

Abstract
Biomaterial-mediated controlled release of soluble signaling molecules is a tissue engineering approach to spatially control processes of inflammation, microvascular remodeling, and host cell recruitment and to generate biochemical gradients in vivo. Lipid mediators, such as sphingosine 1-phosphate (S1P), are recognized for their essential roles in spatial guidance, signaling, and highly regulated endogenous gradients. S1P and pharmacological analogs such as FTY720 are therapeutically attractive targets for their critical roles in the trafficking of cells between blood and tissue spaces both physiologically and pathophysiologically. However, the interaction of locally delivered sphingolipids with the complex metabolic networks controlling the flux of lipid species in inflamed tissue has yet to be elucidated. In this study, complementary in vitro and in vivo approaches are investigated to identify relationships between polymer composition, drug release kinetics, S1P metabolic activity, signaling gradients, and spatial positioning of circulating cells around poly lactic-co-glycolic acid (PLGA) biomaterials. Results demonstrate that biomaterial-based gradients of S1P are short-lived in the tissue due to degradation by S1P lyase, an enzyme that irreversibly degrades intracellular S1P. On the other hand, in vivo gradients of the more stable compound, FTY720 enhance microvascular remodeling by selectively recruiting an anti-inflammatory subset of monocytes (S1P3high) to the biomaterial. Results highlight the need to better understand the endogenous balance of lipid import/export machinery and lipid kinase/phosphatase activity in order to design biomaterial products that spatially control the innate immune environment to maximize regenerative potential.
Graphical abstract
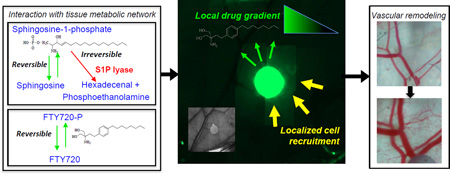
In vivo gradients of sphingosine-1-phosphate receptor targeting compounds
1. Introduction
Soluble chemotactic gradients allow long distance cell communication and guided cell trafficking that are essential for development, homeostasis, pathology, and regeneration [1–3]. Many goals in tissue engineering rely on modulating cellular localization and polarization of cell signaling, including inflammation and foreign body reaction, microvascular growth and remodeling, and recruitment of host stem and progenitor cells to regenerate tissues. Biomaterial-based release of chemotactic molecules and signaling molecules provides a method to spatially control these processes and generate gradients in vivo. While secreted protein signals such as stromal cell derived factor-1α (SDF-1α) [4], fractalkine [5], monocyte chemotactic protein-1 (MCP-1) [6] and vascular endothelial growth factor (VEGF) [7] are well-known chemotactic and growth cues, lipid mediators such as sphingosine-1-phosphate (S1P) have more recently become recognized for their essential roles in spatial guidance, signaling, and ability to modify the sensitivity of cells to other chemokines and growth factors [8–11]. S1P plays a critical role in microvascular physiology and the movement of cells between blood and tissue spaces both physiologically and pathophysiologically [12] making it an attractive signaling axis to target therapeutically. Endogenous S1P gradients are highly regulated and maintained between blood, lymphoid organs, and other tissues. S1P concentration in tissue is maintained relatively low compared to the blood plasma due to greater net activity of the irreversible degradation of S1P through the S1P lyase enzyme intracellularly or reversible degradation through the S1P phosphatases at the cell membrane [13]. Decreased storage and secretion of S1P in tissue relative to blood and plasma results in lower tissue S1P levels [14]. Conversely, low activity of intracellular S1P lyase in the blood maintains the plasma concentration of S1P in the low micromolar range [13, 15]. S1P is the endogenous ligand for five known high-affinity G-protein coupled receptors: S1P receptor 1 through 5 (S1P1–5) [16]. Since many circulating cells express one or more of the S1P receptors (S1PRs), the establishment of S1P gradients between the microvasculature and tissue represents a critical determinant of cell trafficking throughout the body [17]. S1P plays a particularly important role in the trafficking of immune cells by regulating compartmentalization and egress from lymphoid organs, adhesion and migration of circulating monocytes and osteoclastic precursors, pericyte recruitment during embryonic development and regeneration, and the mobilization of hematopoietic stem cells into peripheral blood [8, 17]. S1P gradients play a role in instructing lymphocytes from regions of low S1P concentration within primary and secondary lymphoid organs into blood circulation [18]. In addition to the chemotactic potential of S1P through the S1PRs, S1P also provides spatial guidance cues through the regulation of secondary signaling pathways that may modify the production of other soluble cues or the sensitivity of the cellular response to such signals [8, 10–12].
Recently, our laboratory has shown that release of the S1PR selective drug, FTY720, from degradable polymers enhances recruitment of anti-inflammatory S1P3high monocytes from circulation and localizes these cells within a vascular niche along peri-implant microvessels and enhances the expansion and maturation of regenerative vasculature [12]. Utilizing the dorsal skinfold window chamber as a model of soft tissue inflammatory vascular remodeling, the entire history of a growing vascular network and biomaterial interactions can be scrutinized over time [19, 20]. Surgical removal of dorsal skin and implantation of a biomaterial in the chamber window presents a persistent inflammatory stimulus that significantly and dynamically increases local concentrations of inflammatory and regenerative cytokines such as MCP-1 and SDF-1α [12, 21]. FTY720 increases the secretion of local SDF-1α, strengthening this important chemokine gradient, and enhancing the sensitivity/chemotaxis of anti-inflammatory monocytes to a SDF-1α stimulus [12]. Ongoing discoveries of new selective agonists and antagonists of S1P receptors and sphingolipid metabolizing enzymes suggest exciting new opportunities for tuning lipid chemokine gradients in vivo [22]. The network of sphingolipid metabolism that produces endogenous S1P and preceding sphingolipid metabolites such as sphingosine and ceramide [23] is complex and absolute determination of S1P concentration and that of S1PR targeted drugs following their local delivery is challenging. Many in vitro and in vivo models have been employed to investigate how protein chemokine concentration gradients regulate cell trafficking and tissue remodeling [24–28]; however, alteration of lipid signaling gradients affecting host responses to implanted biomaterials has not been rigorously characterized.
This study takes a combination of in vitro and in vivo approaches to identify relationships between polymer composition, drug release kinetics and the tissue S1P metabolic activity on S1P signaling gradients from biomaterials. Complementary in vitro and in vivo drug release profiles help to define the gradient release profiles of different polymer compositions. The spatial cues conferred to circulating cells for spatial positioning in relation to the biomaterial are also investigated. End-point collection of tissue from the dorsal skinfold window chamber with known spatial relationship to the biomaterial allows the interrogation of in vivo gradients relative to the implant. This model system provides valuable insight into the local perturbations to concentration and production of S1P and how these perturbations may be exploited for tuning growth and remodeling of microvessels around polymeric implants and for spatial positioning of therapeutic cells from circulation. We report that poly lactic-co-glycolic acid (PLGA) release of the S1PR agonist FTY720 produces an in vivo gradient that preferentially recruits anti-inflammatory subset monocytes to the biomaterial film and enhances microvascular remodeling. These studies can inform the design of biomaterial products that spatially control the innate immune environment surrounding the implant to harness endogenous “immuno-regenerative” potential.
2. Materials and methods
2.1. Fabrication of Unloaded and Loaded Polymeric Thin Films
PLGA formulations were purchased from Lakeshore Biomaterials (Birmingham, AL): 1) 50:50 DLG 2A: low molecular weight acid-capped 13.5 kDa (L-A), and 2) 50:50 DLG 5E: high molecular weight ester-capped 71 kDa (H-ME). PLGA films were fabricated unloaded or with S1P, FTY720, FTY720-P, or NBD-FTY720 (Cayman Chemical, Ann Arbor, MI) as previously described [26]. Briefly, drugs were solubilized in dichloromethane in a water bath at 65°C and vortexed until dissolved. Solutions were solvent cast in Teflon molds, stored at −20°C until solvent had evaporated. A biopsy punch was used to generate 1mm diameter thin films. Final loading of drug is 1:400 (w/w) and 1:200 (w/w) drug:polymer ratio. For NBD-FTY720 imaging, NBD-FTY720 was loaded as 10% of the total FTY720 in the polymer. Biopsy punched 1mm films were imaged on an epi-fluorescence Zeiss Axio Imager.D2 microscope. For S1P lyase inhibitor studies 4-deoxypyridoxine (DOP) was loaded in H-ME polymer with or without S1P. Note: Unless specifically stated, all drugs implanted in vivo were encapsulated in H-ME 50:50 PLGA, which is referred to simply as ‘PLGA’.
2.2 Quantification of S1P, Sphingosine, FTY720 or FTY720-P Release from PLGA
2.2.1 Radiolabeled S1P measurements
The in vitro release of [P33]-S1P from PLGA was quantified by scintillation counting of P33 in the bathing solution (specific activity: 3000 Ci/mmol; Perkin-Elmer, Waltham, MA). Bathing solution was simulated body fluid (SBF, pH 7.2; 7.996g NaCl, 0.35 g NaHCO3, 0.3 g KCl, 0.136 g KH2PO4, 0.095 g MgCl2, 0.278 g CaCl2, 0.06 g MgSO4 to 1 L deionized water) with 4% fatty acid free bovine serum albumin (FAF-BSA). At each time point, the release solutions were replaced with fresh solution and combined with 5 mL EcoScint, a biodegradable scintillation solution (National Diagnostics, Atlanta, GA). To calculate the release of S1P from PLGA films in vivo into the tissue space of the dorsal skinfold window chamber, a 1mm tissue biopsy punch through the subreticular dermis was collected immediately adjacent to the edge of the 1mm PLGA film post-mortem. Quantification of [P33]-S1P was performed using a Beckman Coulter LS 6500 liquid scintillation counter. The counts per minute (CPM) were measured and converted to a quantity of total S1P (µg) using the specific activity (CPM/mol).
2.2.2 High-performance Liquid Chromatography-Mass Spectrometry (HPLC-MS) Measurements
Total S1P, sphingosine, FTY720, and FTY720-P were extracted by sphingolipid extraction procedure and quantified using high performance liquid chromatography and mass spectrometry as previously described [29]. The in vitro release of FTY720 and FTY720-P was quantified with HPLC-MS. In FTY720 vs FTY720-P release study, loading was 1:70 (w/w) from 1.5mm diameter films. All other studies were 1mm PLGA films with 1:200 or 1:400 w/w loading as indicated. For release studies, films were incubated in 0.5 mL with 4% FAF-BSA (w/v) and maintained at 37°C with constant agitation. SBF was collected at the desired time-points and replaced with fresh SBF. FTY720 was extracted from the SBF by sphingolipid extraction and HPLC-MS as previously described [29].
2.3 Dorsal Skinfold Window Chamber
2.3.1 Window chamber surgery and imaging
Animal experiments were performed using sterile techniques in accordance with an approved protocol from the University of Virginia Animal Care and Use Committee. Male C57BL/6 mice (Harlan, Indianapolis, IN), age-matched and weighing between 18 and 25 grams, were surgically fitted with dorsal skinfold window chambers (APJ Trading Company, Inc., Ventura, CA) as previously described [30]. Briefly, hair was removed from the dorsal skin and the skin was removed down to the layer of the sub-reticular dermis with sterile surgical scissors in a 12mm diameter circle. Control or drug-loaded films were implanted into each window chamber and sterile cover-glass was fixed above the surgical site. For intra-vital imaging, vessels were maximally dilated by application of Ringer’s solution with 1 mM adenosine in order to determine the effect of PLGA polymer films on the maximal blood column width or luminal diameter of local arterioles. Intra-vital microscopy images were photo-merged into montages of entire vascular windows and analyzed using a combination of Adobe Photoshop CS and ImageJ software as previously described [30]. After the final imaging of the dorsal skinfold window chamber on Day 3 or 7, window chamber tissue was dissected by biopsy punch for sphingolipid extraction. Mice were euthanized prior to tissue harvest of the window chamber area. Blood was collected for sphingolipid extraction and hemavet analysis by cardiac puncture post-mortem.
2.3.2 Adoptive transfer
Adoptive transfer was performed as previously described [12]. Briefly, primary monocytes were isolated from mouse bone marrow, stained for CD45, Ly6C, GR1 (Biolegend; San Diego, CA), and CD11b (eBioscience; San Diego, CA) and sorted by FACS into CD45+CD11b+Ly6ChighGR1high inflammatory monocytes (IM) and CD45+CD11b+Ly6ClowGR1low anti-inflammatory monocytes (AM). Cells were labeled with DiI (1:200 v/v) in serum-free media at 37°C for 20 minutes and then washed three times with PBS prior to injection. Cells were delivered intra-venously 16–18 hours prior to dorsal skin-fold window chamber placement. For adoptive transfer, images were captured in bright-field and rhodamine filter fluorescence. Distance of transferred monocytes from the film was measured in 2D using Zeiss Zen analysis software.
2.3.3. Polymer swelling
Swelling of polymer films was assessed by a change in area of the films after placement in the window chamber from Day 0, 3, and 7 photo montages using ImageJ software.
2.4 Blood cell analysis
Circulating cell populations were analyzed by the Hemavet950FS Hematology analyzer. Briefly, blood was collected from the retro-orbital sinus under isoflurane anesthesia into heparinized tubes and analyzed by Hemavet. Data are represented as percentages of total white blood cells for each cell type.
2.5 Statistical Analyses
All statistical analyses were performed using Minitab 15 statistical software (Minitab, Inc., State College, PA) or Graphpad Prism version 6.0 (La Jolla, CA). Results are presented as mean ± standard error of the mean (SEM). Comparisons used one-way or two-way ANOVA according to experimental design, followed by Tukey’s test for pairwise comparisons. Diameter analysis was performed using a General Linear Model (GLM) ANOVA with an unbalanced nested design, followed by Tukey’s test for pairwise comparisons. Significance was asserted at p < 0.05. Power calculation was performed with α=0.05, power=0.8 to determine statistically significant sample size.
3. Results
3.1 Tunable polymer release kinetics of S1P receptor targeting compounds
In order to generate localized gradients in vivo, PLGA biodegradable polymers were loaded with S1PR targeting compounds as a sustained release vehicle. The molecular weight and end cap chemistry of the PLGA polymers were varied to achieve distinct degradation kinetics. Since PLGA polymers are degraded by hydrolysis of the ester linkages between the lactic and glycolic acid units, water penetration into the polymer matrix is a metric associated with degradation of the polymer [31]. Significant swelling occurred in the hydrophilic low MW-carboxylic acid-capped “L-A” polymer. The more hydrophobic high-MW-methyl-ester-capped “H-ME” polymer did not undergo swelling suggesting slower degradation than the L-A formulation (Figure 1A). PLGA hydrolysis into lactic and glycolic acid can affect the pH of the environment surrounding of the polymer film. After 7 days in vitro the H-ME formulation did not significantly affect pH compared to control; however, L-A films caused a progressive and significant acidification compared to control, culminating at a pH of 2.6 ± 0.1 after 7 days, suggesting an increase in acidic degradation products from the L-A polymer (Figure 1B).
Figure 1. Polymer properties tune the release of S1P receptor compounds.

PLGA polymers of differing molecular weight and end-cap chemistry (Low MW- acid capped polymer (L-A), high-MW ester capped polymer (H-ME)) undergo distinct degradation profiles as is evident by the percent of polymer swelling (A) and change in pH of the surrounding environment (B). In vitro release studies show differential release of S1P from the faster degrading L-A and the slower degrading H-ME (C). Release is related to the amount of drug initially loaded into the polymer (D). (Data are mean with SEM; #, p<0.05 d3 vs d7; *, p<0.05 compared to H-ME at same time-point)
Consistent with the different degradation properties of the polymers, L-A and H-ME films differentially release S1P in vitro. Cumulative S1P release was greater over 7 days in the faster degrading L-A polymer than the H-ME polymer (Figure 1C). Increase in the loading proportion of S1P from 1:400 w/w to 1:200 w/w (S1P:PLGA) results in a greater cumulative release and increased burst release of S1P (Figure 1D). The S1PR agonist FTY720 is also differentially released from L-A and H-ME polymers. In both polymer groups, FTY720 was released with an initial burst in the first hours of incubation in vitro, followed by a delay and a second phase of release between 3 and 5 days (Figure 2A). The burst release was more substantial in the L-A polymer compared to the H-ME polymer. Linear regression analysis was used to determine whether the release of FTY720 from the different polymers was correlated with the swelling of the polymer (Figure 2B). In days 0–3 where the initial burst release occurs, swelling and release were not significantly correlated (R2= 0.35); however, in the second phase from days 4–7, drug release was significantly correlated to polymer swelling with an R2 of 0.88. These findings are consistent with previous observations of a two-phase release from PLGA polymers: a surface burst release and a secondary polymer degradation-based release that depends on polymer hydrolyzation for drug release [31–33]. Fluorescently labeled NBD-FTY720 loaded in H-ME and L-A films distributes differently within the polymer films, which may contribute to the difference in release (Figure 2C).
Figure 2. Two-phase release of FTY720 from PLGA is consistent with non-homogenous distribution of drug in the polymer scaffold.

Polymer composition tunes release kinetics of FTY720 (A). Inset shows close-up of H-ME. Release of FTY720 was correlated with the percent swelling of the polymer film during later time-points but not the first three days (B). Fluorescently labeled FTY720 (NBD-FTY720) was loaded into H-ME or L-A PLGA and 1mm diameter thin films were imaged (C). NBD-FTY720 was visible in the polymer and 20× magnification images demonstrate differential distribution of the drug between the two polymers.
3.2 In vivo release of S1P from PLGA thin films
Utilizing the distinct release profiles of different polymer and loading conditions, we examined the ability to generate an S1P release gradient from PLGA films in vivo. To investigate the total S1P/sphingosine balance in the local tissue, we analyzed tissue S1P and sphingosine by HPLC-mass spectrometry in a 1.5mm diameter biopsy punch surrounding the 1mm implant 3 days after film implantation. Tissue S1P and sphingosine levels in animals treated with the H-ME polymer (black bars) and L-A polymer (white bars) were not different (Figure 3A). No significant difference in percent change of vascular diameter from day 0 to day 3 was observed between the H-ME and L-A polymers releasing S1P after 3 days (Figure 3B). To measure total S1P release, P33-labeled S1P was loaded into polymers (S1P33:L-A, 1:400; S1P33:H-ME, 1:400; and S1P33:H-ME, 1:200) and implanted into the dorsal skinfold window chamber. A 1 mm diameter circle of tissue directly adjacent to the film was excised and analyzed for P33 concentration at 7 days (Figure 3C). Consistent with in vitro release profiles, the L-A polymer trends toward releasing more S1P33 in vivo than the slow degrading H-ME. Doubling the loading ratio of S1P in H-ME polymer also enhances the amount of S1P33 in the tissue surrounding the polymer film (Figure 3C).
Figure 3. Polymer release properties and local metabolism control peri-implant S1P presentation in vivo.

Total tissue concentration of S1P and sphingosine were measured by HPLC-MS/MS in a 1.5 mm diameter tissue biopsy around the film at 3 days (B, n=3–7 per group) (A). Representative dorsal skin-fold window chamber images from un-loaded and S1P loaded polymer films show no significant difference in vascular remodeling from day 0 to day 3 (B). S1P release was measured in a 1mm tissue biopsy that was harvested directly adjacent to the film after 7 days in the dorsal skin-fold window chamber (C, left) and quantified by P33 concentration (C, n=4–12 per group). S1P (D) and sphingosine (E) were measured after release of the S1P lyase inhibitor DOP or a combination of DOP+S1P for 3 days and percent change in arteriolar diameter was quantified after 3 (F) or 7 days (G) (* p<0.05 compared to PLGA control). Representative images from days 0, 3, and 7 (H). White arrowheads indicate arterioles.
3.3 Sphingolipid metabolic environment in tissue controls efficacy of S1P receptor compounds
Homeostatic and pathological S1P cellular and tissue concentrations are maintained in balance by the complex cellular sphingolipid metabolic network. In order to determine whether intracellular degradation of S1P affects the sustained delivery of S1P from polymer films, the S1P lyase inhibitor DOP, was released locally in the dorsal skin tissue from H-ME PLGA films with or without S1P (DOP or DOP+S1P, respectively). As expected, blocking the cellular catabolism of S1P through S1P lyase inhibition increased the local concentration of S1P in the peri-implant tissue compared to S1P alone. S1P release in combination with DOP significantly augmented the local S1P and sphingosine concentrations, indicating that the total pool of tissue sphingolipid is increased by the film releasing S1P (Figure 3D). These data suggest that S1P lyase is an active method of S1P clearance in the peri-implant region (Figure 3D, E). In the absence of activity of the lyase pathway, the increase in local sphingosine concentration may be the result of dephosphorylation of the film-released S1P to sphingosine through S1P phosphatases (Figure 3E). Vascular network remodeling increased in parallel with the increase of S1P in the peri-implant region consistent with the known effects of S1P on vascular remodeling with a trend at day 3 and significant increase at day 7 (Figure 3F–H) [26, 30, 34–37]. White arrows indicate arterioles (Figure 3H). Interference with the S1P lyase can locally stabilize the released S1P and enhance vascular remodeling outcomes (Figure 3F–H).
3.4 Polymer-mediated delivery of an in vivo gradient of FTY720
To investigate the in vivo gradient produced by H-ME PLGA film release of S1P receptor targeting compounds, FTY720 and FTY720-P were released from 1mm diameter thin films in the dorsal skin-fold window chamber for 3 days. Fluorescent NBD-FTY720 was loaded into H-ME films to visualize the release gradient (Figure 4A). Fluorescence intensity dissipates rapidly from the edge of the polymer into the tissue. To achieve more sensitive measurements of the drug gradient, concentric circles of tissue surrounding the implant (Figure 4A) were collected by biopsy punch through the sub-reticular dermis and analyzed for FTY720, FTY720-P, sphingosine, and S1P by HPLC-MS/MS three days after surgery. Total drug release, as measured by the sum of FTY720 (white bar) and FTY720-P (black bar), was greater from the films loaded with FTY720 compared to FTY720-P in both the 1.5mm tissue ring and the 4mm tissue ring (Figure 4B, C). Both FTY720 and FTY720-P produce a gradient of the drugs in the tissue at 3 days (Figure 4D–E). In vitro release studies demonstrate that FTY720 and FTY720-P are released from the film with differing kinetics, with FTY720 being released more rapidly (Figure 4F). The phosphorylated form of the drug, FTY720-P, exhibited less release over 3 days both in vitro and in vivo (Figure 4B, C, F). Tissue sphingosine and S1P concentrations were enhanced closer to the polymer film but no significant difference was observed between FTY720 and FTY720-P releasing films (Figure 4G–H). Analysis of localized arteriolar remodeling surrounding the polymeric implant demonstrates a significant increase in the diameter of arterioles after 3 days as a result of FTY720 release that was not observed with FTY720-P release which may be a product of differing release and gradient (Figure 4I, J).
Figure 4. Polymer release of FTY720 generates a local in vivo gradient.

Tissue biopsies of 1.5 and 4mm diameter centered over the origin of the film (A) were analyzed by HPLC-MS/MS for lipid species of 3 days after surgery and film implantation. NBD-FTY720 film shows release of fluorescent FTY720 in a gradient from the edge of the film outward in vivo (A). FTY720 films release more total compound (white=FTY720, black=FTY720-P) in both the 1.5mm (B) and 4mm (C) tissue regions in vivo. Concentrations of FTY720 (D) and FTY720-P (E) in the tissue at varying distances from the film demonstrate differential gradients between the FTY720 and FTY720-P films. FTY720 and FTY720-P release at different rates in vitro (F). Sphingosine (G) and S1P (H) were measured in the tissue segments illustrated in A. Change in arteriolar diameter with FTY720 or FTY720-P 3 days after surgery (I; n=5–8). Representative images of vascular changes from day 0 to day 3, white arrows indicate arterioles (J). (*p<0.05 compared to FTY720 film)
3.5 Local release of S1PR agonists at the site of injury does not alter lymphocyte circulation
S1P is elevated in the blood after dorsal skinfold inflammatory injury (Figure 5A). Release of FTY720 or FTY720-P from polymer films did not significantly alter the increase in blood S1P. FTY720, but not FTY720-P, was transiently detected in the blood stream after local release (Figure 5B–C). Circulating lymphocytes, monocytes, and neutrophils were measured by a Hemavet analyzer and the percentage of cells out of total white blood cells was not significantly different between groups, although there was an observed trend of decreasing circulating neutrophils from 1–3 days. (Figure 5D–F).
Figure 5. Local release of S1PR agonists at the site of injury does not alter systemic circulating cells.

S1P is elevated in the blood after dorsal skinfold inflammatory injury (A; *p<0.05 compared to day 0). FTY720 was transiently detected in the blood stream but not FTY720-P after local release (B–C). Circulating lymphocytes, monocytes, and neutrophils were not significantly different between groups (% of total white blood cells) (D–F).
3.6 In vivo gradient at site of inflammatory injury positions cells within local tissue
Release of FTY720 from PLGA enhances the recruitment of anti-inflammatory monocytes from circulation to the injured region after 3 days and enhances vascular remodeling [12]. We sought to further investigate the relationship between the in vivo gradient of FTY720 released from a polymer film and the spatial positioning of monocytes relative to the film. Inflammatory (IM) and anti-inflammatory (AM) sub-sets of monocytes were labeled with DiI and adoptively transferred intravenously prior to dorsal skin-fold window chamber surgery and film implantation. On day three, the localization of transferred cells in the region of interest relative to the perimeter of the film was assessed by fluorescence in vivo imaging (Figure 6A–B). Overlay of the bright-field and fluorescence and intra-vital images demonstrate proximity of the labeled cells to the vasculature (Figure 6B) as previously described [12]. FTY720 release did not change the average positioning of IM cells with respect to the film edge, but did significantly reduce the average distance of AM transferred cells to the film (Figure 6 C, D). The proportion of total cells identified within the “peri-implant” region (0–1mm from the film edge) demonstrates that a smaller proportion of the IM labeled cells were concentrated within the first 1mm radius than AM cells (Figure 6E). FTY720 release did not significantly alter the distribution of IM cells surrounding the films (Figure 6A, E), however, FTY720 did significantly enhanced the proportion of AM cells located within 1mm from the edge of the film (Figure 6E). These data suggest that the FTY720 gradient produced from the FTY720 film recruits a selective population of monocytes to peri-implant positions.
Figure 6. In vivo gradient positions cells within local tissue.

Mice were injected i.v. with DiI labeled monocytes of either inflammatory (IM) or anti-inflammatory (AM) profile prior to dorsal skinfold injury and implantation of either H-ME or FTY720 films. Proximity of labeled cells to the film was characterized by intravital microscopy on day 3 (A, B white arrowheads= DiI-labeled cells). Enlarged view of the bright-field and fluorescent channels of a representative peri-implant region shows labeled cells (white arrowheads) were found in proximity to vessel structures (B). The average distance of each cell type from the edge of the film (C, D). A higher proportion of AM cells were found within the peri-implant region 1mm from the edge of the film out of total cells in the region of interest and this effect was enhanced by FTY720 (E). (*p<0.05, H-ME vs FTY720; #p<0.05, IM vs AM)
4. Discussion
Sphingolipids and sphingolipid receptor targeting compounds have been widely investigated as therapeutic agents, however the interaction of locally delivered sphingolipids with the intricate metabolic networks controlling the flux of lipid species in inflamed tissue has yet to be elucidated. Total tissue S1P concentration is determined by a complex combination of metabolism and inter-compartmental transport. Extracellular S1P can be dephosphorylated by three different cell surface lipid phosphate phosphatases [38]. This dephosphorylation appears to be critical for lipid entry into the cell, likely because the polar phosphate group inhibits trans-bilayer movement [39]. Intracellular S1P can be de-phosphorylated to sphingosine by S1P-specific phosphatases [40] or degraded to hexadecenal and ethanolamine phosphate by ER-resident S1P lyase [41]. Alternatively, intracellular S1P can be exported from the cell by transporters such as Spns2 [42]. Using radio-labeled S1P33, released S1P is detectable indirectly by the presence of P33 whether the lipid has been metabolized or not. The differing release properties of H-ME (slow-release) and L-A (fast-release) PLGA polymers in vitro were used to assess whether in vivo local drug concentrations replicate the relative drug release properties of each polymer. Total release in the peri-implant tissue measured by P33 levels suggests that polymer release of S1P in vivo follows the same relative trends as in vitro. The L-A polymer released more than H-ME and there was a dose dependent release from H-ME based on initial loading (Figure 1, 2). The HPLC-MS measurement accounts for both extracellular and intracellular S1P from the tissue. Total tissue S1P measurements indicate that despite differing release of S1P from H-ME and L-A polymers, there is no difference in total tissue S1P or sphingosine between the fast degrading and slow degrading polymers in the peri-implant tissue suggesting that the S1P metabolic processes occur much faster than the release from either polymer (Figure 3). Co-delivery of local S1P lyase inhibitor DOP substantially increases the local tissue S1P concentration as well as the sphingosine concentration (Figure 3). Since the lyase catabolizes S1P into hexadecenal and ethanolamine phosphate rather than sphingosine and phosphate [43], the lyase inhibitor pushes the breakdown of S1P through the reversible phosphatase reaction, thus enhancing the sphingolipid pool in the tissue. S1P lyase is an intracellular enzyme, acting at the ER membrane, so the changes in S1P measured by HPLC-MS after DOP treatment reflect both intracellular and extracellular S1P pools. Consistent with this model, the sphingosine tissue concentration increases in the animals treated simultaneously with DOP and S1P (Figure 3E). These findings suggest a tissue level equilibration of the sphingolipid metabolic environment involving irreversible S1P catabolism. Therefore, endogenous S1P lyase activity is a major barrier to successful delivery of S1P in a localized tissue gradient; when designing a drug release tool for S1P receptor compounds, the interaction of the local metabolic environment with the drug must be considered in addition to the polymer properties.
Targeting S1P receptors in wounded tissue has been shown to enhance vascular remodeling outcomes through angiogenic and arteriogenic processes [26]. Daily injection of S1P into full-thickness dorsal wounds of diabetic mice enhanced the rate and extent of wound closure and stimulated neovascularization in the wound area [44]. Intramuscular S1P injection to the mouse hindlimb resulted in blood flow recovery and dose-dependent increases in intramuscular capillary density [37]. Additionally, sphingosine kinase-1 (SPHK1) transgenic mice, that overexpress the S1P-synthesizing enzyme SPHK1, showed accelerated blood flow recovery and increased capillary density, due to a 1.8-fold increase in intramuscular S1P levels [37]. Taken together, local increases in available S1P, regardless of delivery mode, stimulate therapeutic neovascularization in an ischemic zone. Changes in arteriolar diameter remodeling with release of S1P, DOP alone, or DOP:S1P functionally support the finding of enhanced local S1P concentrations with inhibition of S1P lyase since the extent of remodeling parallels the concentration of S1P in the local tissue (Figure 3).
FTY720 is a potent immunosuppressive compound that has FDA-approved indications for the treatment of relapsing-remitting multiple sclerosis when delivered systemically due to its lymphocyte sequestering activity [45]. However, localized administration of FTY720 has more recently been implicated in recruitment of specific monocyte cell types concurrent with regeneration of vascular and bone structures and therefore the interaction of FTY720 with a local tissue injury environment is important to understand [12, 26–29]. The phosphorylated form of the drug FTY720-P is primarily responsible for receptor mediated signaling and has much lower threshold effective concentrations than FTY720 at all of the S1PRs [45, 46]. FTY720-P, the small molecule analog of S1P is not catabolized by S1P lyase [47] and therefore is more stable in tissue than S1P. Similar to S1P, FTY720-P must first be dephophosphorylated by lipid phosphate phosphatase 3 [48] to enter the cell while FTY720 does not face this barrier [49]. Once inside the cell, FTY720 can be phosphorylated by sphingosine kinase 2 (SPHK2) [50] and subsequently exported from the cell by Spns2 [51]. The ability of the drug to be reversibly phosphorylated allows it to undergo multiple cycles of signaling in the tissue contributing to a sustained gradient. The relative impact of a local gradient of FTY720 compared to FTY720-P has not previously been characterized. The current study compared the nature of a gradient of the two forms of the drug and how the tissue metabolism of the compounds affects the resulting gradient.
Both polymer formulation and drug chemistry affected the release of sphingolipid receptor targeting compounds and present opportunities to tune the release therapeutically. Previous studies suggest an interaction between PLGA and FTY720 [28]. Modification of the hydrophobicity of the polymer influences the drug-polymer interaction (Figure 1, 2). To visualize differences in distribution, NBD-FTY720 was loaded into each polymer. The more hydrophilic L-A polymer appears to contain droplets or aggregates of NBD-FTY720, while the more hydrophobic H-ME polymer has aggregates as well as diffuse distribution of the drug (Figure 2C). This localization suggests that NBD-FTY720 may interact more favorably with a more hydrophobic polymer matrix. The fluorescent modification makes NBD-FTY720 more hydrophilic than un-modified FTY720, which may alter how the drug interacts with the polymer.
FTY720 and FTY720-P release at different rates in vivo and in vitro suggesting a potential for differential interaction with the polymer or differential distribution within the polymer [28]. The critical micelle concentration (CMC) is a measure of water solubility of surfactants; higher CMC correlates with higher partitioning into hydrophilic versus hydrophobic solvents [52]. The addition of the phosphate group to sphingosine to make S1P increases the CMC from about 1 µM for sphingosine to 14 µM for S1P [53]. The CMC of FTY720 is measured at about 75 µM [54] and while the CMC of FTY720-P is not known, based on the sphingosine and S1P relationship, FTY720-P may have an even higher CMC than the unphosphorylated form. Therefore FTY720 is likely more hydrophobic than FTY720-P, which may determine the way in which the drugs interact with the polymer. The discrepancy in release is reflected in the total FTY720 + FTY720-P in the tissue between FTY720 and FTY720-P releasing films (Figure 4 B–C). Interestingly, the FTY720-film produced gradients of both FTY720 and FTY720-P in the tissue (Figure 4D–E) supporting the idea that FTY720 can be phosphorylated by cells in vivo [50]. In the 1.5mm tissue sample, FTY720-films generate a nearly equal tissue concentration ratio of FTY720:FTY720-P; however, FTY720-P-films generate a 3:1 tissue ratio of FTY720:FTY720-P suggesting that the two compounds are metabolized by the tissue differently (Figure 4B). While the reason for this phenomenon is unknown, the differences may reflect the balance of lipid import/export machinery, lipid kinases, and phosphatases that interact with FTY720 versus FTY720-P.
FTY720 is also known to interact with multiple points in the sphingolipid metabolic network including ceramide synthase [55], sphingosine kinase [56], sphingomyelinase [57], and S1P lyase [47]. This makes the net effect of FTY720 on sphingolipid metabolism difficult to predict. Thus, we investigated the impact of FTY720/FTY720-P release on the local S1P and sphingosine concentrations (Figure 4G–H) [47]. While the FTY720-P tissue samples were more variable, the local concentrations of S1P and sphingosine were equivalent in tissue surrounding FTY720 and FTY720-P releasing films suggesting that there is no net difference in the overall impact on S1P metabolism. We cannot exclude the possibility that there is some impact of FTY720 on the local S1P concentration that is already saturated in the FTY720-P animals and therefore no difference is seen between the two gradients. Additionally, the interpretation is complicated by the fact that the FTY720-P films release less total compound and therefore may have a greater effect at a lower total concentration (Figure 4 B, C, F).
The functional difference in arteriolar diameter remodeling is striking. FTY720 films induce significant remodeling while FTY720-P films yield minimal diameter expansion. The differential release properties and unequal gradients of FTY720 and FTY720-P partially confound the evaluation of which molecule is a better candidate for in vivo modulation of sphingolipid signaling and metabolism. The disparity in functional vascular changes between the two compounds could reflect the total concentration of drug in the tissue; however, the different signaling effects, slope/strength of the gradient, or ratio of phosphorylated/un-phosphorylated drug may also contribute to the functional outcome. FTY720-P has been shown to act as a functional antagonist for the S1P receptors, irreversibly internalizing and degrading bound S1P receptors rather then recycling them back to the cell surface as is the case with S1P ligands [58]. Taken together with the dose-dependent effect of local S1P on vascular remodeling (Figure 3F), the concentration of the drug is likely to play a role in the reduced remodeling response seen with release of the active form of FTY720-P (Figure 4I).
S1P signaling is well known for guiding localization and spatial control of many cell types [8, 12, 59]. Depending on the S1PR expression profile of a cell, it may respond differently to an S1P gradient. We have recently demonstrated that a pro-regenerative/anti-inflammatory AM population of monocytes expresses a high level of S1P3, which allows these cells to respond to FTY720 differently than their inflammatory IM counter-part [12]. AM cells are more readily recruited to the dorsal skinfold window chamber in the presence of a FTY720-releasing film compared to PLGA alone. One of the migration cues for AM cells is SDF-1α; FTY720 selectively enhances the migration of AM but not IM toward SDF-1α and the effect requires S1P3 [12]. Results from the current study suggest that local FTY720 administration does not alter circulation of lymphocytes (Figure 5D), a key phenomenon of systemic FTY720 [59]. However, a localized FTY720 gradient does enhance the migration of the AM monocyte population, known to respond to FTY720, to induce directed migration up the FTY720 gradient (Figure 6). The powerful role of S1P gradients in trafficking of endogenous circulating cells provides insight for developing sustained delivery methods for controlling local concentration of S1P receptor targeting drugs and/or sphingolipid metabolizing enzymes for a pro-regenerative therapeutic goal.
5. Conclusion
Delivery of therapeutic compounds from biodegradable polymers such as PLGA can be integrated as a cotherapy with medical devices or transplant tissue, such as stents [60], neuro-electrodes [61], artificial intraocular lens [62], or bone allografts [29], to produce in vivo localized drug delivery. Polymeric delivery provides spatial control of a desired response that cannot be achieved by systemic or bolus drug delivery methods. The manipulation of local S1P gradients in vivo remains an exciting target in regenerative medicine for the recruitment of endogenous stem, progenitor, and effector immune cells to promote tissue repair as well as the spatial positioning of exogenously supplied cells for tissue engineering. The current study assesses the local in vivo gradients of lipid mediators produced in inflamed tissue surrounding a biomaterial releasing S1P or S1P analogs and the coordinated events of vascular remodeling and recruitment of S1P3high monocytes to the in vivo gradient. Results demonstrate that biomaterial-based gradients of S1P are short-lived in the tissue due to degradation by S1P lyase, however stabilization of the gradient by inhibition of S1P catabolism or release of a more stable S1P analog promotes vascular remodeling and local recruitment of regenerative immune cells. Thus, the rational design of biomaterial-mediated delivery of pharmacological gradients should consider spatial control of cell signaling processes and guidance of cell localization in the tissue space of interest.
Acknowledgements
We thank Cynthia Huang and Shaun Tanner for assistance in HPLC-MS and dorsal skinfold window chamber procedure. Sources of support for this study include the National Institutes of Health grants K01AR052352-01A1, R01AR056445-01A2, and R01DE019935-01 to Dr. Botchwey.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Molly E. Ogle, Email: molly.ogle@bme.gatech.edu.
Lauren S. Sefcik, Email: andersol@lafayette.edu.
Anthony O. Awojoodu, Email: A.Awojoodu@gatech.edu.
Nathan F. Chiappa, Email: nchiappa3@gatech.edu.
Kevin Lynch, Email: krl2z@virginia.edu.
Shayn Peirce-Cottler, Email: smp6p@virginia.edu.
Edward A. Botchwey, Email: edward.botchwey@bme.gatech.edu.
References Cited
- 1.Wu D, Lin F. Modeling cell gradient sensing and migration in competing chemoattractant fields. PloS one. 2011;6:e18805. doi: 10.1371/journal.pone.0018805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Y, Irvine DJ. Engineering chemoattractant gradients using chemokine-releasing polysaccharide microspheres. Biomaterials. 2011;32:4903–4913. doi: 10.1016/j.biomaterials.2011.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Y, Irvine DJ. Convolution of chemoattractant secretion rate, source density, and receptor desensitization direct diverse migration patterns in leukocytes. Integrative biology : quantitative biosciences from nano to macro. 2013;5:481–494. doi: 10.1039/c3ib20249f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greenbaum A, Hsu YM, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, et al. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature. 2013;495:227–230. doi: 10.1038/nature11926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shechter R, Miller O, Yovel G, Rosenzweig N, London A, Ruckh J, et al. Recruitment of beneficial M2 macrophages to injured spinal cord is orchestrated by remote brain choroid plexus. Immunity. 2013;38:555–569. doi: 10.1016/j.immuni.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Si Y, Tsou CL, Croft K, Charo IF. CCR2 mediates hematopoietic stem and progenitor cell trafficking to sites of inflammation in mice. The Journal of clinical investigation. 2010;120:1192–1203. doi: 10.1172/JCI40310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen RR, Silva EA, Yuen WW, Mooney DJ. Spatio-temporal VEGF and PDGF delivery patterns blood vessel formation and maturation. Pharmaceutical research. 2007;24:258–264. doi: 10.1007/s11095-006-9173-4. [DOI] [PubMed] [Google Scholar]
- 8.Golan K, Vagima Y, Ludin A, Itkin T, Cohen-Gur S, Kalinkovich A, et al. S1P promotes murine progenitor cell egress and mobilization via S1P1-mediated ROS signaling and SDF-1 release. Blood. 2012;119:2478–2488. doi: 10.1182/blood-2011-06-358614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walter DH, Rochwalsky U, Reinhold J, Seeger F, Aicher A, Urbich C, et al. Sphingosine-1-phosphate stimulates the functional capacity of progenitor cells by activation of the CXCR4-dependent signaling pathway via the S1P3 receptor. Arteriosclerosis, thrombosis, and vascular biology. 2007;27:275–282. doi: 10.1161/01.ATV.0000254669.12675.70. [DOI] [PubMed] [Google Scholar]
- 10.Ishii M, Kikuta J, Shimazu Y, Meier-Schellersheim M, Germain RN. Chemorepulsion by blood S1P regulates osteoclast precursor mobilization and bone remodeling in vivo. The Journal of experimental medicine. 2010;207:2793–2798. doi: 10.1084/jem.20101474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lewis ND, Haxhinasto SA, Anderson SM, Stefanopoulos DE, Fogal SE, Adusumalli P, et al. Circulating monocytes are reduced by sphingosine-1-phosphate receptor modulators independently of S1P3. J Immunol. 2013;190:3533–3540. doi: 10.4049/jimmunol.1201810. [DOI] [PubMed] [Google Scholar]
- 12.Awojoodu AO, Ogle ME, Sefcik LS, Bowers DT, Martin K, Brayman KL, et al. Sphingosine 1-phosphate receptor 3 regulates recruitment of anti-inflammatory monocytes to microvessels during implant arteriogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:13785–13790. doi: 10.1073/pnas.1221309110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spiegel S, Milstien S. The outs and the ins of sphingosine-1-phosphate in immunity. Nature reviews Immunology. 2011;11:403–415. doi: 10.1038/nri2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hla T, Venkataraman K, Michaud J. The vascular S1P gradient-cellular sources and biological significance. Biochimica et biophysica acta. 2008;1781:477–482. doi: 10.1016/j.bbalip.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alewijnse AE, Peters SL. Sphingolipid signalling in the cardiovascular system: good, bad or both? European journal of pharmacology. 2008;585:292–302. doi: 10.1016/j.ejphar.2008.02.089. [DOI] [PubMed] [Google Scholar]
- 16.Sefcik LS, Petrie Aronin CE, Botchwey EA. Engineering vascularized tissues using natural and synthetic small molecules. Organogenesis. 2008;4:215–227. doi: 10.4161/org.4.4.6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Massberg S, von Andrian UH. Novel trafficking routes for hematopoietic stem and progenitor cells. Annals of the New York Academy of Sciences. 2009;1176:87–93. doi: 10.1111/j.1749-6632.2009.04609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olivera A, Spiegel S. Sphingosine-1-phosphate as second messenger in cell proliferation induced by PDGF and FCS mitogens. Nature. 1993;365:557–560. doi: 10.1038/365557a0. [DOI] [PubMed] [Google Scholar]
- 19.Sefcik LS, Wilson JL, Papin JA, Botchwey EA. Harnessing systems biology approaches to engineer functional microvascular networks. Tissue engineering Part B, Reviews. 2010;16:361–370. doi: 10.1089/ten.teb.2009.0611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wieghaus KA, Nickerson MM, Petrie Aronin CE, Sefcik LS, Price RJ, Paige MA, et al. Expansion of microvascular networks in vivo by phthalimide neovascular factor 1 (PNF1) Biomaterials. 2008;29:4698–4708. doi: 10.1016/j.biomaterials.2008.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doyle ME, Perley JP, Skalak TC. Bone marrow-derived progenitor cells augment venous remodeling in a mouse dorsal skinfold chamber model. PloS one. 2012;7:e32815. doi: 10.1371/journal.pone.0032815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davis MD, Clemens JJ, Macdonald TL, Lynch KR. Sphingosine 1-phosphate analogs as receptor antagonists. The Journal of biological chemistry. 2005;280:9833–9841. doi: 10.1074/jbc.M412356200. [DOI] [PubMed] [Google Scholar]
- 23.Alvarez-Vasquez F, Sims KJ, Cowart LA, Okamoto Y, Voit EO, Hannun YA. Simulation and validation of modelled sphingolipid metabolism in Saccharomyces cerevisiae. Nature. 2005;433:425–430. doi: 10.1038/nature03232. [DOI] [PubMed] [Google Scholar]
- 24.Cimetta E, Cannizzaro C, James R, Biechele T, Moon RT, Elvassore N, et al. Microfluidic device generating stable concentration gradients for long term cell culture: application to Wnt3a regulation of beta-catenin signaling. Lab on a chip. 2010;10:3277–3283. doi: 10.1039/c0lc00033g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farahat WA, Wood LB, Zervantonakis IK, Schor A, Ong S, Neal D, et al. Ensemble analysis of angiogenic growth in three-dimensional microfluidic cell cultures. PloS one. 2012;7:e37333. doi: 10.1371/journal.pone.0037333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sefcik LS, Aronin CE, Awojoodu AO, Shin SJ, Mac Gabhann F, MacDonald TL, et al. Selective activation of sphingosine 1-phosphate receptors 1 and 3 promotes local microvascular network growth. Tissue engineering Part A. 2011;17:617–629. doi: 10.1089/ten.tea.2010.0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Das A, Segar CE, Hughley BB, Bowers DT, Botchwey EA. The promotion of mandibular defect healing by the targeting of S1P receptors and the recruitment of alternatively activated macrophages. Biomaterials. 2013;34:9853–9862. doi: 10.1016/j.biomaterials.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Das A, Tanner S, Barker DA, Green D, Botchwey EA. Delivery of S1P receptor-targeted drugs via biodegradable polymer scaffolds enhances bone regeneration in a critical size cranial defect. Journal of biomedical materials research Part A. 2014;102:1210–1218. doi: 10.1002/jbm.a.34779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang C, Das A, Barker D, Tholpady S, Wang T, Cui Q, et al. Local delivery of FTY720 accelerates cranial allograft incorporation and bone formation. Cell and tissue research. 2012;347:553–566. doi: 10.1007/s00441-011-1217-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sefcik LS, Petrie Aronin CE, Wieghaus KA, Botchwey EA. Sustained release of sphingosine 1-phosphate for therapeutic arteriogenesis and bone tissue engineering. Biomaterials. 2008;29:2869–2877. doi: 10.1016/j.biomaterials.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amann LC, Gandal MJ, Lin R, Liang Y, Siegel SJ. In vitro-in vivo correlations of scalable PLGA-risperidone implants for the treatment of schizophrenia. Pharmaceutical research. 2010;27:1730–1737. doi: 10.1007/s11095-010-0152-4. [DOI] [PubMed] [Google Scholar]
- 32.Faisant N, Siepmann J, Benoit JP. PLGA-based microparticles: elucidation of mechanisms and a new, simple mathematical model quantifying drug release. European journal of pharmaceutical sciences : official journal of the European Federation for Pharmaceutical Sciences. 2002;15:355–366. doi: 10.1016/s0928-0987(02)00023-4. [DOI] [PubMed] [Google Scholar]
- 33.Ramchandani M, Robinson D. In vitro and in vivo release of ciprofloxacin from PLGA 50:50 implants. Journal of controlled release : official journal of the Controlled Release Society. 1998;54:167–175. doi: 10.1016/s0168-3659(97)00113-2. [DOI] [PubMed] [Google Scholar]
- 34.Lee H, Goetzl EJ, An S. Lysophosphatidic acid and sphingosine 1-phosphate stimulate endothelial cell wound healing. American journal of physiology Cell physiology. 2000;278:C612–C618. doi: 10.1152/ajpcell.2000.278.3.C612. [DOI] [PubMed] [Google Scholar]
- 35.Kluk MJ, Hla T. Role of the sphingosine 1-phosphate receptor EDG-1 in vascular smooth muscle cell proliferation and migration. Circulation research. 2001;89:496–502. doi: 10.1161/hh1801.096338. [DOI] [PubMed] [Google Scholar]
- 36.Kimura T, Sato K, Malchinkhuu E, Tomura H, Tamama K, Kuwabara A, et al. High-density lipoprotein stimulates endothelial cell migration and survival through S1P and its receptors. Arteriosclerosis, thrombosis, and vascular biology. 2003;23:1283–1288. doi: 10.1161/01.ATV.0000079011.67194.5A. [DOI] [PubMed] [Google Scholar]
- 37.Oyama O, Sugimoto N, Qi X, Takuwa N, Mizugishi K, Koizumi J, et al. The lysophospholipid mediator sphingosine-1-phosphate promotes angiogenesis in vivo in ischaemic hindlimbs of mice. Cardiovascular research. 2008;78:301–307. doi: 10.1093/cvr/cvn002. [DOI] [PubMed] [Google Scholar]
- 38.Roberts R, Sciorra VA, Morris AJ. Human type 2 phosphatidic acid phosphohydrolases. Substrate specificity of the type 2a, 2b, and 2c enzymes and cell surface activity of the 2a isoform. The Journal of biological chemistry. 1998;273:22059–22067. doi: 10.1074/jbc.273.34.22059. [DOI] [PubMed] [Google Scholar]
- 39.Peest U, Sensken SC, Andreani P, Hanel P, Van Veldhoven PP, Graler MH. S1P-lyase independent clearance of extracellular sphingosine 1-phosphate after dephosphorylation and cellular uptake. Journal of cellular biochemistry. 2008;104:756–772. doi: 10.1002/jcb.21665. [DOI] [PubMed] [Google Scholar]
- 40.Le Stunff H, Peterson C, Thornton R, Milstien S, Mandala SM, Spiegel S. Characterization of murine sphingosine-1-phosphate phosphohydrolase. The Journal of biological chemistry. 2002;277:8920–8927. doi: 10.1074/jbc.M109968200. [DOI] [PubMed] [Google Scholar]
- 41.Ikeda M, Kihara A, Igarashi Y. Sphingosine-1-phosphate lyase SPL is an endoplasmic reticulum-resident, integral membrane protein with the pyridoxal 5'-phosphate binding domain exposed to the cytosol. Biochemical and biophysical research communications. 2004;325:338–343. doi: 10.1016/j.bbrc.2004.10.036. [DOI] [PubMed] [Google Scholar]
- 42.Hisano Y, Kobayashi N, Yamaguchi A, Nishi T. Mouse SPNS2 functions as a sphingosine-1-phosphate transporter in vascular endothelial cells. PloS one. 2012;7:e38941. doi: 10.1371/journal.pone.0038941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Serra M, Saba JD. Sphingosine 1-phosphate lyase, a key regulator of sphingosine 1-phosphate signaling and function. Advances in enzyme regulation. 2010;50:349–362. doi: 10.1016/j.advenzreg.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kawanabe T, Kawakami T, Yatomi Y, Shimada S, Soma Y. Sphingosine 1-phosphate accelerates wound healing in diabetic mice. Journal of dermatological science. 2007;48:53–60. doi: 10.1016/j.jdermsci.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 45.Bode C, Graler MH. Immune regulation by sphingosine 1-phosphate and its receptors. Archivum immunologiae et therapiae experimentalis. 2012;60:3–12. doi: 10.1007/s00005-011-0159-5. [DOI] [PubMed] [Google Scholar]
- 46.Hale JJ, Yan L, Neway WE, Hajdu R, Bergstrom JD, Milligan JA, et al. Synthesis, stereochemical determination and biochemical characterization of the enantiomeric phosphate esters of the novel immunosuppressive agent FTY720. Bioorganic & medicinal chemistry. 2004;12:4803–4807. doi: 10.1016/j.bmc.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 47.Bandhuvula P, Tam YY, Oskouian B, Saba JD. The immune modulator FTY720 inhibits sphingosine-1-phosphate lyase activity. The Journal of biological chemistry. 2005;280:33697–33700. doi: 10.1074/jbc.C500294200. [DOI] [PubMed] [Google Scholar]
- 48.Mechtcheriakova D, Wlachos A, Sobanov J, Bornancin F, Zlabinger G, Baumruker T, et al. FTY720-phosphate is dephosphorylated by lipid phosphate phosphatase 3. FEBS letters. 2007;581:3063–3068. doi: 10.1016/j.febslet.2007.05.069. [DOI] [PubMed] [Google Scholar]
- 49.Sensken SC, Bode C, Graler MH. Accumulation of fingolimod (FTY720) in lymphoid tissues contributes to prolonged efficacy. The Journal of pharmacology and experimental therapeutics. 2009;328:963–969. doi: 10.1124/jpet.108.148163. [DOI] [PubMed] [Google Scholar]
- 50.Paugh SW, Payne SG, Barbour SE, Milstien S, Spiegel S. The immunosuppressant FTY720 is phosphorylated by sphingosine kinase type 2. FEBS letters. 2003;554:189–193. doi: 10.1016/s0014-5793(03)01168-2. [DOI] [PubMed] [Google Scholar]
- 51.Hisano Y, Kobayashi N, Kawahara A, Yamaguchi A, Nishi T. The sphingosine 1-phosphate transporter, SPNS2, functions as a transporter of the phosphorylated form of the immunomodulating agent FTY720. The Journal of biological chemistry. 2011;286:1758–1766. doi: 10.1074/jbc.M110.171116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heerklotz H, Seelig J. Correlation of membrane/water partition coefficients of detergents with the critical micelle concentration. Biophysical journal. 2000;78:2435–2440. doi: 10.1016/S0006-3495(00)76787-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sasaki H, Arai H, Cocco MJ, White SH. pH dependence of sphingosine aggregation. Biophysical journal. 2009;96:2727–2733. doi: 10.1016/j.bpj.2008.12.3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Swain J, Mohapatra M, Borkar SR, Aidhen IS, Mishra AK. Study of aqueous phase aggregation of FTY720 (fingolimod hydrochloride) and its effect on DMPC liposomes using fluorescent molecular probes. Physical chemistry chemical physics : PCCP. 2013;15:17962–17970. doi: 10.1039/c3cp53148a. [DOI] [PubMed] [Google Scholar]
- 55.Lahiri S, Park H, Laviad EL, Lu X, Bittman R, Futerman AH. Ceramide synthesis is modulated by the sphingosine analog FTY720 via a mixture of uncompetitive and noncompetitive inhibition in an Acyl-CoA chain length-dependent manner. The Journal of biological chemistry. 2009;284:16090–16098. doi: 10.1074/jbc.M807438200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Billich A, Bornancin F, Devay P, Mechtcheriakova D, Urtz N, Baumruker T. Phosphorylation of the immunomodulatory drug FTY720 by sphingosine kinases. The Journal of biological chemistry. 2003;278:47408–47415. doi: 10.1074/jbc.M307687200. [DOI] [PubMed] [Google Scholar]
- 57.Dawson G, Qin J. Gilenya (FTY720) inhibits acid sphingomyelinase by a mechanism similar to tricyclic antidepressants. Biochemical and biophysical research communications. 2011;404:321–323. doi: 10.1016/j.bbrc.2010.11.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Choi JW, Gardell SE, Herr DR, Rivera R, Lee CW, Noguchi K, et al. FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:751–756. doi: 10.1073/pnas.1014154108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sensken SC, Nagarajan M, Bode C, Graler MH. Local inactivation of sphingosine 1-phosphate in lymph nodes induces lymphopenia. J Immunol. 2011;186:3432–3440. doi: 10.4049/jimmunol.1002169. [DOI] [PubMed] [Google Scholar]
- 60.Pan Ch J, Tang JJ, Weng YJ, Wang J, Huang N. Preparation, characterization and anticoagulation of curcumin-eluting controlled biodegradable coating stents. Journal of controlled release : official journal of the Controlled Release Society. 2006;116:42–49. doi: 10.1016/j.jconrel.2006.08.023. [DOI] [PubMed] [Google Scholar]
- 61.Abidian MR, Kim DH, Martin DC. Conducting-Polymer Nanotubes for Controlled Drug Release. Adv Mater. 2006;18:405–409. doi: 10.1002/adma.200501726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eperon S, Bossy-Nobs L, Petropoulos IK, Gurny R, Guex-Crosier Y. A biodegradable drug delivery system for the treatment of postoperative inflammation. International journal of pharmaceutics. 2008;352:240–247. doi: 10.1016/j.ijpharm.2007.10.054. [DOI] [PubMed] [Google Scholar]