

Abstract
MicroRNAs (miRNAs) are small non-coding RNAs that play important roles in cancer progression through regulating gene expression. Down-regulation of miR-143 has been reported in a number of cancers. However, the biological functions of miR-143 in prostate cancer remain largely unexplored. In this study, we showed that miR-143 expression was reduced in approximately 62.5% of the specimens examined. By loss-of-function and gain-of-function studies in human prostate cancer PC-3 cells, we demonstrated that miR-143 has an inhibitory effect on cell proliferation as evidenced by decreased cell viability, increased cell apoptosis and cell cycle arrest at the G1/S transition. Furthermore, we identified hexokinase 2 (HK2), a metabolic enzyme that executes the first step of aerobic glycolysis, as a target of miR-143 in prostate cancer. Knockdown of HK2 recapitulated the effects of miR-143 and accompanied with decreased glucose metabolism. Taken together, these data indicate that miR-143/HK2 axis plays an important role in the development of prostate cancer and represents a potential therapeutic target for prostate cancer.
Keywords: miR-143, prostate cancer, growth, hexokinase 2
Introduction
Although significant advances have recently been made in the medical management and screening, prostate cancer remain one of the most common malignancies in men worldwide [1,2]. Prostate cancer arises mainly from prostatic intraepithelial neoplasia, a precursor lesion that ultimately progresses to adenocarcinoma and hormone-dependent or hormone-independent metastatic diseases [3]. Multiple alterations in tumor microenvironment are involved in the initiation and development of prostate cancer. Therefore, identifying key signaling events emanating from the microenvironment may contribute to novel therapeutic targets for prostate cancer.
MicroRNAs (miRNAs) are endogenously non-coding RNA molecules of 19 to 25 nucleotides that posttranscriptionally modulate target mRNAs through pairing to complementary sequences [4]. Accumulating evidences suggest that aberrant expression of miRNAs is closely associated with cancer cell proliferation, differentiation, apoptosis and metastasis [5,6]. Down-regulation of miRNAs in tumor tissues can lead to aberrant expression of their targeted genes, which often involved in tumor initiation, development and progression. Therefore, reintroduction of specific miRNA in tumors with lower expression could be considered as a therapeutic strategy.
Recent studies have shown that profiles of miRNA expression differ between normal and tumor tissues [7,8]. Among them, miRNA-143 expression has been found during differentiation of smooth muscle cells and adipocytes in normal circumstances [9]. However, dysregulated expression of miRNA-143 is more commonly observed in cancers, including cervical cancer, colorectal cancer, liposarcoma and B cell malignancies [10-14]. In colorectal cancer, miRNA-143 has been demonstrated that exhibits a growth inhibitory effect by targeting KRAS and DNA methyltransferases 3A (DNMT3A) [11,15]. While in dedifferentiated liposarcoma cells, this effect is associated with decreased expression of BCL2, topoisomerase 2A, protein regulator of cytokinesis 1 and polo-like kinase 1 [13]. However, little is known about the expression pattern and underlying functions of miRNA-143 in prostate cancer.
In this study, we firstly observed that miR-143 was down-regulated in approximately 62.5% prostate cancer tissues examined. Subsequent experiments of cellular functions demonstrated that miR-143 acts as a tumor suppressor by affecting the cell viability, cell apoptosis and cell cycle of prostate cancer. Furthermore, the glycolytic enzyme, hexokinase 2 (HK2), was identified as a direct functional target of miR-143 in prostate cancer.
Materials and methods
Cell culture
Human prostate cancer cell line PC-3 was purchased from Cell Bank of the Chinese Academy of Sciences. Cells were cultured in DMEM specific medium supplemented with 10% (v/v) fetal calf serum at 37°C in a humidified incubator under 5% CO2 condition.
RNA isolation and quantitative real-time PCR
Twenty-four fresh-frozen clinical tissue specimens were recruited with informed consent from Chengdu Military General Hospital, China and this study is approved by the ethics committee of Chengdu Municipality. For detection of miR-143 expression, total RNA extraction was performed using miRNA Extraction Kit (Takara, Japan); reverse transcription of RNA and the detection of mature miR-143 were performed using an All-in-One miRNA quantitative real-time PCR detection kit (GeneCopoeia, China). For detection of HK2 expression, total RNA extracted using Trizol reagent (Takara, Japan), and reversely transcribed through PrimeScript RT-PCR kit (Takara, Japan) according to the protocol. Real-time PCR analyses were performed with SYBR Premix Ex Taq (Takara) on ABI 7300 detection system. Primer sequences used are as follows: HK2: forward: 5’-CTTCTTCACGGAGCTCAACC-3’; reverse: 5’-AAGCCCTTTCTCCATCTCCT-3’; β-actin: forward: 5’-CATGTACGTTGCTATCCAGGC-3’; reverse: 5’-CTCCTTAATGTCACGCACGAT-3’. The relative mRNA expression of HK2 was normalized to β-actin.
Plasmid constructs and luciferase reporter assay
Lentivirus vector expressing antagomir of miR-143 was cloned into the pGreenPuro vector (System Biosciences, Mountain View, CA, USA) and the empty vector was used as control [16]. Virus titers were determined and PC-3 cells were infected with 1 × 106 recombinant lentivirus-transducing units. To construct the HK2 expression plasmids, the full-length construct with wild-type 3’UTR or mutant 3’UTR of HK2 gene was cloned into the PEZ-Lv105 lentivirus vector (GeneCopoeia). For luciferase assays, the full-length wild-type UTR of HK2 was amplified by PCR and cloned into the pMIR-REPORT luciferase reporter vector (Invitrogen, Carlsbad, CA, USA). The mutant of the HK2-3’UTR was constructed by replacing miR-143 binding site with their complimentary sequence. The antisense and sense oligonucleotide sequences are described by Gregersen et al [17]. All constructs were verified by sequencing. Then PC-3 cells were co-transfected with indicated firefly and a Renilla normalization control using X-tremeGENE 9 according to the manufacturer’s protocol (Promega, Madison, WI, USA). The firefly luminescence was measured and normalized to the Renilla luciferase activity. The experiment was performed at least in triplicate.
Transfection
All of the siRNAs and miRNA mimics used in current study were synthesized by GenePharma (Shanghai, China). For transfection, PC-3 cells were transfected with 30 μM siRNA or 20 nM miRNA mimic using siRNA mate according to the manufacturer’s instructions. The HK2 interference efficiency was measured by western blotting.
Cell viability, cell cycle and apoptosis assay
For cell viability assay, 4000 cells per well were seeded into a 96-well plate and cultured overnight. Cell proliferation was evaluated by Cell Counting Kit-8 (CCK-8, Dojindo, Japan) following the manufacturer’s protocol. At 24, 48, 72, 96 and 120 hr, the OD450 absorbance was measured by a Multifunctional Microplate Reader (Tecan). For cell apoptosis assay, cells were stained with propidium iodide (PI) and Annexin V-fluorescein isothiocyanate (BD Bioscience, USA) according the manufacturer’s protocol. The apoptotic cells were measured by flow cytometry and the apoptotic ratio was calculated according to the percentage of early apoptotic cells. For cell cycle analysis, transfected cells were cultured for 24 hr and then fixed into 70% ethanol at -20°C for 24 h. The distribution of cell cycle was monitored by flow cytometric analysis of the DNA content of cell populations stained with propidium iodide. The percentage of cells within G1/S and G2, and M phases was determined by using CELLQUEST software (Becton Dickinson).
Western blotting
Total cell lysates were prepared using RIPA buffer containing protease inhibitor (Beyotime, China). Proteins were separated on a 10% SDS gel and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA). The membranes were then consecutively incubated with specific primary antibodies (HK2 and β-actin, Proteintech) and species-specific secondary antibodies after blocked with 5% (v/v) bovine serum albumin. The immunoreactive proteins were visualized by Odyssey imaging system (LI-COR Biosciences, Lincoln, NE).
Detection of glucose and lactate levels
A Glucose Colorimetric Assay Kit (BioVision) and Lactate Assay Kit (Sigma-Aldrich) were used to detect the intracellular glucose consumption and lactate production in PC-3 cells. The glucose uptake was measure using a standard glucose calibration curve performed under the same condition. The lactate level was detected using a standard lactate calibration curve performed under the same condition. All experiments were repeated at least three times.
Statistical analysis
Data were presented as the means ± SD of three independent experiments. The SPSS software program (version 17.0; IBM Corporation) was used for statistical analysis. Graphical representations were performed with GraphPad Prism 5 (San Diego, CA) software. The significance of the data in this study was determined using the Student t test (2-tailed). P values less than 0.05 were considered statistically significant.
Results
miR-143 expression is down-regulated in human prostate cancer
To determine the expression pattern of miR-143 in prostate cancer, we evaluated miR-143 expression in 24 matched pairs of prostate cancer and non-tumor tissues using quantitative real-time PCR. miR-143 expression was normalized to an endogenous control (U6 RNA). As shown in Figure 1A, miR-143 expression was significantly down-regulated in prostate cancer tissues in relative to corresponding non-tumor tissues (p < 0.001). Besides, down-regulation of miR-143 was observed in 62.5% specimens examined (Figure 1B). However, whether this type of dysregulation has an implication on cellular functions in prostate cancer remains unclear.
Figure 1.
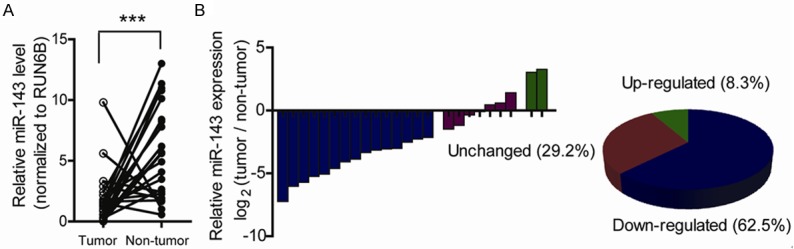
miR-143 expression is down-regulated in human prostate cancer. A. Expression of mature miR-143 in 24 matched pairs of prostate cancer and non-tumor tissues was detected by quantitative real-time PCR and normalized by an endogenous control, U6 RNA. (***P < 0.001). B. Expression of mature miR-143 was distributed by the value of log2 (prostate cancer tissues/non-tumor tissues).
miR-143 exhibits a inhibitory effect on tumor growth in vitro
To investigate the cellular functions of miR-143 in prostate cancer, we performed knockdown and overexpression experiments in PC-3 cells. In PC-3 cells stably transfected with sh-miR-143, increased cell viability and decreased apoptosis ratio were found compared with negative vector cells (Figure 2A, 2B). Meanwhile, miR-143 knockdown also resulted in a markedly decrease in the percentage of cell population in G0/G1 phase and a sharp increase in S phase (Figure 2C), indicating that miR-143 induces cell cycle arrest of PC-3 cells at the G1/S transition. In contrast, when PC-3 cells were transiently transfected with miR-143 mimics, reduced cell viability (Figure 2D) and increased apoptosis ratio (Figure 2E) were observed compared with negative control cells. And expectedly, the G1/S transition was pronounced inhibited after treatment with miR-143 mimics in PC-3 cells (Figure 2F). Collectively, these data above strongly suggest that miR-143 exhibits a growth inhibitory effect in prostate cancer.
Figure 2.
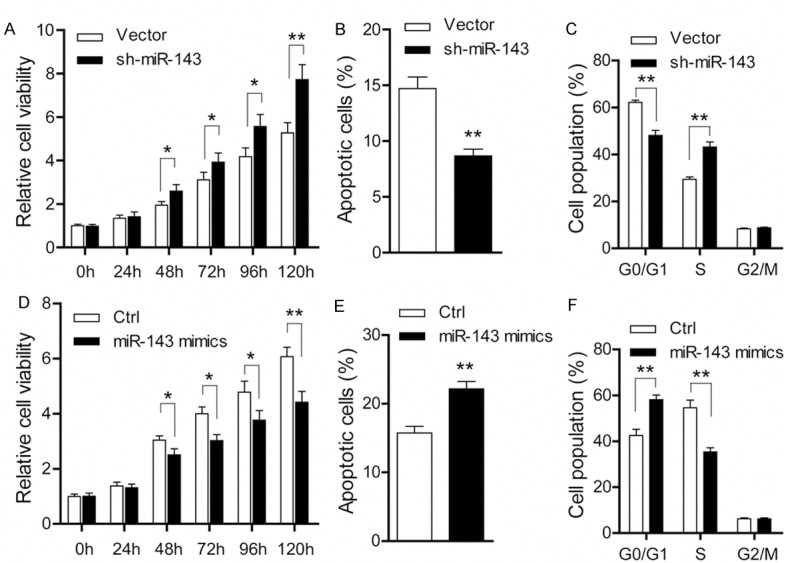
miR-143 exhibits an inhibitory effect on tumor growth in prostate cancer. A. Cell viability detected by CCK8 in PC-3 cells stably transfected with sh-miR-143 or empty vector (vector versus sh-miR-143; *P < 0.05; **P < 0.01). B. Cell apoptosis measured by Annexin V/PI staining in PC-3 cells stably transfected with sh-miR-143 or empty vector (vector versus sh-miR-143; **P < 0.01). C. Cell cycle distribution analyzed by flow cytometry in PC-3 cells stably transfected with sh-miR-143 or empty vector (vector versus sh-miR-143; **P < 0.01). D. Cell viability detected by CCK8 in PC-3 cells treated with miR-143 mimics (control versus miR-143 mimics; *P < 0.05; **P < 0.01). E. Cell apoptosis measured by Annexin V/PI staining in PC-3 cells treated with miR-143 mimics (control versus miR-143 mimics; **P < 0.01). F. Cell cycle distribution analyzed by flow cytometry in PC-3 cells treated with miR-143 mimics (control versus miR-143 mimics; **P < 0.01).
miR-143 modulates prostate cancer cells through targeting HK2
Previous studies have demonstrated several targets of miR-143 in colorectal cancer, such as KRAS, DNA methyltransferases 3A and HK2 [11,15,17]. In current study, by searching the TargetScan (http://www.Targetscan.org/), Pictar (http://pictar.mdcberlin.de/) and miRanda (http://www.microrna.org/) databases, we verified that HK2 is a potential target of miR-143. To confirm this hypothesis in prostate cancer, a luciferase reporter system was constructed. As shown in Figure 3A, the luciferase activity of the wild-type but not the mutant HK2 reporter was significantly reduced by miR-143, indicating that miR-143 can bind directly to the 3’UTR of HK2. Consistent with this, knockdown of miR-143 led to elevated HK2 protein level (Figure 3B), while treatment with miR-143 mimics resulted in decreased HK2 protein level (Figure 3C). Furthermore, mRNA expression level of HK2 evaluated by quantitative real-time PCR was negatively correlated with miR-143 expression (Figure 3D, r = -0.613, p = 0.001). Taken together, above results indicate that HK2 is the downstream target of miR-143 in prostate cancer.
Figure 3.
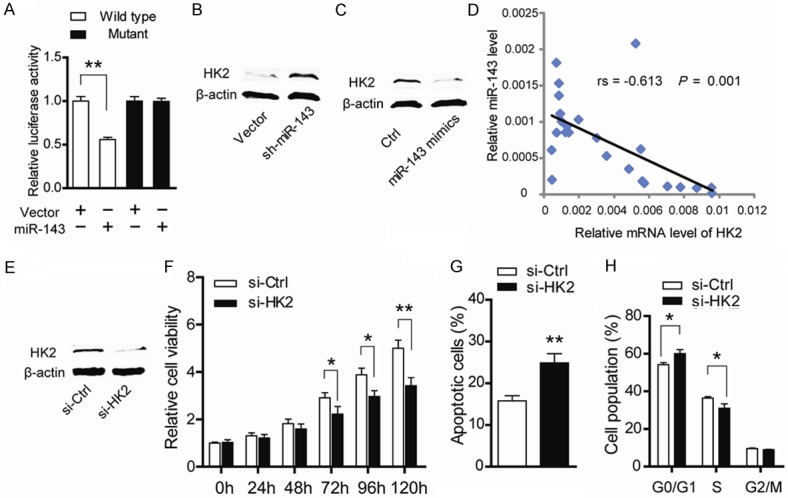
miR-143 modulates prostate cancer cells through targeting HK2. (A) PC-3 cells were transfected with the pGL3-HK2 3’UTR-wild or pGL3-HK2 3’UTR-mutant vector along with miR-143 mimics. The firefly luciferase reporter along with a Renilla luciferase was co-transfected by X-tremeGENE 9. The signals were normalized to Renilla activity. The protein level of HK2 was detected after miR-143 knockdown (B) and miR-143 mimics treatment (C). (D) Spearman correlation analysis of the expression of miR-143 and HK2 in 24 prostate cancer tissues (r = -0.613, p = 0.001). (E) The protein level of HK2 was detected by western blotting after treatment with siRNAs target HK2. (F) Cell viability detected by CCK8 in PC-3 cells treated with HK2 siRNAs (control versus siRNAs; *P < 0.05; **P < 0.01). (G) Cell apoptosis measured by Annexin V/PI staining in PC-3 cells treated with HK2 siRNAs (control versus siRNAs; **P < 0.01). (H) Cell cycle distribution analyzed by flow cytometry in PC-3 cells treated with HK2 siRNAs (control versus siRNAs; *P < 0.05).
Knockdown of HK2 recapitulates the inhibitory effects of miR-143
HK2 catalyzes the first step of anaerobic glycolysis and is highly expressed in many cancer cells but only in a limited number of normal adult tissues [18]. In lung cancer cells, HK2 deletion suppressed glucose-derived ribonucleotides and impaired glutamine-derived carbon utilization in anaplerosis [19]. To determine the role of HK2, a pool of three different siRNAs against HK2 were designed and synthesized. The protein level of HK2 was markedly decreased after targeted siRNAs treatment as demonstrated by western blotting (Figure 3E). Then cellular functions of HK2 in PC-3 cells were evaluated. Consistent with previous reports, suppression of HK2 resulted in growth arrest as evidenced by reduced cell viability (Figure 3F), increased cell apoptosis (Figure 3G) and decelerated G1/S transition (Figure 3H). Taken together, these results suggest that miR-143 down-regulates HK2 to suppress tumor growth in prostate cancer.
Decreased glucose metabolism induced by miR-143/HK2 axis
Given HK2 acts as a key metabolic enzyme in glucose utilization and its crucial roles in tumor initiation and maintenance, we determined whether miR-143-mediated down-regulation of HK2 affects glycolysis in PC-3 cells. Indeed, glucose consumption (Figure 4A) and lactate secretion (Figure 4B) at 24 hr was remarkably reduced in siRNAs-mediated HK2 knockdown cells compared with the negative control cells. When miR-143 expression was suppressed, glycolysis was significantly promoted. In contrast, glycolysis was inhibited by miR-143 mimics treatment (Figure 4C-F). The inhibition observed in glycolysis induced by HK2 siRNAs is comparable to miR-143 mimics treatment, suggesting that decreased glycolysis may contribute to miR-143-mediated effects on tumor growth.
Figure 4.
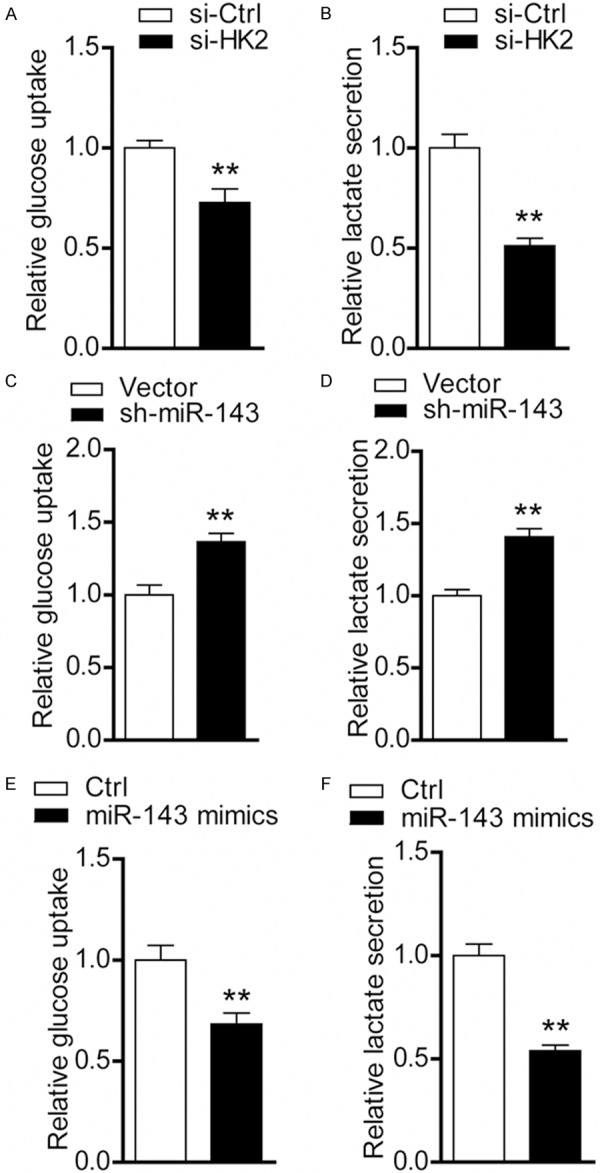
Decreased glucose metabolism induced by miR-143/HK2 axis. Alternations of glucose uptake (A) and lactate production (B) were measured in PC-3 cells after treatment with HK2 siRNAs (control versus siRNAs; **P < 0.01). Glucose consumption (C) and lactate production were measured (D) in PC-3 cells stably transfected with sh-miR-143 or empty vector (vector versus sh-miR-143; **P < 0.01). Glucose consumption (E) and lactate production were measured (F) in PC-3 cells treated with miR-143 mimics (control versus miR-143 mimics; **P < 0.01).
Discussion
In this study, we demonstrated that miR-143 was frequently down-regulated in prostate cancer tissues compared with their corresponding non-tumor tissues. Subsequent loss-of-function and gain-of-function studies showed miR-143 suppressed cancer cell growth. Notably, we confirmed that miR-143 exerts its function in prostate cancer by specifically targeting HK2, a key enzyme involved in glycolysis. In addition, we found that glucose utilization was drastically affected by the miR-143/HK2 axis in prostate cancer.
Previous studies in colorectal cancer have demonstrated that miR-143 is pervasively down-regulated in different ethnic groups [15]. In prostate cancer, down-regulation of several miRNAs, such as miR-16, miR-143, miR-145 and miR-195 has been observed [8,20]. Consistent with previous reports, our data obtained from quantitative real-time PCR validation showed that miR-143 is down-regulated in approximately 62.5% of the specimens examined. However, more clinical specimens are needed to convince the expression profile and possible clinical significance of miR-143 in prostate cancer.
Consideration of the frequent down-regulation of miR-143 in cancers, one may believe that miR-143 may exhibit a tumor-suppressive role in tumor development. Indeed, studies in colorectal cancer, liposarcoma and ours have confirmed the growth inhibitory effect of miR-143. However, the mechanism involved in this effect mediated by miR-143 was largely different, which was mainly due to different targets regulated by miR-143 in different tumors. In colorectal cancer, miR-143 can suppress cancer cell growth through inhibition of KRAS translation, regulating DNMT3A and targeting HK2 [11,15,17]. In liposarcoma cells, BCL2, topoisomerase 2A, protein regulator of cytokinesis 1 and polo-like kinase 1 have been demonstrated as direct targets of miR-143 [13]. Also in the present study, we demonstrated that HK2 was the direct target of miR-143 in prostate cancer as knockdown of HK2 recapitulates the inhibitory effects of miR-143 on PC-3 cancer cell growth. Because many targets except HK2of miR-143 in prostate cancer have been predicted by bioinformatic methods, however, whether the tumor-suppressive effect of miR-143 was mediated by HK2 requires a series of screening processes. In spite of this limitation, HK2 was still a promising target of miR-143 in prostate cancer.
Given HK2 catalyzes the first committed step in glucose metabolism, we hypothesized whether the tumor-suppressive role of miR-143 was mediated by altered glucose metabolism in PC-3 cells. Indeed, cancer cells express high level of HK2, and this distinguishes them from the normal cells [21]. The binding of HK2 to the mitochondrial membrane has been proved to promote the Warburg effect, a phenomenon characterized by accelerated glucose flux and lactate production even in the presence of oxygen [22,23]. Because glycolysis in tumor cells can provide a biosynthetic advantage for cell proliferation [24], it’s reasonable to suggest that miR-143 may regulate the Warburg effect to affect tumor growth. Expectedly, glucose consumption and lactate production was drastically altered by the miR-143/HK2 axis. This finding is consistent with a previous report in colon cancer that decreased lactate secretion was observed after re-introduction of miR-143 in the colon cancer cell line DLD-1 [17].
In conclusion, our study described here is aimed at elucidating the expression pattern and role of HK2 in prostate cancer. And we also demonstrated that miR-143 mediated down-regulation of HK2 accounts for decreased Warburg effect, which ultimately suppresses tumor growth. These data indicate that miR-143/HK2 axis plays an important role in the pathogenesis of prostate cancer and may be a potential therapeutic target for prostate cancer treatment.
Acknowledgements
We are especially grateful to all of the patients participated in this study.
Disclosure of conflict of interest
None.
References
- 1.Thompson IM, Tangen CM. Prostate cancer screening comes of age. Lancet. 2014;384:2004–2006. doi: 10.1016/S0140-6736(14)61008-4. [DOI] [PubMed] [Google Scholar]
- 2.Thompson IM Jr, Cabang AB, Wargovich MJ. Future directions in the prevention of prostate cancer. Nat Rev Clin Oncol. 2014;11:49–60. doi: 10.1038/nrclinonc.2013.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeMarzo AM, Nelson WG, Isaacs WB, Epstein JI. Pathological and molecular aspects of prostate cancer. Lancet. 2003;361:955–964. doi: 10.1016/S0140-6736(03)12779-1. [DOI] [PubMed] [Google Scholar]
- 4.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 5.Ambros V. microRNAs: tiny regulators with great potential. Cell. 2001;107:823–826. doi: 10.1016/s0092-8674(01)00616-x. [DOI] [PubMed] [Google Scholar]
- 6.Kong YW, Ferland-McCollough D, Jackson TJ, Bushell M. microRNAs in cancer management. Lancet Oncol. 2012;13:e249–258. doi: 10.1016/S1470-2045(12)70073-6. [DOI] [PubMed] [Google Scholar]
- 7.Ambs S, Prueitt RL, Yi M, Hudson RS, Howe TM, Petrocca F, Wallace TA, Liu CG, Volinia S, Calin GA, Yfantis HG, Stephens RM, Croce CM. Genomic profiling of microRNA and messenger RNA reveals deregulated microRNA expression in prostate cancer. Cancer Res. 2008;68:6162–6170. doi: 10.1158/0008-5472.CAN-08-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Porkka KP, Pfeiffer MJ, Waltering KK, Vessella RL, Tammela TL, Visakorpi T. MicroRNA expression profiling in prostate cancer. Cancer Res. 2007;67:6130–6135. doi: 10.1158/0008-5472.CAN-07-0533. [DOI] [PubMed] [Google Scholar]
- 9.Esau C, Kang X, Peralta E, Hanson E, Marcusson EG, Ravichandran LV, Sun Y, Koo S, Perera RJ, Jain R, Dean NM, Freier SM, Bennett CF, Lollo B, Griffey R. MicroRNA-143 regulates adipocyte differentiation. J Biol Chem. 2004;279:52361–52365. doi: 10.1074/jbc.C400438200. [DOI] [PubMed] [Google Scholar]
- 10.Lui WO, Pourmand N, Patterson BK, Fire A. Patterns of known and novel small RNAs in human cervical cancer. Cancer Res. 2007;67:6031–6043. doi: 10.1158/0008-5472.CAN-06-0561. [DOI] [PubMed] [Google Scholar]
- 11.Chen X, Guo X, Zhang H, Xiang Y, Chen J, Yin Y, Cai X, Wang K, Wang G, Ba Y, Zhu L, Wang J, Yang R, Zhang Y, Ren Z, Zen K, Zhang J, Zhang CY. Role of miR-143 targeting KRAS in colorectal tumorigenesis. Oncogene. 2009;28:1385–1392. doi: 10.1038/onc.2008.474. [DOI] [PubMed] [Google Scholar]
- 12.Michael MZ, SM OC, van Holst Pellekaan NG, Young GP, James RJ. Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol Cancer Res. 2003;1:882–891. [PubMed] [Google Scholar]
- 13.Ugras S, Brill E, Jacobsen A, Hafner M, Socci ND, Decarolis PL, Khanin R, O’Connor R, Mihailovic A, Taylor BS, Sheridan R, Gimble JM, Viale A, Crago A, Antonescu CR, Sander C, Tuschl T, Singer S. Small RNA sequencing and functional characterization reveals MicroRNA-143 tumor suppressor activity in liposarcoma. Cancer Res. 2011;71:5659–5669. doi: 10.1158/0008-5472.CAN-11-0890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akao Y, Nakagawa Y, Kitade Y, Kinoshita T, Naoe T. Downregulation of microRNAs-143 and -145 in B-cell malignancies. Cancer Sci. 2007;98:1914–1920. doi: 10.1111/j.1349-7006.2007.00618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ng EK, Tsang WP, Ng SS, Jin HC, Yu J, Li JJ, Rocken C, Ebert MP, Kwok TT, Sung JJ. MicroRNA-143 targets DNA methyltransferases 3A in colorectal cancer. Br J Cancer. 2009;101:699–706. doi: 10.1038/sj.bjc.6605195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scherr M, Venturini L, Battmer K, Schaller-Schoenitz M, Schaefer D, Dallmann I, Ganser A, Eder M. Lentivirus-mediated antagomir expression for specific inhibition of miRNA function. Nucleic Acids Res. 2007;35:e149. doi: 10.1093/nar/gkm971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gregersen LH, Jacobsen A, Frankel LB, Wen J, Krogh A, Lund AH. MicroRNA-143 downregulates Hexokinase 2 in colon cancer cells. BMC Cancer. 2012;12:232. doi: 10.1186/1471-2407-12-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilson JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol. 2003;206:2049–2057. doi: 10.1242/jeb.00241. [DOI] [PubMed] [Google Scholar]
- 19.Patra KC, Wang Q, Bhaskar PT, Miller L, Wang Z, Wheaton W, Chandel N, Laakso M, Muller WJ, Allen EL, Jha AK, Smolen GA, Clasquin MF, Robey RB, Hay N. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell. 2013;24:213–228. doi: 10.1016/j.ccr.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Avgeris M, Stravodimos K, Fragoulis EG, Scorilas A. The loss of the tumour-suppressor miR-145 results in the shorter disease-free survival of prostate cancer patients. Br J Cancer. 2013;108:2573–2581. doi: 10.1038/bjc.2013.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mathupala SP, Rempel A, Pedersen PL. Glucose catabolism in cancer cells: identification and characterization of a marked activation response of the type II hexokinase gene to hypoxic conditions. J Biol Chem. 2001;276:43407–43412. doi: 10.1074/jbc.M108181200. [DOI] [PubMed] [Google Scholar]
- 22.Mathupala SP, Ko YH, Pedersen PL. Hexokinase-2 bound to mitochondria: cancer's stygian link to the “Warburg Effect” and a pivotal target for effective therapy. Semin Cancer Biol. 2009;19:17–24. doi: 10.1016/j.semcancer.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–270. [PubMed] [Google Scholar]
- 24.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]