

Abstract
The deregulation of microRNAs has been demonstrated in various tumor processes. Here, we report that microRNA-544 (miR-544) is decreased in cervical cancer tissues compared with normal cervical tissues. To identify the mechanisms involved in miR-544 deregulation, we studied the regulation of miR-544 expression at the transcriptional level. We first identified the transcriptional start site of miR-544 by 5’ rapid amplification of cDNA ends and subsequently determined the miR-544 promoter. We discovered that the transcription factor Krueppel-like factor 4 (KLF4) is involved in the transcriptional regulation of miR-544 through interaction with the miR-544 promoter. In addition, we found that miR-544 directly targets the YWHAZ oncogene and functions as a tumor suppressor in cervical cancer cells. miR-544 is involved in cell cycle regulation and suppresses cervical cancer cell proliferation, colony formation, migration and invasion in a manner associated with YWHAZ downregulation. In summary, our findings demonstrate that KLF4 upregulates miR-544 transcription by activating the miR-544 promoter and that miR-544 functions as a tumor suppressor by targeting YWHAZ. Therefore, miR-544 may be a potential novel therapeutic target and prognostic marker for cervical cancer.
Keywords: microRNA-544, YWHAZ, KLF4, cervical cancer
Introduction
microRNAs (miRNAs) are small, non-coding RNAs that modulate gene expression post-transcriptionally either by inhibiting translation or by causing mRNA degradation through binding to the 3’untranslated regions (3’UTRs) of target mRNAs [1,2]. This interaction leads to the reduced protein expression of miRNA-targeted genes. It has been speculated that miRNAs regulate approximately one-third of all protein-coding genes [3]. Thus, up or downregulation of miRNAs can influence the expression of tumor suppressor genes and oncogenes. Large-scale miRNA expression profiling experiments have been performed in numerous different cancers and have identified a large number of deregulated miRNAs. Recent studies have implicated miRNAs in a wide variety of vital developmental and physiological processes including cell proliferation, differentiation, apoptosis, and organ development [4]. Therefore, it is important to study both the function and regulation of miRNAs. However, most studies have focused on the regulatory functions of miRNAs, and few are directed towards how miRNA genes are themselves transcriptionally regulated. Therefore, the underlying mechanisms of aberrant miRNA regulation are still poorly understood.
Cervical cancer is the second most common cancer in women worldwide, with an estimate global incidence of 529,800 new cases and 275,100 deaths per year [5,6]. Persistent infection of high-risk human papillomavirus (HPV) has been considered as one reason for cervical carcinogenesis [7,8]. The high-risk HPV genome integrates into the host genome and actives the viral oncogenes E6 and E7, leading to malignant transformation of host cells. A large number of aberrantly expressed miRNAs have been identified in cervical cancer and are closely associated with cervical cancer progression [9-11].
In our study, we found that miR-544 is reduced in cervical cancer, while its target gene YWHAZ is increased. In addition, we identified the miR-544 transcription start site (TSS) by 5’ rapid amplification of cDNA ends (5’RACE) and found the miR-544 promoter sequence. We demonstrate that the transcription factor KLF4 is involved in the transcriptional regulation of miR-544 through interaction with the miR-544 promoter region. Furthermore, we revealed that miR-544 is involved in cell cycle regulation and suppresses cervical cancer cell proliferation, colony formation, migration and invasion. Our study provides comprehensive understanding of how a transcription factor regulates miRNAs and suggests a novel therapeutic strategy for YWHAZ-induced tumorigenesis in cervical cancer.
Materials and methods
Cell culture, transfection, tumor specimens and reagents
The human cervical cancer cell lines CaSki and HeLa (ATCC, USA) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco, USA) with 10% fetal bovine serum at 37°C with 5% CO2. The miR-544 mimic, inhibitor and negative control (NC) were synthesized by Ambion. The siRNA against YWHAZ (siRNA-YWHAZ) and negative control (siRNA NC) were synthesized by Genepharma (Shanghai, China) [12]. The sequences were as follows: siRNA-YWHAZ, 5’-AAAGUUCUUGAUCCCCAAUGC-3’ and siRNA NC, 5’-CAGUCGCGUUUGCGACUGG-3’. Transfection was performed using Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer’s instructions. Twenty pairs of human cervical cancer tissues and matched adjacent noncancerous cervical tissues were collected from Shanghai Changhai Hospital (Shanghai, China). All specimens were obtained from informed patients, and the patients provided written consent. The study was approved by the Ethics Committee of Fudan University. Polyclonal antibodies use for western blot analysis were anti-beta-tubulin (Abmart, M20005, 1:2000, China), anti-YWHAZ (Santa Cruz, sc-1019, 1:100, USA), HRP anti-rabbit (KPL, 4751-1516, 1:2000, USA) and HRP anti-mouse (KPL, 0751-1809, 1:2000, USA).
RNA isolation and quantitative real-time PCR (qRT-PCR)
Total RNA was extracted using TRIzol reagent (Ambion, USA). The cDNA was synthesized using the PrimeScriptTM RT reagent Kit (TaKaRa, China) according to the manufacturer’s instructions. The qRT-PCR was performed using SYBR Premix Ex Taq (TaKaRa) and analyzed using the LightCycler 480 II Real Time PCR system (Roche, Germany). The primers used are shown in Table 1. RNU6B and GAPDH were used as internal controls for mature miR-544 and mRNA expression, respectively.
Table 1.
Primer sequences used in this study
| Primer name | primer sequences |
|---|---|
| RT-miR-544 | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACgaactt |
| RT-RNU6B | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAAAAATAT |
| qRT-miR-544-F | gccgcgATTCTGCATTTTTAGC |
| qRT-RNU6B-F | TTCCTCCGCAAGGATGACACGC |
| qRT-miRNA-R | GTGCAGGGTCCGAGGT |
| YWHAZ 3’UTR-1-F-XhoI | ccgctcgagGGATACCCAAGGAGACGA |
| YWHAZ 3’UTR-1-R-NotI | ataagaatgcggccgcCTCAAGTTATTTCCCTGTTA |
| YWHAZ 3’UTR-2-F-XhoI | ccgctcgagTGACATTGGGTAGCATTA |
| YWHAZ 3’UTR-2-R-NotI | ataagaatgcggccgcGTTTGGGATTTCAGTTTCT |
| 1YWHAZ 3’UTR-mut-F1 | TTGACTACAGCTTGCCGAGTGTTCCTTTA |
| 1YWHAZ 3’UTR-mut-R1 | TAAAGGAACACTCGGCAAGCTGTAGTCAA |
| 1YWHAZ 3’UTR-del-F1 | ACCTTTGACTACAGCGTGTTCCTTTAGAC |
| 1YWHAZ 3’UTR-del-R1 | GTCTAAAGGAACACGCTGTAGTCAAAGGT |
| 1YWHAZ 3’UTR-mut-F2 | CTCTGGATAAGGGAGCCAACGGTTCACATT |
| 1YWHAZ 3’UTR-mut-R2 | AATGTGAACCGTTGGCTCCCTTATCCAGAG |
| 1YWHAZ 3’UTR-del-F2 | TTACTCTGGATAAGGACGGTTCACATTC |
| 1YWHAZ 3’UTR-del-R2 | GAATGTGAACCGTCCTTATCCAGAGTAA |
| 2YWHAZ 3’UTR-mut-F | CCTCAGTACTTTAGCCGAAACACCAAACA |
| 2YWHAZ 3’UTR-mut-R | TGTTTGGTGTTTCGGCTAAAGTACTGAGG |
| 2YWHAZ 3’UTR-del-F | TTCCTCAGTACTTAACACCAAACA |
| 2YWHAZ 3’UTR-del-R | TGTTTGGTGTTAAGTACTGAGGAA |
| qRT-YWHAZ-F | CCTCACTCCCGTTTCCG |
| qRT-YWHAZ-R | CAGCACCTTCCGTCTTT |
| qRT-GAPDH-F | ATGGGGAAGGTGAAGGTCG |
| qRT-GAPDH-R | CTGGAAGATGGTGATGGGA |
| miR-544 5’RACE outer | ACACAAAACGGATACACACGGTCA |
| miR-544 5’RACE inner | TGGGTCCAAGACAAGGTTTGATAC |
| miR-544 promoter (-255)-F-XhoI | ccgctcgagGGCTCTGTGAGATGGATGA |
| miR-544 promoter (+15)-F-XhoI | ccgctcgagAGGCTGATTCTGGTGAAGG |
| miR-544 promoter (+256)-F-XhoI | ccgctcgagTTCGGTTTATTGACATGGA |
| miR-544 promoter (+764)-R-HindIII | cccaagcttAAAGGTTAGCACGGAAAAG |
| miR-544 promoter (+918)-R-HindIII | cccaagcttCAAGGTTGGACGGTTAGTA |
| YWHAZ-F-EcoRI | ccggaattcCCACCATGGATAAAAATGAGCTGG |
| YWHAZ-R-KpnI | cggggtaccTTAATTTTCCCCTCCTTCTCC |
| KLF4-F-XhoI | ccgctcgagCCACCATGAGGCAGCCACCTGG |
| KLF4-R-HindIII | cccaagcttTTAAAAATGCCTCTTCATGTGT |
| qRT-KLF4-F | GCCAAAGAGGGGAAGACGAT |
| qRT-KLF4-R | CGCAGGTGTGCCTTGAGATG |
| ChIP qRT-F | ACCTGCCCAGTGCTATTGTTT |
| ChIP qRT-R | CAAAACGGATACACACGGTCA |
| KLF4-miR-544-mut-F1 | ACTCAGCAAGAAGCACGGTATGTGT |
| KLF4-miR-544-mut-R1 | ACACATACCGTGCTTCTTGCTGAGT |
| KLF4-miR-544-mut-F2 | ATGACTTTAGAAGGAATGATGGCCTCTGG |
| KLF4-miR-544-mut-R2 | CCAGAGGCCATCATTCCTTCTAAAGTCAT |
| KLF4-miR-544-mut-F3 | GCCTCTGGGATTATTCCTCTCCTGG |
| KLF4-miR-544-mut-R3 | CCAGGAGAGGAATAATCCCAGAGGC |
| KLF4-miR-544-mut-F4 | GGTGCAGTGTAAATAAGGCCAAAAGC |
| KLF4-miR-544-mut-R4 | GCTTTTGGCCTTATTTACACTGCACC |
| KLF4-miR-544-mut-F5 | TGGGTCCCTGTAATTTGAGCGGGAA |
| KLF4-miR-544-mut-R5 | TTCCCGCTCAAATTACAGGGACCCA |
| KLF4-miR-544-mut-F6 | TTCCTGAGCGAATTATCATCAGCCC |
| KLF4-miR-544-mut-R6 | GGGCTGATGATAATTCGCTCAGGAA |
| KLF4-miR-544-mut-F7 | CGATGGCTGGAACAACGCTCGTGTG |
| KLF4-miR-544-mut-R7 | CACACGAGCGTTGTTCCAGCCATCG |
| KLF4-miR-544-mut-F8 | GCATCTAATTTGCTTCCAGGACCGC |
| KLF4-miR-544-mut-R8 | GCGGTCCTGGAAGCAAATTAGATGC |
| KLF4-miR-544-mut-F9 | CGTACCAAGTTTGTTCAAGGCAGTG |
| KLF4-miR-544-mut-R9 | CACTGCCTTGAACAAACTTGGTACG |
Mutant sequences were underlined.
5’ rapid amplification of cDNA ends (5’RACE)
The TSS of the primary transcript for human miR-544 (from HeLa cells) was determined using a 5’-Full RACE kit (TaKaRa) according to the manufacturer’s instructions. HL60 total RNA, which was provided in the kit and served as a positive control, was used to analyze the 5’ end of human Prohibitin (PHB) gene (PCR product, 750 bp). The primary transcripts were determined by nested PCR using outer and inner primers (Table 1). The PCR products were separated by agarose gel electrophoresis, purified and sequenced.
Dual-luciferase reporter assays and transcription factor analysis
For the YWHAZ 3’UTR reporter assay, the wild type (WT) 3’UTR of YWHAZ, mutant type (Mut) and deletion type (Del) including putative miR-544 binding sites were cloned into the psiCHECK-2 luciferase reporter vector (Promega, USA). HeLa cells were cultured in 96-well plates and co-transfected with the miRNA molecules (miR-544 mimic and NC, 30 nM) and 50 ng of the luciferase reporter constructs. Luciferase activity was measured using the Dual-Luciferase Reporter Assay System on a GloMax 96 Microplate Luminometer (Promega). The results are expressed as a normalized ratio of Renilla to firefly luciferase.
For the miR-544 promoter activity assay, the genomic sequences near the miR-544 TSS were amplified and cloned into the pGL3-Basic reporter vector (Promega). HeLa cells were co-transfected with the pGL3-Basic derived reporter vectors and Renilla luciferase plasmid, pRL-TK and pCDNA-KLF4. The cells were lysed at 48 h after transfection, and luciferase activity was measured using the Dual-Luciferase Reporter Assay System on a GloMax 96 Microplate Luminometer (Promega). The activities of firefly luciferase (PGL3-Basic) were normalized to Renilla luciferase (pRL-TK) activities.
Chromatin immunoprecipitation (ChIP) assays
ChIP assays were performed using the EZ-Magna ChIP One-Day Chromatin Immunoprecipitation Kit (Millipore, USA) according to the instruction manual. Approximately 1×107 HeLa cells were cross-linked with 1% formaldehyde for 10 min, and glycine was used to quench unreacted formaldehyde. Cells were then washed with ice-cold PBS and lysed with cell lysis buffer and then nuclear lysis buffer containing Protease Inhibitor Cocktail II. Chromatin was sheared by sonication to an average size of 200 to 1000 bp. The supernatant was collected in 50-μl aliquots. Each 50-μl aliquot (containing 1×106 cell equivalents of chromatin) was diluted in 450 μl of dilution buffer. “Input” samples [5 μl (1%) of the supernatant] were removed and stored at 4°C. The remaining supernatant was incubated for 4 h with 5 μg of anti-KLF4 (RD, AF3640, USA) or normal IgG (negative control) immunoprecipitating antibody and 20 μl of protein A magnetic beads. Finally, the immunoprecipitate complexes were collected, washed, eluted, and the cross-links were reversed using proteinase K. DNA was purified using spin columns and analyzed by qRT-PCR.
Cell proliferation and colony formation assays
For cell proliferation assays, CaSki and HeLa cells were plated in 96-well plates at 3000 cells per well and transfected with the indicated miRNA mimics (30 nM) and plasmids (100 ng) and maintained in culture media. After 24 h, the cells were incubated with 20 μl MTT (0.5 mg/ml) at 37°C for 4 h. Then, the medium was removed, and 100 μl of DMSO was added to dissolve the precipitated formazan. The absorbance at 490 nm was read using a plate reader (BioTek, USA). For colony formation assays, 200 cells were plated in 24-well plates and incubated for 10 days. The colonies were fixed with methanol and stained with 0.1% crystal violet. Colonies containing more than 50 cells were counted.
Migration and invasion assays
In vitro migration and invasion assays were performed using 8-μm pore size 24-well transwell chambers (Corning, USA). For the migration assays, 5×104 cells were resuspended in 0.1 ml of serum-free DMEM and added to the transwell inserts. For the invasion assays, the transwell inserts were coated with 150 μg of Matrigel (BD, USA), and 1×105 cells in serum-free DMEM were added to the inserts. DMEM with 10% FBS was added to the bottom wells. After incubation for 20 h, the cells on the upper surface of the membrane were removed, and the cells on the lower surface were fixed with methanol and stained with 0.1% crystal violet. The migrated or invaded cells were counted using an 1×51 inverted microscope (Olympus, Japan).
Flow cytometry assays
For cell cycle analysis, cells were collected and fixed in ice-cold 75% ethanol overnight. The fixed cells were washed and resuspended in propidium iodide staining solution containing 50 μg/ml propidium iodide, 50 μg/ml ribonuclease A, and 0.2% Triton X-100 in phosphate-buffered saline for 30 min in the dark. The cells were then analyzed using a FACSCalibur Flow Cytometer (BD, USA). The data were analyzed using the Modfit software.
Statistical analyses
The results are representative of three independent experiments and presented as the mean ± standard deviation (SD). The data were analyzed using Student’s t-test to evaluate significant differences. A P value < 0.05 was considered significant.
Results
miR-544 suppresses YWHAZ expression by targeting the 3’UTR of YWHAZ
Various algorithms (TargetScan, miRanda, and miRecords) were used to predict the potential target genes of miR-544. The results suggested that there are three predicted miR-544 binding sites in the YWHAZ 3’UTR (Figure 1A). To validate this prediction, we cloned the YWHAZ 3’UTR containing the miR-544 target site (wild type, mutant or deletion) into the luciferase reporter vector psiCHECK-2. The predicated binding sites 1 and 2 were cloned together downstream of the luciferase reporter gene (Figure 1A). For the luciferase reporter assays, the luciferase reporter constructs were co-transfected with miR-544 mimic or miRNA mimic NC into HeLa cells. The results indicated that miR-544 decreased the relative luciferase activity with the wild type 3’UTR (WT) compared with the control, whereas the relative luciferase activity was not decreased as sharply in mutant (Mut) and deletion (Del) 3’UTRs (Figure 1B). Furthermore, qRT-PCR and western blot analysis demonstrated that miR-544 dramatically decreased the endogenous mRNA and protein levels of YWHAZ in CaSki and HeLa cells (Figure 1C, 1D). YWHAZ expression levels decreased as the concentration of the transfected miR-544 mimic increased but increased when the cells were transfected with the miR-544 inhibitor. These results indicated that miR-544 downregulated YWHAZ expression by directly binding to its 3’UTR target sites 1, 2 and 3.
Figure 1.
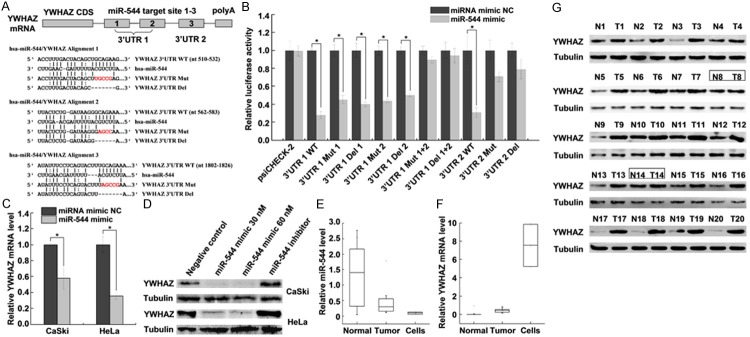
miR-544 downregulates YWHAZ expression by binding to the YWHAZ 3’UTR. A. Predicted binding sites of miR-544 to the 3’UTR of YWHAZ. The wild type (WT), mutated (Mut), and deleted (Del) YWHAZ 3’UTR binding sites are shown. The mutant nucleotides of the miR-544 binding sites are indicated in red. B. Relative luciferase activity of the YWHAZ WT, Mut, and Del reporter in the presence of 30 nM miR-544 mimic or miRNA mimic NC. Renilla luciferase was normalized to firefly luciferase. C. qRT-PCR analysis of YWHAZ mRNA from CaSki and HeLa cells transfected with miR-544 mimic (30 nM) or miRNA mimic NC (30 nM). GAPDH mRNA was used as an internal control. D. Western blot analysis of YWHAZ expression in CaSki and HeLa cells transfected with miR-544 mimic, inhibitor or NC. Tubulin was used as a loading control. E. qRT-PCR analysis of mature miR-544 expression in 20 paired tumor tissues (Tumor) and adjacent normal cervical tissues (Normal) and cervical cancer cell lines (CaSki and HeLa). RNU6B was used as an internal control. F. qRT-PCR analysis of YWHAZ mRNA expression in 20 paired tumor tissues (Tumor) and adjacent normal cervical tissues (Normal) and cervical cancer cell lines (CaSki and HeLa). GAPDH mRNA was used as an internal control. G. Western blot analysis of YWHAZ protein levels in 20 paired tumor tissues (T) and adjacent normal cervical tissues (N). Tubulin was used as a loading control. Two paired samples showing no differences in YWHAZ expression are labeled with boxes. The data represent the mean ± SD from three independent experiments. *P < 0.05.
To determine whether miR-544 expression correlated with YWHAZ mRNA expression levels in cervical cancer, we evaluated miR-544 and YWHAZ mRNA levels in 20 paired cervical cancer tissues and their adjacent normal cervical tissues as well as in cervical cancer cell lines (HPV 16+ CaSki and HPV 18+ HeLa) via qRT-PCR. Compared with normal cervical tissues, miR-544 was decreased in cervical cancer tissues and cervical cancer cells (Figure 1E), whereas YWHAZ mRNA expression was increased in cervical cancer tissues and cervical cancer cells (Figure 1F). Furthermore, western blot analysis indicated that the YWHAZ protein expression was more highly expressed in 18 of 20 paired (90%) tumor tissues (T) compared with normal (N) cervical tissues (Figure 1G). Two paired samples that showed no differences in YWHAZ expression are labeled with boxes. The results suggested that the aberrant decrease of miR-544 is related to the high expression of YWHAZ in cervical cancer.
Identification of the miR-544 promoter
While our results suggested that miR-544 is decreased in cervical cancer, the reasons for this decrease is unknown. To determine the regulation of miR-544 expression at the transcriptional level, we first determined the TSS of miR-544 by 5’RACE assay. As shown in Figure 2A, the 5’RACE PCR products for pri-miR-544 suggested that it was the TSS of miR-544 and was labeled +1 (Figure 2B).
Figure 2.
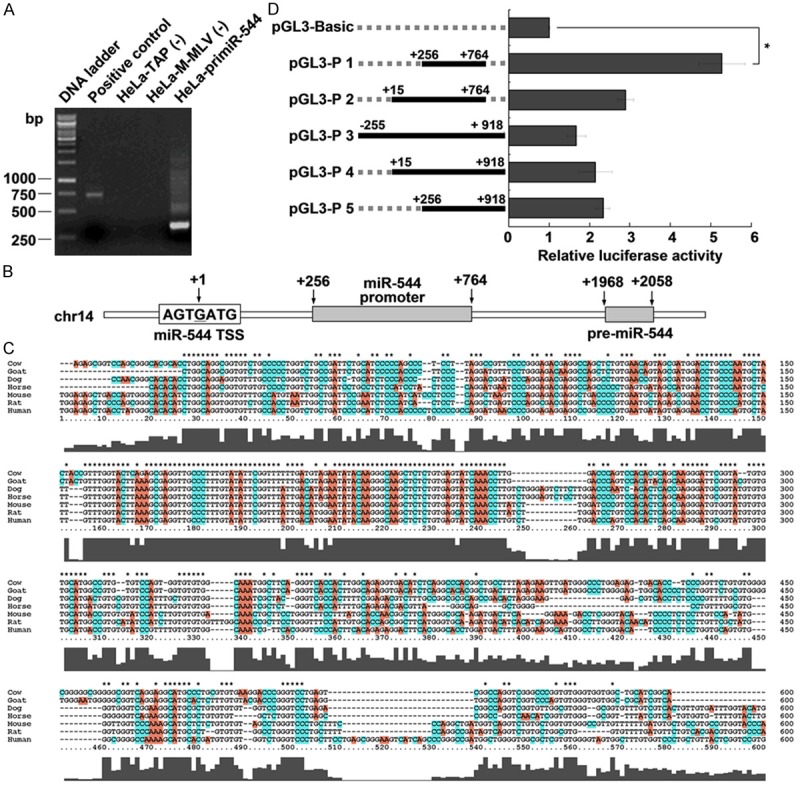
Identification of the miR-544 promoter. A. Agarose gel electrophoresis shows the PCR products of 5’RACE. B. Schematic of the miR-544 promoter location in the genome. The transcription start site of miR-544 is indicated as +1 and the number indicates the relative distance from the miR-544 transcription start site. C. Alignment of the miR-544 promoter sequence between different species. D. Relative luciferase activity of the miR-544 promoter. Luciferase constructs containing the miR-544 promoter (pGL3-P 1 to P 5) and the unmodified construct (pRL-TK) were co-transfected into HeLa cells. Firefly luciferase activity was normalized to Renilla luciferase activity. Data represent the mean ± SD from three independent experiments. *P < 0.05.
Typically, promoter sequences are near TSS and conserved in different species. Therefore, we scanned the sequences including 2 kb upstream and 2 kb downstream of miR-544 TSS with the online tools FirstEF, PromoterScan, softberry-fprom, and Promoter 2.0 Prediction. Unfortunately, no significant results were obtained. Then, we aligned the sequences 2 kb upstream and 2 kb downstream of miR-544 TSS and found a highly conserved region (Figure 2C). We hypothesized that this conserved sequence was most likely to be the promoter of miR-544. Later, we cloned the conserved sequence with different lengths into the luciferase reporter vector pGL3-Basic and evaluated the promoter activities. As shown in Figure 2D, the sequence between +256 to +764 significantly increased luciferase activity (approximately 5 fold). This sequence was considered to be the miR-544 promoter and was used in subsequent experiments.
KLF4 promotes miR-544 transcription by interacting with the miR-544 promoter
To determine which transcription factors might influence miR-544 expression, we performed in silico analysis of the identified promoter region using TRANSFAC (http://www.biobase-international.com/product/transcription-factor-binding-sites) and identified KLF4 as a candidate. KLF4 overexpression was examined by qRT-PCR and western blot, as shown in Figure 3A, 3B, and showed that KLF4 mRNA and protein levels were significantly increased when transfected with pcDNA-KLF4 in CaSki and HeLa cells. We then examined the effect of the transcription factor KLF4 on miR-544 promoter-driven luciferase activity and found KLF4 overexpression in HeLa cells activated miR-544 promoter-driven luciferase activity (Figure 3C). Consistent with the luciferase assay, endogenous mature miR-544 expression levels were increased by KLF4 overexpression (Figure 3D). To determine whether KLF4 regulation of miR-544 can mediate YWHAZ expression, we analyzed YWHAZ expression by qRT-PCR and western blot. As shown in Figure 3E and 3F, YWHAZ mRNA and protein levels were reduced in cells transiently transfected with pcDNA-KLF4, strongly suggesting that KLF4 stimulates miR-544 expression, which, in turn, downregulates YWHAZ expression. In addition, we examined KLF4 mRNA expression levels in 20 paired cervical cancer samples and their adjacent normal cervical samples as well as in cervical cancer cell lines (HPV 16+ CaSki and HPV 18+ HeLa) by qRT-PCR. The result indicated that the KLF4 expression level was decreased in cervical cancer tissues and cells compared with normal cervical tissues (Figure 3G). These results revealed that the low expression of KLF4 may contribute to the decreased expression of miR-544 in cervical cancer.
Figure 3.

KLF4 promotes miR-544 transcription by activating miR-544 promoter. A and B. qRT-PCR and western blot analysis of KLF4 expression in CaSki and HeLa cells transfected with pcDNA-KLF4. C. Relative luciferase activity of the miR-544 promoter in HeLa cells transfected with pcDNA-KLF4. Luciferase constructs containing the miR-544 promoter (pGL3-Promoter) and the unmodified construct (pRL-TK) were co-transfected with pcDNA3.1 or pcDNA-KLF4 into HeLa cells. Firefly luciferase activity was normalized to Renilla luciferase activity. D. qRT-PCR analysis of miR-544 expression in CaSki and HeLa cells with KLF4 overexpression. E and F. qRT-PCR and western blot analysis of YWHAZ expression in CaSki and HeLa cells with KLF4 overexpression. G. qRT-PCR analysis of KLF4 mRNA expression in 20 paired tumor tissues (Tumor) and adjacent normal cervical tissues (Normal) and cervical cancer cell lines (CaSki and HeLa). H. ChIP qRT-PCR assay were performed to detect the in vivo binding of transcription factor KLF4 to miR-544 promoter. HeLa cell chromatin fragments were immunoprecipitated with the specific anti-KLF4 antibody or normal IgG. The immunoprecipitated DNA samples were analyzed by qRT-PCR using primers specific to the miR-544 promoter. I. Relative luciferase activity of the mutant miR-544 promoter constructs in HeLa cells transfected with pcDNA-KLF4. Firefly luciferase activity was normalized to Renilla luciferase activity. The data represent the mean ± SD from three independent experiments. *P < 0.05.
The interaction between KLF4 and the miR-544 promoter was further confirmed by ChIP qRT-PCR assays. As shown in Figure 3H, in vivo binding of KLF4 to the miR-544 promoter region was significantly higher than the control. Promoter scanning using TRANSFAC predicted nine potential KLF4 binding sites in the miR-544 promoter region. To identify KLF4 binding sites in the miR-544 promoter, we performed mutational analysis on the predicted KLF4 binding sites. As shown in Figure 3I, mutation of KLF4 binding site 4, 6 and 8 (mut 4, mut 6, and mut 8) abolished the effects of KLF4 on relative luciferase activity, while mutating other binding sites had no significant effect. According to these results, KLF4 regulates the expression of miR-544 by binding to sites 4, 6 and 8. These results indicated that KLF4 is involved in the transcriptional regulation of miR-544 by directly binding to the miR-544 promoter.
miR-544 suppresses cervical cancer cell proliferation and colony formation and is involved in cell cycle regulation by targeting YWHAZ
To better understand the role of miR-544 in the development of cervical cancer, we performed cell proliferation assays and found that miR-544 led to a significant decrease in the growth of CaSki and HeLa cells (Figure 4A, 4B). However, overexpression of YWHAZ abolished this suppression (Figure 4A, 4B). Similarly, colony formation assays showed that cervical cancer cell colony formation abilities were suppressed in miR-544-transfected cells compared with miRNA NC cells (Figure 4C, 4D). These results indicated that miR-544 exerts a growth suppression function in cervical cancer cells.
Figure 4.

miR-544 suppresses cervical cancer cell proliferation and colony formation and is involved in cell cycle regulation by targeting YWHAZ. A and B. The effect of miR-544 on CaSki and HeLa cell proliferation. miR-544 suppresses cervical cancer cell proliferation, and this can be rescued by over expression of YWHAZ. C and D. The effect of miR-544 on CaSki and HeLa cell colony formation. E and F. Flow cytometry analysis of the miR-544 and YWHAZ induced cell cycle checkpoint in CaSki and HeLa cells. Cervical cancer cells with miR-544 and YWHAZ overexpression were stained with propidium iodide and used for cell cycle analysis. The data represent the mean ± SD from three independent experiments. *P < 0.05.
Meanwhile, we determined whether miR-544 impacts cell cycle progression of cervical cancer cells. Cell cycle analysis indicated that miR-544 overexpression had a higher percentage of G1-phase cells and a lower percentage of S-phase cells (Figure 4E, 4F). However, YWHAZ overexpression induced the opposite effect. YWHAZ overexpression induced a reduction in the percentage of cells in G1 phase and an increase in S phase (Figure 4E, 4F). The results indicated that miR-544 is involved in cell cycle regulation by targeting YWHAZ.
miR-544 suppresses cervical cancer cell migration and invasion in vitro
To further determine whether miR-544 is associated with cervical cancer progression, we analyzed the effect of miR-544 overexpression on migratory and invasive ability. As shown in Figure 5A, 5B, the migratory ability was significantly suppressed when cervical cancer cells were transfected with the miR-544 mimic. Similarly, miR-544 overexpression dramatically suppressed the invasive ability of CaSki and HeLa cells (Figure 5C, 5D). These results demonstrated that miR-544 overexpression contributes to the regulation of cervical cancer cells motility and progression.
Figure 5.

miR-544 suppresses cervical cancer cell migration and invasion. A and B. Transwell migration assays of CaSki and HeLa cells transfected with miR-544 mimic or NC. Representative images and quantification of migrated cells are shown. C and D. Transwell invasion assays of CaSki and HeLa cells transfected with miR-544 mimic or NC. Representative images and quantification of invaded cells are shown. The data represent the mean ± SD from three independent experiments. *P < 0.05.
YWHAZ knockdown by siRNA suppresses cervical cancer cell colony formation, migration and invasion
The function of YWHAZ in cervical cancer cells was analyzed using RNA interference. The results showed that YWHAZ mRNA and protein levels were significantly decreased when cervical cancer cells were transfected with siRNA targeting YWHAZ (Figure 6A, 6B). YWHAZ knockdown significantly suppressed CaSki and HeLa cell colony formation (Figure 6C, 6D). Likewise, YWHAZ knockdown suppressed CaSki and HeLa cell migration and invasion abilities (Figure 6E-H). These results demonstrated that YWHAZ functions as an oncogene and may contribute to the tumorigenesis in cervical cancer.
Figure 6.

YWHAZ knockdown suppresses cell colony formation, migration and invasion. A and B. qRT-PCR and western blot analysis of YWHAZ expression in CaSki and HeLa cells transfected with siRNA NC or siRNA-YWHAZ. C and D. Effect of siRNA-YWHAZ on CaSki and HeLa cells colony formation. Cervical cancer cells were treated with siRNA NC or siRNA-YWHAZ and plated in 6-well plates in triplicate. E and F. Transwell migration assays of CaSki and HeLa cells transfected with siRNA NC or siRNA-YWHAZ. Representative images and quantification of migrated cells are shown. G and H. Transwell invasion assays of CaSki and HeLa cells transfected with siRNA NC or siRNA-YWHAZ. Representative images and quantification of invaded cells are shown. The data represent the mean ± SD from three independent experiments. *P < 0.05.
Discussion
miRNAs are post-transcription regulators of many genes, and their deregulation is related to cancer initiation, development, and progression [4]. Our study demonstrates that miR-544 is decreased in cervical cancer tissues and cells; however, the underlying mechanisms involved in the deregulation of miR-544 are still unknown. Therefore, we explored the regulation of miR-544 expression at the transcriptional level. We identified the miR-544 TSS by 5’RACE and identified the miR-544 promoter sequence downstream of the TSS. Typically, the promoters for RNA polymerase I and II are mostly upstream of the TSS, but some promoters for RNA polymerase III lie downstream of the TSS [13]. It has been reported that miRNA genes can be transcribed by either RNA polymerase II or RNA polymerase III into primary miRNA transcripts [14-16]. Therefore, we speculate that miR-544 may be transcribed by RNA polymerase III, as the promoter sequence lies downstream of the TSS.
The protein product of the miR-544 target gene YWHAZ belongs to the highly conserved 14-3-3 protein family. YWHAZ encodes tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, zeta (14-3-3 ζ). Prior studies have suggested that YWHAZ is a potential oncogene in various human cancers because it is frequently overexpressed [17-20] and affects multiple pathways involved in cancer [12,19,21]. Phosphorylated E6 interacts with YWHAZ directly, and knockdown of YWHAZ expression results in a significant decrease in the levels of HPV-18 E6 in HeLa cells [22]. A defining feature of cervical cancer is the continued expression of E6 and E7 viral oncoproteins [23,24]. Abrogation of E6/E7 function or expression suppresses cell growth and induces senescence and apoptosis [25,26]. Therefore, miR-544 and its target YWHAZ can be considered potential targets for anti-cancer therapy in HPV-induced malignancies (Figure 7).
Figure 7.
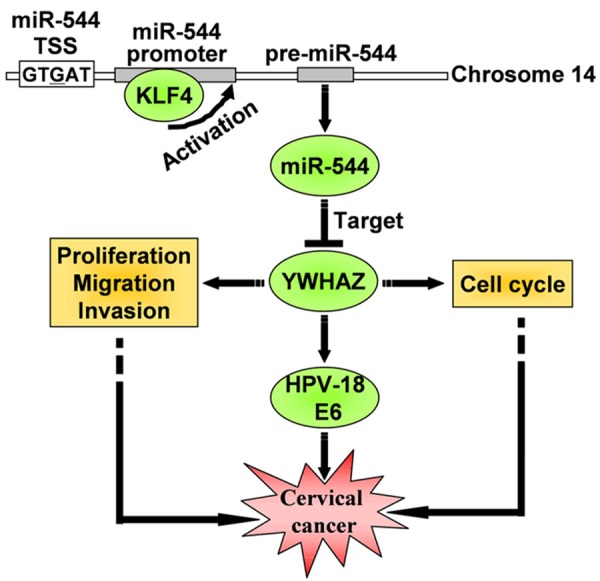
Diagram describing the linear signaling pathway involved in cervical cancer progression.
Krueppel-like factor 4 (KLF4) is a zinc finger transcription factor. Previous studies have indicated that KLF4 expression is reduced in different types of cancers [27,28]. Accumulating clinical evidence also suggests that KLF4 functions as a tumor suppressor gene [29-31]. KLF4 knockdown promotes cell growth, migration and adhesion, and the loss of KLF4 promotes skin tumorigenesis in a mouse model [32]. Our study revealed that KLF4 was decreased in cervical cancer tissues and cells compared with normal cervical tissues. Therefore, the low expression of KLF4 in cervical cancer may contribute to the reduced expression of miR-544 and may also play a crucial role in cervical cancer progression.
In conclusion, miRNAs have emerged as important regulators of diverse biological processes. Aberrant and altered miRNA expression are linked to a multitude of pathological conditions and cancer progression. The cause of the widespread differential expression of miRNAs in pathologic compared with normal cells can be explained by epigenetic mechanisms, miRNA biogenesis machinery and transcriptional regulation of miRNA promoters. Recently, significant advances have been made in miRNA biogenesis and the mode of action and have attracted great attention. Therapeutic approaches based on miRNA-targeted genes may be developed in the future. Therefore, understanding the principles managing miRNA deregulation is essential for the development and successful application of miRNA-based drugs.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (91129730, 21132004 to H. L and 31200988 to W. M), the Specialized Research Fund for the Doctoral Program of High Education (20110071130007), the National Program on the Key Basic Research Project (973 Program, 2009CB825601), and the Shanghai Science and Technology Commission (13DZ2252000).
Disclosure of conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shukla GC, Singh J, Barik S. MicroRNAs: Processing, Maturation, Target Recognition and Regulatory Functions. Mol Cell Pharmacol. 2011;3:83–92. [PMC free article] [PubMed] [Google Scholar]
- 3.Mallanna SK, Rizzino A. Emerging roles of microRNAs in the control of embryonic stem cells and the generation of induced pluripotent stem cells. Dev Biol. 2010;344:16–25. doi: 10.1016/j.ydbio.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 5.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 6.Bray F, Ren JS, Masuyer E, Ferlay J. Global estimates of cancer prevalence for 27 sites in the adult population in 2008. Int J Cancer. 2013;132:1133–1145. doi: 10.1002/ijc.27711. [DOI] [PubMed] [Google Scholar]
- 7.Narisawa-Saito M, Kiyono T. Basic mechanisms of high-risk human papillomavirus-induced carcinogenesis: roles of E6 and E7 proteins. Cancer Sci. 2007;98:1505–1511. doi: 10.1111/j.1349-7006.2007.00546.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schiffman M, Castle PE, Jeronimo J, Rodriguez AC, Wacholder S. Human papillomavirus and cervical cancer. Lancet. 2007;370:890–907. doi: 10.1016/S0140-6736(07)61416-0. [DOI] [PubMed] [Google Scholar]
- 9.Lee JW, Choi CH, Choi JJ, Park YA, Kim SJ, Hwang SY, Kim WY, Kim TJ, Lee JH, Kim BG, Bae DS. Altered MicroRNA expression in cervical carcinomas. Clin Cancer Res. 2008;14:2535–2542. doi: 10.1158/1078-0432.CCR-07-1231. [DOI] [PubMed] [Google Scholar]
- 10.Zhu X, Er K, Mao C, Yan Q, Xu H, Zhang Y, Zhu J, Cui F, Zhao W, Shi H. miR-203 suppresses tumor growth and angiogenesis by targeting VEGFA in cervical cancer. Cell Physiol Biochem. 2013;32:64–73. doi: 10.1159/000350125. [DOI] [PubMed] [Google Scholar]
- 11.Tang T, Wong HK, Gu W, Yu MY, To KF, Wang CC, Wong YF, Cheung TH, Chung TK, Choy KW. MicroRNA-182 plays an onco-miRNA role in cervical cancer. Gynecol Oncol. 2013;129:199–208. doi: 10.1016/j.ygyno.2012.12.043. [DOI] [PubMed] [Google Scholar]
- 12.Neal CL, Yao J, Yang W, Zhou X, Nguyen NT, Lu J, Danes CG, Guo H, Lan KH, Ensor J, Hittelman W, Hung MC, Yu D. 14-3-3zeta overexpression defines high risk for breast cancer recurrence and promotes cancer cell survival. Cancer Res. 2009;69:3425–3432. doi: 10.1158/0008-5472.CAN-08-2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krebs JE, Lewin B, Goldstein ES, Kilpatrick ST. Lewin’s essential genes. Jones & Bartlett Publishers; 2013. p. 505. [Google Scholar]
- 14.Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23:4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borchert GM, Lanier W, Davidson BL. RNA polymerase III transcribes human microRNAs. Nat Struct Mol Biol. 2006;13:1097–1101. doi: 10.1038/nsmb1167. [DOI] [PubMed] [Google Scholar]
- 16.Winter J, Jung S, Keller S, Gregory RI, Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol. 2009;11:228–234. doi: 10.1038/ncb0309-228. [DOI] [PubMed] [Google Scholar]
- 17.Neal CL, Yu D. 14-3-3zeta as a prognostic marker and therapeutic target for cancer. Expert Opin Ther Targets. 2010;14:1343–1354. doi: 10.1517/14728222.2010.531011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin M, Morrison CD, Jones S, Mohamed N, Bacher J, Plass C. Copy number gain and oncogenic activity of YWHAZ/14-3-3zeta in head and neck squamous cell carcinoma. Int J Cancer. 2009;125:603–611. doi: 10.1002/ijc.24346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi JE, Hur W, Jung CK, Piao LS, Lyoo K, Hong SW, Kim SW, Yoon HY, Yoon SK. Silencing of 14-3-3zeta over-expression in hepatocellular carcinoma inhibits tumor growth and enhances chemosensitivity to cis-diammined dichloridoplatium. Cancer Lett. 2011;303:99–107. doi: 10.1016/j.canlet.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 20.Matta A, Bahadur S, Duggal R, Gupta SD, Ralhan R. Over-expression of 14-3-3zeta is an early event in oral cancer. BMC Cancer. 2007;7:169. doi: 10.1186/1471-2407-7-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nishimura Y, Komatsu S, Ichikawa D, Nagata H, Hirajima S, Takeshita H, Kawaguchi T, Arita T, Konishi H, Kashimoto K, Shiozaki A, Fujiwara H, Okamoto K, Tsuda H, Otsuji E. Overexpression of YWHAZ relates to tumor cell proliferation and malignant outcome of gastric carcinoma. Br J Cancer. 2013;108:1324–1331. doi: 10.1038/bjc.2013.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boon SS, Banks L. High-risk human papillomavirus E6 oncoproteins interact with 14-3-3zeta in a PDZ binding motif-dependent manner. J Virol. 2013;87:1586–1595. doi: 10.1128/JVI.02074-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Androphy EJ, Hubbert NL, Schiller JT, Lowy DR. Identification of the HPV-16 E6 protein from transformed mouse cells and human cervical carcinoma cell lines. EMBO J. 1987;6:989–992. doi: 10.1002/j.1460-2075.1987.tb04849.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smotkin D, Wettstein FO. Transcription of human papillomavirus type 16 early genes in a cervical cancer and a cancer-derived cell line and identification of the E7 protein. Proc Natl Acad Sci U S A. 1986;83:4680–4684. doi: 10.1073/pnas.83.13.4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goodwin EC, DiMaio D. Repression of human papillomavirus oncogenes in HeLa cervical carcinoma cells causes the orderly reactivation of dormant tumor suppressor pathways. Proc Natl Acad Sci U S A. 2000;97:12513–12518. doi: 10.1073/pnas.97.23.12513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshinouchi M, Yamada T, Kizaki M, Fen J, Koseki T, Ikeda Y, Nishihara T, Yamato K. In vitro and in vivo growth suppression of human papillomavirus 16-positive cervical cancer cells by E6 siRNA. Mol Ther. 2003;8:762–768. doi: 10.1016/j.ymthe.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 27.Wei D, Gong W, Kanai M, Schlunk C, Wang L, Yao JC, Wu TT, Huang S, Xie K. Drastic down-regulation of Kruppel-like factor 4 expression is critical in human gastric cancer development and progression. Cancer Res. 2005;65:2746–2754. doi: 10.1158/0008-5472.CAN-04-3619. [DOI] [PubMed] [Google Scholar]
- 28.Katz JP, Perreault N, Goldstein BG, Actman L, McNally SR, Silberg DG, Furth EE, Kaestner KH. Loss of Klf4 in mice causes altered proliferation and differentiation and precancerous changes in the adult stomach. Gastroenterology. 2005;128:935–945. doi: 10.1053/j.gastro.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 29.Dang DT, Chen X, Feng J, Torbenson M, Dang LH, Yang VW. Overexpression of Kruppellike factor 4 in the human colon cancer cell line RKO leads to reduced tumorigenecity. Oncogene. 2003;22:3424–3430. doi: 10.1038/sj.onc.1206413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao W, Hisamuddin IM, Nandan MO, Babbin BA, Lamb NE, Yang VW. Identification of Kruppel-like factor 4 as a potential tumor suppressor gene in colorectal cancer. Oncogene. 2004;23:395–402. doi: 10.1038/sj.onc.1207067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang WT, Zheng PS. Kruppel-like factor 4 functions as a tumor suppressor in cervical carcinoma. Cancer. 2012;118:3691–3702. doi: 10.1002/cncr.26698. [DOI] [PubMed] [Google Scholar]
- 32.Li J, Zheng H, Yu F, Yu T, Liu C, Huang S, Wang TC, Ai W. Deficiency of the Kruppel-like factor KLF4 correlates with increased cell proliferation and enhanced skin tumorigenesis. Carcinogenesis. 2012;33:1239–1246. doi: 10.1093/carcin/bgs143. [DOI] [PMC free article] [PubMed] [Google Scholar]