

Abstract
The incorporation of cells and sensitive compounds can be better facilitated without the presence of UV or other energy sources that are common in the formation of biomedical hydrogels such as poly(ethylene glycol) hydrogels. The formation of hydrogels by the step-growth polymerization of maleimide- and thiol-terminated poly(ethylene glycol) macromers via Michael-type addition is described. The effects of macromer concentration, pH, temperature, and the presence of biomolecule gelatin on gel formation were investigated. Reaction kinetics between maleimide and thiol functional groups were found to be rapid. Molecular weight increase over time was characterized via gel permeation chromatography during step-growth polymerization. Swelling and degradation results showed incorporating gelatin enhanced swelling and accelerated degradation. Increasing gelatin content resulted in the decreased storage modulus (G’). The in vitro release kinetics of FITC-labeled dextran from the resulting matrices demonstrated the potential in the development of novel in situ gel-forming drug delivery systems. Moreover, the resulting networks were minimally adhesive to primary human monocytes, fibroblasts and keratinocytes thus providing an ideal platform for further biofunctionalizations to direct specific biological response.
Keywords: poly(ethylene glycol) hydrogel, gelatin, semi-interpenetrating network, Michael-type addition, cell adhesion
INTRODUCTION
Synthetic hydrogels are an important type of biomaterials with a wide range of applications in controlled drug delivery, implants, wound dressings, biosensors, and tissue scaffolds. In general, in situ forming hydrogels are prepared through various gelation methods, such as photopolymerization, ionic interaction, and chemical crosslinking.1-3 Among these methods, photopolymerization is one of the most widely used techniques, for example to fabricate poly(ethylene glycol) (PEG)-based hydrogels. However, photopolymerization requires the addition of highly toxic photoinitiators and the usage of UV light, which is clinically unfavorable. Thus, attentions have been focused on the chemical crosslinking method. The Michael addition is a facile reaction between nucleophiles and activated olefins and alkynes.4
The thiol-Michael addition to electron-deficient carbon-carbon double bonds carries many advantages such as requiring small amount of catalysts, displaying rapid reaction rates in benign reaction conditions (no heat or light needed)5, and the absence of byproducts. Functional groups that carry the reactive unsaturated structure include (meth)acrylates, maleimides, α,β-unsaturated ketones, fumarate esters, acrylonitrile, cinnamates, and crotonates.6 The thiol-Michael reaction has been extensively studied in small molecule synthesis, polymer modification, protein derivatization, tissue engineering scaffolds, and drug delivery devices.4,5 Hubbell and colleagues developed a series of degradable hydrogels for protein delivery formed via Michael-type addition reaction between acrylated star PEG polymer and dithiol.7-9 Yuan and Kopećek et al reported an enzyme-based hybrid hydrogel synthesized from maleimide terminated four-arm PEG and adenylate kinase via thiol-maleimide addition.10 Hyaloronic acid based hydrogels were also synthesized via thiol-Michael reaction for stem cell differentiation11,12, cartilage repair13, and bone regeneration14. The mild reaction condition, the rapid cure and high conversions under physiological environment are of great significance for biological applications such as the incorporation of bioactive macromolecules and cells.
In this study, we demonstrated a step-growth polymerization via thiol-maleimide (SH-Mal) Michael-type addition in the formation of a physically entangled PEG network under mild aqueous conditions. The reaction is conducted between two linear PEG polymers modified with different end-functional groups. When the polymerization degree increases and the polymer chain concentration reaches a critical value, the polymer coils diffuse in each other and get tied into each other.15 Therefore, the gel formation is not due to the formation of crosslinks but rather the physical chain entanglements, which makes it an interesting system different from classical crosslinked PEG hydrogels. The effects of macromer concentration, pH, temperature, and the presence of biomolecule gelatin on gel formation were investigated. The storage and loss moduli (G’ and G”) were evaluated by small strain oscillatory shear experiments to gain insight into the structure and mechanical properties of the newly formed hydrogel network. Reaction kinetics between macromer terminated maleimide and thiol functional groups were characterized. In our previous studies, gelatin was incorporated into the growing PEG networks to form a semi-interpenetrating network (sIPN) and provided a substrate for biofunctionalization, i.e. the gelatin backbone was conjugated with bioadhesive peptide sequences such as Arg-Gly-Asp and Pro-His-Arg-Ser-Asn tethered by a heterbifunctional PEG linker.16 The presence of labile gelatin also provides a system to explore the extent of macromolecular thiol-maleimide reaction in a heterogeneous mixture of biomolecules.
MATERIALS AND METHODS
Materials
Bihomofunctional poly(ethylene glycol)-maleimide (PEG-(Mal)2, Mw ~2000) was purchased from Nanocs (NY, USA). Poly(ethylene glycol)-diol (Mw, ~2000), Fluorescein isothiocyanatedextran (FD-40, Mw ~40,000), gelatin (Type A, ~175 bloom), triethylamine (TEA), triethanolamine (TEOA), p-toluenesulfonyl chloride (TsCl), lithium aluminum hydride (LiAlH4), and sodium hydrosulfide (NaSH) were obtained from Sigma-Aldrich (USA). Dichloromethane (CH2Cl2), tetrahydrofuran (THF), toluene, diethyl ether, and anhydrous magnesium sulfate (MgSO4) were purchased from Fisher Scientific (USA). Polyethylene glycol diacrylate (PEGdA) was synthesized from polyethylene glycol (PEG; MW 2000; Sigma-Adrich) following an established procedure.17 All chemicals were used as received unless otherwise specified.
Synthesis of PEG-dithiol
PEG dithiol was synthesized via a two-step reaction per a previously described procedure with modifications (Fig. 1).18 10.0 g of PEG diol (Mw 2000, 5 mM) was dissolved in 50 mL of dry CH2Cl2. 7.2 g of TsCl was dissolved in 20 mL of CH2Cl2 and added dropwise to the PEG-diol solution while stirring. 1.4 mL of TEA (10 mM) was subsequently added to the mixed solution. The reaction was kept at room temperature and stirred overnight. The crude mixture was filtered twice, concentrated via rotavap, dissolved in 50 mL of toluene, and filtered. Filtrates were collected and precipitated in cold diethyl ether while stirring. The precipitates were collected and dried under vacuum. PEG-(Tos)2: 9.6 g, yield 96%. 1H NMR (CDCl3): δ2.45, s, CH3-aromatic; δ 3.65, m, PEG backbone; δ7.5, d, 2Hs ortho to sulfonate; δ7.8, d, 2Hs meta to sulfonate.
FIGURE 1.

Synthetic scheme of PEG dithiol. Adapted from Ref 18.
PEG-(Tos)2 (5 g, 2.3 mM) dissolved in 100 mL of ddH2O was treated with NaSH hydrate (1.4 g, ~ 25 mM). The reaction was stirred for 5 h at room temperature and then 1 h at 60 °C. The crude mixture was neutralized with concentrated H2SO4, and extracted with CH2Cl2. The organic layer was collected and dried over anhydrous MgSO4 and precipitated in dry cold diethyl ether. The precipitated product was collected and dried in vacuum overnight (2.8 g, yield 56%). 1H NMR (D2O): δ2.68, t, −SH; δ2.76 −OCH2CH2SH; δ2.92, t, −OCH2CH2SH; δ3.65, m, PEG backbone. The products obtained from the above reaction were treated with LiAlH4 in 20 mL of dry THF for 1 h and then 20 mL of ddH2O was added to stop the reaction. Reduced PEG-(SH)2 was extracted with CH2Cl2, dried with anhydrous MgSO4, and precipitated in cold diethyl ether. The final products were dried in vacuum overnight and stored in argon at −20 °C. (WARNING: this reaction is extremely dangerous to perform due to the generation of H2S during neutralization. Alternative synthesis method is to be used in future work using potassium thioacetate instead of NaSH.19)
PEG hydrogel and sIPN formation
For the preparation of gels, PEG-(SH)2 and PEG-(Mal)2 were dissolved in various buffer solutions in separate glass vials. Polymer weight percent (wt%) was varied by varying the total amount of solvents (Table I). Then, the two solutions were quickly mixed via vortexing (~30 s) to form the network (Fig. 2). The time of gelation was determined by the test tube inverting method, i.e. observation of the stop of flow when inverting the vial horizontally.20 pH effect was investigated using pH6.5 TEOA PBS, pH7.5 TEOA PBS, rand pH8.0 TEOA PBS as the solvent for PEG-(SH)2, while pH7.4 PBS was used to solvate PEG-(Mal)2 (Table II). Temperature effect on formulation with 18.4% total polymer was also investigated at 25 °C and 37 °C. Gelatin solutions of various concentrations were prepared at 37 °C with PEG-(SH)2 and PEG-(Mal)2 (Table III). Prior to mixing, all solutions were placed in the 37 °C water bath. The gelatin solution was quickly mixed with PEG-(SH)2 solution and subsequently mixed with PEG-(Mal)2 solution via vortexing. The fully mixed prepolymer solution was placed in a 37 °C incubator to cure.
Table I.
Various macromer concentrations employed in PEG hydrogel formulation
| Formulation # | PEG-(SH)2 (g) | PEG-(Mal)2 (g) | pH 7.5 PBS w/0.3M TEOA | pH 7.4 PBS (μl) | Total polymer % w/w | Gelation time |
|---|---|---|---|---|---|---|
| 1 | 0.05 | 0.042 | 100 | 100 | 31 | ~ 40 min |
| 2 | 0.05 | 0.042 | 200 | 200 | 18.4 | ~ 90 min |
| 3 | 0.05 | 0.042 | 300 | 300 | 13 | > 20 h |
FIGURE 2.

Synthetic scheme of poly(ethylene glycol) hydrogel via macromeric thiol-maleimide addition.
Table II.
pH effect on gelation time
| pH | PEG-(SH)2 (g) | PEG-(Mal)2 (g) | PBS w/ 0.3M TEOA | Gelation time |
|---|---|---|---|---|
| 6.5 | 0.05 | 0.042 | 400 | > 24 h |
| 7.5 | 0.05 | 0.042 | 400 | ~ 70 min |
| 8.0 | 0.05 | 0.042 | 400 | ~ 90 min |
Table III.
Various gelatin concentration employed in the sIPN formation
| # | PEG-(SH)2 (g) | PEG-(Mal)2 (g) | Gelatin (g) | 1× PBS (ml) | pH 7.5 TEOA (ml) | Total polymer w/w% | Gelation Time |
|---|---|---|---|---|---|---|---|
| 1 | 0.05 | 0.042 | 0.02 | 0.4 | 0.2 | 15.7 | − (*) |
| 2 | 0.05 | 0.042 | 0.03 | 0.4 | 0.2 | 17 | ~ 1 d |
| 3 | 0.05 | 0.042 | 0.06 | 0.4 | 0.2 | 20 | ~ 1 d |
formulation #1 showed slight fluidity after 3 days of curation.
Fourier transform infrared spectroscopy (FTIR)
FTIR spectra were recorded on a Bruker Equinox 55 ATR-FTIR spectrometer (single reflection, ZnSe crystal, 20 scans per spectrum) in a N2 atmosphere. Samples were dissolved in THF to minimize the influence of water on the absorption spectrum and spectra were recorded at predetermined time points up to 20 min. A decrease in the IR absorption peak at 695 cm−1 was observed and attributed to the =C-H bending vibration of the maleimide group. The conversion of C=C bonds (=C-H bend, 695 cm−1) on the maleimide group was determined using C=O stretch (1724 cm−1) as the internal reference. Absorption peaks at 695 cm−1 and 1724 cm−1 were integrated using OPUS version 4.2 software provided with the spectrometer. The degree of reacted maleimide groups at a specific time was calculated from the peak area, A. Thus, the conversion of unsaturated maleimide groups, α, was calculated by the following equation:
| Eq 1 |
where Ai is the area of the vibration band at time t.21
Gel permeation chromatography (GPC)
The GPC analysis was performed to characterize the step-growth polymerization over time during hydrogel formation. The chromatographic system consisted of a single phase HPLC pump (Waters 1515) with a column heater and a refractive index detector (Waters 2414). Separation was accomplished using three TSK-GEL columns (Sigma-Aldrich) in series: G5000PWXL (pore size = 1000 μm, 30 cm × 7.8 mm), G3000PWXL (pore size = 200 μm, 30 cm × 7.8 mm), G2500PWXL (pore size < 200 μm, 30 cm × 7.8 mm). The system was calibrated using a series of PEO/PEG standards (232 – 932,000 Da). All samples were run at 0.7 mL/min at 37 °C in a mobile phase consisting of 80% (v/v) 0.1 M NaNO3 with 20% (v/v) acetonitrile. Chromatogram analysis was performed using PeakFit software (v4.12, SeaSolve Software, Inc.) to fit Gaussian peaks based on features within the chromatorgram. The integral area of each peak was determined and the percent area under the curve was then calculated. Samples subject to GPC analysis were prepared in a diluted condition, i.e. 0.05 g PEG-(SH)2 and 0.042 g PEG-(Mal)2 were dissolved in 2 mL of 1X PBS and allowed to react for 20 h, 2 d and one month. The reactants were diluted with the eluent to about 2 mg/mL and filtered through 0.22 μm membrane filter (Millipore, USA) before analysis.
Physical characterization: swelling, surface hydrophilicity and rheological characterization
The hydrogel and sIPN samples via thiol-maleimide addition were synthesized and dried in vacuum overnight. Photopolymerized PEG hydrogel and sIPN with 10% gelatin were prepared as previously described16 and tested for comparison. The dried gels were accurately weighed prior to the swelling study. The gel disks were then immersed in 10 mL of pH 7.4 PBS at 37 °C. At predetermined time points, hydrogels were carefully removed from the petri dishes, blotted with Kim-wipes® to remove the excess water on the hydrogel surfaces, and weighed. Equilibrium weight swelling ratio (Qs) was defined as follows:
| Eq 2 |
where wt is the gel weight at the time t, and wd is the dried gel weight. The hydrogel surface hydrophilicity was quantified with a modified, computerized, underwater air-captured surface contact angle system (VCA2500; AST products, Inc. Billerica, MA). Fully swollen hydrogels were secured to a glass slide with Superglue® and placed in a water chamber. Air bubbles were placed on the exposed and submerged surface of the hydrogel via syringe. Two angles per bubble for three bubbles were measured for each sample to obtain the static contact angle.
Storage modulus (G′) and loss modulus (G″) on nonswollen and swollen gels were measured with an ARES-LS2 2000ex rheometer (TA Instruments) in parallel plate geometry with a 8 mm diameter plate at 25 °C ± 0.1 °C, a frequency range of 0.1 - 10 Hz, and a constant 5% strain. Hydrogel samples were prepared to yield discs of 8 mm diameter and 1 mm thickness. Swollen samples were equilibrated in 1× PBS overnight and cut with razor blade to yield discs of 8 mm diameter and 1 mm thickness. The water from the hydrogel surface was carefully blotted before measurement and each sample was covered with a thin layer of silicone oil to minimize water evaporation effect. Samples include PEG SH-Mal hydrogel and PEG SH-Mal sIPN (Table IV).
Table IV.
(i) PEG SH-Mal hydrogel formulations for rheological characterization
| Formula | PEG-(Mal)2 (g) | PEG-(SH)2 (g) | 0.3M TEOA/PBS (μL) | Total polymer weight (w/w%) |
|---|---|---|---|---|
| 1 | 0.042 | 0.05 | 100 | 48 |
| 2 | 0.042 | 0.05 | 138 | 40 |
| (ii) PEG SH-Mal sIPN formulations rheological characterization | |||||
|---|---|---|---|---|---|
| Formula | PEG-(Mal)2 (g) | PEG-(SH)2 (g) | Gelatin (g) | 0.3M TEOA in PBS (μL) | Total polymer weight (w/w%) |
| 1 | 0.042 | 0.05 | 0.002 | 140 | 40.5 |
| 2 | 0.042 | 0.05 | 0.004 | 180 | 35.3 |
In vitro controlled release
FD-40 (Mw, ~40 KDa) was selected as the model solute for in vitro release study. The stock solution was prepared by dissolving FD-40 in pH7.4 PBS at a concentration of 2 mg/mL. FD-40 was loaded prior to gelation, and two PEG SH-Mal formulations were included, i.e. 13% total polymer and sIPN with 20% total polymer. After complete polymerization, each disk was placed in 5 mL of pH7.4 phosphate buffer saline at 37 °C. At given time points, 1 mL of release sample was collected and replaced with 1 mL of fresh media. The concentration of FD-40 release samples were determined by fluorescence spectroscopy at an excitation wavelength of 485 nm and an emission wavelength of 520 nm. FD-40 showed a linearity range between 3.9 – 62.5 μg/mL (Y=468.86X+1617.3, R2 = 0.9916).
Cell culture and adhesion assay
Primary monocytes were isolated from healthy human volunteers using an established density gradient non-adhesion method with modifications.22 Cells were statically seeded on various hydrogel samples in tissue culture polystyrene (TCPS, Becton Dickinson Labware, NJ) wells at a concentration of 106 cells/mL suspended in RPMI 1640 media with 10% autologous serum. Neonatal human dermal fibroblasts were obtained from Lonza (NJ, USA) and cultured in fibroblast growth medium-2 (FGM-2, Lonza) containing FBM, 0.1% insulin, 0.1% recombinant human fibroblast growth factor-B, 0.1% GA-1000, and 2% fetal bovine serum (FBS; Atlantic Biologics, Miami, FL, USA). Fibroblasts for adhesion assay were derived from the same cell stock at passage 3 at a seeding density of 4 × 104 cells/mL. Normal human epidermal keratinocytes at passage 3 from neonatal foreskin (NHEK; Lonza, Allendale, NJ, USA) were cultured in keratinocyte growth medium (KGM; Lonza) supplemented with 5% FBS and seeded at a concentration of 4 × 104 cells/mL. All cell cultures were maintained at 37 °C and 5% CO2.
Samples were cold-sterilized with 75% ethanol per standard procedures and equilibrated in culture medium prior to cell seeding. At 2, 24, 96, and 168 h thereafter, samples were washed twice with respective culture medium to remove non-adherent cells, and resupplied with fresh culture medium and allowed to further incubate. TCPS was employed as a positive surface control, and photopolymerized PEG hydrogel was used as an additional surface comparison. Adherent cells on all surfaces were imaged using a computer-assisted video analysis system (Metamorph v4.1) coupled to an inverted microscope (Nikon, Eclipse TE300). The adherent cells were quantified and expressed as number of cells/mm2 surface area. Five images per sample were taken at random fields of view. Adherent cell density was determined per mm2 surface area using the image analysis software ImageJ (NIH). Additionally, Live/dead® stain (4 μM calcein AM/4 μM EthD-1, Invitrogen) was used to evaluate fibroblast and keratinocyte viability and morphology.
Cytotoxicity via MTT assay
Hydrogel extracts were prepared by adding PEG SH-Mal hydrogel fragments to specific culture medium at a concentration of 10 mg/mL and incubated at 37 °C for 24 h.23 After this period, the hydrogel extract was sterilized via filtration (0.22 μm, Millipore, USA). Primary human monocytes, neonatal human dermal fibroblasts, and normal human epidermal keratinocytes were used to evaluate the cytotoxic effects of hydrogel extracts. 100 μL of the cell suspension (Monocytes, 5 × 104 cells/well; Fibroblasts, 3000 cells/well; Keratinocytes, 8000 cells/well), were seeded in flat-bottomed 96-well micro-culture plates (Costar, USA). The 96-well plates were incubated at 37 °C in a 5% CO2 humidified incubator. After reaching 70 - 80% confluence, the medium was discarded and replaced with 100 μL of hydrogel extract. The serum free culture medium was used as negative control. Samples and controls were tested in sextuplet. These plates were then incubated for 24 and 48 h, respectively, before the viability was determined via standard MTT assay. At the end of culturing time, 20 μL of MTT solution (5 mg/mL) was added to each well. After 4 h incubation at 37 °C in a 5% CO2 humidified incubator, supernatant was removed carefully and 100 μL of dimethyl sulfoxide (DMSO) was added into each well to dissolve the intracellular blue formazan product. The optical density of each well was read using a microplate reader (Bio-Tek Elx800) at 570 nm with a reference wavelength of 650 nm. The percentage cell viability of samples compared to the control was calculated from the following equation:
Statistical analysis
All data are represented as mean ± standard deviation (S.D.) of samples in at least three independent experiments. Cell density data were analyzed by unpaired Student-t test using SigmaStat version 2.03. A value of p < 0.05 was considered statistically significant.
RESULTS AND DISCUSSIONS
Formation of PEG hydrogel and sIPN via thiol-maleimide Michael-type addition
Combining aqueous solutions of PEG-(Mal)2 and PEG-(SH)2 resulted in the formation of an entangled hydrogel system (Fig. 2). The effects of buffer system, macromer concentration, and reaction temperature were explored. Gelation time ranged from minutes to days (Table I and II) and was significantly affected by pH (Table II). More rapid gel formation was observed under basic conditions, most likely due to the favored condition for thiol groups to form nucleophiles.24
However, the rates of thiol-Michael reaction and oxidation, i.e. the formation of disulfide products, were both sensitive to pH.25 With an increase in pH value, oxidization was accelerated and the content of free −SH decreased.26 Therefore, pH7.4 phosphate buffer saline and 0.3M TEOA were selected as the solvent for further studies on the macromer concentration effect and sIPN formation studies. Increasing macromer concentration led to a significant decrease in the gelation time, which might create more polymer entanglements during gel formation. While macromer concentration had a significant influence on gelation time, temperature effect was less significant between room temperature and 37 °C (data not shown). The presence of gelatin perturbed the macromeric thiol-maleimide reaction and the subsequent gelation time (Table III). Particularly, the presence of 3.3% w/v gelatin completely abrogated gelation after several days at 37 °C and room temperature. As shown in Table II, PEG hydrogel of the same macromer concentration (13% w/w) showed complete gelation after 20 h. 3% w/v gelatin solution gelled within minutes at room temperature.27 However, the incorporation of gelatin into PEG networks at the concentration of 3.3% w/v displayed sol-gel properties different from either single component, implying that the formation of semi-interpenetrating network created a dynamic gel network at room temperature. Though gelatin solutions with concentrations ranging from 3% - 10% w/v remained in a solution form at 37 °C, more physical entanglements might be created within PEG networks during sIPN formation that led to the gel formation at 37 °C. Thus, a higher gelatin concentration may contribute to the complete gelation of sIPN at 37 °C.
PEG-maleimide and PEG-thiol reaction kinetics and network physical properties
The conversion of PEG terminal maleimide groups was monitored by the characteristic absorption bands at 1724 cm−1 (ν, C=O in maleimide) and 695 cm−1 (ν, =C-H in maleimide) (Fig. 3). The decrease of the latter band was due to the addition reaction between –SH and –C=C– group. In the present study, the area under peaks was integrated and the 1724 cm−1 peak was used as the internal reference, which provided a kinetic conversion profile for the PEG terminal thiol- maleimide addition reaction. As shown in Fig. 3, the conversion of the C=C group reached steady state around 10 min, indicating a near complete conversion of the C=C group.
FIGURE 3.
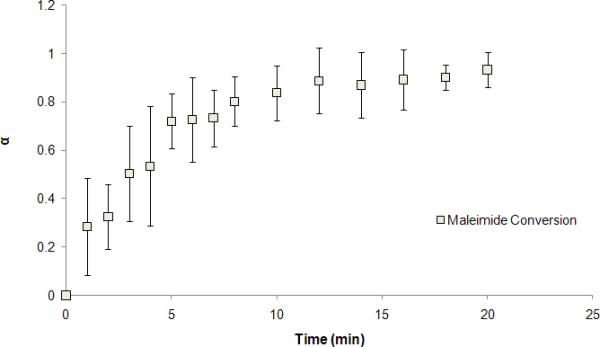
PEG terminal maleimide conversion over time in THF via thiol-maleimide addition.
GPC chromatograms (Fig. 4A) showed a wide molecular weight distribution of the polymer products via thiol-maleimide addition over the time range from 20 h to 1 month. Peak integration analysis and molecular weight calibration revealed a major peak shift towards the higher molecular weight (Table V). In addition, the higher polymer concentration will also likely result in a higher degree of conversion further increasing the step-growth molecular weight increase. The hydrogel samples subject to swelling study were proven to degrade to small molecular weight products after about six months (Fig. 4B). The degradation is mostly likely due to the hydrolysis of maleimide- in the polymer. Taken together, the network formation that was observed in the study is due to the step-growth of PEG polymer chains that resulted in chain entanglement and hydrogel formation.
FIGURE 4.

GPC Chromatograms of PEG polymers via thiol-maleimide addition obtained at 20 h, 2 d, and 1 month (A) and degraded PEG SH-Mal hydrogels (B).
Table V.
Estimated molecular weight of major species in the chromatogram (Fig. 4)
| Reaction time | Peak retention time | %AUC | Mw (approx) |
|---|---|---|---|
| 20 h | 30.00 | 18.1 | 25911 |
| 31.80 | 21.4 | 11319 | |
| 34.20 | 23.5 | 4085 | |
| 2 d | 26.00 | 13.1 | 247753 |
| 30.20 | 11.9 | 23540 | |
| 35.20 | 7.4 | 2714 | |
| 1 month | 25.01 | 13.6 | 499950 |
| 29.95 | 74.2 | 26545 | |
| 35.09 | 11.2 | 2839 | |
PEG SH-Mal hydrogels synthesized via thiol-maleimide addition displayed consistent and increasing swelling through 7 days, with a maximum average weight swelling ratio of (23 ± 3) (Fig. 5). In comparison, photopolymerized PEG hydrogels displayed a significantly lower maximum average weight swelling ratio of (5.4 ± 0.7) (p < 0.05). No significant mass loss of PEG SH-Mal hydrogel was observed during a 7-day period, indicating the relative robustness of the hydrogel structure. However, the amide linkage is likely to undergo hydrolysis over a long time period, and complete degradation was observed during six-month period as mentioned previously. The weight swelling ratio of PEG SH-Mal hydrogel reached a steady state around 48 h, which indicated an equilibrium swelling state of the hydrogel. However, photopolymerized PEG hydrogel reached the steady state at a more rapid swelling rate around 2 h. Compared to photopolymerized PEG hydrogels, the greater swelling ratio and more hydrophilic surface properties of PEG SH-Mal hydrogels are probably due to the less rigid polymer chain entanglements formed via thiol-maleimide addition. The incorporation of gelatin in gels resulted in different swelling and degradation properties. sIPN SH-Mal with 10% gelatin showed a maximum average swelling weight ratio of (18 ± 1) around 4 h (Fig. 5), while SH-Mal sIPN with 5% gelatin reached maximum average swelling weight ratio of (15 ± 3.4) in about 24 h (Fig. 5). Thus, increasing gelatin concentration may potentially increase the swelling ratio. However, the maximum weight swelling ratio is not significantly different between SH-Mal sIPN and PEG SH-Mal hydrogel (p > 0.05). The photopolymerized sIPN showed significantly smaller maximum weight swelling ratio than photopolymerized hydrogel (p < 0.05). Gelatin has such sol-gel transformation properties related to temperature changes, and thus it is expected that gelatin incorporated in the semi-interpenetrating network may undergo dissolution at 37°C thus creating more free volumes within the polymeric matrices that would contribute to the enhanced swelling effect. As shown in Fig. 5, sIPNs synthesized via either thiol-maleimide addition or photopolymerization showed mass loss after 24 h, and the PEG SH-Mal sIPNs became physically unstable to handle for measurement after 48 h.
FIGURE 5.

Equilibrium weight swelling ratio of PEG hydrogels and sIPNs via thiol-maleimide addition and photopolymerization over time. (n=3)
In the underwater air-captured surface contact angle analysis, a larger surface contact angle indicates a higher surface hydrophilicity.28,29 PEG hydrogels synthesized via click chemistry showed a significantly larger surface contact angle than photopolymerized PEG hydrogels (p < 0.05), indicating a higher degree of hydrophilicity (Table VI). The surface hydrophilic nature of the material is associated with the surface structure. As is discussed in the previous section, hydrogels synthesized via thiol-maleimide addition may result in more dangling ends compared to crosslinked gel system. Thus, less stiff surface structure is expected compared to photopolymerized PEG hydrogel. The observation is in good agreement with empirical rules, i.e. the more stiff the gel, the higher the hydrophobicity, and the less degree of the permeability and gel swelling.30,31
Table VI.
Underwater air-captured surface contact angle
| Formulation | Contact angle (Degree, mean ± S.D.) | |
|---|---|---|
| Left | Right | |
| PEG Mal-SH hydrogel (18%, w/w) | 152 ± 1.9 | 152 ± 2.6 |
| PEG Mal-SH + gelatin sIPN (17%, w/w) | 159 ± 4.2 | 160 ± 3.5 |
| Photopolymerized PEG hydrogel (10%, w/w) | 141 ± 4.4 | 141 ± 4.9 |
n=3, with three air bubbles on each sample with two angles per air bubble.
PEG hydrogels and sIPNs were subject to dynamic frequency sweep to gain insight into the mechanical characteristics and structure of the physically entangled network. The storage modulus (G’) and loss modulus (G”) were monitored over frequency ranges from 0.1 to 10 Hz at 25 °C. PEG SH-Mal hydrogels with G’ values independent of the tested frequencies were proven to have the characteristic viscoelastic properties of gels (Fig. 6A). Results also showed significantly higher storage modulus (G’) than loss modulus (G”) for all formulations, indicating that these hydrogels are highly elastic. Non-swollen PEG SH-Mal hydrogel showed much higher G’ (~5.8 KPa) and G” (~2.6 KPa) at the polymer concentration of 48% than at 40%, indicating higher polymer concentration corresponds to higher storage modulus (Fig. 6). However, there are no significant differences in storage modulus (G’) between different formulations in the swollen hydrogels (Fig. 6A). The similar storage modulus values may be due to the high water capacity of these hydrogels, which could be a specific characteristic of such non-crosslinked physical entanglements. Additionally, the storage modulus (G’) value was decreased by up to 10-fold after equilibrium swelling. Increasing gelatin content resulted in the lower storage modulus (G’) and lower loss modulus (G”) (Fig. 6C). Unlike swollen PEG SH-Mal hydrogels that displayed no significant differences in storage modulus (G’) (Fig. 6B), swollen PEG SH-Mal sIPNs with lower gelatin content showed slightly higher storage modulus (G’) (Fig. 6D), indicating incorporating gelatin into the PEG matrix creates more structural complexities to the current system. In summary, the storage modulus of PEG SH-Mal hydrogels/sIPNs can be adjusted by varying total polymer concentration and gelatin concentration.
FIGURE 6.
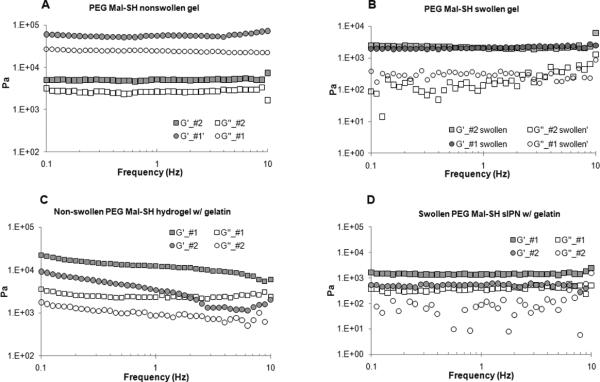
Storage modulus (G’) and loss modulus (G”) at 25 °C as a function of frequency for non-swollen PEG SH-Mal hydrogels (A), swollen PEG SH-Mal hydrogels (B), non-swollen PEG SH-Mal sIPNs (C), swollen PEG SH-Mal sIPNs (D).
In vitro controlled release kinetics
The release kinetics of FD-40 showed a biphasic release profile (Fig. 7). FD-40 release from PEG SH-Mal hydrogel displayed a significant burst release with near 80% cumulative release at the 1-h point, which may be due to the great swelling effect of click hydrogels and the rapid release of surface associated solutes. In contrast, PEG SH-Mal sIPN showed a less significant burst effect. A near zero-order release was observed from 0 to 4 h, which corresponded well with previous release studies on the release kinetics of photopolymerized sIPN-based delivery systems.32 Although swelling results showed comparable maximum weight swelling ratio between click hydrogel and sIPN, the different release kinetics of FD-40 from both formulations indicated that the incorporation of gelatin greatly delayed the FD-40 release by potentially creating more physical entanglements thus more hydrodynamic hindrance to solute diffusion. It is expected that the release kinetics can be tailored by varying the amount of gelatin incorporated in the network as well as the PEG-to-gelatin weight ratio.
FIGURE 7.

In vitro release kinetics of FD-40 from PEG SH-Mal hydrogel and PEG SH-Mal sIPN with 10% gelatin at 37 °C.
Cell adhesion and cytotoxicity
Adherent monocyte density was significantly different amongst material surfaces. The monocyte morphology on TCPS remained relatively round up through 96 h but was larger with more cytoplasmic spreading by 168 h (Fig. 8). Monocytes on PEG SH-Mal hydrogels remained round through 168 h with a slightly noticeable increase in size by the end of the culture. Additionally, adherent monocytes on the PEG SH-Mal hydrogel were in small clusters throughout the culture duration. Monocytes on the photopolymerized PEG hydrogels increased most dramatically in size by 96 h and had been shown in an earlier study to be most phenotypically different than TCPS.17 Adherent density on TCPS decreased most dramatically with the largest decrease between 24 and 96 h (Fig. 9A). Adherent density on PEG SH-Mal hydrogels remained relatively constant through the culture with a slight decrease between 24 and 96 h (Fig. 9A). Adherent density on photopolymerized PEG hydrogels decreased over time but to a lesser degree than on TCPS (Fig. 9A), which was consistent to our previous studies.17 By 24 h, TCPS and photopolymerized PEG hydrogels supported similar levels of adhesion (p > 0.05) whereas at 96 h and 168 h photopolymerized PEG hydrogels had significantly higher levels of adhesion than either of the other two materials tested (p < 0.05). This sustained adhesion may be due to the limited and selective protein adsorption mediated by the hydrophilic hydrogel. Extensive studies had shown that protein molecules selectively adsorb onto hydrophobic surfaces via hydrophobic interactions33 and as a result, the adsorption of adhesion mediating proteins can potentially promote cell adhesion onto more hydrophobic surfaces. Thus, the adhesion results might imply the relative surface hydrophilicity in the following order: TCPS < Photopolymerized PEG hydrogel < PEG SH-Mal hydrogel, which is in good agreement with the contact angle measurement.
FIGURE 8.

Optical micrographs of adherent monocytes on PEG SH-Mal hydrogel (A, B), photopolymerized PEG hydrogel (C, D), and TCPS (E, F) at 24 h (A, C, E) and 7 d (B, D, F) (20× magnification).
FIGURE 9.

Adherent cell density on TCPS, photopolymerized PEG hydrogel and PEG SH-Mal hydrogel. Three cell types were included, primary human monocyte (A), keratinocyte (B), and fibroblast (C). Cells were observed at 10× magnification. Five images per sample were taken at random fields of view. †, p < 0.05 vs. the cell adhesion density on TCPS at the same time point; ‡, p < 0.05 vs. the cell adhesion density on photopolymerized PEG hydrogel at the same time point. All data presented as average ± standard deviation (n=3).
Adherent keratinocyte and fibroblast densities on TCPS increased between the 2, 24, and 96-h time points (Fig. 9B, 9C). In contrast, adherent cell density on photopolymerized PEG hydrogels and PEG SH-Mal hydrogels remained significantly lower than on TCPS through the course of the experiment. At 96 h, photopolymerized PEG hydrogels showed a significantly higher adherent cell density than PEG SH-Mal hydrogels (p < 0.05). The differences in cell adhesion pattern may also be due to the differences in protein adhesion as discussed above. Photopolymerized PEG hydrogels may have more rigid and robust structure, while PEG SH-Mal hydrogels seem to have a more flexible and dynamic structure.
Though PEG SH-Mal hydrogels showed a significantly lower adherent cell density compared to TCPS, cells were shown to have normal morphology on the bottom of the TCPS surfaces that contained the PEG SH-Mal hydrogel sample (Fig. 10), indicating the material leachables and any potential degradation products were not cytotoxic. Additionally, cytotoxicity evaluation via MTT assay was conducted on three cell types that were used in the cell adhesion study, including primary human monocytes, fibroblasts, and keratinocytes. The results showed fibroblasts and monocytes treated with PEG SH-Mal hydrogel extract maintained 90 – 100% viability compared to control groups at both 24 and 48 h in a serum-starved condition (Fig. 11). Keratinocytes at 24 h showed around 70% viability compared to the control and above 75% normalized viability at 48 h (Fig. 11). Both the presence of potential keratinocyte toxic material and the serum-depleted condition could contribute to this decrease. According to GB/T 16886.5-2003 (ISO 10993-5:1999), samples with cell viability larger than 75% can be considered as noncytotoxic. Thus, the hydrogel extract was of low toxicity to primary monocytes and fibroblasts.
FIGURE 10.

Microscopic images of adherent fibroblasts and keratinocytes on TCPS surface in the presence of PEG SH-Mal hydrogels at 24, 48 and 96 h (20× Magnification). (A) Fibroblast optical microscopy images (left-hand side) and fluorescent microscopy images (right-hand side, green, live stained cells; red, dead stained cells); (B) Keratinocyte optical microscopy images (left-hand side) and fluorescent microscopy images (right-hand side, green, live stained cells; red, dead stained cells).
FIGURE 11.

Normalized cell viability of primary human monocytes, fibroblasts, and keratinocytes treated with hydrogel extracts on 24 and 48 h. Control: cells treated with serum-depleted medium only. (n=6)
CONCLUSIONS
A noncrosslinked PEG-based hydrogel was successfully synthesized via a metal-free thiolmaleimide reaction. Buffer pH, macromer concentration, and the presence of biomolecule gelatin showed significant effects on gel formation. Release studies using FD-40 indicated that the hydrogels has the potential as an in situ forming drug delivery matrix. Preliminary cell adhesion and cytotoxicity studies demonstrated that the networks were minimally adhesive to primary human monocytes, fibroblasts, keratinocytes and the material leachables were of low cytotoxicity thus providing an ideal platform for further biofunctionalization to direct specific biological response.
ACKNOWLEDEGMENTS
This work was supported by NIH Grant R01 EB6613. The authors would like to thank Evan Joyce, Whitney Johnson, Yiwei Ma, and Kedi Xu for assistance with cell culture, and proofreading.
References
- 1.Nguyen KT, West JL. Photopolymerizable hydrogels for tissue engineering applications. Biomaterials. 2002;23:4307–4314. doi: 10.1016/s0142-9612(02)00175-8. [DOI] [PubMed] [Google Scholar]
- 2.Wang ZC, Xu XD, Chen CS, Yun L, Song JC, Zhang XZ, Zhuo RX. In situ formation of thermosensitive PNIPAAm-based hydrogels by Michael-type addition reaction. ACS Appl Mater Interfaces. 2010;2:1009–1018. doi: 10.1021/am900712e. [DOI] [PubMed] [Google Scholar]
- 3.Bae SJ, Suh JM, Sohn YS, Bae YH, Kim SW, Jeong B. Thermogelling poly(caprolactone-bethylene glycol-b-caprolactone) aqueous solutions. Macromolecules. 2005;38:5260–5265. [Google Scholar]
- 4.Mather BD, Wiswanathan K, Miller KM, Long TE. Michael addition reactions in macromolecular design for emerging technologies. Prog Polym Sci. 2006;31:487–531. [Google Scholar]
- 5.Chan JW, Hoyle CE, Lowe AB, Bowman M. Nucleophile-initiated thiol-Michael reactions: effect of organocatalyst, thiol, and ene. Macromolecules. 2010;43:6381–6388. [Google Scholar]
- 6.Dondoni A. The emergence of thiol-ene coupling as a click process for materials and bioorganic chemistry. Angew Chem Int Ed Engl. 2008;47:8995–8997. doi: 10.1002/anie.200802516. [DOI] [PubMed] [Google Scholar]
- 7.Rizzi SC, Ehrbar M, Halstenberg S, Raeber GP, Schmoekel HG, Hagenmüller H, Müller R, Weber FE, Hubbell JA. Recombinant protein-co-PEG networks as cell-adhesive and proteolytically degradable hydrogel matrices. Part II: Biofunctional characteristics. Biomacromolecules. 2006;7:3019–3029. doi: 10.1021/bm060504a. [DOI] [PubMed] [Google Scholar]
- 8.Rizzi SC, Hubbell JA. Recombinant protein-co-PEG networks as cell-adhesive and proteolytically degradable hydrogel matrices. Part I: Development and physicochemical characteristics. Biomacromolecules. 2005;6:1226–1238. doi: 10.1021/bm049614c. [DOI] [PubMed] [Google Scholar]
- 9.Elbert DL, Pratt AB, Lutolf MP, Halstenberg S, Hubbell JA. Protein delivery from materials formed by self-selective conjugate addition reactions. J Control Release. 2001;76:11–25. doi: 10.1016/s0168-3659(01)00398-4. [DOI] [PubMed] [Google Scholar]
- 10.Yuan W, Yang J, Kopecková P, Kopecek J. Smart Hydrogels Containing Adenylate Kinase: Translating Substrate Recognition into Macroscopic Motion. J Am Chem Soc. 2008;130:15760–15761. doi: 10.1021/ja805634x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu SQ, Tian Q, Hedrick JL, Po Hui JH, Ee PL, Yang YY. Biomimetic hydrogels for chondrogenic differentiation of human mesenchymal stem cells to neocartilage. Biomaterials. 2010;31:7298–7307. doi: 10.1016/j.biomaterials.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 12.Kim J, Park Y, Tae G, Lee KB, Hwang CM, Hwang SJ, Kim IS, Noh I, Sun K. Characterization of low-molecular-weight hyaluronic acid-based hydrogel and differential stem cell responses in the hydrogel microenvironments. J Biomed Mater Res A. 2009;88:967–975. doi: 10.1002/jbm.a.31947. [DOI] [PubMed] [Google Scholar]
- 13.Jin R, Moreira Teixeira LS, Krouwels A, Dijkstra PJ, van Blitterswijk CA, Karperien M, Feijen J. Synthesis and characterization of hyaluronic acid-poly(ethylene glycol) hydrogels via Michael addition: An injectable biomaterial for cartilage repair. Acta Biomater. 2010;6:1968–1977. doi: 10.1016/j.actbio.2009.12.024. [DOI] [PubMed] [Google Scholar]
- 14.Kim J, Kim IS, Cho TH, Lee KB, Hwang SJ, Tae G, Noh I, Lee SH, Park Y, Sun K. Bone regeneration using hyaluronic acid-based hydrogel with bone morphogenic protein-2 and human mesenchymal stem cells. Biomaterials. 2007;28:1830–1837. doi: 10.1016/j.biomaterials.2006.11.050. [DOI] [PubMed] [Google Scholar]
- 15.Eskandari M, Brey E, Cinar A. A Gaussian model for substrates of entangled cross-linked poly(ethylene glycol) in biomedical applications. Biotechnol Bioeng. 2011;108:435–445. doi: 10.1002/bit.22889. [DOI] [PubMed] [Google Scholar]
- 16.Chung AS, Gao Q, Kao WJ. Macrophage matrix metalloproteinase-2/-9 gene and protein expression following adhesion to ECM-derived multifunctional matrices via integrin complexation. Biomaterials. 2007;28:285–298. doi: 10.1016/j.biomaterials.2006.08.038. [DOI] [PubMed] [Google Scholar]
- 17.Schmidt DR, Kao WJ. Monocyte activation in response to polyethylene glycol hydrogels grafted with RGD and PHSRN separated by interpositional spacers of various lengths. J Biomed Mater Res A. 2007;83:617–625. doi: 10.1002/jbm.a.31270. [DOI] [PubMed] [Google Scholar]
- 18.Harris JM, Herati RS. Synthesis of polyethylene glycol thiol. Polymer preprints (American Chemical Society, Division of Polymer Chemistry) 1991;32:154–155. [Google Scholar]
- 19.Woghiren C, Sharma B, Stein S. Protected thiol-polyethylene glycol: a new activated polymer for reversible protein modification. Bioconjugate Chem. 1993;4:314–318. doi: 10.1021/bc00023a002. [DOI] [PubMed] [Google Scholar]
- 20.Shim WS, Kim JH, Kim K, Kim YS, Park RW, Kim IS, Kwon IC, Lee DS. pH- and temperature-sensitive, injectable, biodegradable block copolymer hydrogels as carriers for paclitaxel. Int J Pharm. 2007;331:11–8. doi: 10.1016/j.ijpharm.2006.09.027. [DOI] [PubMed] [Google Scholar]
- 21.Becalde IB, Recalde D, García-Lopera R, Gómez CM. FTIR isothermal cure kinetics and morphology of dicyanate ester resin/polysulfone blends. Eur Polym J. 2005;41:2635–2643. [Google Scholar]
- 22.Seager DJ, Lutz M, Hama S, Cruz D, Castrillo A, Lazaro J, Phillips R, Premack B, Berliner J. Method for large scale isolation, culture and cryopreservation of human monocytes suitable for chemotaxis, cellular adhesion assays, macrophage and dendritic cell differentiation. J Immunol Methods. 2004;288:123–34. doi: 10.1016/j.jim.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 23.Liu TY, Lin YL. Novel pH-sensitive chitosan-based hydrogel for encapsulating poorly water soluble drugs. Acta Biomater. 2010;6:1423–1429. doi: 10.1016/j.actbio.2009.10.010. [DOI] [PubMed] [Google Scholar]
- 24.Kade MJ, Burke DJ, Hawker CJ. The power of thiol-ene chemistry. J Polym Sci Part A: Polym Chem. 2010;48:743–751. doi: 10.1002/pola.23917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Niu G, Zhang H, Song L, Cui X, Cao H, Zheng Y, Zhu S, Yang Z, Yang H. Thiol/acrylate-modified PEO-PPO-PEO triblocks used as reactive and thermosensitive copolymers. Biomacromolecules. 2008;9:2621–2628. doi: 10.1021/bm800573e. [DOI] [PubMed] [Google Scholar]
- 26.Niu G, Du F, Song L, Song L, Zhang H, Yang J, Cao H, Zheng Y, Yang Z, Wang G, Yang H, Zhu S. Synthesis and characterization of reactive poloxamer 407s for biomedical applications. J Control Release. 2009;138:49–56. doi: 10.1016/j.jconrel.2009.04.026. [DOI] [PubMed] [Google Scholar]
- 27.Normand V, Muller S, Ravey JC, Parker A. Gelation kinetics of gelatin: a master curve and network modeling. Macromolecules. 2000;33:1063–1071. [Google Scholar]
- 28.Hou Y, Schoener CA, Regan KR. Photo-cross-linked PDMS star-PEG hydrogels: synthesis, characterization, and potential application for tissue engineering scaffolds. Biomacromolecules. 2010;11:648–656. doi: 10.1021/bm9012293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andrade JD, Gregonis DE, King RN, Ma SM. Contact angles at the solid–water interface. J Colloid Interface Sci. 1992;3:488–494. [Google Scholar]
- 30.Lin CC, Anseth KS. PEG hydrogels for the controlled release of biomolecules in regenerative medicine. Pharm Res. 2009;26:631–643. doi: 10.1007/s11095-008-9801-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hamilton WC. A technique for the characterization of hydrophilic solid surfaces. J Colloid Interface Sci. 1971;2:219–222. [Google Scholar]
- 32.Fu Y, Kao WJ. Drug release kinetics and transport mechanisms from semi-interpenetrating networks of gelatin and poly(ethylene glycol) diacrylate. Pharm Res. 2009;26:2115–2124. doi: 10.1007/s11095-009-9923-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dunne G, McMillan ND, O'Rourke B, Morrin D, O'Neill M, Seidel S, McDonell L, Scully P. Experimental tensiometric protein adsorption studies. Colloids and Surfaces A: physiochemical and engineering aspects. 2010;354:364–367. [Google Scholar]