

Abstract
Recursive splicing is a process in which large introns are removed in multiple steps by resplicing at ratchet points - 5′ splice sites recreated after splicing1. Recursive splicing was first identified in the Drosophila Ultrabithorax (Ubx) gene1 and only three additional Drosophila genes have since been experimentally shown to undergo recursive splicing2,3. Here, we identify 197 zero nucleotide exon ratchet points in 130 introns of 115 Drosophila genes from total RNA sequencing data generated from developmental time points, dissected tissues, and cultured cells. The sequential nature of recursive splicing was confirmed by identification of lariat introns generated by splicing to and from the ratchet points. We also show that recursive splicing is a constitutive process, that depletion of U2AF inhibits recursive splicing, and that the sequence and function of ratchet points are evolutionarily conserved in Drosophila. Finally, we identified four recursively spliced human genes, one of which is also recursively spliced in Drosophila. Together these results indicate that recursive splicing is commonly used in Drosophila, occurs in human and provides insight into the mechanisms by which some large introns are removed.
Recursive splicing was first identified in the Drosophila melanogaster Ultrabithorax (Ubx) gene1. The 73 kb intron within Ubx houses two alternative microexons (mI and mII) which both contain the consensus 5′ splice site sequence GTAAGA immediately downstream of the 3′ splice sites. In addition, this intron contains a ratchet point, a zero nucleotide exon consisting of juxtaposed 3′ and 5′ splice sites. It has been shown that rather than being removed in a single step, the 73 kb Ubx intron is removed in four steps in which the upstream constitutive exon is spliced to exon mI, and subsequently re-spliced to exon mII, the ratchet point, and finally the downstream constitutive exon. A previous genome-wide computational search for potential ratchet points conserved between D. melanogaster and D. pseudoobscura predicted 160 potential ratchet points in 124 introns of 106 genes2. Of these, only 7 ratchet points in three genes (kuzbanian (kuz), outspread (osp), and frizzled (fz)) have been reported to be experimentally validated2,3.
We generated 10.9 billion uniquely mapped reads of rRNA-depleted, paired-end, strand-specific RNA sequence from 183 D. melanogaster individual RNA samples comprising 35 dissected tissue samples, 24 untreated and 11 ecdysone treated cell lines, 30 distinct developmental stages and males and females of four strains from the D. melanogaster Genetic Reference Panel4 (Supplementary Table 1). The majority of these RNA samples were previously used to generate poly(A)+ RNA sequence data5,6. As the current libraries were prepared without poly(A) selection, they contain a mixture of mRNA, pre-mRNA and nascent RNA. Co-transcriptional splicing can be observed in total, nuclear, or nascent RNA-seq data by the sawtooth pattern of read density across introns in the 5′ to 3′ direction of transcription7 (Fig. 1a). While visually inspecting these data on a genome browser, we noticed several large introns that lacked internal annotated exons yet possessed sawtooth patterns of read density suggestive of co-transcriptional splicing, including the introns from Ubx (Fig. 1b), kuz, osp, and fz that were previously shown to undergo recursive splicing. We hypothesized that such sawtooth patterns could be indicative of recursive splicing and performed a genome-wide search for ratchet points supported by the RNA-Seq data.
Figure 1. Identification and validation of recursive splice sites in Drosophila.

a, Schematic diagram of nascent pre-mRNA transcripts during co-transcriptional splicing and the corresponding read density that would be observed in total RNA-Seq data. Note the sawtooth pattern created by the 5′ to 3′ gradient of RNA-Seq read density from the exon to the downstream ratchet point and splice site. b, Example of total RNA-Seq data for the Ubx gene which is known to contain three recursive splice sites. Also shown are the splice junction reads supporting recursive splicing at each site. c, Example of five recursive splice sites identified in luna. Shown are the recursive junctions identified and the overall RNA-Seq read density from all samples (blue). d, RT-PCR validation of the luna ratchet points (red dots) using primers in the upstream constitutive exon and flanking the putative ratchet points (UP). The RP primers are expected to yield RT-PCR products if the constitutive exon is spliced to the ratchet point. The URP primers, which are upstream of each ratchet point, serve as negative controls.
To identify potential zero nucleotide exon-type ratchet points, we parsed the RNA-Seq alignments to identify novel splice junctions where the reads mapped to an annotated 5′ splice site and an unannotated 3′ splice site, and the genomic sequence at the 3′ splice site junction was AG/GT (Extended Data Fig. 1a). We also aligned the total RNA-Seq data to a database of splice junctions between annotated exons and all potential ratchet points (AG/GT sequences) in the downstream intron that did not correspond to annotated 3′ splice sites. We then identified ratchet point junctions where reads mapped without any mismatches, with at least three distinct offsets, and with an overhang of at least eight nt (Extended Data Fig. 1b). We then visually inspected each ratchet point independently identified by both methods on the genome browser, removing candidates that did not display an obvious sawtooth pattern of read density or which clearly corresponded to an unannotated exon.
We identified a total of 197 ratchet points in 130 introns of 115 genes (Supplementary Table 2). Two of these ratchet points were missed by our computational approaches, but identified during the course of manual inspection on the browser, were validated and included in the remainder of these analyses. This provides the first experimental verification of 91 of the 160 ratchet points computationally predicted by Burnette et al. based on comparative genomics2 (Supplementary Table 3). Of the 69 unverified ratchet points predicted by Burnette et al., 34 correspond to previously unannotated exons, 23 lacked convincing sawtooth patterns, 7 did not pass our recursive junction thresholds, and five could not be identified in the current assembly of the genome. Though it is difficult to conclude that these are not true ratchet points, we have not included them in our subsequent analysis as their supporting evidence is inconclusive. The other 106 (53.8%) of the ratchet points we identified are described here for the first time.
Most genes (100) contain only one recursively spliced intron, though 15 genes contain two. The number of ratchet points in an intron ranges from one to six (Extended Data Fig. 2a). The recursively spliced introns range in size from 11,341 bp to 132,736 bp with an average size of 45,164 bp. The introns containing recursive splice sites are enriched in large introns (97% of all introns are smaller than the smallest recursive intron), not all large introns contain recursive splice sites (Extended Data Fig. 2b). In fact, only 6% of introns larger than the smallest recursive intron are recursively spliced. The segments of the introns removed by recursive splicing range from 2,596 bp to 63,580 bp with an average size of 17,953 bp (+/− 9,039 bp) and median size of 16,368 bp (Extended Data Fig. 2c). The luna gene contains an 108 kb intron with five ratchet points, such that the intron is removed in six stepwise recursive splicing events (Fig. 1d). The five ratchet points are supported by the sawtooth pattern of read density across the intron, reads that map to the exon-ratchet point splice junctions (Fig. 1c), and have been validated by RT-PCR and Sanger sequencing (Fig. 1d). In total, RT-PCR and Sanger sequencing validated 24 ratchet points from 14 genes in Drosophila S2 cells (Extended Data Fig. 3).
Ratchet points are zero nucleotide exons, and therefore do not exist in the mRNA. However, direct evidence of recursive splicing can be obtained by identifying lariat introns – byproducts of all splicing reactions that contain a 2′-5′ linkage between the first nucleotide of the intron and the branchpoint. Because reverse transcriptase can occasionally traverse the branchpoint, reads corresponding to the 5′ splice site-branchpoint junction may be present in the total RNA-seq data (Fig. 2a). To identify putative recursive lariat introns, we generated a set of potential 5′ splice site-branchpoint junctions for all recursively spliced introns, and all possible permutations, and aligned the total RNA-seq reads to them (Methods). Though rare, we identified 46 reads that mapped uniquely to 27 recursive lariats introns in 20 genes (Supplementary Table 4). Directed RT-PCR and sequencing experiments independently verified 14 recursive lariats in 9 genes (Extended Data Table 1) for a total of 41 recursive lariats introns in 26 genes. Ten of the lariat introns detected correspond to the first segment of the recursive introns and are also supported by standard splice junction reads. However, the remaining lariat introns detected correspond to internal segments further supporting the sequential nature of recursive splicing. For example, couch potato (cpo) contains an intron that is removed in three recursive splicing events involving two ratchet points. We obtained evidence for all three lariats from both the total RNA-Seq data and directed RT-PCR sequencing experiments (Fig. 2b). This analysis also identified the putative branchpoints used for these recursive splicing events. All but five of these branchpoints reside from −42 to −19 upstream of the 3′ splice site with a peak at −29 (Fig. 2b,c). Six of the 3′ splice sites appear to use two different branchpoints. We observed that 81% have an A at the branchpoint, while 12%, 5%, and 2% have a T, C, or G, respectively (Fig. 2d).
Figure 2. Identification of recursive lariat introns in Drosophila.

a, RNA-Seq reads (red and orange indicate the first and second half of an individual read) that traverse a 5′ splice site-branchpoint junction would align to the linear intron as out-of order split-reads. b, Example of recursive lariat introns in cpo. Shown are the recursive junctions identified (blue), the lariat junction reads (orange), and the overall RNA-Seq read density from all samples (red). A magnification of each branch point region is also shown along with the conservation among 16 insects. The positions of the branch points are indicated by the vertical arrows. c, Distribution of the distance of the recursive lariat intron branch points from the 3′ splice sites. d, Sequence logo of the recursive lariat intron branch point sequences.
The nucleotide sequences of ratchet points resemble juxtaposed 3′ and 5′ splice sites (Fig. 3a) and the regions immediately flanking the ratchet points are much more highly conserved than those flanking non-ratchet point AG|GT sequences in the same introns (Fig. 3b). However, the ratchet points have a more prominent pyrimidine tract, and a significantly (P=<0.0001) higher frequency of a TT dinucleotide at positions −5 and −6 relative to the 3′ splice site when compared to introns genome-wide. Whereas only 43.76% (30,151/68,898) of all introns have Ts at positions −5 and −6, 99.5% (196/197) of ratchet points do. The only ratchet point lacking a TT dinucleotide at positions −5 and −6 is in CG15360 which has a C at position −6 that is conserved in other Drosophila species. Intriguingly, the majority of C. elegans 3′ splice sites have this sequence8 and it has been shown that the large U2AF subunit (encoded by U2af50 in Drosophila) interacts with these bases. Thus, the strong preference for the TT dinucleotide at positions −5 and −6 of Drosophila ratchet points could represent high affinity U2AF binding sites so that the ratchet points are efficiently recognized.
Figure 3. Characteristics of Drosophila ratchet points.

a, Sequence logos of 5′ splice sites, 3′ splice sites, ratchet points, and non-ratchet point AG/GT sequences located in the same introns as ratchet points (top to bottom). b, Sequence conservation of ratchet points. Average PhastCons scores of ratchet points (green) and non-ratchet points (blue). Solid line indicates the average PhastCons score, shaded regions indicate the 95% confidence interval. Normalized recursive junction (c) reads and percent non-recursive junctions (d) observed in untreated S2 cells and cells treated with the indicated dsRNAs.
To test this hypothesis, we sequenced total RNA from untreated S2 cells as well as cells treated with dsRNA to knock down expression of lariat debranching enzyme (ldbr) as a control, U2af38, U2af50, or both U2af38 and U2af50 (Extended Data Table 2). We observed ~20 recursive junction reads per million mapped reads (corresponding to 119 and 100 distinct ratchet points) in untreated controls and ldbr depleted cells (Fig. 3c). Depletion of U2af38 or U2af50 alone reduced the frequency of recursive junction reads 3–4-fold (corresponding to 81 and 64 distinct ratchet points) (Fig. 3c). Strikingly, depletion of both U2af38 and U2af50 resulted in a complete absence of detectable recursive junctions reads (Fig. 3c), though similar fractions of non-recursive junction reads were observed in all samples (Fig. 3d). Depletion of U2AF may so strongly impact recursive splicing, but not non-recursive splicing, because recursive junction reads can only be generated from nascent RNA while non-recursive junction reads can be generated from stable mRNAs. Additionally, exon or intron definition may not be possible for zero nucleotide exons in Drosophila due to the combination of large introns and non-existent exons. This would eliminate many of the cooperative interactions normally involved in splice site recognition making recursive splicing particularly sensitive to decreased U2AF levels. Though additional work will be required to fully elucidate the role for U2AF in recursive splicing, this result strongly suggest that U2AF is required for efficient recognition of ratchet points in S2 cells.
To determine if recursive splicing is evolutionarily conserved, we generated rRNA-depleted, stranded RNA-Seq data from mixed D. simulans, D. sechellia, D. yakuba, D. pseudoobscura, and D. virilis adults (Extended Data Table 3). We aligned these data to the corresponding reference genomes and searched for splice junction reads whose 3′ splice sites mapped to positions orthologous to the identified D. melanogaster ratchet points. Despite having 2 orders of magnitude fewer reads from these species, 131 of the 197 D. melanogaster ratchet points (66.5%) were identified in at least one of the five other Drosophila species, 69 of which were identified in at least two species (Extended Data Table 4 and Supplementary Table 5). Together these observations demonstrate that the nucleotide sequence and function of ratchet points are conserved among Drosophila species indicating that recursive splicing is evolutionarily conserved.
We also searched for zero nucleotide exons in human by generating and analyzing >1.1 billion reads of total RNA data from 20 tissues (Extended Data Table 5). Our analysis pipeline identified 76 putative ratchet points, but upon further inspection all but five ratchet points in four genes were eliminated because they either lacked an obvious sawtooth pattern of read density or corresponded to unannotated exons. The five ratchet points we identified in PDE4D, HS6ST3, CADM2, and ROBO2 (Supplementary Fig. 1, Supplementary Table 6) were independently identified by Sibley et al.9 who also demonstrated that recursive splicing in humans involve recursive exons rather than true zero nucleotide exons as in Drosophila. This suggests that though recursive splicing occurs in both Drosophila and human, the precise nature of recursive splicing differs between these organisms. Nonetheless, the presence of ratchet points in both Drosophila Hs6st and its human ortholog HS6ST3 indicates that recursive splicing is either very ancient or evolved independently.
The host genes containing recursively spliced introns are expressed in a broad spectrum of developmental timepoints, tissues, and cell types – the recursive host genes are expressed at FPKM>1 in 72%, 93% and 83% of cell lines, developmental time points, and tissues, respectively. However, host gene expression levels are quite dynamic throughout development and 63% have their peak expression in nervous system tissues (Fig. 4a), consistent with GO enrichments in development and neural functions (Supplementary Table 7).
Figure 4. Expression characteristics of recursively spliced Drosophila genes.

a, Heatmap representation of Z-scores of mRNA expression levels of the recursively spliced genes among the samples examined. Male (M), female (F), imaginal discs (ID), ovaries (OV), accessory gland (A), testes (T), digestive tract (Dig), salivary gland (Saliv). b, Distribution of the Spearman correlations of mRNA expression levels and recursive indexes of each ratchet point for the cell line (red), developmental (green), and tissue (blue) samples. c, Example of the correlation of mRNA expression levels and recursive indexes for four ratchet points in Antp in the developmental (green), and tissue (blue) samples.
Several lines of evidence suggest that recursive splicing is constitutive – specifically, when the host gene is transcribed, it is recursively spliced. First, we have been unable to detect lariat introns that would be generated by ratchet point skipping or the direct splicing of the flanking constitutive exons without recursive splicing. In our directed RT-PCR experiments, we failed to amplify lariats generated by ratchet point skipping using primers that successfully amplified lariats from individual recursive segments in the same intron. We also were unable to identify skipping events in the total RNA-Seq data though we did identify lariats in non-recursive introns as large as 84,027 bp in our total RNA-Seq data (data not shown). Second, we calculated a recursive index for each ratchet point (the number of ratchet point junction reads/mapped reads) and observed generally strong correlations between the recursive index and the gene expression level for most genes (Fig. 4b). For example, there is a strong positive correlation between gene expression and recursive splicing for all four ratchet points in the Antennapedia (Antp) gene (Fig. 4c). The correlation between gene expression and recursive splicing is strongest among the tissue samples and weakest among the cell lines, which have the highest and lowest number of mapped reads, respectively (Extended Data Fig. 4), indicating that low correlation is related to sequencing depth. Together, these results strongly suggest that recursive splicing is constitutive, though it remains possible that regulated or alternative ratchet points may be identified in the future.
Recent studies have demonstrated strong associations between chromatin marks and particular features of gene architecture, including intron-exon boundaries. Of particular note, H3K4me3(ref. 10), H3K79me2(ref. 11), and H3K36me3(ref. 12) have been shown to specifically transition near intron-exon boundaries in humans. We inspected ChIP-seq data obtained from whole larvae to determine whether any chromatin marks are associated with ratchet points (Extended Data Fig. 5). None of the chromatin marks we examined are specifically associated with ratchet points, yet the recursive splice sites are associated with chromatin marks that would be expected given their position relative to canonical exons.
Here we provide experimental evidence that 130 Drosophila introns, 26 times the number previously known, are removed in multiple, sequential steps by recursive splicing, rather than by a single splicing event. We also identified five ratchet points in four human genes, including one case of orthologous Drosophila and human genes, indicating that recursive splicing evolved long ago. We do note, however, that recursive splicing in Drosophila involves true zero nucleotide exons but that Sibley et al.9 have demonstrated that recursive splicing in human involves recursive exons pointing to mechanistic and perhaps functional differences in this process between flies and human. The ratchet points involved in recursive splicing are highly conserved and share sequence similarity with one another. While recursive splicing clearly occurs in Drosophila, its function and mechanism remains elusive, though we provide evidence that U2AF is required for recursive splicing. It also remains unknown why some Drosophila introns are recursively spliced and others are not. Further investigation will be necessary to determine whether recursive splicing is required for the function of the host gene in Drosophila and how the upstream exons re-engage in subsequent splicing reactions.
METHODS
RNA collections
The D. melanogaster RNA samples used for this study were previously described5,6. RNA was isolated from D. simulans, D. sechellia, D. yakuba, D. pseudoobscura, and D. virilis mixed adults using Trizol. Total RNA from 20 human tissues was obtained from Clonetech (Catalog #636643). RNA from the RNAi experiments was extracted from Drosophila S2 cells treated with 20 μg of dsRNA for 5 days. The following darn sequences were used for the RNAi experiments: ldbr (AAGCTAGGAGATGCTGAATCTTCCTCTTCCAGCAGCAGCAGTGAAGATGAAGACG AGGAAAGGGAGAAGGTAAAGAAAGCTGCTCCTGTACCTCCACCATCCAAATCTGTT CCCGTGACCAAGTTTCTGGCTCTCGACAAATGCCTGCCACGTCGTGCTTTCCTGCA AGTGGTAGAGGTACCCAGTGACCCCATCGAAGGCACTCCCCGCCTGGAATACGAC GCAGAGTGGCTAGCCATCTTGCACAGTACAAATCACTTGATTTCAGTGAAGGAGAA TTATTATTACCTGCCCGGAAAAAAGGCGGGAGAGTTTACAGAGCGATCAAACTTTA CCCCCACTGAAGAAGAACTAGAAGCAGTGACCGCAAAGTTTCAGAAACTTCAAGTC CCCGAGAACTTTGAGCGCACAGTGCCAGCTTTCGATCCCGCGGAGCAGTCTGATT ATAAGCACATGTTTGTGGATCAACCCAAGGTTCAACTAAACCCCCAGAGCAATACG TTCTGTGCCACTCTGGGTATAGACGATC), U2af38 (AGATGCAAGAACACTACGACAATTTTTTCGAGGACGTGTTCGTAGAGTGCGAGGA CAAGTACGGGGAAATCGAGGAGATGAACGTGTGCGACAACCTAGGCGACCATCTG GTCGGCAATGTGTACATCAAATTCCGTAACGAGGCTGATGCGGAAAAGGCGGCAA ACGATTTGAACAACCGGTGGTTCGGTGGTCGACCGGTGTACTCGGAACTATCGCC GGTGACCGACTTCCGCGAGGCTTGCTGTCGGCAGTACGAGATGGGCGAATGTAC CCGCTCCGGCTTCTGCAACTTCATGCACTTGAAGCCCATCTCGCGTGAGCTGCGA AGGTACCTCTACTCCCGCCGCCGTCGTGCCCGCTCCCGTTCCCGATCCCCTGGAC GCCGTCGCGGCTCCCGCAGCAGGTCCCGATCCCCGGGTCGAAGAGGAGGCGGC AGAGGCGACGGTGTCGGCGGAGGAAACTACTTGAACAAC) and U2af50 (CCGAGGAGGAAATGATGGAGTTCTTCAACCAACAGATGCATTTAGTTGGGCTCGC CCAGGCGGCCGGCAGTCCCGTCTTGGCATGCCAAATTAACTTGGACAAAAACTTT GCTTTCCTCGAATTCCGATCGATTGATGAAACCACCCAGGCCATGGCATTCGATGG CATCAATTTGAAGGGGCAGAGCTTAAAGATTAGGCGTCCGCACGATTACCAGCCC ATGCCGGGTATAACAGATACGCCGGCAATTAAGCCCGCTGTTGTTTCCAGTGGAG TTATTTCGACAGTGGTTCCGGACTCGCCTCACAAAATCTTCATCGGAGGTCTACCA AACTATCTGAATGACGATCAGGTTAAGGAACTGCTTTTGTCGTTTGGCAAGCTACG AGCCTTCAACCTGGTTAAGGATGCCGCTACTGGGTTGAGTAAGGGTTATGCTTTCT GTGAATATGTCGATCTTAGCATCACAG).
RNA sequencing
Total RNA-Seq libraries were prepared using Illumina TruSeq Stranded Total RNA Sample Prep Kits as described by the manufacturer. Libraries were quantitated by analysis on an Agilent Bioanalyzer or TapeStation and sequenced on an Illumina HiSeq2000 to generate paired-end 100 bp reads or an Illumina NextSeq500 to generate paired-end 76 bp reads (for the RNAi experiments).
Alignments
Total RNA strand-specific paired-end sequence data from D. melanogaster was aligned to the D. melanogaster genome (Release 5, dm3) lacking chromosome U extra, guided by the modENCODE annotation MDv1(ref. 5) using TopHat13 version 1.4.1 with the following settings: -p 8 -z0 -a 6 -m 2 --min-intron-length 28 -I 200000 -g 1 --library-type fr-firststrand -x 60 -n 2. The D. simulans, D. sechellia, D. yakuba, D. pseudoobscura, and D. virilis RNA-Seq datasets were aligned to the D. simulans (droSim1), D. sechellia (droSec1), D. yakuba (droYak2), D. pseudoobscura (dp4), and D. virilis (droVir3) genomes, respectively, using the same method and parameters, but without a reference transcriptome annotation. The human RNA-Seq datasets were aligned to hg19 using the same method and parameters and Gencode v19 as a reference annotation.
Computational identification of ratchet points
Parsing TopHat alignments to identify ratchet points
We identified sets of novel splice junctions from the TopHat alignments which share the same 5′ splice site. Ratchet point junctions were kept from a sample if the 3′ splice site of the junction is an AG|GT, the 3′ splice site was unannotated in the previous modENCODE annotation5, and the distance to the previous splice junction and the next splice junction is longer than 2 kbp.
de novo identification of ratchet points
We generated a set of potential ratchet point junctions by joining 95 nt of each exon to the 95 nt downstream of every unannotated 3′ splice site (AG|GT) in the downstream intron. We aligned reads 1 and 2 of the total RNA reads independently to the database of all possible ratchet point junctions using Bowtie14 version 0.12.7 with the following options: -v 2 -k 5 -M 5 --best. As the paired-end reads were aligned separately, post-processing was used to enforce constraints on gene-strand and alignment-strand that are a consequence of the stranded protocol. For each potential ratchet-point junction, we tabulated the coordinates of the genomic regions that comprise the ratchet-point junction sequences, the intron(s) and gene(s) the ratchet-site is derived from, the number of alignments to the ratchet-point junction, the average number of mismatches per alignment, detailed offset and mismatch information, the alignment offset entropy5, and the number of distinct offsets for only perfect alignments with >= 8 nt overhang. This latter parameter is intended to be a robust and conservative measure of alignment diversity. Ratchet points contained in introns that overlap no other distinct introns or any exons, were filtered to require >= 3 perfect alignments to 3 distinct offsets & overhang>=8 (or 2 perfect alignments to 2 distinct offsets & overhang>=8 AND >= 10 general alignments with <= 2 mismatch & overhang>=5). Ratchet points in introns that do overlap other introns or exons, were filtered using slightly more-restrictive criteria and required at least 5 zero-mismatch ratchet-junction alignments with distinct offsets).
Verification of potential ratchet points
We next compared the lists of potential ratchet points individually identified by analysis of the TopHat alignments and by alignment to all potential ratchet point junctions. This resulted in a list of 356 potential ratchet points that were then individually examined on the genome browser to verify their identity. To facilitate this analysis we merged the bedGraphs from all of the TopHat alignments into one positive- and one negative-strand-specific bedGraph file. For each intron containing potential rachet points, we calculated a robust linear regression of the read density of each segment of the recursive introns from the merged bedGraph profile using the robustfit feature of MATLAB. This required masking out repeatMasker regions and overlapping annotated features, both of which confound the regression process, and calculating the robust regression lines based on the remaining unmasked portions of the recursive segment. We used MATLAB to generate a “flip-book” of browser-like images of each potential recursive intron with bedGraph and local robust regression plots superimposed to aid in the manual inspection of each ratchet point for verification.
The merged bedGraphs were loaded into the genome browser along with tracks of all 356 potential ratchet point splice junctions. In addition, we loaded the FlyBase 5.45 annotation, which was the most recently version of FlyBase at the time of this analysis, as well as the most recent modENCODE annotation (MDv3)6 to identify ratchet points that corresponded to exons identified more recently than the modENCODE annotation (MDv1)5 used to seed the alignments. Finally, we loaded in the modENCODE CAGE data6 to assess whether any potential ratchet points corresponded to previously unannotated promoters, which could also give rise to a sawtooth pattern of RNA-Seq read density. Potential ratchet points were removed if the 3′ ratchet point junction corresponded to an annotated exon, of if the sawtooth pattern of read density was not apparent on the browser or from the local robust regression plots. During the course of this manual inspection we identified two ratchet points in luna and mbl that were not identified in this computational analysis. Both of these were present in introns that contained other computationally-identified ratchet points. We identified these based on their strong pattern of sawtooth read density in both he browser and the local robust regression plots and the fact that they had conserved AG/GT sequences at the ratchet point junctions. These were missed computational because they did not pass the stringent filters used. Both of these were experimentally validated and included in the analysis of all ratchet points. In total, the final list of ratchet points consists of 197 ratchet points (Supplementary Table 2). The same approach was used to manually review the putative ratchet points identified in the human RNA-Seq data.
Validation of recursive splice sites by RT-PCR
500 ng of total RNA isolated from S2 cells with Trizol was used to synthesize cDNA using SuperScript® II Reverse Transcriptase (RT) kits according to the manufacturers protocol. PCR amplification was performed using Phusion® High-Fidelity DNA Polymerase (NEB) according to the manufacturer’s instructions using specific primers with the following amplification program: 98°C for 3 min, followed by 40 cycles 98°C for 10 sec, 55°C for 30 sec, and 72°C for 20 sec. Ratchet points were first confirmed by gel electrophoresis followed by Sanger sequencing.
Validation of recursive splice sites by cross-species RNA sequencing
The coordinates of the D. melanogaster ratchet points, and 50 nt on either side, were lifted over to the D. simulans (droSim1), D. sechellia (droSec1), D. yakuba (droYak2), D. pseudoobscura (dp4), and D. virilis (droVir3) genomes using the UCSC liftover tool on galaxy. The TopHat alignments of the D. simulans, D. sechellia, D. yakuba, D. pseudoobscura, and D. virilis were searched for splice junction reads whose 3′ end mapped within the lifted over coordinates and which had an AG/GT at the recursive junction.
Identification of recursive lariat introns and branchpoint analysis
To generate potential junctions between the 5′ splice site and branchpoints of intron lariats, we used a custom perl script to fused the last 94 nt of an intron to the first 94 nt of the intron. We used 94 nt of each portion of the intron to enforce a minimum of a 6 nt overhang when aligning 100 nt reads. Because the precise location of the branchpoints are not know ahead of time, and because a previous study in human has shown that most known branchpoints occur between 18 and 35 nt upstream of the 3′ splice site15, we generated 100 possible lariat junctions by sliding a window of the 3′ end of the intron in the 5′ direction at 1 nt intervals, and fusing each to the first 94 nt of the intron. We generated the potential lariat junction databases for each segment of all recursive introns, as well as all possible permutations of these segments. For example, for an intron with two ratchet points, we generated potential lariat junctions for the first, second and third recursive segments as well as the introns from the first 5′ splice site to the second ratchet point (segments 1 and 2), from the 5′ splice site of the first ratchet point to the last 3′ splice site (segments 2 and 3), and from the first 5′ splice site to the last 3′ splice site (segments 1, 2 and 3).
Bowtie14 version 0.12.7 was used to generate an index of the potential recursive lariat junctions and to align all of the total RNA-Seq reads, where each mate pair was aligned separately, with the following parameters:-v 2 -p 8 --all —quiet. In total, 72,712 alignments were reported. These were then filtered for reads that mapped uniquely to the lariat junction database, then sorted by the number of nucleotides overlapping the lariat junction. Randomly selected reads with various extents of overlap were aligned to the D. melanogaster genome using BLAT to determine whether or not they mapped elsewhere in the genome. From this we determined that all reads that overlapped the lariat junction with fewer than 14 nt also aligned elsewhere in the genome. We therefore used BLAT to align all reads reported from the Bowtie alignment and discarded those which mapped elsewhere resulting in a total of 46 reads. We identified the approximate locations of the branch points based on the coordinates of the 94 nt segment of the 3′ end of the intron that the reads aligned to.
Validation of recursive lariats
Thirty-eight recursively spliced genes were selected for lariat analysis, choosing genes that were highly expressed in the nervous system. These genes contain 95 distinct segments corresponding to 52 ratchet points. To detect splice lariats for these ratchet points, “outward-facing” PCR primer pairs were designed to amplify through branch-points. The PCR primers contained overhangs so that Illumina clustering, indexing, and sequencing oligonucleotides could be added in a subsequent nested PCR. The same primers designed for ratchet point lariat amplification were also used in different combinations to attempt to amplify the branchpoint lariats that would be generated by skipping one or more ratchet points.
Total RNA was extracted from whole D. melanogaster (Bloomington Drosophila Stock Center strain #2057) using TRIZOL reagent (Invitrogen, Grand Island, NY), followed by cDNA synthesis using Superscript II (Invitrogen) primed with random hexamers. The primer pairs described above were used for the first-round PCR, after which products were visualized on an agarose gel. For 13 genes, no product was detected for any of the sub-introns targeted and these genes were not analyzed further. For the other 25 genes, which contained a total of 64 sub-introns, we obtained a PCR product for at least one sub-intron. For these 25 genes we also used the primers in combinations to amplify any potential lariats created if splicing skipped ratchet points in a total of forty-one combinations. Regardless of whether or not a product was visualized we prepared sequencing libraries from all reactions in an effort to capture any low-level amplicons. Nested PCR was performed to add the Illumina sequencing oligonucleotides to the first-round PCR products. The amplicon libraries were pooled, purified, and size selected to select amplicons between 300 to 1000 bases in length. The pooled amplicon library was then sequenced on an Illumina MiSeq using a V3, 600-cycle kit to produce 200 by 400 bp paired-end reads.
Reads were filtered to remove mis-primed sequences and then aligned to the target genes using BLAT. Mapped reads were manually reviewed on the genome browser and a lariat considered to be “confirmed” if a portion of a read aligned precisely to the 5′ splice site (ratchet point or exon) and to a second region within 100 bp upstream of the 3′ splice site (ratchet point or exon) in a continuous manner. This analysis confirmed 14 of the 64 recursive segments examined. Importantly, none of the primer combinations used to detect ratchet point skipping yielded any reads corresponding to a lariat. The few sequence reads that were obtained for these controls were PCR artifacts that primarily contained Illumina sequencing oligonucleotides and had no homology to the target genes.
Sequence logos were generated with WebLogo16.
Comparison of gene expression and recursive splicing
The total number of reads mapping to each ratchet point junction was tabulated for each library and then summed across biological and/or technical replicates for each biological sample. The recursive index was calculated as the number of ratchet point junction read per mapped reads per billion reads ((ratchet point junction reads/mapped reads) × 1,000,000,000). The RNA-seq data generated for this study is from total RNA, a mixture of mRNA, nascent RNA, and pre-mRNA. Due to the complexity of RNA types and intronic reads it is difficult to accurately quantitate gene expression levels when using most existing software. We therefore used the expression values calculated from the corresponding poly(A)+ RNA-Seq data that was previously generated5,6 from the same RNA samples. Comparisons of the recursive index and gene expression levels were performed using R (http://www.r-project.org). GO analysis was performed using Funcassociate 2.0(ref. 17).
Analysis of chromatin marks
Visualizations of chromatin marks at recursively spliced genes were generated using custom R scripts (http://www.r-project.org). We obtained ChIP-seq scores (http://encode-x.med.harvard.edu/data_sets/chromatin/) and Affymetrix tiling array gene expression scores (http://intermine.modencode.org/) generated from L3 larvae via the modENCODE projects. For each feature of gene architecture illustrated, mean ChIP-seq scores were calculated for non-overlapping bins of 200 bp in length.
As expected, transcription-associated marks were specific to genes actively transcribed in larvae (Supplementary Fig. 5b). At these active genes, we observed low levels of H3K4me3 near recursive splice sites compared to first exons (Supplementary Fig. 5b), which suggests that the saw-tooth patterns observed by total RNA-seq were not due to cryptic transcription initiation or unannotated promoters, but rather, co-transcriptional splicing. We also observed lower levels of H3K36me3 near recursive splice sites compared to downstream exons (Supplementary Fig. 5b). Since the degree of H3K36me3 has been shown to increase with each internal exon in humans18, the low levels of H3K36me3 seen at recursive splice may reflect the fact that recursive splices are typically located in 5′ introns, and thus, preceded by few internal exons (Supplementary Fig. 5c). Indeed, recursive splice sites were associated with high levels of H3K79me2 exons, which is typical of long 5′ introns in humans11.
Extended Data
Extended Data Figure 1. Two approaches for identifying recursive splice sites.

a, Identification of recursive splice sites by parsing alignments. RNA-Seq reads were mapped to the genome using TopHat in a manner that allowed for novel splice junctions to be predicted. The alignments were then parsed for splice junction reads where the 5′ splice site mapped to an annotated 5′ splice site, but the 3′ splice site was unannotated. b, de novo identification of recursive splice sites. A database was generated in which each annotated 5′ splice site was spliced to all AG/GT sequences in an intron that did not correspond to an annotated 3′ splice site. All RNA-Seq reads were aligned to this database and the alignments parsed to find cases where reads mapped perfectly with at least 3 distinct offsets and at least an 8 nt overhang.
Extended Data Figure 2. Characteristics of Drosophila ratchet points.
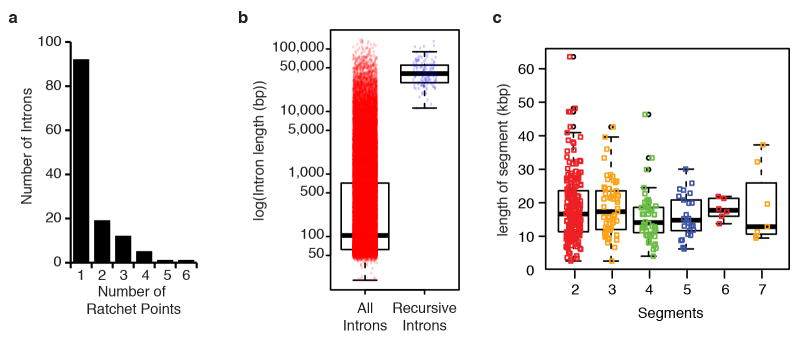
a, Distribution of the number of ratchet points per recursive intron. b, Size distribution (log10(bp)) of all (red) and recursive (blue) introns. c, Size distribution (in kbp) of the individual intron segments removed by recursive splicing binned by the number of segments per intron.
Extended Data Figure 3. RT-PCR validations of Drosophila recursive splicing events.

RT-PCR validation of ratchet points (red dots) from the indicated genes using primers in the upstream constitutive exon and flanking the putative ratchet points. The RP primers are expected to yield RT-PCR products if the constitutive exon is spliced to the ratchet point. The URP primers, which are upstream of each ratchet point, serve as negative controls. The identity of all RT-PCR products were verified by Sanger sequencing. Though the URP control RT-PCR reactions yielded a product for hppy RP1 and pum RP2, we were not able to generate sequence from them and therefore consider them to be amplification artifacts.
Extended Data Figure 4. Number of mapped reads per sample used for gene expression analysis.

Extended Data Figure 5. Chromatin marks associated with recursive splice sites.

a, Examples of chromatin marks at the luna gene locus, which contains 5 recursive splice sites (red triangles) within a single long intron. b, Heatmaps show relative ChIP-seq enrichment for H3K4me3 (top, red), H3K79me2 (middle, green), and H3K36me3 (bottom, blue), within 2 kb of the indicated gene features from 171 genes containing at least one ratchet point. Heatmaps are centered around gene features, which include the transcription start site of the first exon (First exon, arrow), the 5′ ss of the exon upstream of the recursive splice site (Upstream exon, black rectangle), the ratchet point (red triangle), the 3′ ss of the exon downstream of the recursive splice site (downstream exon, black rectangle), and the poly(A) site of the last exon (last exon, red octogon); the average exon of each gene feature is drawn to scale. Genes are sorted from top to bottom by decreasing expression level. For genes containing more than one ratchet point, the first, upstream, downstream, and last exons are represented multiple times. c, Histogram illustrating the intron positions the ratchet points reside in based on RefSeq annotations.
Extended Data Table 1. Summary of recursive intron lariats identified by directed RT-PCR and sequencing.
For each recursive lariats confirmed by RT-PCR and sequenced, the following information is provided: gene, segment, coordinate of the putative branchpiont, distance upstream of the 3′ splice the branch point is located, the sequence surrounding the branchpoint, whether the lariat intron is newly validated with respect to the total RNA analysis described in Supplementary Table 4a.
| Gene | Segment | Coordinate of putative branchpoint | Distance upstream of 3′ splice site | Sequence surrounding bp | Identified in Total RNA-Seq Data |
|---|---|---|---|---|---|
| Sdc | segment3 | 2R:17299944 | 21 | CATCTCACTCATAAATGTGTT | No |
| cpo | segment1 | 3R:13803044 | 34 | AAGGTAACTAATATGATTTTT | Yes |
| cpo | segment2 | 3R:13815266 | 31 | CCAAATGCTAATTTTATACTT | Yes |
| cpo | segment3 | 3R:13832619 | 37 | AGCAATCATCTAACGATTCTC | Yes |
| bun | segment2 | 2L: 12458256 | 32 | CAACATACTTACAGAACCTTT | No |
| CG7029 | segment2 | 3R:18592273 | 55 | GTTTGTGCTCACAGAGTCTGC | No |
| nuf | segment2 | 3L:14223246 | 29 | ATATAGACTTATCAGTTCTCT | No |
| CG31637 | segment2 | 2L:6525343 | 23 | GAGTATTCTAACAAGTTTCTC | Yes |
| dally | segment1 | 3L:8843654 | 26 | CTAAATCTGTGCTTAATTTCT | No |
| dally | segment2 | 3L:8855584 | 45 | AATTTGCACCATCGCATAACT | Yes |
| dally | segment3 | 3L:8870223 | 29 | ATCCAAGCTCATCTCCTCTTT | Yes |
| Mmp2 | segment2 | 2R:5503040 | 33 | TAGCATGCTGATATCATGTTT | No |
| osp | segment2 | 2L: 14656399 | 30 | AACCAAACTAATTTTTCTACC | No |
| osp | segment1 | 2L:14677529 | 41 | ACATCTTCTTACTAAATTATT | No |
Extended Data Table 2. Summary of Total RNA-Seq Data from ldbr and U2AF RNAi experiments.
For each datasets the following information is included: Sample, Total reads, Mapped reads (from TopHat alignments), % mapped.
| Sample | S2 Untreated | Idbr dsRNA | U2af38 dsRNA | U2af50 dsRNA | U2af38 & U2af50 dsRNA |
|---|---|---|---|---|---|
| Total reads | 183,451,394 | 146,237,470 | 174,626,770 | 162,012,182 | 179,417,376 |
| Mapped reads | 66,597,777 | 54,762,604 | 56,120,630 | 43,620,982 | 61,437,507 |
| % mapped | 36.30% | 37.45% | 32.14% | 26.92% | 34.24% |
| # junctions | 44,860 | 42,736 | 52,228 | 46,184 | 50,883 |
| junctions/1 M reads | 673.60 | 780.39 | 930.64 | 1058.76 | 828.21 |
| # total junc reads | 7,402,459 | 4,925,448 | 5,528,462 | 3,935,048 | 5,999,468 |
| % junction reads | 11.12% | 8.99% | 9.85% | 9.02% | 9.77% |
| # novel junction reads | 2,369,949 | 1,575,057 | 1,337,830 | 919,616 | 1,273,867 |
| % novel junctin reads | 3.56% | 2.88% | 2.38% | 2.11% | 2.07% |
| # annotated junc reads | 5,032,510 | 3,350,391 | 4,190,632 | 3,015,432 | 4,725,601 |
| % annotated junction reads | 7.56% | 6.12% | 7.47% | 6.91% | 7.69% |
| #novel junction cases | 22,688 | 20,977 | 29,211 | 24,279 | 28,193 |
| novel/total junctions | 0.51 | 0.49 | 0.56 | 0.53 | 0.55 |
| # annotated junc cases | 22,172 | 21,759 | 23,017 | 21,905 | 22,690 |
| annotated/total junctions | 0.49 | 0.51 | 0.44 | 0.47 | 0.45 |
| #RPs on both strands | 119 | 100 | 81 | 64 | 0 |
| RP/million reads | 1.79 | 1.83 | 1.44 | 1.47 | 0.00 |
| Total RP junc reads | 1449 | 985 | 377 | 259 | 0 |
| RP junc reads/million | 21.76 | 17.99 | 6.72 | 5.94 | 0.00 |
Extended Data Table 3. Summary of Total RNA-Seq Data from Related Drosophila Species.
For each datasets the following information is included: Species, Total reads, Mapped reads (from TopHat alignments), % mapped.
| Species | Total reads | Mapped reads | % mapped |
|---|---|---|---|
| D. simulans | 92,949,544 | 45,797,569 | 49.27% |
| D. sechellia | 38,674,020 | 19,974,783 | 51.65% |
| D. yakuba | 46,002,798 | 18,300,604 | 39.78% |
| D. pseudoobscura | 45,274,524 | 14,457,616 | 31.93% |
| D. virilis | 46,675,210 | 18,544,567 | 39.73% |
| Total | 269,576,096 | 117,075,139 |
Extended Data Table 4. Summary of ratchet points experimentally validated in other Drosophila species.
For each species analyzed, the following information is provided: Species, Total leftovers (the number of D. melanogaster ratchet point coordinates that were successfully lifted over to the other species genome coordinates), number of ratchet junction reads (the number of reads that mapped to the AG/GT sequence of the lifted over ratchet points), distinct ratchets (the number of distinct ratchet points identified by the mapped reads), % validated (the percent of lifted over ratchet points with at least one mapped read).
| Species | Total liftovers | number of ratchet junction reads | distinct ratchet points | % validated |
|---|---|---|---|---|
| D. sechellia | 163 | 93 | 74 | 45.40% |
| D. simulans | 166 | 106 | 87 | 52.41% |
| D. yakuba | 150 | 52 | 43 | 28.67% |
| D. pseudoobscura | 78 | 15 | 15 | 19.23% |
| D. virilis | 40 | 18 | 18 | 45.00% |
Extended Data Table 5. Summary of Total RNA-Seq Data from Human Tissues.
For each datasets the following information is included: Sample, Total reads, Total Aligned Reads, Uniquely Aligned Reads, % Uniquely Aligned.
| Tissue | Total Reads | Total Aligned Reads | Unique Aligned Reads | % Uniquely Aligned |
|---|---|---|---|---|
| adrenal-gland | 75,843,326 | 66,482,852 | 59,958,580 | 79.06% |
| brain-cerebellum | 70,868,098 | 64,382,088 | 60,087,066 | 84.79% |
| brain-whole | 75,684,718 | 63,391,490 | 59,015,902 | 77.98% |
| fetal-brain | 77,532,892 | 65,028,630 | 58,748,974 | 75.77% |
| fetal-liver | 69,092,806 | 47,109,928 | 38,162,898 | 55.23% |
| heart | 79,200,464 | 70,490,860 | 65,388,604 | 82.56% |
| kidney | 65,109,248 | 57,411,382 | 52,917,602 | 81.28% |
| liver | 67,086,550 | 58,934,448 | 50,433,480 | 75.18% |
| lung | 66,115,012 | 58,503,662 | 51,846,946 | 78.42% |
| placenta | 70,985,882 | 62,429,622 | 56,803,382 | 80.02% |
| prostate | 68,767,818 | 62,197,480 | 56,967,700 | 82.84% |
| salivary-gland | 77,811,154 | 73,634,924 | 55,954,594 | 71.91% |
| skeletal-muscle | 75,606,186 | 63,492,280 | 59,413,946 | 78.58% |
| small-intestine | 72,492,290 | 62,291,298 | 57,178,572 | 78.88% |
| spleen | 82,675,794 | 62,553,250 | 55,318,774 | 66.91% |
| stomach | 73,018,660 | 62,932,016 | 56,174,974 | 76.93% |
| thymus | 72,695,752 | 67,467,912 | 60,657,380 | 83.44% |
| thyroid | 75,794,702 | 68,200,264 | 63,259,560 | 83.46% |
| trachea | 70,890,274 | 58,922,766 | 53,022,564 | 74.80% |
| uterus | 72,876,872 | 61,974,024 | 56,835,240 | 77.99% |
| total | 1,460,148,498 | 1,257,831,176 | 1,128,146,738 |
Supplementary Material
Acknowledgments
This work was supported by NHGRI grant U54HG006994 to S.E.C. (PI) and B.R.G. (co-PI) and R01GM095296 to B.R.G.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions: B.R.G and S.E.C. supervised data production. S.O., A.O., M.B., and S.G. performed experiments. M.D., X.W., A.P., M.B., S.G., and B.R.G. performed computational analysis. B.R.G. wrote the paper with input from all authors.
Author Information: Sequences are available from www.encodeproject.org and the Short Read Archive under accession numbers SRP056962, SRP056965, and SRP056969 and those listed in Supplementary Table 1
The authors declare no competing financial interests.
References
- 1.Hatton AR, Subramaniam V, Lopez AJ. Generation of alternative Ultrabithorax isoforms and stepwise removal of a large intron by resplicing at exon-exon junctions. Mol Cell. 1998;2:787–796. doi: 10.1016/s1097-2765(00)80293-2. [DOI] [PubMed] [Google Scholar]
- 2.Burnette JM, Miyamoto-Sato E, Schaub MA, Conklin J, Lopez AJ. Subdivision of large introns in Drosophila by recursive splicing at nonexonic elements. Genetics. 2005;170:661–674. doi: 10.1534/genetics.104.039701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Conklin JF, Goldman A, Lopez AJ. Stabilization and analysis of intron lariats in vivo. Methods. 2005;37:368–375. doi: 10.1016/j.ymeth.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 4.Mackay TF, et al. The Drosophila melanogaster Genetic Reference Panel. Nature. 2012;482:173–178. doi: 10.1038/nature10811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Graveley BR, et al. The developmental transcriptome of Drosophila melanogaster. Nature. 2011;471:473–479. doi: 10.1038/nature09715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown JB, et al. Diversity and dynamics of the Drosophila transcriptome. Nature. 2014;512:393–399. doi: 10.1038/nature12962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oesterreich FC, Bieberstein N, Neugebauer KM. Pause locally, splice globally. Trends Cell Biol. 2011;21:328–335. doi: 10.1016/j.tcb.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 8.Hollins C, Zorio DA, MacMorris M, Blumenthal T. U2AF binding selects for the high conservation of the C. elegans 3′ splice site. RNA. 2005;11:248–253. doi: 10.1261/rna.7221605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sibley CR, et al. Recursive splicing in long vertebrate genes. Nature. 2015 doi: 10.1038/nature14466. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bieberstein NI, Carrillo Oesterreich F, Straube K, Neugebauer KM. First exon length controls active chromatin signatures and transcription. Cell Rep. 2012;2:62–68. doi: 10.1016/j.celrep.2012.05.019. [DOI] [PubMed] [Google Scholar]
- 11.Huff JT, Plocik AM, Guthrie C, Yamamoto KR. Reciprocal intronic and exonic histone modification regions in humans. Nat Struct Mol Biol. 2010;17:1495–1499. doi: 10.1038/nsmb.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kolasinska-Zwierz P, et al. Differential chromatin marking of introns and expressed exons by H3K36me3. Nat Genet. 2009;41:376–381. doi: 10.1038/ng.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taggart AJ, DeSimone AM, Shih JS, Filloux ME, Fairbrother WG. Large-scale mapping of branchpoints in human pre-mRNA transcripts in vivo. Nat Struct Mol Biol. 2012;19:719–721. doi: 10.1038/nsmb.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berriz GF, Beaver JE, Cenik C, Tasan M, Roth FP. Next generation software for functional trend analysis. Bioinformatics. 2009;25:3043–3044. doi: 10.1093/bioinformatics/btp498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tilgner H, et al. Nucleosome positioning as a determinant of exon recognition. Nature Structure & Molecular Biology. 2009;16:996–1001. doi: 10.1038/nsmb.1658. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.