monitored for up to 15 years for delayed adverse events in accordance with FDA Guidance.
A third (34%) of the HLA-A2 positive patients who were screened expressed NY-ESO-1 and/or LAGE-1 mRNA by PCR and were therefore eligible for enrollment. LAGE-1 expression frequency was approximately twice that of NY-ESO-1. Supplementary Table 1 summarizes demographics and pre-transplant characteristics. All patients had symptomatic myeloma with active disease, representing an advanced stage population including 5 (25%) with prior ASCT and 12 (60%) with cytogenetic abnormalities, including 7 (35%) categorized as high-risk [t(4;14), del17p13, or complex]. Peripheral blood mononuclear cells obtained by steady-state apheresis were transduced with lentiviral vector encoding the affinity-enhanced NY-ESOc259 TCR and expanded using anti-CD3/28 antibody conjugated paramagnetic microbeads. After autologous stem cell collection, patients were conditioned with high-dose melphalan (140-200 mg/m2) followed 2 days later by autologous stem cell infusion. On day +2 after stem cell transplantation, patients received a mean of 8 billion total CD3 T-cells (range 1-10 billion) with an average transduction efficiency of 33% (range 18% – 49%). A mean of 2.4 billion NY-ESOc259-engineered CD3 T-cells (range 0.45-3.9 billion) were infused, which were comprised of >90% CD62L expressing cells, including a variable percentage of CD4 and CD8 T cells (Supplementary Table 2).
T-cell persistence, trafficking, and function
T-cell expansion, persistence and trafficking to marrow were evaluated using a qualified Q-RT-PCR assay. Peak numbers of engineered T-cells were detected at study day 14, and median persistence per microliter blood at days 7, 14, 28 and 42 post-ASCT was 104 (range 16-741), 489 (range 63-1850), 107 (range 3-881), and 51 (range 1-237), respectively (Fig. 2a). Peak percentages of engineered cells in leukocytes were found on day 7 (median 47%; range 10%-80%), during the white blood cell (WBC) nadir (Fig. 2b), and declined thereafter to a median of 3% (range <1%-11%) and 1% (range <1%-9%) on days 28 and 180, respectively.
Figure 2. Persistence and function of gene-modified cells in blood and marrow.

(a). The total number of gene-modified cells from infusion to 1 year on study is shown, as measured by Q-RT-PCR. The red line represents the median across patients. The limit of detection is approximately at 2.5 cells/microliter. (b). The percent of gene marking in blood over time is shown, using the left vertical axis. The red bold line represents the average WBC count using the right vertical axis. Standard deviation is represented by the error bars. (c). The total number of vector copies per microgram of DNA in bone marrow is represented for patients with two or more marrow collections post infusion. The percent of cells marked was calculated assuming a copy number of 1 per cell. The limit of detection for the assay is 10 copies and is represented by the red dotted line. (d). NY-ESO TCR-positive CD8 T-cells were evaluated for functionality in the cell product (MP) and at multiple timepoints post infusion by measuring production of IFN-γ, granzyme B and CD107a in response to antigen loaded T2 target cells. Non-antigen loaded T2 target cells were used as a negative control for all samples and background was subtracted from the values shown. Subsets of IFN-γ positive cells with +/− expression of granzyme B and CD107a are represented by the various colors in each histogram. Data from four responding patients with durable persistence are shown.
A subset of patients (13/20) underwent marrow biopsies at multiple timepoints post-transplant; engineered cells were detected in marrow from day 7 through day 180 (Fig. 2c). The near 100% marking in marrow biopsy at day 7 is inconsistent with flow and histochemistry, and is higher than in blood. This suggests enrichment of gene-marked cells at the site of antigen (marrow), and is possibly due in part, to preferential infiltration of marrow with engineered cells containing multiple integration events. This data suggests that multiple integration events may improve T cell reactivity to the targeted antigen. A majority of patients (15/20) underwent marrow biopsy for response assessment at day 100; 14/15 had detectable engineered cells, with 9/15 with >1% and 14/15 with >0.1% marking (Supplementary Fig. 2). NY-ESOc259 expressing T-cells were detected by dextramer staining in blood and marrow through 2 years of follow-up (Supplementary Fig. 3). Of 10 patients who reached two years of follow-up, 9 continued to have detectable engineered cells in the peripheral blood (Supplementary Table 3). Serum cytokine analysis was performed in all patients. A significant elevation of IL-6 was detected in all patients (median 22-fold increase; range 8- to 2272-fold) within 7-28 days post infusion, which overlapped with the period of maximum T cell expansion (Supplementary Table 4). Four responding patients with high levels of engineered T cells were evaluated by flow cytometry for cytokine production (IFN-γ) and cytotoxic potential (granzyme B production and CD107α surface expression) in response to peptide loaded targets. The data show that polyfunctional T cells that were generated during manufacturing engrafted in the patients where they remained functional in blood for up to a year after infusion (Fig. 2d).
Immunohistochemical analysis of marrow from one of 4 patients who had day +7 marrow biopsies demonstrated that residual CD138+ myeloma was associated with CD8 T-cell infiltration (Fig. 3a). A substantial fraction of the infiltrating CD3+ cells (11.2%) were engineered (Fig. 3b). As per protocol, patients who relapsed with NYESO-1/LAGE-1 antigen-positive disease were permitted to receive a second gene-modified T cell infusion without a second ASCT. In one case (pt 253) the marrow was densely packed with CD138+ myeloma cells before the second infusion. CD138+ myeloma cell numbers decreased at D+24 after reinfusion in association with an increase in CD8+ T-cells in that compartment. Low dose lenalidomide (10 mg/day) was started on day +24. Two weeks later on day +38, the marrow biopsy demonstrated a considerable increase in the abundance of CD8+ T-cells concomitant with a marked decrease in marrow plasmacytosis with extensive CD138+ myeloma cell necrosis (Fig. 3c; also see the legend to Supplementary Fig. 4 for a detailed clinical summary). Notably, while engineered T-cells were detected in marrow at the D+38 timepoint, deep-sequencing-based T cell receptor spectratyping revealed the presence in the marrow biopsies of two dominant TCR clonotypes that were distinct from the Vβ13.1 clonotype expressed by the NY-ESOc259 engineered cells (see Supplementary Fig. 4). Collectively, these results suggest that anti-myeloma-specific T-cell activity mediated by infusion of engineered autologous T-cells, and potentially enhanced by immunomodulatory treatment with lenalidomide, may result in antigenic spreading and the development of a secondary endogenous anti-tumor immune response.
Figure 3. Tumor and T-cell infiltration in marrow.

Core marrow samples were collected from patient 258 at day 7. (a). Marrow was stained for the plasma cell marker CD138 which is expressed on normal plasma and myeloma cells, and for CD8 to evaluate T-cell infiltration. (b). Mononuclear cells were isolated from fresh marrow aspirates, and stained with anti-CD3 antibody to detect T-cells, and NY-ESO TCR specific peptide/HLA-A2 dextramer reagent to detect gene-modified cells. The dextramer FMO is shown as a negative control. Gene-modified cells in the controls and test panel are denoted by the box, and the number to the left of the box represents the overall percent positive cells from the CD3 positive fraction. (c). Patient 253 received a second infusion of NY-ESO TCR T-cells in the absence of ASCT, after progressing following ASCT and first infusion. Core marrow biopsies were collected just prior to 1 infusion, and at day 24 and 38 after infusion, and were stained for the CD138 plasma cell/myeloma tumor marker, and for CD8 T-cell infiltration. Bars shown in (a) and (b) represent 20 microns.
A representative example of the myeloma tumor targeting that we observed in patients with advanced and refractory disease is provided by the clinical course of patient 250. This 72 year old patient had a prior ASCT with a 2 year response duration, after which he relapsed with a high-risk 4;14 translocation. After additional treatment (using both bortezomib and lenalidomide), he developed progressive disease (PD) with high serum free kappa light chain levels, extensive osteolytic bone lesions associated with large adjacent soft tissue masses, and a biopsy-proven pancreatic plasmacytoma. He attained a stringent complete response (CR) (Fig. 4a), accompanied by expansion and then durable persistence of engineered T-cells in marrow and blood, with approximately 1% of CD3+ lymphocytes expressing the transgenic receptor at one year (Fig. 4a, b). This expansion and persistence was accompanied by normalization of marrow and clearance of the pancreatic plasmacytoma (Fig. 4c and d). The patient later relapsed and at relapse, he patient developed progression in the abdominal mesentery and pelvic soft tissue. Notably, the patient's marrow remained free of myeloma cells and expression of NY-ESO-1/LAGE-1 was absent until his death from disease progression at 2 years post engineered T cell infusion, suggesting ongoing immune surveillance and antigen escape. Of note, figure 2d demonstrates the polyfunctionality and cytotoxic potential of the gene-modified T-cells isolated from this patient, which persisted for at least 1 year after infusion.
Figure 4. Clinical response in patient 250 correlates with engineered T cell expansion.

(a). The number of NY-ESO specific T-cells per microliter of blood is shown for patient 250, and is overlaid with the corresponding tumor burden assessed at days 42 - 180 post infusion. The kappa/lambda light chain ratio is shown on the right axis; n.b. that when ratio becomes normalized, plot is superimposed on X-axis. (b). The levels of TCR expression on CD8 T-cells over time were measured by peptide/HLA-A2 dextramer in marrow and blood; samples were gated on CD3+ lymphocytes. The percent double positive cells is represented by the number in the upper right hand corner. (c). H&E stain of a marrow biopsy pre and post treatment, demonstrating normalization of cellularity at two months. Note the magnification is slightly higher in the left panel. (d). Abdominal CT scan showing a pancreatic plasmacytoma (confirmed by biopsy) at baseline, and loss of the tumor two months after treatment. Bars in panel (c) are 10 microns and bars in panel (d) are 4 centimeters.
Evidence for NY-ESO-1/LAGE-1 tumor antigen targeting
To evaluate antigen-specific anti-tumor activity of the engineered T-cells, we performed qRT-PCR analysis on marrow specimens collected from patients, and quantitatively assessed transcript levels for NY-ESO-1 and LAGE-1, as well as CD138 as a measure of myeloma/plasma cell burden. Relative to levels at enrollment, loss of NY-ESO-1 and LAGE-1 transcripts was observed in 12/15 patients at day 100, and in 11/13 at day 180. At day 100, 3/15 patients had detectable levels of NY-ESO-1 and LAGE-1 transcripts, an observation that accompanied a drop of engineered cells in the peripheral blood to very low or undetectable levels and preceded relapse in two cases (Fig. 5). Four patients had an increase in CD138 transcripts in the absence of NY-ESO-1/LAGE-1 transcripts, suggesting that the treatment was potentially selecting for tumor escape subclones that lacked target tumor antigen. Patient 256 and 212 were in confirmed complete responses, despite elevated CD138 transcripts, which may reflect polyclonal plasma cell recovery. Taken together, these data suggest induction of robust and, in a subset of patients, persistent tumor antigen-specific immune responses. Two patients (252 and 260) with aggressive relapsed and refractory disease prior to study treatment are also instructive with respect to antigen-specific targeting (Supplemental Note).
Figure 5. CD138, LAGE-1 and NY-ESO-1 expression in marrow.
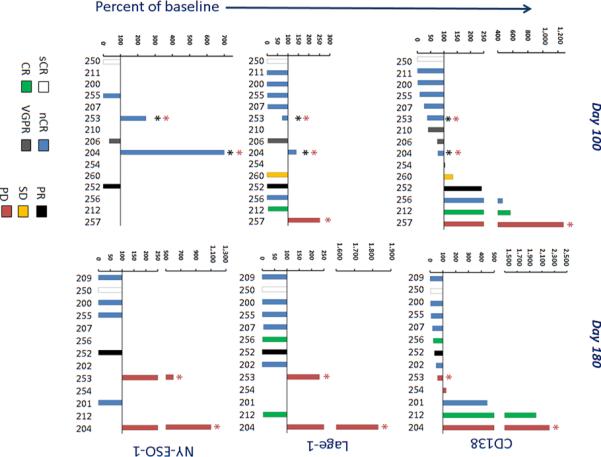
Transcripts for the plasma cell and myeloma marker CD138 (a), and the tumor antigens LAGE-1 (b) and NY-ESO-1 (c) were evaluated by QRT-PCR in all marrow collections performed post infusion, and the percent change from baseline values was calculated at day 100 (left panels) and 180 (right panels). The numbers along the x-axis are patient numbers and correspond to numbers in other figures. Bars are colored to reflect the clinical response of each patient at day 100, which is the pre-specified timepoint for response assessment in the study. Note Loss of engineered T cells (red asterisk) and relapse (black asterisk) is indicated.
These patients had partial myeloma responses following ASCT and NY-ESOc259-engineered T cell infusion which were ongoing at 2 years and 9 months respectively without lenalidomide maintenance. Serial followup marrow examinations showed that the residual myeloma cells were NY-ESO-1/LAGE-1-negative by PCR, suggesting that the engineered T cells selectively eliminated the antigen-positive myeloma fraction which may have been responsible for the more aggressive clinical courses that were observed in these patients before transplant. Additional details on these 2 patients are provided in the Supplemental data.
Clinical Outcomes
There were no treatment related fatalities. All 7 serious adverse events (SAEs) resolved (Supplementary Table 5). Seventeen (17) adverse events occurred which were at least probably related to the treatment, all of grade 3 or lower (Supplementary Table 6). Skin rash with lymphocytosis occurred in 3/20 patients. Some patients had a diarrheal syndrome that occurred later than expected for melphalan-induced mucositis, and in 3/20 patients was confirmed to be autologous graft versus host disease (aGVHD) (Supplementary Fig. 5). Analysis of engineered T-cells in inflamed and normal colonic tissue and peripheral blood was performed in patients who developed aGVHD. While engineered T-cells were present in inflamed tissue, they were proportionately lower at sites of inflammation compared to adjacent non-inflamed tissues suggesting that they were not driving the event (Supplementary Table 7). Furthermore we have previously observed aGVHD in the gut and skin after post-ASCT adoptive transfers of activated but non-gene modified T cells 14.
Overall and progression-free survival (OS and PFS) functions were estimated using the Kaplan-Meier method (Figs. 6a and 6b). With a median follow-up of 21.1 months (the corresponding 95% CI: 12.7-22.9 months), 10/20 patients (50%) were alive and progression-free, 15/20 (75%) patients were alive, and 5/20 (25%) died after disease progression. All 20 patients were assessed for clinical response at day 100, which was prior to lenalidomide maintenance received by a subset of patients (Supplementary Table 1). Of the 20 patients, 14/20 (70%) patients had a near complete response (nCR, defined as myeloma monoclonal band only detectable by sensitive immunofixation assay) or CR while 2 patients had a very good partial response (VGPR, ≥ 90% reduction in paraprotein levels), 2 had a partial response (PR, 50-90% reduction), 1 had stable disease (SD, < 50% reduction), and 1 had progressive disease (PD). Nine patients experienced gradual improvements in tumor markers and/or disease response between the first and second disease assessment at day 100 (Supplementary Fig. 6), consistent with delayed immune-mediated antitumor effects as reported in other trials 38. Three patients (206, 256, and 257) had ongoing responses at the time of transplant from additional therapy given post enrollment. We note that as of March, 2014, the estimated median PFS time was 19.1 months, the corresponding 95% CI lower bound was 8.5 months, and the upper bound has not been reached yet. As of April, 2015 with a median follow up of 30.1 months the median PFS has remained the same at 19.1 months, and the median OS has increased to 32.1 months (Supplementary Fig. 7). A swimmer plot shows the duration and depth of response for each patient, and associated levels of T cell persistence at major timepoints (Fig. 6c). Progression in 8 of 10 cases correlated with a loss of persisting gene-modified cells in the peripheral blood, and the myeloma was antigen-positive upon relapse. In no cases was an antigen-positive tumor relapse observed in the presence of engineered cells at ≥ 6 cells/μl (Supplementary Table 8); as noted above, 2 patients relapsed with NY-ESO-1/LAGE-1 antigen-negative subclones.
Figure 6. Clinical responses and durability.

(a). OS and PFS functions estimated by the Kaplan-Meier approach for the 20 patient cohort. Times-to-event are truncated as 24 months. Surviving (censored) patients are represented by tick marks; the number of patients available for assessment at each 6 month interval is indicated below the graph. (b). Swimmer plot showing the duration of clinical response (black bars) and survival following progression (hatched bars). The depth of each response as measured at day 100 on study is represented by the triangles overlying each bar at that time point. The number of gene-marked cells per microliter of blood, as determined by Q-RT-PCR, is noted above each patient bar for day 100, 180 and 360 post-infusion.
An association between LAGE-1, NY-ESO-1, CD138 and T-cell persistence in the blood was estimated using mixed-effects modeling. Notably, there was a significant inverse correlation between target antigen levels and gene-modified T cells. Between days 0 through 180 post-transplant the T-cell persistence in blood was inversely correlated with the level of NY-ESO-1 expression in the marrow (p=0.022), and marginally correlated with LAGE-1 (p=0.098) (Supplementary Table 9). In contrast, there was no relationship over time between T-cell persistence in blood and CD138 expression in marrow.
Inferences regarding patients’ survival experience were made based on the joint modelling for longitudinal and time-to-event (OS and PFS) data. The statistical model revealed that baseline levels of NY-ESO-1, LAGE-1, CD138, cytogenetic abnormalities, engineered cell dose, prior SCT status, and age, did not affect OS. Neither T-cell persistence in blood or marrow were predictive of OS. However, longitudinal expression of NY-ESO-1 and LAGE-1 was associated with OS, and the corresponding p-values for their effects in the model are 0.037 and 0.055, respectively. There was a marginal effect of the CD138 expression (p-value = 0.110). All three biomarkers had positive coefficients (estimated by joint model), hence, the hazard of death increases with a higher level of these biomarkers (Supplementary Table 10).
PFS was also jointly modeled with time-changing biomarkers and the results were concordant with the OS results. The NY-ESO-1, LAGE-1 and CD138 levels may be predictive of the time-to-progression, but the p-values did not reach significance with values of 0.094, 0.065, and 0.160, respectively for PFS (Supplementary Table 11), perhaps reflecting the small sample size. Thus the effect of NY-ESO-1, LAGE-1 and CD138 levels in the marrow was more pronounced in OS than in PFS, but the same pattern was observed. Higher levels of these markers over time may predict a worse prognosis.
Discussion
We show in this study that infusion of ex-vivo expanded NY-ESOc259 TCR-engineered T-cells were well-tolerated without significant safety concerns, and that engineered T cells expanded in vivo, trafficked to disease tissue, persisted and manifested durable target-specific anti-tumor activity. The observation of safety is a significant finding, and may be related to physiological signaling and antigen specificity provided by the TCR. In contrast, T-cells engineered to express a CD19-directed chimeric-antigen-receptor (CAR) have been associated with severe adverse events attributable in part to grade 3 or higher CRS 39, an IL-6 mediated macrophage activation syndrome 40, or sepsis 41. Complications associated with CRS or neurotoxicity have led to deaths in the CD19 CAR studies 42. We report that severe CRS did not occur in our study, despite the elevated levels of IL-6 detected. These levels are comparable to what has been observed in CD19 CAR T cell studies for patients without severe CRS but are generally lower than the peak levels measured for patients who developed severe CRS21. It is important to note that there are a constellation of post-ASCT adverse events that may mask clinical diagnosis of mild cases of CRS. In our other ongoing solid tumor NY-ESOc259T studies, we have observed multiple cases of mild CRS43. High antigen burden may increase the risk of CRS for both CAR and TCR approaches. However, a potential risk that may be unique to engineered TCRs is enhanced recognition of non-target peptides, as observed in a prior study where the wild-type and affinity-enhanced TCR recognized a contractile protein in cardiac tissue 44,45.
Persistence of engineered T-cells and trafficking to tumor sites are increasingly being reported as biomarkers predictive of clinical responses 46, and durable persistence may be important to maintain clinical responses in at least a subset of patients. Previous studies with engineered T cells have not demonstrated persistence and expression beyond one month 26,30. In this study, long term persistence was detectable in a majority of patients and continued expression of the TCR was observed up to two years post infusion, suggesting that significant gene silencing was not occurring, which is a novel finding and is in contrast to clinical data utilizing retroviral vector gene transfer 26,47. Notably, this is the first published study of lentiviral vector mediated TCR gene expression in humans. Additionally, the method of T cell manufacturing, which selects for younger T cells and also prolonged expansion also is likely a significant factor in durable persistence 48, and studies using CAR modified T cells have demonstrated similar durations of persistence in oncology and HIV clinical studies 21,49. Robust trafficking to marrow, the principal site of tumor, was also observed. Relapse was associated with a loss of gene-modified T-cells, which suggests that methods for sustaining long-term persistence of engineered T-cells in a larger fraction of patients may improve treatment durability.
Several lines of evidence support on target activity of the engineered T-cells. First, expression levels of NY-ESO-1/LAGE-1 decreased post-transplant in all patients, with progressive improvement over time in a subset of patients, and remained very low except in those patients who had loss of gene-modified T-cells. Second, a subset of patients with partial responses exhibited only antigen negative myeloma after treatment and another subset of patients developed antigen negative myeloma on relapse. Third, expression of NY-ESO-1 correlated inversely and significantly to higher levels of engineered T-cells while no association of T cell persistence with CD138 expression (more reflective of burden of normal plasma cells and/or antigen negative myeloma cells) was observed. Future approaches should consider targeting more than one antigen in order to avoid emergence of antigen escape variants and allow treatment of patients presenting with NY-ESO-1/LAGE-1 negative tumors. Targeting more than one tumor antigen combined with the development of tumor specific TCRs restricted to additional common HLA antigens, should enable this therapy to be available to most patients.
70% of patients on study achieved CR (nCR or CR), which compares favorably with the expected response frequencies following ASCT or double sequential (tandem) ASCT where response rates are typically less than 40% in patients without high risk disease 50,51. This is encouraging given the predisposition of patients on our study to inferior outcomes due to an enrollment requirement for high-risk or advanced disease, a considerable frequency of prior ASCT, and the known association of cancer testis antigen expression with poor prognosis. Interestingly, LAGE-1 expression was identified by microarray analysis to also correlate with bortezomib resistance 52. In our small data set, we observed a significant inverse association between expression of LAGE-1 and NY-ESO-1 mRNA expression over time and OS. It must be acknowledged that the use of high-dose chemotherapy in our trial, as with lymphodepleting chemotherapy in virtually all trials of gene-modified T cells, makes it difficult to rigorously ascribe clinical activity to the NY-ESO-TCR-T cells without a randomized study.
Taken together, our data support the continued development of NY-ESO-1/LAGE-1 TCR-engineered T-cells for treatment of myeloma, and that methods for enhancing long term survival and function of the cells may improve treatment durability. Such strategies may include repeat infusions and/or incorporation of immune-modulatory agents such as checkpoint inhibitors and maintenance lenalidomide 53-55. The striking clinical response that occurred in patient 253 after the second infusion of T-cells in concert with the emergence of non-transduced T-cell TCR clonotypes suggests that such approaches may also induce secondary tumor-specific immune responses to myeloma through antigen spreading. Future strategies to augment antigen spreading to cancer neoantigens may further enhance durable clinical activity 56.
Online Methods
The clinical study is registered at clinicaltrials.gov (NCT01352286). Briefly, the study is a phase I/IIa single-arm trial which enrolled 24 patients, 20 of whom received ASCT followed by gene-modified T cells. Based on published data 50 the expected nCR/CR rate following first ASCT in patients without high risk disease was estimated to be 36%50. We hypothesized that the CR rate in this trial would be 66% (2/3 patients). Using the one-sided Chi-square test, we had 85.8% power to detect this difference at a significant level of 5%.
Fourteen patients (70%) were Caucasian, 6 patients (30%) were African-American or Hispanic. Ten patients (50%) were female and the median age at enrollment was 59 (range: 46-72). Patients were required to have symptomatic myeloma with active and/or high-risk disease and adequate cardiac, renal and pulmonary function. Patients in complete remission were excluded unless they had high-risk cytogenetics. Patients received a median of 3 lines of prior therapy (range: 1-5); 5 patients (25%) had relapsed after an earlier ASCT. Twelve patients (60%) had cytogenetic abnormalities including 7 (35%) with confirmed high-risk abnormalities [t(4;14), del17p13, complex]. Administration of lenalidomide (10 mg/day) was recommended starting at day 100 post-transplant.
To be eligible for the treatment phase, HLA-A*0201 positive patients first consented to a marrow examination to obtain myeloma cells to test and confirm NY-ESO-1 or LAGE-1 expression by RT-PCR. All participants gave written informed consent in accordance with the Declaration of Helsinki. Study approval was obtained from the Institutional Review Boards (IRBs) of the University of Maryland and the University of Pennsylvania, the FDA and the NIH Recombinant DNA Advisory Committee (RAC).
A flow diagram depicting the trial design is shown (Fig. 1). Briefly, after confirmation of HLA-A*0201 status and expression of NY-ESO-1 or LAGE-1 in the myeloma cells, patients received an intramuscular injection of Prevnar-13® - the pneumococcal conjugate vaccine (PCV) – into the non-dominant deltoid muscle. PCV served as a marker of antigen-specific immune reconstitution. About 10 days after this immunization all patients had steady-state apheresis to collect approximately 1 × 108 mononuclear cells per kilogram body weight. Patients proceeded to stem cell mobilization using cyclophosphamide at a dose of 1.5–3.0 g/m2 followed by G-CSF (10 μg/kg).
Approximately 4 weeks following the apheresis, patients received high-dose therapy with melphalan (200 mg/m2 or 140 mg/m2 if age > 70 years) followed by infusions of autologous stem cells (≥ 2 × 106 CD34+ cells/kg body weight) at day 0. The engineered T cells were infused on day +2. Supportive care measures included antibiotic prophylaxis and administration of G-CSF starting on day +5. Three additional pneumococcal conjugate vaccine immunizations (Prevnar-13®) were given at days +14, +42 and +90. Lenalidomide maintenance at 10 mg per day was started at day +100 post-transplant for patients with sufficient marrow recovery (A total of 13/20 patients received maintenance lenalidomide for at least 3 months or more). Optional bone marrow aspirates for assessment of T-cell trafficking were collected at days +7, +21, +42, +100 and +180 post-transplant.
Clinical response assessments
Myeloma assessments were performed at days +42, +100, +180 and every 3 months thereafter while patients remained on study. Disease response was assessed in accordance with the International Uniform Response Criteria for myeloma assessment 57 with the additional category of near complete response (nCR) defined as disease that is detected only by immunofixation, less than 5% plasma cells in the marrow, and no increase in size or number of lytic bone lesions. This method of assessment is consistent with the methods employed by the Bone Marrow Transplantation Clinical Trials Network at the time of the conduct of this study and is consistent with methods used in recently published studies. The category of nCR is relevant to studies of adoptive T cell therapy where the early immune reconstitution can lead to oligoclonal banding that can confound accurate assessment of disease clearance58.
Cell Manufacturing
Engineered T cells were manufactured initially at the Cell and Vaccine Production Facility at the University of Pennsylvania (Philadelphia, PA), and later the process was transferred to a commercial contractor, Progenitor Cell Therapy (PCT, Allendale, NJ). Comparability studies were conducted and submitted to the Food and Drug Administration to demonstrate comparability of the product at PCT. Engineered T cells were manufactured from CD25 depleted CD4 and CD8 T cells that were activated and expanded using anti-CD3/28 antibody conjugated paramagnetic microbeads (Life Technologies, Carlsbad, CA) as previously described 48. T cells were transduced at a multiplicity of infection of 1 transducing unit (TU)/cell. The manufacturing process took 9-12 days, and an additional 7-10 days were required to complete release testing. An overview of the process is shown in Figure 1B.
Vector
The lentiviral vector is a self-inactivating vector derived from HIV-1 as described in Dull et al, 1998 59, and containing the WPRE element. The transgene is expressed off of the EF1α promoter. Lentiviral vector was produced at the City of Hope (Duarte, CA) using transient transfection with four plasmids expressing the transfer vector, Rev, VSV-G and gag/pol, in 293T cells. Supernatant was collected at multiple timepoints, clarified, treated with benzonase, and concentrated by tangential flow filtration and centrifugation. Transduction potency was measured on primary T cells.
Assays for NY-ESO-1/LAGE-1 Expression and Persistence, Trafficking and Function of Gene-modified T-cells
Research sample processing, freezing, and laboratory analyses were performed in the Translational and Correlative Studies Laboratory at the University of Pennsylvania, which operates under principles of Good Laboratory Practice with established SOPs and/or protocols for sample receipt, processing, freezing, and analysis. Assay performance and data reporting conforms with MIATA guidelines 60. Toward the end of the study, assays were transferred to Cambridge Biomedical (Cambridge, MA), a commercial laboratory also operating in compliance with Good Laboratory Practices.
Sample draws and processing
Samples (peripheral blood, bone marrow) were collected in lavender top (K2EDTA,) tubes (Becton Dickinson). Samples drawn at the University of Pennsylvania were delivered to the laboratory within 2 hours of draw; samples drawn at the University of Maryland were delivered to the laboratory by overnight shipment at room temperature and in insulated containers. Samples were processed within 30 minutes of receipt according to established laboratory SOPs. Peripheral blood and marrow mononuclear cells were purified, processed, and stored in liquid nitrogen.
Q-PCR analyses
Whole-blood samples were collected in lavender top (K2EDTA) BD vacutainer tubes (Becton Dickinson). Genomic DNA was isolated from whole blood or marrow using QIAamp DNA blood midi kits (Qiagen) and established laboratory SOPs, quantified by spectrophotometer, and stored until testing at −80°C. Q-PCR analyses were performed in bulk using ABI Taqman technology and a validated assay to detect the WPRE sequence present in the lentivirus backbone, using 200 ng genomic DNA/time-point for peripheral blood and marrow samples and using the methodology described 61 including normalizing for amplification of input DNA using amplifications with the CDKN1A primer/probe set (NF). The WPRE primers were as follows: 5’ WPRE.227.Forward: 5' CGCAACCCCCACTGGTT 3’ (nt 227-244 of the WPRE gene) an anti-sense primer with the sequence: WPRE.289.R: 5'AAAGCGAAAGTCCCGGAAA 3’ (nt 270-289 of the WPRE gene). These primers amplify a 62-nt long sequence from the WPRE coding sequence that can be detected using the FAM ™ labeled probe 5'FAM-TTGCCACCACCTGTC 3’. Calculated values were adjusted by a factor of 0.5 (EF) to account for amplification efficiency of this primer/probe combination and based on flow-cytometric analysis of the infused cell product. Values from this analysis are reported as average transgene copies/cell, calculated according to the formula: Average transgene copies/cell = copies plasmid detected by Q-PCR/input DNA (ng) × 0.0063 ng DNA/cell × CDKN1A NF × EF. The limit of detection for the assay is <50 copies / μg genomic DNA. The white blood cell count was used as an amplifier to calculate the number of gene modified cells in the blood. An assumption of one vector copy per cell was assumed based upon the multiplicity of infection of 1 used for cell transduction; however the gene modified cell population is likely to contain subset of cells with multiple integration events.
RT-Q-PCR analysis
Total RNA was isolated directly from whole marrow using Ribopure TM blood kits (Ambion), and cDNA synthesis was performed using iScript cDNA synthesis kits (Biorad). cDNA was used directly in Q-PCR assays to detect and quantify the relative abundance of LAGE-1 (Hs00535628_m1), NYESO-1 (Hs00265824), and CD138 (Hs00896423_m1) transcripts, using the inventoried and recommended ABI-based primer/probes (indicated in parentheses). Each data-point (sample, standard curve) was evaluated in triplicate with a positive Ct value in 2/3 replicates with % CV less than 15% required for all reported values. A parallel amplification reaction to control for the quality and quantity of interrogated cDNA was performed using a primer/probe combination specific for the housekeeping gene PP1B, and an inventoried ABI Taqman assay (Hs00168719_m1). cDNA generated using RNA isolated from the melanoma cell line A375 was used as a reference sample. Data were reported as the relative quantity (RQ) value.
TCR clonotype analyses
TCR clonotype analysis was performed by Adaptive Biotechnologies (Seattle, WA) and high-throughput next-generation sequencing of the TCR CDR3 region using the Illumina HiSeq/MiSeq platform-based immunoSEQ assay v.5. For these analyses, approximately 1,000 ng (approximately 16,000 genome equivalents) of genomic DNA isolated from marrow samples were subjected to combined multiplex PCR and sequencing followed by algorithmic analyses to quantify individual TCR Vβ CDR3 sequences in samples essentially as described in 62.
Multiparametric flow cytometry
Cells were evaluated by flow cytometry either fresh after Ficoll-Paque processing or, if frozen, after overnight rest at a density of 2×106 cells/ml. Multi-parametric immunophenotyping was performed using approximately 1×106 total cells/condition. Cells were acquired using an Accuri C6 cytometer equipped with a Blue (488) and Red (633 nm) laser. Compensation values were established using single antibody stains and BD compensation beads (Becton Dickinson) and were calculated manually. Data were analyzed using C-Flow software analysis package (version 1.0.264.9, Accuri cytometers)
Flow cytometry detection reagents
The following antibodies were used for basic T cell identification: CD3-PE, CD8-FITC, CD14-PE-Cy7CD16-PE-Cy7, CD19-PE-Cy7, all from Becton Dickinson. To detect transduced NYESO-1 TCR expressing cells, APC-conjugated dextramer reagents specific for the HLA-A*0201 / SLLMWITQV complex (Immudex, Copenhagen Denmark) were utilized at the manufacturer recommended concentrations.
Statistical Analyses
Joint modelling for longitudinal and time-to-event data (OS and PFS) was used to make inferences. The assumption was made that intermittently missing time-dependent biomarkers were missing at random. This was especially important for analysis of parameters involving marrow after infusion since these collections were optional. The likelihood for the survival model was derived under the assumption that the baseline hazard follows a Weibull failure time model with frailty components 63. A numerical integration technique, the Gauss-Hermite quadrature method, was applied to evaluate the likelihood. All biomarker values were transformed using a square root transformation to decrease variability and to smooth distribution. Plausible associations between NY-ESO-1, Lage-1, CD138 and T-cell persistence in blood or marrow were estimated using a mixed-effects models approach. Survival curves were also estimated with the method of Kaplan and Meier, calculated from the date of transplantation to the date of death from any cause, censored at date of last contact. The estimated parameters were presented as median time -to -event with a corresponding 95% confidence interval (CI). The values of laboratory correlative measurements were transformed by taking a square root to decrease variability and to smooth the corresponding distributions. Data were analyzed within the R statistical environment (R ×64, 3.03); some statistical analyses were also performed in SAS (v.9.3, SAS Institute Inc., Cary, NC).
Antibodies and pentamers
The CD3, CD4, CD8, TNFα, Ki67, and GrB were purchased from BD Biosciences. The CD107a, HLA-DR, IFNγ, CCR7, CD45RA and IL-2 were purchased from BioLegend. The dead cell exclusion marker (Live/Dead Aqua) was purchased from InVitrogen. The NY-ESO-1 Pro5® MHC-pentamer conjugated to PE (A*02:01 SLLMWITQC) was purchased from ProImmune (Oxford, UK).
T2 cells pulsing with peptide
T2 cells were cultured at 1×10e6 cells/mL in RPMI-10% FBS in the presence of the NY-ESO-1 (9V) peptide (SLLMWITQV, Peptide Protein Research) at 1 μM, or with vehicle only (DMSO 0.027% v/v). After an overnight incubation at 37°C, 5%CO2, non-pulsed or pulsed T2 cells were centrifuged and the supernatants were discarded. Non-pulsed T2 and pulsed T2 cells were resuspended in RPMI-10% FBS and were used in the ICS assay.
ICS assay with pentamer staining
On the day of the assay, cryopreserved PBMC from patients enrolled in the clinical trial, as well as PBMC from a healthy donor control and NY-ESO-1 TCR transduced cells were thawed and counted. Pentamer staining was performed before the 6-hour stimulation period.
Briefly, PBMC (1.5×10e6 cells per well) first incubated with the NY-ESO-1 pentamer in 50 μL of 2% FBS for 10 minutes at room temperature. The cells were then washed in RPMI-10% FBS, and the NY-ESO-1 pulsed T2 cells (7.5×10e5 cells per well) were added in RPMI-10% FBS. The CD107a antibody was added and the cells were incubated at 37°C and 5% CO2 for one (1) hour. The Golgi Plug/Golgi Stop reagent was added and the incubation was resumed for an additional 5 hours at 37°C and 5% CO2.
The cells were washed and were surface stained at 4°C for 30 min (Live/Dead Aqua, CD4, CD8, CCR7, HLA-DR, and CD45RA) followed by fixation/permeabilization and intracellular staining (Ki67, TNFα, IL-2, CD3, GrB and IFNγ) at 4°C for 30 min. Labeled cells were acquired on a LSR II flow cytometer using the FACS DiVa software (BD Bioscience).
Supplementary Material
Acknowledgements
We thank the staff of the Clinical Cell and Vaccine Production Facility and the Translational and Correlative Sciences Laboratory at the University of Pennsylvania, apheresis centers and nurses of the BMT programs of the University of Maryland Greenebaum Cancer Center and the Abramson Cancer Center for outstanding clinical care provide to our patients. We also thank the courageous and visionary patients who agreed to participate in this study.
Grant Support
This work was supported in part by a grant from the National Institutes of Health to A.P.R. and M.K. (R01-CA166961) and a Senior Investigator Award to A.P.R. from the Multiple Myeloma Research Foundation (MMRF), and by a sponsored research grant from Adaptimmune to M.K. and C.H.J.
Footnotes
Author contribution statement. A.P.R., E.A.S., C.H.J., M.K., and G.K.B. designed and carried out the study and wrote the manuscript. O.G. and S.K.S. performed statistical analysis. D.T.V., A.Z.B., S.Y., N.H., J.Y., A.G. and B.W. treated patients on study. T.H. provided clinical safety oversight. S.L., J.F., I.K., S.K.S., S.K., M.G., S.B., L.M. and D.W. performed correlative studies. J.E.B., A.D.B., A.B.G., N.J.P., H.K.T., and B.K.J. developed the NY-ESO TCR. N.K., L.R., S.W. and S.P. were clinical coordinators for the study. D.L.S., and B.L.L. performed cell manufacturing.
Competing Financial Interests
This study was funded in part by Adaptimmune Ltd, and several authors are employed by Adaptimmune.
References
- 1.Tricot G, et al. Graft-versus-myeloma effect: proof of principle. Blood. 1996;87:1196–1198. [PubMed] [Google Scholar]
- 2.Alyea E, et al. T-cell--depleted allogeneic bone marrow transplantation followed by donor lymphocyte infusion in patients with multiple myeloma: induction of graft-versus-myeloma effect. Blood. 2001;98:934–939. doi: 10.1182/blood.v98.4.934. [DOI] [PubMed] [Google Scholar]
- 3.Lokhorst HM, et al. The occurrence of graft-versus-host disease is the major predictive factor for response to donor lymphocyte infusions in multiple myeloma. Blood. 2004;103:4362–4364. doi: 10.1182/blood-2003-11-3862. [DOI] [PubMed] [Google Scholar]
- 4.Barlogie B, et al. Superiority of tandem autologous transplantation over standard therapy for previously untreated multiple myeloma. Blood. 1997;89:789–793. [PubMed] [Google Scholar]
- 5.Attal M, et al. A prospective, randomized trial of autologous bone marrow transplantation and chemotherapy in multiple myeloma. Intergroupe Francais du Myelome. The New England journal of medicine. 1996;335:91–97. doi: 10.1056/NEJM199607113350204. [DOI] [PubMed] [Google Scholar]
- 6.Child JA, et al. High-dose chemotherapy with hematopoietic stem-cell rescue for multiple myeloma. The New England journal of medicine. 2003;348:1875–1883. doi: 10.1056/NEJMoa022340. [DOI] [PubMed] [Google Scholar]
- 7.Porrata LF, et al. Early lymphocyte recovery predicts superior survival after autologous hematopoietic stem cell transplantation in multiple myeloma or non-Hodgkin lymphoma. Blood. 2001;98:579–585. doi: 10.1182/blood.v98.3.579. [DOI] [PubMed] [Google Scholar]
- 8.Porrata LF, Markovic SN. Timely reconstitution of immune competence affects clinical outcome following autologous stem cell transplantation. Clinical and experimental medicine. 2004;4:78–85. doi: 10.1007/s10238-004-0041-4. [DOI] [PubMed] [Google Scholar]
- 9.Dhodapkar MV, Krasovsky J, Olson K. T cells from the tumor microenvironment of patients with progressive myeloma can generate strong, tumor-specific cytolytic responses to autologous, tumor-loaded dendritic cells. Proc Natl Acad Sci U S A. 2002;99:13009–13013. doi: 10.1073/pnas.202491499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noonan K, et al. Activated marrow-infiltrating lymphocytes effectively target plasma cells and their clonogenic precursors. Cancer research. 2005;65:2026–2034. doi: 10.1158/0008-5472.CAN-04-3337. [DOI] [PubMed] [Google Scholar]
- 11.Rapoport AP, et al. Restoration of immunity in lymphopenic individuals with cancer by vaccination and adoptive T-cell transfer. Nature medicine. 2005;11:1230–1237. doi: 10.1038/nm1310. [DOI] [PubMed] [Google Scholar]
- 12.Rapoport AP, et al. Combination immunotherapy using adoptive T-cell transfer and tumor antigen vaccination on the basis of hTERT and survivin after ASCT for myeloma. Blood. 2011;117:788–797. doi: 10.1182/blood-2010-08-299396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rapoport AP, et al. Combination Immunotherapy after ASCT for Multiple Myeloma Using MAGE-A3/Poly-ICLC Immunizations Followed by Adoptive Transfer of Vaccine-Primed and Costimulated Autologous T Cells. Clin Cancer Res. 2014;20:1355–1365. doi: 10.1158/1078-0432.CCR-13-2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rapoport AP, et al. Rapid immune recovery and graft-versus-host disease-like engraftment syndrome following adoptive transfer of Costimulated autologous T cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15:4499–4507. doi: 10.1158/1078-0432.CCR-09-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stadtmauer EA, et al. Transfer of influenza vaccine-primed costimulated autologous T cells after stem cell transplantation for multiple myeloma leads to reconstitution of influenza immunity: results of a randomized clinical trial. Blood. 2011;117:63–71. doi: 10.1182/blood-2010-07-296822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosenblatt J, et al. Vaccination with Dendritic Cell/Tumor Fusions following Autologous Stem Cell Transplant Induces Immunologic and Clinical Responses in Multiple Myeloma Patients. Clin Cancer Res. 2013;19:3640–3648. doi: 10.1158/1078-0432.CCR-13-0282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nature medicine. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aleksic M, et al. Different affinity windows for virus and cancer-specific T-cell receptors: implications for therapeutic strategies. Eur J Immunol. 2012;42:3174–3179. doi: 10.1002/eji.201242606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Purbhoo MA, et al. Quantifying and imaging NY-ESO-1/LAGE-1-derived epitopes on tumor cells using high affinity T cell receptors. Journal of immunology. 2006;176:7308–7316. doi: 10.4049/jimmunol.176.12.7308. [DOI] [PubMed] [Google Scholar]
- 20.Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases--elimination, equilibrium and escape. Current opinion in immunology. 2014;27:16–25. doi: 10.1016/j.coi.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maude SL, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. The New England journal of medicine. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davila ML, et al. Chimeric antigen receptors for the adoptive T cell therapy of hematologic malignancies. International journal of hematology. 2014;99:361–371. doi: 10.1007/s12185-013-1479-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee DW, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2014 doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morgan RA, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson LA, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–546. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burns WR, Zheng Z, Rosenberg SA, Morgan RA. Lack of specific gamma-retroviral vector long terminal repeat promoter silencing in patients receiving genetically engineered lymphocytes and activation upon lymphocyte restimulation. Blood. 2009;114:2888–2899. doi: 10.1182/blood-2009-01-199216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y, et al. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nature biotechnology. 2005;23:349–354. doi: 10.1038/nbt1070. [DOI] [PubMed] [Google Scholar]
- 28.Robbins PF, et al. Single and dual amino acid substitutions in TCR CDRs can enhance antigen-specific T cell functions. Journal of immunology. 2008;180:6116–6131. doi: 10.4049/jimmunol.180.9.6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robbins PF, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:917–924. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robbins PF, et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T cell receptor: Long term follow up and correlates with response. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014 doi: 10.1158/1078-0432.CCR-14-2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hunder NN, et al. Treatment of metastatic melanoma with autologous CD4+ T cells against NYESO-1. The New England journal of medicine. 2008;358:2698–2703. doi: 10.1056/NEJMoa0800251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuan J, et al. CTLA-4 blockade enhances polyfunctional NY-ESO-1 specific T cell responses in metastatic melanoma patients with clinical benefit. Proc Natl Acad Sci U S A. 2008;105:20410–20415. doi: 10.1073/pnas.0810114105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Baren N, et al. Genes encoding tumor-specific antigens are expressed in human myeloma cells. Blood. 1999;94:1156–1164. [PubMed] [Google Scholar]
- 34.Jungbluth AA, et al. The cancer-testis antigens CT7 (MAGE-C1) and MAGE-A3/6 are commonly expressed in multiple myeloma and correlate with plasma-cell proliferation. Blood. 2005;106:167–174. doi: 10.1182/blood-2004-12-4931. [DOI] [PubMed] [Google Scholar]
- 35.Condomines M, et al. Cancer/testis genes in multiple myeloma: expression patterns and prognosis value determined by microarray analysis. Journal of immunology. 2007;178:3307–3315. doi: 10.4049/jimmunol.178.5.3307. [DOI] [PubMed] [Google Scholar]
- 36.Atanackovic D, et al. Cancer-testis antigens are commonly expressed in multiple myeloma and induce systemic immunity following allogeneic stem cell transplantation. Blood. 2007;109:1103–1112. doi: 10.1182/blood-2006-04-014480. [DOI] [PubMed] [Google Scholar]
- 37.van Rhee F, et al. NY-ESO-1 is highly expressed in poor-prognosis multiple myeloma and induces spontaneous humoral and cellular immune responses. Blood. 2005;105:3939–3944. doi: 10.1182/blood-2004-09-3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoos A, et al. Improved endpoints for cancer immunotherapy trials. Journal of the National Cancer Institute. 2010;102:1388–1397. doi: 10.1093/jnci/djq310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee DW, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124:188–195. doi: 10.1182/blood-2014-05-552729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barrett DM, Teachey DT, Grupp SA. Toxicity management for patients receiving novel T-cell engaging therapies. Current opinion in pediatrics. 2014;26:43–49. doi: 10.1097/MOP.0000000000000043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brentjens RJ, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Corrigan-Curay J, et al. T-cell immunotherapy: looking forward. Mol Ther. 2014;22:1564–1574. doi: 10.1038/mt.2014.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Merchant Melinda S., M.C.C., Stadtmauer Edward A., Tap William D., D'Angelo Sandra P., Grupp Stephan A., Holdich Tom, Binder-Scholl Gwendolyn, Jakobsen Bent K, Odunsi Kunle, Rapoport Aaron, Mackall Crystal. American Society of Clinical Oncology. Illinois; Chicago: 2015. Genetically engineered NY-ESO-1 specific T cells in HLA-A201+ patients wtih advanced cancers. [Google Scholar]
- 44.Cameron BJ, et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Science translational medicine. 2013;5:197ra103. doi: 10.1126/scitranslmed.3006034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Linette GP, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122:863–871. doi: 10.1182/blood-2013-03-490565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kalos M, June CH. Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity. 2013;39:49–60. doi: 10.1016/j.immuni.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stein S, et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nature medicine. 2010;16:198–204. doi: 10.1038/nm.2088. [DOI] [PubMed] [Google Scholar]
- 48.Levine BL, et al. Effects of CD28 costimulation on long-term proliferation of CD4+ T cells in the absence of exogenous feeder cells. Journal of immunology. 1997;159:5921–5930. [PubMed] [Google Scholar]
- 49.Scholler J, et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Science translational medicine. 2012;4:132ra153. doi: 10.1126/scitranslmed.3003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krishnan A, et al. Autologous haemopoietic stem-cell transplantation followed by allogeneic or autologous haemopoietic stem-cell transplantation in patients with multiple myeloma (BMT CTN 0102): a phase 3 biological assignment trial. The lancet oncology. 2011;12:1195–1203. doi: 10.1016/S1470-2045(11)70243-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sonneveld P, et al. Bortezomib-based versus nonbortezomib-based induction treatment before autologous stem-cell transplantation in patients with previously untreated multiple myeloma: a meta-analysis of phase III randomized, controlled trials. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31:3279–3287. doi: 10.1200/JCO.2012.48.4626. [DOI] [PubMed] [Google Scholar]
- 52.Richardson P. Novel strategies in the treatment of relapsed/refractory multiple myeloma. From the Multiple Myeloma Research Foundation. Oncology. 2003;17:1063–1065. [PubMed] [Google Scholar]
- 53.Armand P, et al. Disabling immune tolerance by programmed death-1 blockade with pidilizumab after autologous hematopoietic stem-cell transplantation for diffuse large B-cell lymphoma: results of an international phase II trial. J Clin Oncol. 2013;31:4199–4206. doi: 10.1200/JCO.2012.48.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Galustian C, et al. The anti-cancer agents lenalidomide and pomalidomide inhibit the proliferation and function of T regulatory cells. Cancer immunology, immunotherapy : CII. 2009;58:1033–1045. doi: 10.1007/s00262-008-0620-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ramsay AG, et al. Chronic lymphocytic leukemia T cells show impaired immunological synapse formation that can be reversed with an immunomodulating drug. The Journal of clinical investigation. 2008;118:2427–2437. doi: 10.1172/JCI35017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 57.Rajkumar SV, et al. Consensus recommendations for the uniform reporting of clinical trials: report of the International Myeloma Workshop Consensus Panel 1. Blood. 2011;117:4691–4695. doi: 10.1182/blood-2010-10-299487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mark T, et al. Atypical serum immunofixation patterns frequently emerge in immunomodulatory therapy and are associated with a high degree of response in multiple myeloma. British journal of haematology. 2008;143:654–660. doi: 10.1111/j.1365-2141.2008.07374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dull T, et al. A third-generation lentivirus vector with a conditional packaging system. Journal of virology. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Janetzki S, et al. “MIATA”-minimal information about T cell assays. Immunity. 2009;31:527–528. doi: 10.1016/j.immuni.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kalos M, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Science translational medicine. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Robins HS, et al. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood. 2009;114:4099–4107. doi: 10.1182/blood-2009-04-217604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rizopoulos D. Joint Models for Longitudinal and Time-to-Event Data. Chapman & Hall/CRC; 2012. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.