

Abstract
Hydrophilic coordinating chiral ionic liquids with an amino alcohol substructure were developed and efficiently applied to the asymmetric reduction of ketones. Their careful design and adaptability to the desired reaction conditions allow for these chiral ionic liquids to be used as the sole source of chirality in a ruthenium-catalyzed transfer hydrogenation reaction of aromatic ketones. When used in this reaction system, these chiral ionic liquids afforded excellent yields and high enantioselectivities.
Keywords: Ionic liquids, Self-assembly, Asymmetric catalysis, Hydrogenation, Ruthenium, Ketones
Introduction
Because of the significant role that enantiopure compounds have in biological applications, there is tremendous interest in research with regard to their stereospecific synthesis. The stereoselective reduction of carbonyls and imines are among the most important transformations in asymmetric synthesis, as chiral alcohols can be widely used as starting materials for further synthetic transformations.[1] Consequently, a variety of options for this reaction have been investigated in search of the best conditions with respect to conversion, enantioselectivity, atom efficiency, and sustainability.
Among catalytic procedures that have emerged in recent years, asymmetric transfer hydrogenations (ATH) are a particularly attractive strategy, as they provide efficient access to chiral alcohols by using small and nonhazardous organic molecules as the hydrogen donor.[2] The use of water-soluble sodium formate as a hydrogen donor is particularly attractive, as it provides not only an inexpensive and nonreversible hydrogen source but also the opportunity to run the reaction in aqueous medium.[3] Driven by the trend of modifying organic reaction procedures to have greener conditions, the replacement of volatile organic solvents with water is presently a rapidly growing area of research.[4] In addition to the environmental benefits, water has been shown to influence reaction rates and the selectivity of many organic transformations.[5] Consequently, considerable effort has been put into the development of new water-soluble catalysts and ligands for ATH reactions.[3,6]
Still, many known asymmetric reactions have drawbacks such as the comparatively high price of the chiral ligands as well as catalyst leaching, and, therefore, the optimization and improvement of these methods is a constant matter of investigation.[7] Chiral ionic liquids (CILs) represent an innovative approach to deal with some of the drawbacks, as they can be used as a sole source of chirality in asymmetric reactions.[8] Their unique structures allow for the activity of the ligand or catalyst in hand to be fine-tuned to the given reaction conditions.[9] The adaptation of properties, especially solvation behavior, is difficult with conventional catalysts, which makes chiral ionic liquids particularly attractive in this field of study. With the employment of chiral ionic liquids, there is also the possibility of recycling the catalyst (Figure1).
Figure 1.

Concept and benefits of coordinating chiral ionic liquids with tunable properties (IL-pre = ionic liquid precursor).
In addition, the ionic structure, high degree of organization, and hydrogen-bonded supramolecular network inherent to ionic liquids have already been proposed as features of chiral solvents that might allow for a significant transfer of chirality in an asymmetric synthesis as well as for its ease of separation from the reaction mixture.[10] Hence, it was the wide field of asymmetric organocatalysis that gave access to these highly enantioselective reactions that were catalyzed by chiral ionic liquids, along with the extra benefit of recycling the chiral catalyst.[11] Soon applications in other asymmetric reactions followed, and successful examples for the use of chiral ionic liquids as a catalyst, solvent, or ligand included reactions as diverse as asymmetric hydrogenations,[12] aza-Baylis–Hillman reactions,[13] sulfoxide oxidations,[14] and alkylation reactions.[15]
Inspired by the outstanding progress with ruthenium-catalyzed asymmetric transfer hydrogenation reactions in the past years, a new approach that featured hydrophilic chiral ionic liquids was explored. The modular design of chiral ionic liquids with variable anions is a clear advantage, as chiral ligands with adaptable solubility can be designed for the desired reaction conditions in aqueous asymmetric transfer hydrogenations that use sodium formate as the hydrogen source. Herein, we present the design, synthesis, and application of the highly coordinating amino alcohol derived chiral ionic liquids that, on the basis of their tunable properties, can be applied as the sole chirality source in asymmetric transfer hydrogenations under aqueous conditions.[16]
Results and Discussion
As demonstrated in the literature, chiral amino alcohols are increasingly considered for asymmetric transfer hydrogenations.[17] In contrast to chiral diamines, which are preferably used as ligands in this reaction,[2] amino alcohols can often be directly obtained from the chiral pool, and, hence, they provide an inexpensive and attractive alternative for the often expensive chiral diamine ligands. Furthermore, amino alcohol based ligands in ligand-accelerated RuII-catalyzed ATH reactions have result in higher reaction rates than those that have used other chiral ligands such as diamines, 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP), or phosphorus-based ligands. Excellent selectivities were also observed.[17c,18] Previously, we reported the design of highly coordinating chiral ionic liquids with a tertiary amino alcohol substructure to be applied as recyclable ligands in asymmetric diethylzinc additions.[15] Consequently, the concept of coordinating chiral ionic liquids with an amino alcohol structure was further adapted and expanded to provide hydrophilic chiral ligands grafted onto a cationic head group for future applications in transfer hydrogenations under aqueous conditions.
However, the application in asymmetric transfer hydrogenations requires a secondary amine functionality to form a six-membered transition state with the carbonyl substrate (Figure2). A small set of chiral ionic liquids with the secondary amino alcohol substructure were therefore developed, which relied on grafting an ionic liquid precursor to an already established chiral ligand followed by a selective alkylation to form an ionic liquid (Scheme 1). We selected three different chiral primary amino alcohols as starting materials. Norephedrine (1), 2-amino-1,2-diphenylethanol (6), and 2-amino-1-indanol (9) were treated with pyridine-2-carbaldehyde in the presence of activated molecular sieves and subsequently reduced with sodium borohydride to obtain the chiral ionic liquid precursors. As already shown in the case of chiral ionic liquid mediated asymmetric alkylations, we chose a pyridinium system as the ionic liquid head group, as it avoids the presence of acidic protons that are found in common imidazolium cations, which have an unpredictable influence on transition-metal catalysis because of carbene formation.[19] The enantiopure N,N,O-tridentate ligands were further converted into the ionic liquid through quarternization with n-butyl bromide at 50 °C under solvent-free conditions. The obtained halide ionic liquids are highly hydrophilic and provide water solubility for the active ruthenium complex.
Figure 2.
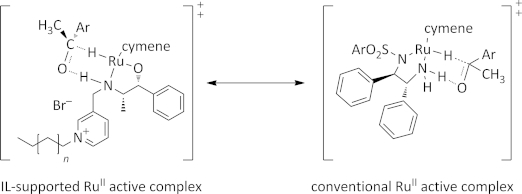
Design of chiral ionic liquid-supported complex vs conventional complex (IL = ionic liquid).
Scheme 1.

Synthesis of amino alcohol derived chiral ionic liquids for asymmetric transfer hydrogenation.
Additionally, to investigate the influence of the self-assembly of the catalyst complex in water, different alkyl chain lengths were installed at the pyridinium moiety in the case of norephedrine-derived chiral ionic liquids 3–5. The different alkyl chain lengths might further influence their catalytic performance as ligands in the ATH reaction. We have already shown that the self-assembly of ionic liquids in water can result in increased reaction rates.[20]
The successful selective alkylation of the pyridine nitrogen atom is the key step in the preparation of these highly coordinating chiral ionic liquids. In contrast to the alkylation of tertiary N,N,O-ligands such as the methylated ephedrine derivative, the alkylation of secondary amino alcohols 1, 6, and 9 with n-butyl bromide did not occur selectively at the pyridine nitrogen atom. The quaternization of 2, 7, and 10 with n-butyl bromide at 50 °C gave a mixture of the mono- and dialkylated derivatives. However, the pyridinium cation was still formed preferentially, and the ratio of the mono-/dialkylated species was approximately 2:1, as calculated by 1H NMR analysis (Figure3). When the amount of n-butyl bromide was decreased to 1 equiv. and the reaction temperature was lowered to 40 °C, a double alkylation was still observed. The mono- and dialkylated species in the mixture were eventually separated by preparative HPLC, and the isolated yields of the desired final chiral ionic liquids were still in an acceptable range of 45–64 %. Preparative HPLC was performed under reversed-phase conditions by using methanol and water as the mobile phase on a C18 column.
Figure 3.

1H NMR spectra after alkylation of substrate 7, which afforded a mixture of mono- and dialkylated species (top). Purification by preparative HPLC afforded the monoalkylated species (bottom).
To evaluate these new hydrophilic chiral ionic liquids in an asymmetric transfer hydrogenation reaction, the reduction of acetophenone 12 by using 5 mol-% [Ru(p-cymene)Cl2]2 as the catalyst with sodium formate as the hydrogen source was chosen as the model system. Initially, we evaluated the reaction for the ideal conditions with respect to temperature and concentration of acetophenone in water (Scheme 2).
Scheme 2.

Asymmetric transfer hydrogenation reaction of acetophenone in the presence of a chiral ionic liquid.
Varying the concentration of chiral ionic liquid 3 revealed that a range from 0.5 to 2 m was suitable to observe the complete conversion of acetophenone 12 after 48 h with good enantioselectivities of 68–75 % ee (enantiomeric excess). More dilute conditions resulted in a significant decrease in the reaction rate (Table 1, Entries 1–6).
Table 1.
Optimization of reaction conditions[a]
| Entry | T [°C] | Conc. of3[m] | % Conversion[b]/isol. yield[c] | % ee[d][e] |
|---|---|---|---|---|
| 1 | 50 | 2 | 97 | 73 (R) |
| 2 | 50 | 1 | >99/99 | 68 (R) |
| 3 | 50 | 0.5 | 98/97 | 71 (R) |
| 4 | 50 | 0.25 | 86 | 68 (R) |
| 5 | 50 | 0.1 | 51 | 46 (R) |
| 6 | 50 | 0.05 | 27 | 66 (R) |
| 7 | 30 | 0.5 | 85 | 75 (R) |
| 8 | 40 | 0.5 | >99/97 | 75 (R) |
| 9 | 60 | 0.5 | >99/96 | 63 (R) |
| 12 | 70 | 0.5 | 76 | 40 (R) |
Performed with acetophenone (2 mmol), sodium formate (10 mmol), chiral ionic liquid 3 (12 mol-%), and RuII catalyst (5 mol-%) for 48 h.
Conversion determined by GC analysis.
Isolated yields after flash column chromatography.
Determined by HPLC analysis using a DAICEL Chiralcel IB column.
Absolute configuration determined by optical rotation data and comparing with literature values.
Changing the reaction temperature was observed to have a strong influence on the enantioselectivity, as a higher reaction temperature gave lower enantiomeric excess values. Because the reaction reached completion after 24 h in most of the cases, an increase in the temperature to over 50 °C was not necessary. Eventually, the ideal reaction conditions were identified as a 0.5 m solution of acetophenone 12 in degassed water at 50 °C, and these conditions were employed to investigate the influence of different hydrophilic chiral ionic liquids. As can be seen from Table 2, the best results were obtained with the ephedrine-derived hydrophilic chiral ionic liquids. Aminodiphenylethanol-derived system 8 also performed quite well, whereas indanol-based chiral ionic liquid 11 gave a lower conversion and selectivity.
Table 2.
Variation of chiral ionic liquids for asymmetric transfer hydrogenation reaction[a]
| Entry | CIL | Core structure | % Conv.[b]/isol. yield[c] | % ee[d][e] |
|---|---|---|---|---|
| 1 | 3 | ephedrine, n = 4 | 94/93 | 71 (R) |
| 2 | 4 | ephedrine, n = 8 | 98/98 | 72 (R) |
| 3 | 5 | ephedrine, n = 12 | >99/98 | 71 (R) |
| 4 | 8 | aminodiphenylethanol | 83/80 | 47 (S) |
| 5 | 11 | indanol | 28/22 | 28 (R) |
| 6[f] | 3 | ephedrine, n = 4 | 61 | 63 (R) |
| 7[f] | 3 | ephedrine, n = 4 | 25 | 38 (R) |
Performed with acetophenone (2 mmol), sodium formate (10 mmol), chiral ionic liquid (12 mol-%), and RuII catalyst (5 mol-%) for 24 h.
Conversion determined by GC analysis.
Isolated yields after flash column chromatography.
Determined by HPLC analysis using a DAICEL Chiralcel IB column.
Absolute configuration determined by optical rotation data and comparing with literature values.
Reaction was carried out at 60 °C with recycled catalyst (1 and 2×).
In a further experiment, the reaction with the chiral ionic liquid supported Ru catalyst 3 was compared to that with the neutral ephedrine ligand 2, that is, the product prior to the quaternization step. Although there was no difference in the enantioselectivity of the two systems, the reaction did proceed significantly faster with the salt form of the ligand. In the case of ephedrine-derived ionic liquid 3, the reaction reached complete conversion after 7 h, whereas the same reaction with neutral ephedrine ligand 2 only reached 60 % conversion after this period of time, thereby emphasizing the merit of a hydrophilic chiral ionic liquid. The influence of different chain lengths however seems to have a limited effect in the reaction, as ephedrine-derived chiral ionic liquids 3, 4, and 5 with different alkyl chain lengths gave similar results (Table 2, Entries 1–3). This is surprising, given the fact that dynamic light scattering (DLS) experiments clearly indicate the presence of ruthenium metallomicelles for N-dodecylpyridinium bromide 5, which were not observed with the CILs of shorter alkyl chain length (Figure4).
Figure 4.

Dynamic light scattering of chiral ionic liquid 5 and chiral ionic liquid based ruthenium metallomicelles. Reagents and conditions: chiral ionic liquid 5 (0.02 mmol) with and without [Ru(p-cymene)Cl2]2 (0.01 mmol) in H2O (4 mL).
In principle, the concept of hydrophilic ionic liquids also holds potential for recycling the precious chiral ruthenium species, as the ionic liquid bound catalyst is immobilized in the bulk aqueous layer. We consequently investigated recycling of the active catalyst in asymmetric transfer hydrogenation and extracted the obtained chiral alcohol derivative from the aqueous layer containing the chiral ionic liquid with small amounts of degassed n-hexane. After phase separation, the ruthenium-containing aqueous layer could indeed be recycled for use in a consecutive asymmetric transfer hydrogenation reaction (Table 2, Entries 6 and 7). However, losses to both the conversion and enantioselectivity occurred in the third run, which indicates that the recycling protocol under these conditions suffers from catalyst leaching and still needs to be optimized.
To investigate the substrate scope, a series of prochiral ketones were chosen that contained both electron-withdrawing and electron-rich substituents. With the exception of propiophenone, moderate to good conversions and good selectivities of >60 % ee were obtained by using chiral ionic liquid 3 (Table 3).
Table 3.
Investigation of substrate scope[a]
| Entry | Substrate | % Conv.[b]/isol. yield[c] | % ee[d][e] |
|---|---|---|---|
| 1 | propiophenone | 34/30 | 65 (R) |
| 2 | 2-nitroacetophenone | 90/86 | 26 (R) |
| 3 | 3-nitroacetophenone | 93/89 | 70 (R) |
| 4 | 4-nitroacetophenone | 89/87 | 68 (R) |
| 5 | 4-bromoacetophenone | 77/76 | 73 (R) |
| 6 | 4-chloroacetophenone | 98/95 | 67 (R) |
| 7 | 4-methoxyacetophenone | 64/62 | 60 (R) |
| 8 | 1-indanone | 76/71 | 85 (S) |
| 9 | 1-acetonaphthone | 86/85 | 71 (S) |
Performed with acetophenone (2 mmol), sodium formate (10 mmol), chiral ionic liquid 3 (12 mol-%), and RuII catalyst (5 mol-%) for 24 h.
Conversion determined by GC analysis.
Isolated yields after flash column chromatography.
Determined by HPLC analysis using a DAICEL Chiralcel IB column.
Absolute configuration determined by optical rotation data and comparing with literature values.
Conclusions
Chiral ionic liquids can be efficiently synthesized from naturally occurring enantiopure starting materials and can demonstrate strong chiral interactions. This characteristic in combination with the tunable nature of ionic liquids reveals new solutions for asymmetric synthesis. Herein, we showed that amino alcohol derived chiral ionic liquids provide an attractive alternative to conventional ligands in asymmetric ruthenium-catalyzed transfer hydrogenations. The modular design based on the chiral pool derived amino alcohol norephedrine allowed the formation of highly coordinating chiral ionic liquids, and the adaptation of its solubility to the given reaction conditions could be obtained by the choice of anion. When employed as a sole source of chirality in asymmetric transfer hydrogenation reactions, the chiral ionic liquids provided excellent enantioselectivities and yields. Our current research focuses on the development of improved recycling strategies for ionic liquid bound transition-metal catalysts that are immobilized in the aqueous layer as well as on more detailed investigations towards the merit of surface-active chiral ionic liquids, which is an active area of research for our group.
Experimental Section
General Methods: All reagents were purchased from commercial suppliers and used without further purification unless otherwise noted. CH2Cl2, Et2O, and MeOH that were intended for water-free reactions were predistilled and dried on Al2O3 columns (PURESOLV, Innovative Technology). Column chromatography was performed on a Büchi Sepacore Flash System (2 × Büchi Pump Module C-605, Büchi Pump Manager C-615, Büchi UV Photometer C-635, Büchi Fraction Collector C-660) with mixtures of petroleum ether (PE)/ethyl acetate (EtOAc) or MeOH/CH2Cl2 as the eluents. Preparative HPLC was performed on a Shimazu preparative HPLC that was equipped with a SDP20A PDA detector by using a C18(2) column (250 × 21.20 cm ID) and MeOH/H2O as the eluent at a flow rate of 20 mL min–1. TLC analysis was carried out with precoated aluminum-backed plates that were purchased from Merck (silica gel 60 F254). The 1H and 13C NMR spectroscopic data were recorded with a Bruker AC 200 (200 MHz) or a Bruker Advance UltraShield 400 (400 MHz) spectrometer, and CDCl3 or CD3OD was used as the NMR solvent. The chemical shifts (δ) are reported in ppm by using tetramethylsilane as the internal standard, and coupling constants (J) are reported in Hertz [Hz]. The following abbreviations are used to explain the signal multiplicities: s (singlet), d (doublet), t (triplet), q (quartet), quin (quintet), sext (sextet), m (multiplet), br. s (broad singlet). Infrared spectra were recorded on a Perkin–Elmer Spectrum 65 FT IR spectrometer that was equipped with a Specac MK II Golden Gate Single Reflection ATR (attenuated total reflectance) unit. Optical rotations were measured on an Anton Paar MCP500 polarimeter at the specified conditions. Thermal stabilities were determined on a Netzsch TGA in a range of 25 to 500 °C with a heating rate of 10 °C min–1. Decomposition temperatures (T5 %onset) were reported from the onset to a 5 wt.-% loss of mass. Melting points above room temperature were measured on a Kofler hot-stage microscope or on an automated melting point system OPTI MELT of Stanford Research Systems. Elemental analysis was performed at Vienna University, Department of Physicochemistry - Laboratory for Microanalysis, Währingerstraße 42, 1090 Vienna. GC analysis was conducted with a Thermo Finnigan Focus GC/DSQ II that was equipped with a flame ionization detector (FID) detector and used a standard capillary column BGB5 (30 m × 0.32 mm ID) at a flow rate of 2.0 mL min–1 and a split ratio of 20 with helium as the carrier gas. The enantiomeric excess values were determined by HPLC analysis on a DAIONEX UPLC that was equipped with a photodiode array (PDA) plus detector (190–360 nm) and by using a DAICEL IB column (250 × 4.60 mm) as the stationary phase, mixtures of n-heptane/iPrOH as the solvent, and flow rates of 0.5–1.0 mL min–1.
(1R,2S)-1-Phenyl-2-[(pyridin-3-ylmethyl)amino]propan-1-ol (2): Freshly distilled pyridine-3-carbaldehyde (0.31 mL, 3.31 mmol) was added to a mixture of (1R,2S)-norephedrine (1, 0.50 g, 3.31 mmol) and activated molecular sieves (3 Å, 1.50 g) in anhydrous methanol (50 mL), and the resulting mixture was heated at reflux for 14 h until TLC analysis showed full conversion of the starting material. Sodium borohydride (0.14 g, 3.64 mmol) was then added in small portions, and the mixture was stirred at room temperature until the TLC plate showed complete conversion of the starting material. The reaction mixture was filtered through silica and diluted with distilled H2O. Methanol was removed under reduced pressure, and the aqueous layer was extracted with CH2Cl2. The product was further purified by MPLC (100 g silica, CH2Cl2/MeOH, 6:1) to give product 2 (0.65 g, 2.1 mmol, 80 % yield) as a colorless solid. Data was in accordance with the literature.[15] 1H NMR (400 MHz, CD3OD): δH = 8.46–8.40 (m, 2 H, H-pyridine), 7.78–7.73 (m, 1 H, H-pyridine), 7.41–7.25 (m, 5 H, H-Ar), 4.70 (d, J = 5.00 Hz, CH-OH), 3.82 (dd, J1 = 5.28 Hz, J2 = 32.50 Hz, 2 H, CH2-NH), 2.92–2.79 (m, 1 H, CH-CH3), 1.04 (d, J = 6.46 Hz, 3 H, CH3) ppm.
1-Butyl-3-({[(1R,2S)-1-hydroxy-1-phenylpropan-2-yl]amino}methyl)pyridin-1-ium Bromide (3): Compound 2 (1.14 g, 4.71 mmol) and freshly distilled n-butyl bromide (0.51 mL, 4.71 mmol) were mixed in a round-bottom flask, which was sealed, and the resulting mixture was stirred at 60 °C for 24 h. The excess amount of n-butyl bromide was evaporated, and the brown oil was washed (2×) with anhydrous ethyl acetate. The remaining volatile materials were removed under reduced pressure at 60 °C to give the crude product, which was purified by preparative HPLC (MeOH/H2O, 50:50) to give compound 3 (0.89 g, 2.58 mmol, 55 % yield) as a light yellow oil. 1H NMR (400 MHz, CD3OD): δH = 8.92 (s, 1 H, H-pyridine), 8.85 (d, J = 6.00 Hz, 1 H, H-pyridine), 8.46 (d, J = 8.05 Hz, 1 H, H-pyridine), 8.03–7.96 (m, 1 H, H-pyridine), 7.40–7.25 (m, 5 H, H-Ar), 4.72 (d, J = 4.80 Hz, CH-OH), 4.60 (t, J = 7.54 Hz, 2 H, CH2-N), 4.08 (dd, J1 = 6.60 Hz, J2 = 37.28 Hz, 2 H, CH2-NH), 2.98–2.86 (m, 1 H, CH-CH3), 2.07–1.91 (m, 2 H, CH2-CH2-N), 1.42 (sext, J = 7.44 Hz, 2 H, CH2-CH2-CH2-N), 1.08–0.98 (m, 6 H, CH3, CH3-CH2-CH2-CH2-N) ppm. 13C NMR (100 MHz, CD3OD): δC = 143.4 (2 d, C-Ar), 142.5 (d, C-Ar), 141.3 (d, C-Ar), 141.2 (s, C-Ar), 126.6 (s, C-Ar), 126.5 (2 d, C-Ar), 126.0 (d, C-Ar), 124.9 (2 d, C-Ar), 73.4 (d, CH-OH), 60.0 (t, CH2-NH), 57.2 (d, CH-CH3), 45.4 (t, CH2-N+), 31.6 (t, CH2-CH2-CH2-CH3), 17.6 (t, CH2-CH2-CH2-CH3), 12.2 (q, CH2-CH2-CH2-CH3), 11.0 (q, CH-CH3) ppm. [α]D20 = +3.52 (c = 0.79, MeOH). T5 %onset: 213 °C. IR:  max = 3276 (O–H), 1633 (C–C), 1593 (C–C), 1449 (C=C), 1116 (NH–CH3), 702 (C–H-Ar) cm–1. C19H27BrN2O (379.34): calcd. C 60.16, H 7.17, N 7.38; calcd. (including 0.69 × H2O) C 58.25, H 7.30, N 7.15; found C 58.27, H 6.99, N 7.38.
max = 3276 (O–H), 1633 (C–C), 1593 (C–C), 1449 (C=C), 1116 (NH–CH3), 702 (C–H-Ar) cm–1. C19H27BrN2O (379.34): calcd. C 60.16, H 7.17, N 7.38; calcd. (including 0.69 × H2O) C 58.25, H 7.30, N 7.15; found C 58.27, H 6.99, N 7.38.
3-({[(1R,2S)-1-Hydroxy-1-phenylpropan-2-yl]amino}methyl)-1-octylpyridin-1-ium Bromide (4): By following the procedure for compound 3, compound 2 (0.46 g, 1.89 mmol) and n-octyl bromide (0.33 mL, 1.89 mmol) afforded the crude product as a viscous brown oil, which was purified by preparative HPLC (MeOH/H2O, 50:50) to give 4 (0.35 g, 0.92 mmol, 49 % yield) as a light yellow oil. 1H NMR (400 MHz, CD3OD): δH = 8.94 (s, 1 H, H-pyridine), 8.86 (d, J = 5.98 Hz, 1 H, H-pyridine), 8.48 (d, J = 8.05 Hz, 1 H, H-pyridine), 8.03–7.96 (m, 1 H, H-pyridine), 7.40–7.25 (m, 5 H, H-Ar), 4.89 (d, J = 4.89 Hz, 1 H, CH-OH), 4.59 (t, J = 7.54 Hz, 2 H, CH2-N), 4.09 (dd, J1 = 6.10 Hz, J2 = 36.82 Hz, 2 H, CH2-NH), 3.00–2.88 (m, 1 H, CH-CH3), 2.04–2.01 (m, 2 H, CH2-CH2-N), 1.38–1.30 (m, 10 H, CH2-CH2-CH2-CH2-CH2-N), 1.06 (d, J = 6.60 Hz, 3 H, CH3), 0.93–0.86 (m, 3 H, CH3-alkyl-N) ppm. 13C NMR (100 MHz, CD3OD): δC = 148.6 (2 d, C-Ar), 147.7 (d, C-Ar), 146.5 (d, C-Ar), 141.4 (s, C-Ar), 134.5 (s, C-Ar), 129.6 (2 d, C-Ar), 129.0 (d, C-Ar), 127.0 (2 d, C-Ar), 71.5 (d, CH-OH), 63.6 (t, CH2-NH), 61.6 (d, CH-CH3), 43.3 (t, CH2-N+), 33.9 [t, (CH2)10-CH2], 32.4 [t, (CH2)9-CH2], 30.2/30.1 [2 t, (CH2)7-CH2, (CH2)8-CH2], 27.7 [t, (CH2)6-CH2], 20.5 [t, (CH2)5-CH2], 20.1 [t, (CH2)4-CH2], 15.6 [t, (CH2)3-CH2], 14.4 [q, (CH2)12-CH3], 10.1 (q, CH-CH3) ppm. [α]D20 = = +6.75 (c = 0.77, MeOH). T5 %onset: 200 °C. IR:  max = 3294 (N–H), 2925 (O–H), 1632 (C–C), 1535 (C–C), 1498 (C=C), 1197 (NH–CH3), 702 (C–H-Ar) cm–1. C23H35BrN2O (435.45): calcd. C 63.44, H 8.30, N 6.43; calcd. (including 4.71 × H2O) C 53.95, H 8.76, N 5.24; found C 53.97, H 8.53, N 4.97.
max = 3294 (N–H), 2925 (O–H), 1632 (C–C), 1535 (C–C), 1498 (C=C), 1197 (NH–CH3), 702 (C–H-Ar) cm–1. C23H35BrN2O (435.45): calcd. C 63.44, H 8.30, N 6.43; calcd. (including 4.71 × H2O) C 53.95, H 8.76, N 5.24; found C 53.97, H 8.53, N 4.97.
1-Dodecyl-3-({[(1R,2S)-1-hydroxy-1-phenylpropan-2-yl]amino}methyl)pyridin-1-ium Bromide (5): By following the procedure for compound 3, compound 2 (1.14 g, 4.70 mmol) and n-dodecyl bromide (1.13 mL, 4.70 mmol) afforded the crude product as a viscous brown oil, which was purified by preparative HPLC (MeOH/H2O, 50:50) to give 5 (1.05 g, 2.77 mmol, 59 % yield) as a light yellow oil. 1H NMR (400 MHz, CD3OD): δH = 8.89 (s, 1 H, H-pyridine), 8.84 (d, J = 6.06 Hz, 1 H, H-pyridine), 8.45 (d, J = 8.15 Hz, 1 H, H-pyridine), 8.02–7.95 (m, 1 H, H-pyridine), 7.40–7.25 (m, 5 H, H-Ar), 4.69 (d, J = 4.89 Hz, CH-OH), 4.58 (t, J = 7.53 Hz, 2 H, CH2-N), 4.05 (dd, J1 = 6.90 Hz, J2 = 37.66 Hz, 2 H, CH2-NH), 2.94–2.81 (m, 1 H, CH-CH3), 2.04–1.97 (m, 2 H, CH2-CH2-N), 1.38–1.30 (m, 18 H, CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-CH2-N), 1.06 (d, J = 6.65 Hz, 3 H, CH3), 0.93–0.87 (m, 3 H, CH3-alkyl-N) ppm. 13C NMR (100 MHz, CD3OD): δC = 147.0 (2 d, C-Ar), 146.1 (d, C-Ar), 144.9 (d, C-Ar), 140.2 (s, C-Ar), 128.2 (2 d, C-Ar), 128.0 (s, C-Ar), 127.5 (d, C-Ar), 125.7 (2 d, C-Ar), 70.4 (d, CH-OH), 62.2 (t, CH2-NH), 62.1 (d, CH-CH3), 52.2 (t, CH2-N+), 45.5 [2 t, (CH2)11-CH2], 31.5 [t, (CH2)9-CH2], 31.2 [t, (CH2)8-CH2], 28.8/28.7 [2 t, (CH2)6-CH2, (CH2)7-CH2], 25.9 [t, (CH2)5-CH2], 22.3 [t, (CH2)4-CH2], 13.0 [t, (CH2)3-CH2], 9.08 [t, (CH2)2-CH2], 7.8 [q, (CH2)12-CH3], 6.3 (q, CH-CH3). [α]D20 = +0.58 (c = 0.97, MeOH). T5 %onset: 225 °C. IR:  max = 3305 (N–H), 2922 (O–H), 1632 (C–C), 1634 (C–C), 1498 (C=C), 1198 (NH–CH3), 702 (C–H-Ar) cm–1. C27H43BrN2O (491.56): calcd. C 65.97, H 8.82, N 5.70; found C 65.89, H 8.52, N 6.01.
max = 3305 (N–H), 2922 (O–H), 1632 (C–C), 1634 (C–C), 1498 (C=C), 1198 (NH–CH3), 702 (C–H-Ar) cm–1. C27H43BrN2O (491.56): calcd. C 65.97, H 8.82, N 5.70; found C 65.89, H 8.52, N 6.01.
(1S,2R)-1,2-Diphenyl-2-[(pyridin-3-ylmethyl)amino]ethanol (7): By following the procedure for compound 2, (1S,2R)-2-amino-1,2-diphenylethanol (6, 2.00 g, 9.33 mmol), freshly distilled pyridine-3-carbaldehyde (0.88 mL, 9.33 mmol), activated molecular sieves (3 Å, 5.00 g), and sodium borohydride (0.35 g, 9.33 mmol) in anhydrous methanol (100 mL) were employed to give the product, which was further purified by MPLC (200 g silica, CH2Cl2/MeOH, 40:1 + Et3N) to give 7 (2.52 g, 8.28 mmol, 89 % yield) as a colorless solid. Crystallization from toluene gave colorless crystals. Data was in accordance with the literature.[15] 1H NMR (400 MHz, CD3OD): δH = 8.38 (d, J = 4.78 Hz, 1 H, H-pyridine), 8.28 (s, 1 H, H-pyridine), 7.42–7.38 (m, 1 H, H-pyridine), 7.18–7.10 (m, 11 H, H-Ar), 4.71 (d, J = 6.06 Hz, 1 H, CH-OH), 3.81 (d, J = 6.06 Hz, 1 H, CH-NH), 3.60 (d, J = 13.67 Hz, 1 H, CH2-NH), 3.43 (d, J = 13.69 Hz, 1 H, CH2), 2.80 (br. s, 1 H, OH), 1.68 (br. s, 2 H, NH2) ppm.
1-Butyl-3-({[(1S,2R)-2-hydroxy-1,2-diphenylethyl]amino}methyl)pyridin-1-ium Bromide (8): By following the procedure for compound 3, compound 7 (0.40 g, 1.31 mmol) and n-butyl bromide (0.14 mL, 1.31 mmol) gave the crude product as a viscous yellow oil, which was was purified by preparative HPLC (MeOH/H2O, 80:20) to yield product 8 (0.31 g, 0.79 mmol) as a light yellow liquid. 1H NMR (200 MHz, CD3OD): δH = 9.33 (s, 1 H, H-pyridine), 9.06 (d, J = 5.76 Hz, 1 H, H-pyridine), 8.11 (d, J = 7.89 Hz, 1 H, H-pyridine), 7.86–7.79 (m, 1 H, H-pyridine), 7.17–7.06 (m, 5 H, H-Ar), 5.09 (d, J = 3.91 Hz, CH-OH), 4.74 (t, J = 7.25 Hz, 2 H, CH2-N), 3.97 (d, J = 4.00 Hz, CH-NH), 3.86 (s, 2 H, CH2-NH), 2.91 (br. s, 1 H, NH), 2.98–2.86 (m, 1 H, CH-CH3), 2.00–1.85 (m, 2 H, CH2-CH2-N), 1.40–1.29 (m, 2 H, CH2-CH2-CH2-N), 0.92 (t, J = 7.33 Hz, 3 H, CH3-CH2-CH2-CH2-N) ppm. 13C NMR (50 MHz, CD3OD): δC = 146.5 (d, C-Ar), 144.4 (d, C-Ar), 143.5 (s, C-Ar), 140.9 (d, C-Ar), 130.0 (s, C-Ar), 129.2 (s, C-Ar), 129.0 (d, C-Ar), 128.8 (d, C-Ar), 128.7 (d, C-Ar), 128.3 (d, C-Ar), 78.3 (d, CH-OH), 69.9 (d, CH-N), 62.8 (t, CH2-CH2-CH2), 55.7 (t, CH2), 34.4 (t, CH2-CH2-CH2), 20.5 (t, CH2-CH2-CH2), 13.8 (q, CH3-CH2-CH2) ppm. [α]D20 = +9.66 (c = 1.02, MeOH). T5 %onset: 218 °C. IR:  max = 3250 (N–H), 2707 (O–H), 1598 (C–C), 1357 (C=C), 1191 (NH–CH3), 702 (C–H-Ar) cm–1. C24H29BrN2O (441.41): calcd. C 65.31, H 6.62, N 6.35; calcd. (including 2.62 × H2O) C 61.25, H 6.91, N 5.95; found C 61.29, H 7.07, N 5.81.
max = 3250 (N–H), 2707 (O–H), 1598 (C–C), 1357 (C=C), 1191 (NH–CH3), 702 (C–H-Ar) cm–1. C24H29BrN2O (441.41): calcd. C 65.31, H 6.62, N 6.35; calcd. (including 2.62 × H2O) C 61.25, H 6.91, N 5.95; found C 61.29, H 7.07, N 5.81.
(1S,2R)-2-[(Pyridin-3-ylmethyl)amino]-2,3-dihydro-1H-inden-1-ol (10): By following the procedure for compound 2, (1S,2R)-aminoindanol (9, 0.51 g, 3.41 mmol), pyridine-3-carbaldehyde (0.32 mL, 3.41 mmol), activated molecular sieves (3 Å, 2.00 g), and sodium borohydride (0.13 g, 3.41 mmol) were employed to give the crude product, which was purified by MPLC (CH2Cl2/MeOH, 6:1) to afford 10 (0.74 g, 3.08 mmol, 59 % yield) as colorless crystals; m.p. 90–93 °C. 1H NMR (200 MHz, CD3OD): δH = 8.64 (d, J = 2.00 Hz, 1 H, H-pyridine), 8.55 (dd, J1 = 1.59 Hz, J2 = 4.81 Hz, 1 H, H-pyridine), 7.78 (td, J1 = 1.84 Hz, J2 = 7.45 Hz, 1 H, H-pyridine), 7.34–7.25 (m, 5 H, H-Ar), 4.48–4-42 (m, 1 H, CH-NH), 4.14 (d, J = 5.28 Hz, CH-OH), 4.04 (s, 2 H, CH2-NH), 3.15–2.92 (m, 2 H, CH2-indol), 2.85 (br. s, 1 H, NH) ppm. 13C NMR (50 MHz, CD3OD): δC = 149.6 (d, C-pyridine), 148.8 (d, C-pyridine), 142.0 (s, C-Ar), 140.9 (s, C-pyridine), 136.0 (d, C-pyridine), 135.2 (s, C-Ar), 128.2 (C-pyridine), 126.8 (d, C-Ar), 125.7 (d, C-Ar), 123.9 (d, C-Ar), 123.6 (d, C-Ar), 71.0 (d, CH-OH), 65.3 (d, CH-NH), 49.8 (t, CH2-NH), 39.7 (t, CH2-indanol) ppm. [α]D20 = –9.84 (c = 0.89, MeOH). T5 %onset: 255 °C. IR:  max = 3300 (N–H), 2916 (O–H), 1737 (C–C), 1456 (C=C), 729 (C–H-Ar) cm–1. C15H16N2O (240.31): calcd. C 74.97, H 6.71, N 11.66; found C 75.00, H 6.55, N 11.66.
max = 3300 (N–H), 2916 (O–H), 1737 (C–C), 1456 (C=C), 729 (C–H-Ar) cm–1. C15H16N2O (240.31): calcd. C 74.97, H 6.71, N 11.66; found C 75.00, H 6.55, N 11.66.
1-Butyl-3-({[(1S,2R)-1-hydroxy-2,3-dihydro-1H-inden-2-yl]amino}methyl)pyridin-1-ium Bromide (11): By following the procedure for compound 3, compound 10 (0.81 g, 3.35 mmol) and n-butyl bromide (0.36 mL, 3.35 mmol) gave 11 (0.78 g, 2.06 mmol, 61 % yield) as a viscous brown oil. 1H NMR (200 MHz, CD3OD): δH = 9.17 (s, 1 H, 8.64, H-pyridine), 8.94 (d, J = 6.06 Hz, 1 H, H-pyridine), 8.70 (d, J = 8.03 Hz, 1 H, H-pyridine), 8.11–8.08 (m, 1 H, H-pyridine), 7.53–7.25 (m, 5 H, H-Ar), 4.70–4.63 (m, 3 H, CH2-N+, CH2a-NH), 4.38–4.30 (m, 3 H, CH-OH, CH2b-NH, CH-NH), 3.10–2.92 (m, 2 H, CH2-indanol), 2.03 (m, J = 7.58 Hz, 2 H, CH2-CH2-CH2-CH3), 1.50–1.38 (m, 2 H, CH2-CH2-CH2-CH3), 1.02 (t, J = 7.23 Hz, 3 H, CH2-CH2-CH2-CH3) ppm. 13C NMR (50 MHz, CD3OD): δC = 149.6 (d, C-pyridine), 148.8 (d, C-pyridine), 142.0 (s, C-Ar), 140.9 (s, C-pyridine), 136.0 (d, C-pyridine), 135.2 (s, C-Ar), 129.4 (C-pyridine), 128.9 (d, C-Ar), 127.8 (d, C-Ar), 126.4 (d, C-Ar), 126.0 (d, C-Ar), 72.8 (d, CH-OH), 66.8 (d, CH-NH), 62.9 (t, CH2-N+), 40.5 (t, CH2-NH), 34.4 (t, CH2-indanol), 20.5 (t, CH2-CH2-CH3), 13.8 (q, CH3) ppm. [α]D20 = –2.04 (c = 0.47, MeOH). T5 %onset: 231 °C. IR:  max = 3264 (N–H), 2929 (O–H), 1637 (C–C), 1634 (C–C), 1456 (C=C), 729 (C–H-Ar) cm–1. C19H25BrN2O (377.33): calcd. C 60.48, H 6.68, N 7.42; calcd. (including 1.47 × H2O) C 56.54, H 6.97, N 6.94; found C 56.48, H 6.45, N 6.90.
max = 3264 (N–H), 2929 (O–H), 1637 (C–C), 1634 (C–C), 1456 (C=C), 729 (C–H-Ar) cm–1. C19H25BrN2O (377.33): calcd. C 60.48, H 6.68, N 7.42; calcd. (including 1.47 × H2O) C 56.54, H 6.97, N 6.94; found C 56.48, H 6.45, N 6.90.
General Procedure for the Enantioselective Transfer Hydrogenation of Ketones. Application of Amino Alcohol Chiral Ionic Liquids: Inside a glove box, the chiral ionic liquid (0.02 mmol) and the Ru catalyst (0.01 mmol) were weighed, added to a predried Schlenk flask, and then dissolved in water (4 mL), which was freeze-dried prior to use. The active catalyst complex was stirred at 40 °C for 30 min, and then the ketone (2.00 mmol) and sodium formate (10.00 mmol) were added under an inert atmosphere. The reaction mixture was stirred under the given conditions for 24 h. Upon completion of the reaction, the mixture was extracted with n-hexane, and the combined extracts were dried with Na2SO4 and filtered through a short plug of silica to remove the excess ruthenium particles. The filtrate was concentrated to yield the product as a colorless liquid. The alcohols were purified by column chromatography [PE with diethyl ether (EE) as a gradient), and the product was analyzed by 1H NMR, GC, and HPLC. The analytical data were in accordance with the literature.
(R)-1-Phenyl-1-ethanol:[21] 1H NMR (200 MHz, CDCl3): δH = 7.30–7.18 (m, 5 H, H-Ar), 4.83 (q, J = 6.45 Hz, 1 H, CH-OH), 1.70 (br. s, 1 H, OH), 1.42 (d, J = 6.45 Hz, CH3-CH) ppm.
(R)-1-Phenyl-1-propanol:[22] 1H NMR (200 MHz, CDCl3): δH = 7.7–7.17 (m, 5 H, H-Ar), 4.50 (t, J = 6.50 Hz, 1 H, CH-OH), 1.91 (br. s, 1 H, OH), 1.75–1.61 (m, 2 H, CH2-CH3), 0.83 (t, J = 7.43 Hz, CH3-CH) ppm.
(R)-1-(2-Nitrophenyl)-1-ethanol:[23] 1H NMR (200 MHz, CDCl3): δH = 7.65 (td, J1 = 5.39 Hz, J2 = 1.27 Hz, 2 H, H-Ar), 7.57 (td, J1 = 3.87, J2 = 1.18 Hz, 1 H, H-Ar), 7.31 (t, J = 7.73 Hz, 1 H, H-Ar), 5.37 (q, J = 6.26 Hz, 1 H, CH-OH), 2.41 (br. s, 1 H, OH), 1.49 (d, J = 6.46 Hz, CH3-CH) ppm.
(R)-1-(3-Nitrophenyl)-1-ethanol:[24] 1H NMR (200 MHz, CDCl3): δH = 8.07–8.06 (m, 1 H, H-Ar), 8.06–8.02 (m, 1 H, H-Ar), 7.66–7.62 (m, 1 H, H-Ar), 7.46 (t, J = 7.92 Hz, 1 H, H-Ar), 4.97–4.93 (m, 1 H, CH-OH), 2.11 (br. s, 1 H, OH), 1.47 (d, J = 6.45 Hz, CH3-CH) ppm.
(R)-1-(4-Nitrophenyl)-1-ethanol:[24] 1H NMR (200 MHz, CDCl3): δH = 8.11 (d, J = 8.70 Hz, 2 H, H-Ar), 7.46 (d, J = 8.70 Hz, 2 H, H-Ar), 5.00–4.88 (m, 1 H, CH-OH), 2.18 (br. s, 1 H, OH), 1.43 (d, J = 6.46 Hz, CH3-CH) ppm.
(R)-1-(4-Bromophenyl)-1-ethanol:[23] 1H NMR (200 MHz, CDCl3): δH = 7.38 (d, J = 8.49 Hz, 2 H, H-Ar), 7.15 (d, J = 8.39 Hz, 2 H, H-Ar), 4.78 (q, J = 6.39 Hz, 1 H, CH-OH), 2.04 (br. s, 1 H, OH), 1.38 (d, J = 6.46 Hz, CH3-CH) ppm.
(R)-1-(4-Chlorophenyl)-1-ethanol:[23] 1H NMR (200 MHz, CDCl3): δH = 7.22 (s, 4 H, H-Ar), 4.76 (q, J = 6.43 Hz, 1 H, CH-OH), 1.97 (br. s, 1 H, OH), 1.38 (d, J = 6.46 Hz, CH3-CH) ppm.
(R)-1-(4-Methoxyphenyl)-1-ethanol:[25] 1H NMR (200 MHz, CDCl3): δH = 7.21 (d, J = 8.63 Hz, 2 H, H-Ar), 6.79 (d, J = 8.74 Hz, 2 H, H-Ar), 4.76 (q, J = 6.39 Hz, 1 H, CH-OH), 1.88 (br. s, 1 H, OH), 1.39 (d, J = 6.46 Hz, CH3-CH) ppm.
(S)-1-Indanol:[26] 1H NMR (200 MHz, CDCl3): δH = 7.36–7.32 (m, 1 H, H-Ar), 7.22–7.15 (m, 3 H, H-Ar), 5.17 (t, J = 6.19 Hz, 1 H, CH-OH), 3.07–3.01 (m, 1 H, H-cyclopent), 2.82–2.66 (m, 1 H, H-cyclopent), 2.50–2.33 (m, 1 H, H-cyclopent), 2.01–1.89 (m, 1 H, H-cyclopent), 1.92 (s, 1 H, H-cyclopent) ppm.
(S)-1-(2′-Naphthyl)ethanol:[25] 1H NMR (200 MHz, CDCl3): δH = 8.07–8.04 (m, 1 H, H-Ar), 7.83–7.59 (m, 3 H, H-Ar), 7.46–7.37 (m, 3 H, H-Ar), 5.63–5.60 (m, 1 H, CH-OH), 1.84 (br. s, 1 H, OH), 1.57 (d, J = 6.54 Hz, 3 H) ppm.
Supporting Information (see footnote on the first page of this article): Copies of the 1H and 13C NMR spectra for all new chiral ionic liquids and their precursors and copies of the 1H NMR spectra for the enantioenriched alcohols.
Acknowledgments
Financial support from the Austrian Science Fund (FWF) (project number P25504-N28) is gratefully acknowledged.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1a.Palmer MJ, Wills M. Tetrahedron: Asymmetry. 1999;10:2045–2061. [Google Scholar]
- 1b.Jacobsen EN, Pfaltz A. In: Comprehensive Asymmetric Catalysis I–III. Yamamoto H, editor. Vol. 3. New York: Springer; 1999. [Google Scholar]
- 1c.Anderson PG. In: Modern Reduction Methods. Munslow IJ, editor. Weinheim, Germany: Wiley-VCH; 2008. [Google Scholar]
- 2.Hashiguchi S, Fujii A, Takehara J, Ikariya T, Noyori R. J. Am. Chem. Soc. 1995;117:7562–7563. [Google Scholar]
- 3.Wu X, Xiao J. Chem. Commun. 2007:2449–2466. doi: 10.1039/b618340a. [DOI] [PubMed] [Google Scholar]
- 4.Cornils B. In: Applied Homogeneous Catalysis with Organometallic Compounds. 2nd ed. Herrmann WA, editor. Vol. 3. Weinheim, Germany: Wiley-VCH; 2002. Developments. [Google Scholar]
- 5a.Dwars T, Paetzold E, Oehme G. Angew. Chem. Int. Ed. 2005;44:7174–7199. doi: 10.1002/anie.200501365. Angew. Chem.2005, 117, 7338. [DOI] [PubMed] [Google Scholar]
- 5b.Engberts JBFN. In: Methods and Reagents for Green Chemistry: An Introduction. Tundo P, Perosa A, Zecchini F, editors. Hoboken: John Wiley & Sons, Inc.; 2007. pp. 159–170. [Google Scholar]
- 6a.Rhyoo HY, Park H-J, Chung YK. Chem. Commun. 2001:2064–2065. doi: 10.1039/b106130p. [DOI] [PubMed] [Google Scholar]
- 6b.Li L, Wu J, Wang F, Liao J, Zhang H, Lian C, Zhu J, Deng J. Green Chem. 2007;9:23–25. [Google Scholar]
- 7.Yoshimura M, Tanaka S, Kitamura M. Tetrahedron Lett. 2014;55:3635–3640. [Google Scholar]
- 8a.Bica K, Gaertner P. Eur. J. Org. Chem. 2008:3235–3250. [Google Scholar]
- 8b.Ding J, Armstrong DW. Chirality. 2005;17:281–292. doi: 10.1002/chir.20153. [DOI] [PubMed] [Google Scholar]
- 8c.Baudequin C, Bregeon D, Levillain J, Guillen F, Plaquevent J-C, Gaumont A-C. Tetrahedron: Asymmetry. 2005;16:3921–3945. [Google Scholar]
- 8d.Winkel A, Reddy PVG, Wilhelm R. Synthesis. 2008:999–1016. [Google Scholar]
- 9.Rios-Lombardia N, Busto E, Gotor-Fernandez V, Gotor V, Porcar R, Garcia-Verdugo E, Luis SV, Alfonso I, Garcia-Granda S, Menendez-Velazquez A. Chem. Eur. J. 2010;16:836–847. doi: 10.1002/chem.200901623. [DOI] [PubMed] [Google Scholar]
- 10.Chen S, Zhang S, Liu X, Wang J, Wang J, Dong K, Sun J, Xu B. Phys. Chem. Chem. Phys. 2014;16:5893–5906. doi: 10.1039/c3cp53116c. [DOI] [PubMed] [Google Scholar]
- 11a.Luo S-P, Xu D-Q, Yue H-D, Wang L-P, Yang W-L, Xu Z-Y. Tetrahedron: Asymmetry. 2006;17:2028–2033. [Google Scholar]
- 11b.Vasiloiu M, Rainer D, Gaertner P, Reichel C, Schroeder C, Bica K. Catal. Today. 2013;200:80–86. [Google Scholar]
- 11c.Luo S, Mi X, Zhang L, Liu S, Xu H, Cheng J-P. Tetrahedron. 2007;63:1923–1930. [Google Scholar]
- 11d.Luo S, Zhang L, Mi X, Qiao Y, Cheng J-P. J. Org. Chem. 2007;72:9350–9352. doi: 10.1021/jo7020357. [DOI] [PubMed] [Google Scholar]
- 11e.Luo S, Zhang L, Cheng J-P. Chem. Asian J. 2009;4:1184–1195. doi: 10.1002/asia.200900080. [DOI] [PubMed] [Google Scholar]
- 12a.Schulz PS, Mueller N, Boesmann A, Wasserscheid P. Angew. Chem. Int. Ed. 2007;46:1293–1295. doi: 10.1002/anie.200604406. Angew. Chem.2007, 119, 1315. [DOI] [PubMed] [Google Scholar]
- 12b.Wagner V, Schulz PS, Wasserscheid P. J. Mol. Liq. 2014;192:177–184. [Google Scholar]
- 12c.Schneiders K, Boesmann A, Schulz PS, Wasserscheid P. Adv. Synth. Catal. 2009;351:432–440. [Google Scholar]
- 12d.Ferlin N, Courty M, Van Nhien AN, Gatard S, Pour M, Quilty B, Ghavre M, Haiss A, Kuemmerer K, Gathergood N, Bouquillon S. RSC Adv. 2013;3:26241–26251. [Google Scholar]
- 13.Gausepohl R, Buskens P, Kleinen J, Bruckmann A, Lehmann CW, Klankermayer J, Leitner W. Angew. Chem. Int. Ed. 2006;45:3689–3692. doi: 10.1002/anie.200600327. Angew. Chem.2006, 118, 3772–3775. [DOI] [PubMed] [Google Scholar]
- 14.Bigi F, Nimal Gunaratne HQ, Quarantelli C, Seddon KR. C. R. Chim. 2011;14:685–687. [Google Scholar]
- 15.Vasiloiu M, Leder S, Gaertner P, Mereiter K, Bica K. Org. Biomol. Chem. 2013;11:8092–8102. doi: 10.1039/c3ob41635f. [DOI] [PubMed] [Google Scholar]
- 16.Gonzalez L, Altava B, Bolte M, Burguete MI, Garcia-Verdugo E, Luis SV. Eur. J. Org. Chem. 2012:4996–5009. [Google Scholar]
- 17a.Brandt P, Roth P, Andersson PG. J. Org. Chem. 2004;69:4885–4890. doi: 10.1021/jo030378q. [DOI] [PubMed] [Google Scholar]
- 17b.Han M-L, Hu X-P, Huang J-D, Chen L-G, Zheng Z. Tetrahedron: Asymmetry. 2011;22:222–225. [Google Scholar]
- 17c.Alonso DA, Nordin SJM, Roth P, Tarnai T, Andersson PG, Thommen M, Pittelkow U. J. Org. Chem. 2000;65:3116–3122. doi: 10.1021/jo991914a. [DOI] [PubMed] [Google Scholar]
- 17d.Williams GD, Pike RA, Wade CE, Wills M. Org. Lett. 2003;5:4227–4230. doi: 10.1021/ol035746r. [DOI] [PubMed] [Google Scholar]
- 18a.Takehara J, Hashiguchi S, Fujii A, Inoue S-i, Ikariya T, Noyori R. Chem. Commun. 1996:233–234. [Google Scholar]
- 18b.Fujii A, Hashiguchi S, Uematsu N, Ikariya T, Noyori R. J. Am. Chem. Soc. 1996;118:2521–2522. [Google Scholar]
- 19a.Ta L, Axelsson A, Bijl J, Haukka M, Sunden H. Chem. Eur. J. 2014;20:13889–13893. doi: 10.1002/chem.201404288. [DOI] [PubMed] [Google Scholar]
- 19b.Enders D, Niemeier O, Henseler A. Chem. Rev. 2007;107:5606–5655. doi: 10.1021/cr068372z. [DOI] [PubMed] [Google Scholar]
- 20a.Bica K, Gaertner P, Gritsch PJ, Ressmann AK, Schroeder C, Zirbs R. Chem. Commun. 2012;48:5013–5015. doi: 10.1039/c2cc31503c. [DOI] [PubMed] [Google Scholar]
- 20b.Li J, Tang Y, Wang Q, Li X, Cun L, Zhang X, Zhu J, Li L, Deng J. J. Am. Chem. Soc. 2012;134:18522–18525. doi: 10.1021/ja308357y. [DOI] [PubMed] [Google Scholar]
- 21.Wu X, Li X, McConville M, Saidi O, Xiao J. J. Mol. Catal. A. 2006;247:153–158. [Google Scholar]
- 22.Rachwalski M, Jarzyński S, Leśniak S. Tetrahedron: Asymmetry. 2013;24:421–425. [Google Scholar]
- 23.Vitale P, D'Introno C, Perna FM, Perrone MG, Scilimati A. Tetrahedron: Asymmetry. 2013;24:389–394. [Google Scholar]
- 24.Barros-Filho BA, Nunes FM, Lemos TLG, de Mattos MC, de Gonzalo G, Gotor-Fernández V, Gotor V. J. Mol. Catal. B. 2010;65:37–40. [Google Scholar]
- 25.Ito J-i, Teshima T, Nishiyama H. Chem. Commun. 2012;48:1105–1107. doi: 10.1039/c1cc16057e. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki T, Ghozati K, Katoh T, Sasai H. Org. Lett. 2009;11:4286–4288. doi: 10.1021/ol9016436. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information