

Abstract
Many self-reactive B cells exist in the periphery in a rapidly reversible state of unresponsiveness referred to as anergy. Reversibility of anergy indicates that chronically occupied BCR must transduce non-durable regulatory signals that maintain unresponsiveness. Consistent with such a mechanism, studies of immunoglobulin transgenic, as well as naturally occurring polyclonal autoreactive B cells demonstrate activation of the inositol 5-phosphatase SHIP-1 in anergic cells, and low affinity chromatin autoantigen-reactive B cells have been shown to require expression of this phosphatase to maintain anergy. However, it has been reported that anergy of B cells recognizing high affinity soluble antigen may not require SHIP-1, and is instead mediated by upregulation of the inositol 3-phosphatase PTEN. To further explore this apparent difference in mechanism we analyzed the effect of B cell-targeted SHIP-1 deletion on immune tolerance of high affinity anti-HEL B cells in mice expressing soluble HEL (MD4.ML-5). We report that SHIP-1 functions to dampen responses of naïve and low-dose antigen-primed B cells in vitro, and is required for induction of B cell tolerance. Thus, while anergy of B cells reactive with low affinity and likely polyvalent chromatin antigens is maintained by activation of inhibitory signaling circuitry involving SHIP-1, anergy of B cells recognizing soluble self antigen with high affinity also requires increased activity of SHIP-1.
1. Introduction
The B cell repertoire is finely tuned to enable generation of protective anti-pathogen immunity while avoiding production of potentially harmful antibodies to self and presentation of autoantigen peptides to T cells. Underscoring the magnitude this challenge are findings that >70% of newly produced B cells in bone marrow express autoreactive antigen receptors (BCR) [1]. Elimination of immature cells specific for high avidity autoantigens that induce strong antigen receptor signals is accomplished by receptor editing, which changes the receptors’ specificity [2, 3]. In the event that high avidity self-reactivity is retained, i.e. editing fails, cells are eliminated by apoptotic death in a process termed clonal deletion [4]. A more challenging situation exists in the case of cells that recognize low avidity antigens. Even if receptor affinity is very high, low antigen avidity can limit signaling sufficiently to render cells ignorant of antigen in their environment or be induced to enter a state of unresponsiveness referred to as anergy [5, 6]. Anergy is fragile, being readily reversed by removal of antigen from receptors, and thus must require continuous transduction of anergy-enforcing signals through BCRs [7, 8]. Indeed, available evidence indicates that these regulatory signaling mechanisms can be compromised by autoimmunity risk alleles [9]. The signals that emanate from antigen receptors and enforce anergy are poorly understood.
Three effectors have emerged as potential mediators of anergy-enforcing signals, the SH2-containing Inositol 5-Phosphatase SHIP-1 [10], Phosphatase and Tensin Homolog PTEN[11], and the SH2-containing Tyrosine Phosphatase SHP-1[12]. While SHIP-1 is activated in anergic cells and is critical for maintenance of anergy in the anti-chromatin immunoglobulin transgenic model ARS/A1 it has been suggested that in the MD4.ML-5 model, wherein BCR bind a protein antigen, HEL, with very high affinity, anergy does not require SHIP-1 activation [13], but rather is maintained by upregulation of PTEN [14]. Consistent with this possibility, while B cell-targeted SHIP-1 deficiency in mice possessing a polyclonal repertoire resulted in production of anti-chromatin autoantibodies, these mice did not make autoantibodies against protein autoantigens, suggesting SHIP-1 is not necessary to silence cells reactive to these antigens [10].
PTEN and SHIP-1 regulate the PI3-kinase pathway by removing specific phosphate groups from PI(3,4,5)P3. However, there is an important functional distinction. PTEN attacks its substrate PI(3,4,5)P3 at the 3 position of the inositol ring, generating the PI(4,5)P2 substrate of phospholipase C important in positive signaling. SHIP-1 attacks the 5 position of the inositol ring generating PI(3,4)P2, a feedback activator of SHIP-1 and stimulator of pathways involving the adaptors TAPP1 and TAPP2 that are thought to inhibit Akt [15]. SHIP-1 also associates with the rasGAP adaptor Dok-1[16]. Thus while both PTEN and SHIP could have negative function by depleting PI(3,4,5)P3 needed for BCR signaling, both have additional functions that could be important in maintaining anergy. A better understanding of their function in the context of anergy may provide avenues for therapeutic intervention in autoimmunity.
It seems unlikely that two mutually exclusive mechanisms would evolve to maintain anergy in B cells. However, one might imagine that additional enforcing mechanisms might be required to counteract increased signal strength associated with high affinity autoantigen binding. Therefore we set out to explore whether SHIP-1 is required for maintenance of anergy in the high affinity MD4/ML-5 anti HEL model. Using B cell-targeted SHIP-1 knockouts in the context of a variety of in vitro and in vivo approaches we demonstrate that SHIP-1 functions in feedback regulation of antigen-induced BCR signaling in naïve B cells and is required for maintenance of tolerance of chronically stimulated cells. Thus SHIP-1 is required for induction and maintenance of B cell anergy in both low affinity Ars/A1 and high affinity MD4.ML-5 models despite increased expression of PTEN in the latter [14]. These findings are consistent with our previous report that lupus-like autoimmunity with early death is characteristic of C57BL/6 mice in which B cells lack SHIP-1 [10].
2. Materials and Methods
2.1 Mice
MD4 (HyHEL10-Ig transgene) and ML-5 (soluble HEL transgene) were crossed to obtain MD4.ML-5 double transgenic mice [17]. SHIPfl/fl [18] were crossed with mb1cre/wt (cre driven by the mb1 promoter) [19] to generate mice that delete SHIP specifically in all B cells. The SHIPfl/fl.mb1cre/wt mice were then crossed to MD4 and MD4.ML-5 transgenic mice. ML-5 mice were bred with C57BL/6 mice to obtain wt and HEL transgenic recipient mice. All mice used in this study were on the C57BL/6 background. Bone marrow chimeric mice were created by reconstituting lethally irradiated (960 rads) WT C57BL/6 and ML-5 mice with 3×106 bone marrow cells from MD4.mb1cre/wt or MD4.SHIPfl/fl.mb1cre/wt donor mice via tail vein injection. Experiment was repeated twice with at least three mice per experimental group. All animals were bred and housed in the Biological Resource Center at National Jewish Health. All experiments with mice were performed in accordance with the regulations and with the approval of National Jewish Health (Denver, CO) Institutional Animal Care and Use Committee (IACUC).
2.2 B Cell Isolation
Splenocytes were isolated by forcing spleens through 70 micron screens, red blood cells were lysed in a hypotonic solution of ammonium potassium chloride. Cells were washed in IMDM with 2% FCS. In adoptive transfer experiments, “Untouched” B cells were isolated from splenocytes by depletion of CD43+ cells with anti-CD43-conjugated magnetic beads as per manufacturers protocol. (MACS anti-mouse CD43; Miltenyi Biotec). B cell purity was routinely >97% and fewer than 1% of these cells were plasmablasts based on B220low and CD138+ phenotype.
2.3 Flow Cytometry
Cells were resuspended in PBS containing 1% FCS and 0.05% sodium azide and incubated with an optimal dilution of fluorochrome conjugated antibodies. Antibodies employed included those specific for B220 (RA3-6B2, BD), IgM (b-7-6), IgD (11-26c, Southern Biotech), IgMa (RS3.1), SHIP-1 (polyclonal rabbit antibody against the C-terminal 100aa), CD138 (281-2, BD), CD23(EBVCS-5, Biolegend), CD21 and D1.3 anti-HEL recognizing a distinct, non-hindering HEL epitope [20]. The D1.3 hybridoma was obtained from Richard Willson (Univ. of Houston ) via National Cell Culture Center (www.nccc.com/). Antibodies to SHIP-1, CD21, IgMa, IgM and HEL (D1.3) were produced in our own laboratory and were directly conjugated to Alexa (Invitrogen) or DyLight (Pierce) fluorochromes, according to the manufacturer’s protocol. For detection of plasmablasts, splenocytes were first stained with B220-FITC and CD138PE, then fixed and permeabilized with BD Cytofix/Cytoperm™ and stained with HEL-DyLight 650 (HEL from Sigma reconstituted and conjugated to DyLight-650). Cells that were highly stained with HEL-DyLight 650 were routinely found to be CD138 positive (data not shown). Cells were analyzed on a Cyan (Beckman Coulter) or LSRII flow cytometer (BD) with data analysis using FlowJo (Tree Star).
2.4 Analysis of calcium mobilization
For measurements of relative intracellular free calcium concentration in follicular B cells ([Ca2+]i), B220-FITC, CD23-PE-Cy7, CD21-Alexa 647-stained splenocytes were loaded at room temperature with Indo-1 acetoxymethyl (Indo1-AM) (Molecular Probes) as described [7] and resuspended at 5×106cells/ml in IMDM with 1% fatty acid free BSA. Samples were transferred to 37°C 15 minutes prior to analysis. Analysis was initiated and the baseline FL-5/FL-4 ratio was established. After 30 sec cells were stimulated with F(ab′)2 rabbit anti-mouse IgM (Zymed) or HEL (Sigma) for 2.5 minutes. Relative mean intracellular calcium ion concentration [Ca2+]i was measured over time using an LSR II flow cytometer (BD) and data analyzed using FlowJo software (Tree Star). Integrated calcium response was determined by calculating the area under the curve delineated by the baseline of each sample at T=30 seconds and relative mean fluorescence ratio over time.
2.5 Receptor occupancy
To establish background and 100% occupied receptor controls for each sample, 1–3×106 MD4 splenocytes in 200 uL PBS were incubated with saturating concentrations of HEL (200 ng/ml PBS HEL (Sigma) or PBS alone on ice for 15 min. They were then fixed using PFA at a final concentration of 2% volume/volume, then washed and stained with anti-B220-PerCP, anti-IgMa-FITC, anti-IgD-PE and D1.3-DyLight 650 anti-HEL. Staining was analyzed on an LSR II flow cytometer (BD). Mean fluorescence intensity (MFI) of HEL-preincubated B cells provided a 100% occupied measure, T cells yielded a measurement of background intensity. MFI of PBS-pretreated B cells provided a measure of occupied BCR with T cells again controlling for background. Parenthetically, the 0% occupied naïve B cell control staining was equivalent to that of T cells (data not shown). To measure the proportion of receptors occupied by HEL in experimental samples, each cell population was stained as described above and their MFI minus MFI of background control divided by that of the 100% occupied internal control minus the MFI of background control for the same population. This value was multiplied by 100 to obtain the percentage of receptors occupied by HEL (Figure 1).
Figure 1. Measurement of antigen receptor occupancy.
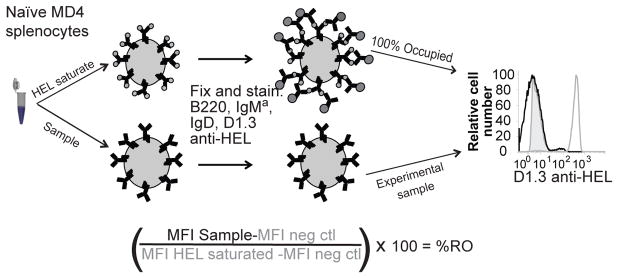
Surface HEL was detected by staining cells with anti-HEL mAb D1.3, which recognizes a HEL epitope that is not hindered by HyHEL10 (MD4) BCR, and measured by flow cytometry. Percent of receptors occupied was calculated based on staining relative to HEL-saturated (100%) and unstained (0%) controls.
2.6 In vitro antigen stimulation
Splenocytes were incubated for 2 hours at room temperature with 0–30 ng/mL HEL. Cells were maintained in these concentrations of HEL during all subsequent staining and washing procedures, and throughout the calcium mobilization assay.
2.7 Enzyme-Linked Immunosorbent Assay (ELISA)
For detection of IgM anti-HEL in serum of mice, 96-well microtiter plates (Costar) were coated with HEL (10 μg/mL) in PBS, followed by incubation with blocking buffer solution (2 mg/mL bovine serum albumin (BSA). Mouse serum was diluted serially in PBS and 100 μL incubated in the 96 well plates overnight at 4°C. Between all steps the plates were washed 4 times with PBS-0.05% Tween-20. IgM anti-HEL was detected with an HRP-conjugated goat anti-mouse IgM (μ chain (Southern Biotechnology). For detection of anti-HEL IgM:HEL complexes microtiter plates were coated with 10 μg/mL D1.3 anti-HEL in carbonate buffer, then washed and incubated as above. Complexes were detected using HRP-conjugated goat anti-mouse IgM (μ chain-specific) (Southern Biotechnology). The ELISAs were developed with TMB single solution (Invitrogen) and the reaction was stopped with 1N H2PO4 (Sigma). The OD was determined at 405nm using a VERSAMax plate reader (Molecular Devices) and data analyzed using PRISM software.
2.8 ELISPOT assay for quantification of cells secreting anti-hen egg lysozyme antibodies
For ELISPOT analysis, 96-well plates were coated with 10 ug/ml hen egg lysozyme (HEL)/well, incubated at RT for 2 h, and blocked with 2% BSA in PBS for 2 h. Plates were then washed three times with PBS + 0.05% Tween, with the final wash consisting of IMDM with 10% FCS. Freshly isolated splenocytes were washed and resuspended in IMDM with 10% FCS. 2-fold serial dilutions were made starting with 1/100th of a spleen in the first well. Samples were incubated at 37°C for 6 h. The cells were then lysed by washing three times with PBS + 0.05% Tween, letting each wash sit for 10 min at RT. Plates were then incubated with RS3.1-biotin (anti-IgMa; 1:2000) in PBS at 4°C overnight. The plates were incubated with Streptavidin-AP (as directed by provider; Southern Biotech Cat. No. 7100–04) in PBS at RT for 1–2 hours. Between each incubation plates were washed four times with PBS + 0.05% Tween. Plates were developed with Elispot developing solution (25 mM 5-bromo-chloro-3-indolyl phosphate p-toluidine,100 mM NaCl, 100 mM Tris, 10 mM MgCl2 [pH 9.5]) for 1 h. The reaction was stopped by washing the plate three times with double-distilled H2O. ASC frequency was calculated based on number of spots at a cell dilution in the linear range [21].
2.9 B cell adoptive transfer
106 splenic B cells isolated by depletion of CD43+ cells were labeled with Cell Trace Violet (Invitrogen) according to manufacturers protocol, then resuspended in 200 μl PBS and injected i.v. into non-irradiated C57BL/6 or ML-5 mice. Mice were rested for three days to allow B cell tolerance to develop, before spleens were harvested and analyzed.
2.10 Statistical Methods
Student’s unpaired t-test was used to determine statistically significance of differences between sample groups. Error bars represent the standard error of the mean.
3. Results
3.1 The SH2-containing inositol 5-phosphatase SHIP-1 negatively regulates BCR signaling in naïve, low dose antigen-primed and antigen desensitized B cells
To first address the role of SHIP-1 in regulation of BCR signaling in a reductionist in vitro setting we assessed the effect of B cell-targeted SHIP-1 deletion on antigen-induced calcium mobilization responses of ex vivo MD4 anti-HEL B cells. We compared effects of SHIP-1 knockout on responses of naïve B cells and B cells that had been incubated with varied concentrations of antigen at 37°C for two hours. B cell-targeted SHIP-1 knockout was accomplished using mb1 promoter driven CRE in floxed SHIP-1, anti-HEL (MD4) immunoglobulin transgenic mice. Antigen receptor occupancy of control and pre-incubated populations was assessed by staining cells with the D1.3 anti-HEL monoclonal antibody that binds an epitope distinct from MD4 (Figure 2A) [20]. Receptor occupancy (RO) increased as antigen exposure increased with 1ng HEL/2×106 cells/ml leading to 14% RO, 3ng HEL/ml leading to 41% RO and 10ng HEL/ml leading to 86% RO in SHIP-1-sufficient MD4 B cells. IgD expression remained unchanged at all levels antigen exposure, while IgM was decreased slightly following exposure to antigen at the highest antigen concentration, yielding 86% RO (Figure 2A). Receptor occupancy and IgD expression were unaffected by SHIP-1 deficiency, but IgM expression was reduced in SHIP-1 deficient cells as has been previously reported [13] (Figure 2A).
Figure 2. SHIP regulates BCR-mediated signaling in naïve, antigen primed and anergic B cells.
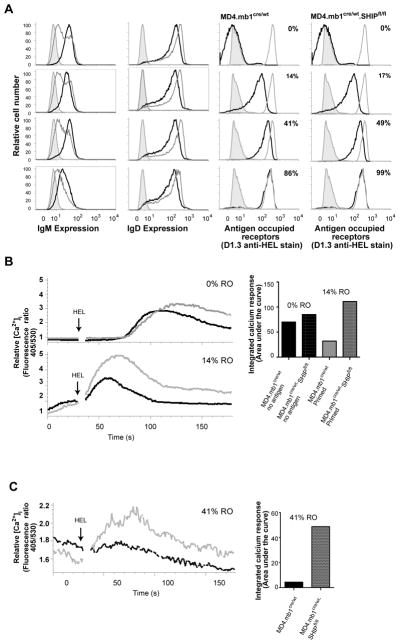
Immediately ex vivo MD4 splenocytes were incubated for two hours at 37°C with various concentrations of HEL (0–30ng/mL) before; A. Levels of IgM and IgD expression by SHIP-1 sufficient (black traces left panels) or SHIP-1 deficient (gray traces left panels) B cells were assessed relative to proportion of BCR occupied by HEL (RO) (black line on right panels). Receptor occupancy was determined as described in section 2.5. Also shown in left panels is negative control T cell staining (gray shaded histograms). In right panels staining of zero (T cells, gray shaded) and 100% receptor-saturated B cells (gray) controls are also shown. B. B cells that had been incubated with concentrations of HEL that achieved 0% or 14% receptor occupancy were Indo1-AM loaded and intracellular free calcium monitored before and after stimulation with 15 ng/mL HEL. Responses of SHIP sufficient (black) and SHIP deficient (gray) B cells are shown. Integrated calcium response (right panel) was derived from determination of the area above the baseline (at time of stimulation) of each sample. C. Cells incubated with sufficient antigen to achieve 41% RO were Indo1-AM loaded before analysis of HEL-induced calcium mobilization as above; SHIP sufficient (black), SHIP deficient (gray). Integrated calcium mobilization scaled to relative [Ca2+]i is shown at right. Experiment was repeated more than four times pooling B cells from three mice per experimental group.
Absence of SHIP-1 led to enhanced late phase calcium mobilization responses by naïve MD4 follicular B cells (B220+CD23+CD21+) to HEL stimulation (Figure 2B). MD4 B cells primed by treatment with HEL under conditions leading to occupancy of 14% of BCR, described above, responded more rapidly to antigen stimulation, and absence of SHIP-1 in these cells led to a robust increase in the amplitude and duration of this response. Analysis of the integrated area under the curve revealed a five-fold increase in the calcium response of SHIP-1 deficient as compared to SHIP-1 sufficient 14% RO B cells.
Anergic B cells from MD4.ML-5 mice are known to maintain around 40–50% RO and yet the remaining 50–60% of receptors that are unoccupied do not mediate significant calcium mobilization upon further antigen stimulation [22, 23]. Consistent with these observations, in vitro incubation of naïve MD4 B cells with HEL at concentrations that resulted in 41% receptor occupancy (Figure 2A) rendered cells unresponsive to subsequent HEL stimulation (Figure 2C). However, equivalently pretreated SHIP-1-deficient MD4 B cells mounted a significantly increased calcium mobilization upon HEL stimulation. The response of 49% RO SHIP-1 deficient cells was increased by relative to SHIP-1 sufficient cells suggesting loss of anergy.
These findings indicate that SHIP-1 functions as a negative regulator of BCR signaling in naïve follicular B cells, as well as in cells that have encountered sufficient antigen to prime for a subsequent response. Considered in the context of central tolerance, increased responsiveness of naïve SHIP-1 deficient B cells suggests that absence of SHIP-1 might lead to more efficient receptor editing and clonal deletion (Figure 2B). Conversely, failure of MD4 B cells exposed to higher doses of antigen to maintain unresponsiveness suggests that absence of SHIP-1 may compromise anergy in vivo (Figure 2C).
3.2 B cell-targeted SHIP-1 deficiency leads to autoimmunity in MD4.ML-5 mice
To explore the B cell intrinsic role of SHIP-1 in induction and maintenance of B cell tolerance in vivo we first analyzed the effect of mb1 promoter driven CRE deletion of SHIP-1 on tolerance in HEL anti-HEL (MD4.ML-5) mice. In these mice SHIP-1 expression is decreased by >90% in mature splenic B cells and this depletion occurs early in B cell development due to activation of CRE expression at the pro B cell stage of development [10]. Characterization of SHIP-1 gene-targeted Ars/A1 mice revealed a 70% reduction of B cells in spleens relative to controls [24]. A recent report showed a similar reduction of peripheral B cells in C57BL/6 mice in which SHIP-1 is ablated in B cells [14]. This finding is suggestive of enhanced central tolerance, which is consistent with the observed enhanced BCR signaling of naïve follicular B cells lacking SHIP-1 (Figure 2B). Consistent with previous reports of anergy-based tolerance, B cells from control MD4.ML-5.mb1cre/wt mice expressed >90% reduced levels of cell surface IgM and normal levels of IgD (Figure 3A), exhibited blunted BCR-mediated calcium signaling responses (Figure 3B) and showed >40% BCR occupancy by HEL (Figure 3F). These animals did not spontaneously produce anti-HEL antibody (Figure 3C) or anti-HEL antibody secreting cells detectable by ELISPOT (Figure 3D). Finally, IgM anti-HEL immune complexes were not detected in peripheral blood, consistent with lack of antibody production. In contrast, ex vivo B cells from MD4.ML-5.mb1cre/wt.SHIP-1fl/fl mice expressed only slightly reduced (<5%) mIgM levels (Figure 3A) and mounted very robust calcium mobilization responses following antigen stimulation (Figure 3B), both consistent with loss of anergy. These mice spontaneously produced high levels of antibody secreting cells (Figure 3D) and serum anti-HEL IgM (Figure 3C), some of which was detected in immune complexes (Figure 3E). Interestingly, these B cells exhibited 22% receptor occupancy (Figure 3F), a level that would not be expected to induce or sustain anergy (Figure 2)[22].
Figure 3. B cells from SHIP-deficient MD4.ML-5 mice are ignorant of HEL.

A. IgM and IgD expression by ex-vivo splenic B cells from SHIP-sufficient MD4.ML-5 (black), SHIP-deficient MD4.ML-5 (gray), MD4 (dashed black), SHIP-1 deficient (dashed gray) and T cells (gray shaded) is shown. B. Splenic B cells from SHIP-sufficient MD4.ML-5 (black), SHIP-deficient MD4.ML-5 (gray) and MD4 SHIP-sufficient control mice (dashed black) were Indo1-AM loaded and stained with non-stimulating antibodies specific to B220, CD21 and CD23. Follicular B cells were identified by gating on B220+, CD23hi, CD21mid cells and intracellular free calcium of these cells was monitored before and after stimulation with 15 ng/mL HEL. Receptor occupancy of each population was assessed immediately ex vivo and is noted adjacent to trace. C. Anti-HEL antibody levels were determined by ELISA of serum from SHIP-sufficient MD4.ML-5 (square), SHIP-deficient MD4.ML-5 (circle) and C57BL/6 (triangle) mice. D. Anti-HEL antibody secreting cells (ASC) were measured by ELISPOT analysis of splenocytes from MD4.ML-5 mice in which B cells were SHIP-sufficient (squares) or SHIP-deficient (circles). Shaded area reflects the lower limit of detection of the assay. E. IgMa anti-HEL:HEL complexes were measured by ELISA of serum from SHIP-sufficient MD4.ML-5 (squares), SHIP-deficient MD4.ML-5 (circles) and C57Bl/6 mice (triangles). F. BCR occupancy by HEL in splenic B cells from SHIP-sufficient MD4.ML-5 (left panel) or SHIP-deficient MD4.ML-5 (right panel) mice. Shown are histograms illustrating occupancy of experimental populations (gray), compared to internal 0% occupancy (gray shaded) and 100% occupancy (black) controls. Bar graph below shows quantification of RO distribution for all mice in experiment; SHIP-sufficient (black), SHIP-deficient (gray). Characterization was repeated twice with at least three mice per experimental group.
Taken together, these data demonstrate that loss of SHIP-1 in MD4.ML-5 B cells leads to loss of tolerance. However, an interpretation of loss of anergy per se is clouded by the fact that MD4.ML-5.mb1cre/wt.SHIP-1fl/fl mice produce anti-HEL antibody that by neutralizing HEL reduces BCR occupancy sufficiently to prevent induction/maintenance of anergy. Thus while it is clear that B cells must express SHIP-1 to maintain tolerance, this breech cannot with certainty be ascribed to anergy.
3.3 MD4.ML-5 bone marrow chimeric mice require SHIP-1 to maintain B cell tolerance
To explore the role of SHIP-1 in maintenance of tolerance in an alternate model, we constructed bone marrow chimeras in which lethally irradiated WT or ML-5 mice were reconstituted with MD4.mb1cre/wt.SHIP-1fl/fl or control MD4.mb1cre/wt bone marrow cells. After allowing 12 wks for reconstitution we analyzed parameters as described in figure 3. In ML-5 recipients, SHIP-1 sufficient MD4 B cells were anergic based on reduced mIgM expression while IgD expression was unchanged (Figure 4A), reduced calcium mobilization following antigen stimulation (Figure 4B), and lack of anti-HEL antibody production (Figures 4C, D and E). Consistent with induction of anergy, antigen receptor occupancy exceeded 40%. In ML-5 recipients of MD4.mb1cre/wt. SHIP-1fl/fl bone marrow, SHIP-1 deficient B cells expressed variably reduced mIgM (Figure 4A), but despite this underwent robust calcium mobilization responses following antigen stimulation (Figure 4B). Differentiation of these cells to antibody secreting cells was evident (Figure 4D), and antibody was detectable in blood (Figure 4C and E). Analysis of the timecourse of appearance of anti-HEL in peripheral blood revealed that by 5 weeks following bone marrow transfer, 60% of the ML-5 mice that received MD4.mb1cre/wt. SHIP-1fl/fl bone marrow were producing detectable anti-HEL and this increased to 100% by 10 weeks (data not shown). While these data were consistent with loss of anergy, analysis of BCR occupancy again revealed that restoration of responsiveness was associated with levels of antigen receptor occupancy too low to maintain anergy (Figure 4F).
Figure 4. Lack of B cell anergy in SHIP-deficient MD4-->ML-5 bone marrow chimeric mice reflects ignorance.

Splenocytes and serum were isolated from chimeric ML-5 mice 12wk after transfer of MD4 bone marrow. A. IgM and IgD expression by SHIP-sufficient (black), SHIP-deficient (gray) B cells, as well as positive control MD4 B cells (dashed black) and negative control T cells (gray shaded). B. Cells were Indo1-AM loaded and B cell intracellular free calcium monitored before and after stimulation with 15 ng/mL HEL; responses of SHIP-1-sufficient (black), SHIP-1-deficient (gray) and MD4 SHIP-sufficient control B cells (dashed black) are shown. C. Serum IgM anti-HEL concentrations were determined by ELISA. Shown are antibody levels in mice containing SHIP-sufficient (square) and SHIP-deficient (circle) B cells. D. Splenic ASC were measured by ELISPOT. Shown are ASC in spleens of mice in which B cells were SHIP sufficient (squares) or SHIP deficient (circles). Shaded area reflects the 100 ASC/spleen limit of detection of the assay. E. IgMa anti-HEL:HEL immune complexes were measured by ELISA in serum from chimeric mice in which B cells were SHIP sufficient (squares) or SHIP deficient (circles). F. HEL occupancy of BCR on ex vivo SHIP-sufficient (black) and SHIP-deficient (gray) B cells. Experiment was repeated twice with 3–5 mice per experimental group.
3.4 B cell intrinsic SHIP-1 is required for induction of anergy following adoptive transfer of MD4 B cells into autoantigen-sufficient ML-5 recipients
Experiments using in vivo models in which SHIP-1 deficient MD4 B cells are exposed to autoantigen throughout their development clearly demonstrate a requirement for B cell intrinsic SHIP-1 to induce and/or maintain tolerance. However, due to the confounding effects of high affinity autoantibody production and resultant neutralization of antigen it is unclear whether this reflects failure of central tolerance, failure to induce anergy or failure to maintain anergy once induced. It has been shown that transfer of MD4 anti-HEL B cells into HEL-producing ML-5 mice leads to induction of anergy as indicated by failure to generate anti-HEL secreting cells in response to HEL-HRBC immunization [8, 22]. This experimental paradigm afforded an opportunity to examine requirements for SHIP-1 for in vivo induction of anergy under conditions of constant autoantigen availability.
Proliferation reporter dye-labeled splenic B cells from MD4.mb1cre/wt. SHIP-1fl/fl or MD4.mb1cre/wt control mice were transferred into ML-5 or WT recipients and three days later serum anti-HEL antibody and parameters of B cell activation and differentiation assessed. Anti-HEL antibody levels were negligible in all ML-5 mice at three days following transfer (Figure 5A). As shown in figure 5, following transfer to ML-5, SHIP-1 sufficient MD4 B cells underwent proliferation, but did not differentiate into antibody secreting cells (Figure 5C). SHIP-1-deficient MD4 B cells proliferated to a similar degree, but a large proportion of these cells also differentiated as indicated by intracellular anti-HEL staining, dilution of proliferation dye and generation of anti-HEL antibody secreting cells (Figure 5C, 5D, 5E). The cytoplasmic HEL population is CD138 positive (data not shown). These data indicate that while SHIP-1 sufficient MD4 B cells are initially activated by exposure to antigen in ML-5 mice, this response is rapidly aborted as cells become anergic. However, SHIP-1-deficient MD4 cells respond to antigen in ML-5, undergoing proliferation and, in addition, differentiation to become antibody secreting cells. Thus it would appear that in the absence of intrinsic SHIP-1 MD4 anti-HEL B cells are not rendered anergic by exposure to high affinity autoantigen (HEL) in vivo.
Figure 5. SHIP-deficient mature MD4 B cells are not rendered tolerant upon adoptive transfer to ML-5 recipients.

Isolated B cells from SHIP-1-sufficient or SHIP-1-deficient MD4 mice were adoptively transferred to ML-5 recipients. Three days later the status of transferred B cells and recipients was assessed. A. Serum IgM anti-HEL was measured by ELISA; shown are titration data from recipients of SHIP-1-sufficient (square) and SHIP-1-deficient (circle) animals, as well as serum antibody levels in an unimmunized MD4 mouse (triangle). B. B cell antigen receptor occupancy by HEL was determined as previously described; shown are percent of antigen receptors on transferred MD4 cells that are HEL occupied in recipients of SHIP-sufficient (black) and SHIP-deficient (gray) B cells. C. For detection of proliferation, B cells were labeled with cell tracker violet (CTV) before transfer and dye dilution assayed three days after transfer. Shown is proliferation of SHIP-sufficient MD4 B cells transferred into ML-5 (solid black), SHIP-deficient MD4 B cells transferred to ML-5 (solid gray), compared to SHIP-sufficient MD4 transferred to C57BL/6 (dashed black), SHIP-deficient MD4 transferred into C57BL/6 (dashed gray) and unlabeled T cells (shaded gray) adjacent bar graph shows quantification of proliferated IgMa B cells from SHIP-sufficient (black) and SHIP-deficient (gray). D. Differentiated anti-HEL B cells were identified as B220+, IgMa+, intracellular anti-HELhigh and CTVneg. Shown are frequencies of differentiated anti-HEL B cells in recipients of SHIP-1-sufficient (black), SHIP-1-deficient (gray) MD4 B cells. E. IgMa Anti-HEL antibody secreting cells were detected by ELISPOT as described. Shown are ELISPOTs per spleen in indicated donor recipient combinations. Statistical analyses noted are two separate comparisons; first between SHIP-1 sufficient versus deficient donor cells transferred into ML-5 recipients and secondly comparing C57B/6 and ML-5 recipients of SHIP-1 deficient donor cells. Experiment was repeated two times with four or five mice per experimental group.
4. Discussion
Elucidation of the mechanisms operative in inducing and maintaining B cell anergy is critical for understanding how autoimmune diseases originate. In this study we utilized a B cell-specific knockout to examine the role of SHIP-1 in the HEL anti-HEL (MD4.ML-5) mouse model of B cell anergy. We studied the impact of SHIP-1 deficiency in four experimental conditions: an in vitro system in which mature ex vivo B cells were exposed to self-antigen and their subsequent responsiveness assessed; and in vivo approaches using 1) MD4.ML-5 mice, 2) bone marrow chimeric MD4-->ML-5 mice, and 3) adoptive transfer of mature MD4 B cells into ML-5 mice. Results in all models extend conclusions drawn in studies of the Ars/A1 low affinity anti-chromatin anergy model by demonstrating that SHIP-1 is also required for induction and maintenance of B cell anergy in a model in which BCR have high affinity for a protein autoantigen, HEL, which does not stimulate innate immunity. Importantly, we also report the unexpected finding that SHIP-1-sufficient mature MD4 cells proliferate in vivo in response to initial exposure to autoantigen but do not differentiate to become antibody secreting cells, thus the manifestation of anergy. This deficit in differentiation is overcome by SHIP deficiency, as SHIP-1-deficient B cells are able to both proliferate and differentiate upon in vivo exposure to autoantigen.
Due to the very high affinity of HyHEL10 BCR/antibodies produced by the MD4 mouse, use of four experimental approaches was important to obtain clear picture of the function of SHIP-1 in this model. In the in vivo experiments described in figure 3 and 4, SHIP-1-deficient anti-HEL B cells were clearly not rendered anergic, but more careful scrutiny revealed that there was insufficient occupancy of antigen receptors on these cells to induce anergy. These mice produced high levels of anti-HEL IgM antibody in response to endogenous HEL, and some of this antibody occurred in immune complexes in association with HEL. The inevitable conclusion is that tolerance was defeated by lack of SHIP-1 in B cells, but no firm conclusion could be drawn regarding whether this reflected loss of anergy; peripheral B cells in these mice were essentially ignorant due to low effective autoantigen concentration. Therefore we were confined to use of strategies in which loss of anergy could be assessed in the absence of confounding anti-HEL antibody. Two approaches provided this possibility. The first, described in figure 2, involved in vitro exposure of naïve anti-HEL B cells to their autoantigen at concentrations sufficient to occupy a proportion of receptors (~40%) shown previously to induce anergy, followed by analysis of responses to stimulation of available BCR. These studies revealed that in the absence of SHIP-1, these levels of receptor occupancy do not result in BCR desensitization, a surrogate measure of anergy. A complementary approach is shown in figure 5 in which naïve MD4 anti-HEL B cells were transferred to HEL-sufficient recipients. Previous studies have shown that B cells are rendered anergic following such transfers [25, 26]. SHIP-1-deficient B cells did not become anergic following transfer, rather they mounted a robust immune response to the endogenous autoantigen.
Our previous characterization of the effect of B cell-targeted SHIP-1 deletion revealed that SHIP-1 is required to induce and/or maintain tolerance in C57BL/6 mice. These mice, possessing a wild-type BCR repertoire, develop lupus-like autoimmune disease characterized by production of high levels of anti-chromatin autoantibodies, and exhibit a lifespan of less than one year [10]. This may reflect breach of ignorance or anergy, or failed induction of central tolerance in chromatin-reactive cells in the natural polyclonal repertoire [5]. However, in view of a recent report that loss of SHIP-1 leads to enhanced central tolerance, this seems most likely to reflect breach of anergy or ignorance [13]. Supporting the latter, we recently showed that B cell-targeted loss of SHIP-1 in anti-DNA BCR transgenic Ars/A1 mice leads to failure to silence B cells by anergy, despite evidence of continued B cell exposure to autoantigen [10]. Finally, we found that SHIP-1 and its adaptor Dok-1 are tyrosine phosphorylated in both anergic Ars/A1 and MD4.ML-5 B cells [10]. These observations led us to strongly suspect that SHIP-1 is involved in the maintaining anergy in both of these phenotypically similar anergy models, and in wild-type C57BL/6 mice. This seemed somewhat surprising given a previous report that expression of the inositol phosphatase PTEN is increased in anergic B cells in MD4.ML-5, and is required to maintain anergy in this model [14]. PTEN is not upregulated in the anergic Ars/A1 B cells [10] but loss of PTEN leads to loss of anergy in this model (Getahun and Cambier, submitted). Both SHIP-1 and PTEN act to reduce PI(3,4,5)P3 required for activation of PLCγ, BTK, PDK1, rasGRP and AKT during BCR signaling [27]. Based on findings described here it appears that in MD4.ML-5 anergic B cells dual mechanisms are activated to reduce PI(3,4,5)P3 levels, thus inhibiting the PI3-kinase pathway signaling, while only increased SHIP-1 function is induced in the lower affinity Ars/A1 model. Further studies will be required to determine why these models differ in this regard, but it seems most likely due to differing affinity of the BCR for antigen. “Stronger” anergy enforcing BCR signals driven by higher affinity autoantigens may uniquely activate transcription or post-transcriptional mechanisms leading to increased PTEN expression.
The initial motivation for the studies described here was provided by a report that concluded that SHIP-1 is not required for B cell anergy in the MD4.ML-5 model [13]. Seeking to examine the effect of BCR signal strength on B cell development and negative selection, the Bolland group utilized loss of SHIP-1 to increase PI3-kinase pathway amplitude. They showed that loss of SHIP-1 enhanced central tolerance induction and increased antigen sensitivity of B cells in the periphery, while also causing spontaneous down-regulation of membrane IgM expression. They then studied bone marrow chimeras of SHIP-1-deficient MD4 mice and HEL-expressing ML-5 mice, and concluded that anergy remains intact in SHIP-1 deficient B cells. This interpretation was based solely on the fact that membrane IgM was downregulated. Studies reported here indicate that this conclusion was erroneous, and underscore the fact that anergy is not mediated by mIgM modulation, and further that mIgM modulation is not a reliable surrogate measure of anergy.
The experiments conducted in this study yielded an additional interesting finding that has not been reported previously. We found that mature naïve SHIP-1 sufficient MD4 B cells transferred into an antigen rich ML-5 environment proliferate but do not differentiate to antibody forming cells. This is surprising in view of early studies indicating that proliferation in response to this putative thymus dependent antigen requires cognate T cell help [25, 26, 28], yet in ML-5 mice HEL specific T cells are chronically exposed to systemic HEL and thus are reportedly tolerant [29].
It is possible that this T cell tolerance is leaky and thus supports B cell proliferation but not differentiation, or that HEL-induced MD4 B cell proliferation, but not differentiation, is T cell independent. These findings are interesting in view of early studies suggesting that ambient cytokines may provide sufficient “help” to achieve naïve B cell proliferation but not differentiation [30]. There seemingly are no absolutes. Finally, abortive B cell proliferation in response to antigen is reminiscent of the situation in T cells where adoptive transfer of T cells into recipients expressing their cognate antigen led to robust proliferation followed by onset of tolerance [31]. Thus under certain circumstances autoreactive B cells first encountering their autoantigen in the periphery may proliferate prior to onset of unresponsiveness. This has interesting implications regarding the sites of SHIP-1 regulatory function in anergy. As we have previously shown, in addition to BCR signaling SHIP-1 regulates responses to chemokines and presumably other ligands whose signaling requires PI3-kinase activation, including those required for B cell differentiation into plasma cells.
Highlights.
SHIP-1 is required to induce B cell anergy to hen egg lysozyme in the high affinity MD4.ML-5 immunoglobulin transgenic anergy model.
Low dose antigen exposure primes B cells for subsequent responses to antigen.
SHIP-1 regulates responses of both naïve and antigen experienced B cells to antigen.
Upon transfer to mice expressing soluble HEL, SHIP-1 sufficient HEL-specific transgenic MD4 B cells proliferate but do not differentiate into plasmablasts; SHIP-1 deficiency leads to both proliferation and differentiation.
Acknowledgments
These studies were funded by NIH grants RO1 AI 077597-05 and PO1 AI 022295-22. We thank Richard Wilson for providing D1.3 anti-HEL antibody.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant Autoantibody Production by Early Human B Cell Precursors. Science. 2003;301:1374–7. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- 2.Gay D, Saunders T, Camper S, Weigert M. Receptor editing: an approach by autoreactive B cells to escape tolerance. J Exp Med. 1993;177:999–1008. doi: 10.1084/jem.177.4.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Halverson R, Torres RM, Pelanda R. Receptor editing is the main mechanism of B cell tolerance toward membrane antigens. Nat Immunol. 2004;5:645–50. doi: 10.1038/ni1076. [DOI] [PubMed] [Google Scholar]
- 4.Nemazee DA, Burki K. Clonal deletion of B lymphocytes in a transgenic mouse bearing anti-MHC class I antibody genes. Nature. 1989;337:562–6. doi: 10.1038/337562a0. [DOI] [PubMed] [Google Scholar]
- 5.Merrell KT, Benschop RJ, Gauld SB, Aviszus K, Decote-Ricardo D, Wysocki LJ, et al. Identification of Anergic B Cells within a Wild-Type Repertoire. Immunity. 2006;25:953–62. doi: 10.1016/j.immuni.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 6.Cambier JC, Gauld SB, Merrell KT, Vilen BJ. B-cell anergy: from transgenic models to naturally occurring anergic B cells. Nature Reviews Immunology. 2007;7:633–43. doi: 10.1038/nri2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gauld SB, Benschop RJ, Merrell KT, Cambier JC. Maintenance of B cell anergy requires constant antigen receptor occupancy and signaling. Nature Immunology. 2005;6:1160–7. doi: 10.1038/ni1256. [DOI] [PubMed] [Google Scholar]
- 8.Goodnow CC, Brink R, Adams E. Breakdown of self-tolerance in anergic B lymphocytes. Nature. 1991;352:532–6. doi: 10.1038/352532a0. [DOI] [PubMed] [Google Scholar]
- 9.Cambier JC. Autoimmunity risk alleles: hotspots in B cell regulatory signaling pathways. J Clin Invest. 2013;123:1928–31. doi: 10.1172/JCI69289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Neill Shannon K, Getahun A, Gauld Stephen B, Merrell Kevin T, Tamir I, Smith Mia J, et al. Monophosphorylation of CD79a and CD79b ITAM Motifs Initiates a SHIP-1 Phosphatase-Mediated Inhibitory Signaling Cascade Required for B Cell Anergy. Immunity. 2011;35:746–56. doi: 10.1016/j.immuni.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nature Reviews Molecular Cell Biology. 2012 doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 12.Pao LI, Lam KP, Henderson JM, Kutok JL, Alimzhanov M, Nitschke L, et al. B cell-specific deletion of protein-tyrosine phosphatase Shp1 promotes B-1a cell development and causes systemic autoimmunity. Immunity. 2007;27:35–48. doi: 10.1016/j.immuni.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 13.Leung W-H, Tarasenko T, Biesova Z, Kole H, Walsh ER, Bolland S. Aberrant antibody affinity selection in SHIP-deficient B cells. European Journal of Immunology. 2013;43:371–81. doi: 10.1002/eji.201242809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Browne CD, Del Nagro CJ, Cato MH, Dengler HS, Rickert RC. Suppression of Phosphatidylinositol 3,4,5-Trisphosphate Production Is a Key Determinant of B Cell Anergy. Immunity. 2009;31:749–60. doi: 10.1016/j.immuni.2009.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Landego I, Jayachandran N, Wullschleger S, Zhang T-t, Gibson IW, Miller A, et al. Interaction of TAPP adapter proteins with phosphatidylinositol (3,4)-bisphosphate regulates B-cell activation and autoantibody production. European Journal of Immunology. 2012;42:2760–70. doi: 10.1002/eji.201242371. [DOI] [PubMed] [Google Scholar]
- 16.Tamir I, Stolpa JC, Helgason CD, Nakamura K, Bruhns P, Daeron M, et al. The RasGAP-binding protein p62dok is a mediator of inhibitory FcgammaRIIB signals in B cells. Immunity. 2000;12:347–58. doi: 10.1016/s1074-7613(00)80187-9. [DOI] [PubMed] [Google Scholar]
- 17.Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink RA, et al. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature. 1988;334:676–82. doi: 10.1038/334676a0. [DOI] [PubMed] [Google Scholar]
- 18.Karlsson MC, Guinamard R, Bolland S, Sankala M, Steinman RM, Ravetch JV. Macrophages control the retention and trafficking of B lymphocytes in the splenic marginal zone. J Exp Med. 2003;198:333–40. doi: 10.1084/jem.20030684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hobeika E, Thiemann S, Storch B, Jumaa H, Nielsen PJ, Pelanda R, et al. Testing gene function early in the B cell lineage in mb1-cre mice. Proceedings of the National Academy of Sciences. 2006;103:13789–94. doi: 10.1073/pnas.0605944103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lavoie TB, Drohan WN, Smith-Gill SJ. Experimental analysis by site-directed mutagenesis of somatic mutation effects on affinity and fine specificity in antibodies specific for lysozyme. J Immunol. 1992;148:503–13. [PubMed] [Google Scholar]
- 21.Hardy IR, Anceriz N, Rousseau F, Seefeldt MB, Hatterer E, Irla M, et al. Anti-CD79 antibody induces B cell anergy that protects against autoimmunity. J Immunol. 2014;192:1641–50. doi: 10.4049/jimmunol.1302672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goodnow CC, Crosbie J, Jorgensen H, Brink RA, Basten A. Induction of self-tolerance in mature peripheral B lymphocytes. Nature. 1989;342:385–91. doi: 10.1038/342385a0. [DOI] [PubMed] [Google Scholar]
- 23.Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature. 1997;386:855–8. doi: 10.1038/386855a0. [DOI] [PubMed] [Google Scholar]
- 24.O’Neill SK, Getahun A, Gauld SB, Merrell KT, Tamir I, Smith MJ, et al. Monophosphorylation of CD79a and CD79b ITAM motifs initiates a SHIP-1 phosphatase-mediated inhibitory signaling cascade required for B cell anergy. Immunity. 2011;35:746–56. doi: 10.1016/j.immuni.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fulcher DA, Basten A. Reduced life span of anergic self-reactive B cells in a double-transgenic model. J Exp Med. 1994;179:125–34. doi: 10.1084/jem.179.1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cooke MP, Heath AW, Shokat KM, Zeng Y, Finkelman FD, Linsley PS, et al. Immunoglobulin signal transduction guides the specificity of B cell-T cell interactions and is blocked in tolerant self-reactive B cells. J Exp Med. 1994;179:425–38. doi: 10.1084/jem.179.2.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dal Porto JM, Gauld SB, Merrell KT, Mills D, Pugh-Bernard AE, Cambier J. B cell antigen receptor signaling 101. Mol Immunol. 2004;41:599–613. doi: 10.1016/j.molimm.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 28.Parker DC. T cell-dependent B cell activation. Annu Rev Immunol. 1993;11:331–60. doi: 10.1146/annurev.iy.11.040193.001555. [DOI] [PubMed] [Google Scholar]
- 29.Akkaraju S, Ho WY, Leong D, Canaan K, Davis MM, Goodnow CC. A range of CD4 T cell tolerance: partial inactivation to organ-specific antigen allows nondestructive thyroiditis or insulitis. Immunity. 1997;7:255–71. doi: 10.1016/s1074-7613(00)80528-2. [DOI] [PubMed] [Google Scholar]
- 30.Noelle RJ, Snow EC, Uhr JW, Vitetta ES. Activation of antigen-specific B cells: role of T cells, cytokines, and antigen in induction of growth and differentiation. Proc Natl Acad Sci U S A. 1983;80:6628–31. doi: 10.1073/pnas.80.21.6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kearney ER, Pape KA, Loh DY, Jenkins MK. Visualization of peptide-specific T cell immunity and peripheral tolerance induction in vivo. Immunity. 1994;1:327–39. doi: 10.1016/1074-7613(94)90084-1. [DOI] [PubMed] [Google Scholar]
- 32.Brauweiler A, Merrell K, Gauld SB, Cambier JC. Cutting Edge: Acute and chronic exposure of immature B cells to antigen leads to impaired homing and SHIP1-dependent reduction in stromal cell-derived factor-1 responsiveness. J Immunol. 2007;178:3353–7. doi: 10.4049/jimmunol.178.6.3353. [DOI] [PubMed] [Google Scholar]