

Abstract
Once thought of as a vestigial organelle, the primary cilium is now recognized as a signaling hub for key cellular pathways in vertebrate development. The recent renaissance in cilia studies significantly improved our understanding of how cilia form and function, but little is known about how ciliogenesis is initiated and how ciliary proteins enter cilia. These important ciliary events require transition fibers (TFs) that are positioned at the ciliary base as symmetric nine-bladed propeller fibrous structures. Up until recently, TFs have been the most underappreciated ciliary structures due to limited knowledge about their molecular composition and function. Here, we highlight recent advances in our understanding of TF composition and the indispensable roles of TFs in regulating the initiation of ciliogenesis and the selective import of ciliary proteins.
Introduction
Microtubule-based cilia fulfill important sensory functions in most eukaryotic cells and are critical in vertebrate embryonic development and tissue homeostasis [1, 2]. Cilia dysfunction is correlated with an expanding spectrum of human genetic diseases (collectively termed ciliopathies) [3, 4]. Since cilia are ubiquitous on cell surfaces, most ciliopathies occur as syndromic disorders that affect many vital organs during development, including the central nervous system (CNS), eyes, cardiovascular system, kidney, liver, limbs, bones, and fat storage tissue. Cilia dysfunction might affect as many as 100 human disorders [4, 5]. Ciliopathies are probably the fastest growing category within human disease family: ~60 new causal loci were identified in last decade, and more are suspected [6].
Despite the physiological and clinical relevance of cilia, our understanding of how cilia form remains poor, and several key questions remain to be answered. For example, how is ciliogenesis initiated? At a morphological level, the mother centriole must dock to the membrane to initiate ciliogenesis. In different cell types, the mother centriole first attaches to vesicles, presumably Golgi-derived, in smooth muscle and endothelial cells [7] or the cell membrane in some epithelial cells [8, 9]. Distal appendages (DAs), the fibrous structure at the distal end of the mother centriole, might mediate the centriole-membrane docking [10]. During the docking, the mother centriole transforms into the basal body, and its DAs mature into TFs. After that, the ciliary axoneme begins to elongate with the assistance of intraflagellar transport (IFT) machinery, which moves bidirectionally inside cilia to transport ciliary proteins essential for cilia formation, maintenance, and signaling [11–13].
As another question, how are cilia functionally separated from the cytosol? Unlike other membrane-enclosed cellular organelles, the cilium is open to the cytoplasm at its base. The ciliary base needs to control the selective entry of ciliary proteins and, thus, functionally separates the cilium from the cell body and makes it a discrete sensing organelle. The morphology of the ciliary base is highly conserved [14]: a basal body with fibrous apparatuses TFs and basal feet, and the transition zone (TZ, the proximal part of the axoneme that contains Y-links) (Fig. 1). TFs form a 9-bladed propeller-like structure, and their anchoring points with the membrane define the border between the plasma membrane and the ciliary membrane [10]. Above the TFs, the Y-links of the TZ connect axonemal microtubules to the ciliary membrane [15]. The distinct locations of TFs and the TZ make them good candidates for the enigmatic ciliary gate or “ciliary pore complex” that regulates the selective import of the ciliary proteins [13, 14].
Figure 1. Schematic diagram of a cilium.
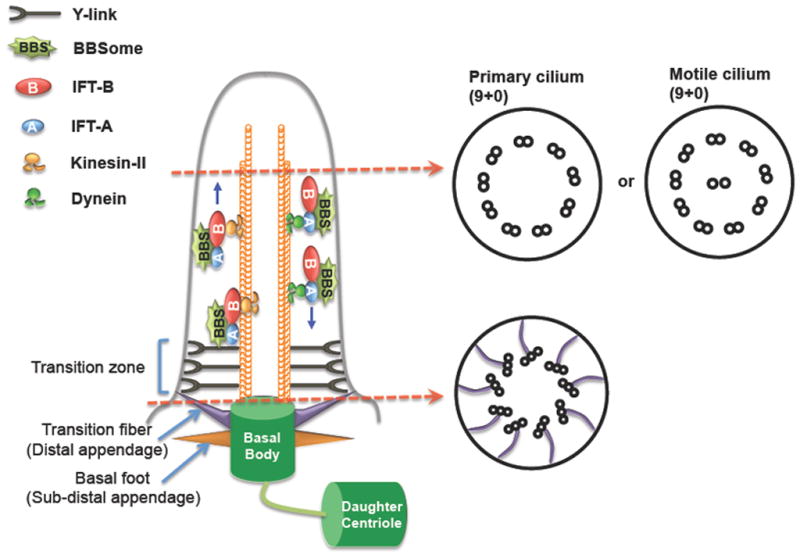
All cilia have a microtubule-based core structure, called the axoneme, which projects from the basal body and is tightly surrounded by the ciliary membrane. Based on the motility, cilia can be divided into primary cilia (non-motile cilia) and motile cilia. The axoneme of primary cilia typically consists of nine microtubule doublets (9+0), typical motile cilia have an extra pair of microtubule singlet in the center of the ring of nine outer doublets (9+2). The basal body is transformed from the mother centriole during ciliogenesis. At the ciliary base, there are two structurally distinct sub-regions: TFs and the TZ. TFs are analogous to DAs of the mother centriole and form a 9-bladed propeller-like structure linking the basal body to the ciliary membrane. Basal feet (analogous to sub-DAs) locate below TFs on the basal body. Above TFs is the TZ that is characterized by the Y-links connecting axoneme microtubules to the ciliary membrane. Extension, maintenance and function of cilia require intraciliary transport machinery IFT, which is composed of IFT-A, IFT-B, BBSome and motors. IFT moves bidirectionally along the ciliary axoneme to transport cargos into or out of cilia. TFs, transition fibers; TZ, transition zone; DAs, distal appendages; IFT, Intraflagellar transport; Sub-DAs, sub-distal appendages.
For many years, many roles were proposed for TFs in ciliogenesis initiation [10] and cilia import [13, 16, 17]. However, only recently, characterization of the molecular identities of DAs/TFs made it possible to elucidate and confirm the roles of TFs at a molecular level. Here, we review the insights that establish and expand our views of TFs as the indispensable structures in the context of cilia.
TF composition and assembly
So far, five proteins have been identified as genuine DA/TF components. These include CEP164, CEP83 (CCDC41), CEP89 (CCDC123), SCLT1 (sodium channel and clathrin linker 1) and FBF1 (Fas (TNFRSF6) binding factor 1) [18–23]. CEP83 was reported to regulate the TF targeting of the other four proteins, and SCLT1 specifically affects the localization of CEP164 and FBF1 (Fig. 2) [20]. However, the strict role of CEP83 in regulating the TF localization of CEP164 has been questioned [23]. Also Cby (Chibby) and TTBK2 (Tau tubulin kinase 2), two newly identified CEP164 interactors, localize to TFs and regulate ciliogenesis in different types of mammalian cells [24, 25]. In super-resolution studies, the CEP164 ring is actually larger and more proximal than the Cby ring at the distal end of the mother centriole, suggesting that Cby attaches apically on TFs [24].
Figure 2. The assembly of distal appendages/transition fibers.
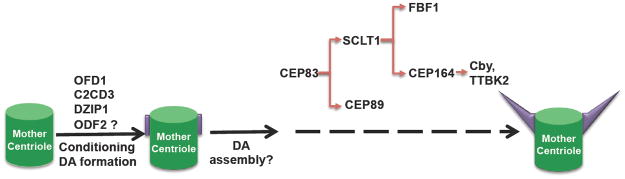
OFD1, C2CD3, DZIP1, and ODF2 condition the distal end of the mother centriole for DA formation. Then different structural/functional components of DAs/TFs are recruited in a sequential manner. DAs, distal appendages; TFs, transition fibers.
The key structural TF components have not been determined due to the fact that the electron density of TFs in mammalian cells is too low for conclusive EM studies. TTBK2 and Cby are likely effectors of CEP164 that are recruited to TFs during ciliogenesis [24, 25]. Since TFs exist in all ciliated organisms, the core structural component(s) should be evolutionarily conserved. However, of the other five components, only the homolog of FBF1 can be found in the genome of non-vertebrate ciliated organism (Table 1), and depletion of DYF-19, the worm homolog of FBF1, does not affect TF biogenesis [21]. These observations suggest that either some of the identified TF components are only mammalian-specific TF structural component(s), or more likely, the seven identified candidates are just the functional components of TFs, and the key structural components of TFs remain to be identified.
Table 1.
Function and associated diseases of reported transition fiber related proteins
| Protein name | Conservation in ciliated model organisms* | Mutation-related diseases | TF formation | Function | References | |
|---|---|---|---|---|---|---|
| TFs proteins | CEP164 | Hs, Mm, Dr, Dm | Nephronophthisis | No | Basal body docking | [18, 20, 24, 25,32, 67] |
| CEP83 | Hs, Mm, Dr, | Nephronophthisis | No | Basal body docking | [20, 23, 68] | |
| SCLT1 | Hs, Mm, Dr, | Orofaciodigital syndrome | No | [20, 69] | ||
| CEP89 | Hs, Mm, Dr, Dm | Vesicle formation | [20, 22, 33] | |||
| FBF1 | Hs, Mm, Dr, Dm, Ce | No | IFT entry | [20, 21] | ||
| TTBK2 | Hs, Mm, Dr, Dm,Ce | Spinocerebellar ataxia | No | Ciliogenesis initiation | [25, 36] | |
| Chibby | Hs, Mm, Dr, Dm | No | Vesicle formation, basal body docking | [24] | ||
| TF-related proteins | OFD1 | Hs, Mm, Dr, Dm? | Oral-facial-digital syndrome | Yes | Inhibit centriole elongation | [27] |
| C2CD3 | Hs, Mm, Dr, Dm, Ce? | Oral-facial-digital syndrome | Yes | Promote centriole elongation | [28, 29] | |
| ODF2 | Hs, Mm, Dr, Dm, | Yes | Formation of both distal appendages and sub-distal appendages | [26, 31] | ||
| DZIP1 | Hs, Mm, Dr, Dm, | Yes | Formation of both distal appendages and sub-distal appendages | [30] |
Note: Hs, Homo sapiens; Mm, Mus musculus; Dr, Danio rerio; Dm, Drosophila melanogaster; Ce, Caenorhabditis elegans.
, Conservation data are based on literatures and NCBI BLASTP.
?, Potential homolog with low sequence similarity
Several proteins have been implicated in the proper formation of DAs on mother centrioles. These include OFD1 (oral-facial-digital syndrome 1), C2CD3 (C2 calcium-dependent domain containing 3), ODF2 (outer dense fiber 2), and DZIP1 (DAZ-interacting zinc finger protein 1). Yet none of them appears to be the TF component [26–31] (Fig. 2). OFD1 localizes to both centrioles and centriolar satellites and seems to have different roles in different locations. In murine OFD1-lacking ES cells, centriole distal ends elongate abnormally and show severe defects in distal appendage formation and ciliogenesis [27]. Conversely, autophagy-dependent depletion of OFD1 from centriolar satellites can actually promote ciliogenesis independent of the role of centriolar OFD1 [32]. C2CD3, another OFD syndrome protein, might physically interact with OFD1, regulate DA formation, but functionally antagonize OFD1 in centriolar elongation [28, 29]. ODF2 and DZIP1 are required for the assembly of both sub-distal appendages (sub-DAs) and DAs [26, 30, 31]. Therefore, although the mechanisms underlying TF malformation are not clear when these proteins are depleted, they are probably secondary to other centriole anomalies, such as a malformed distal end or altered outer wall structure of the centriole. Notably, ODF2’s regulation of DA formation was challenged: when ODF2 was knocked down, none of DA components was missing from the ciliary base [20]. Whether the discrepancy is due to the efficiency of RNAi knockdown or differences in cell types is unknown.
TFs and ciliogenesis initiation
As seen in transmission EM studies, the earliest event in ciliogenesis is the docking of the mother centriole to the primary cilia vesicle (PCV) or the apical membrane [7–9]. Most recent studies have focused on the roles of TF components in centriole-vesicle-docking. Depleting CEP164, CEP83, CEP89, or Cby leads to defects in either centriole-vesicle-docking or PCV formation [20, 23, 24, 33, 34]. Intriguingly, a group of small vesicles called the distal appendages vesicles (DAVs) was recently suggested to first anchor to the mother centriole. Then the membrane shaping proteins EHD1 and a SNARE membrane fusion regulator SNAP29 regulate DAV-mediated PCV formation (Fig. 3) [35]. How the DAVs and the PCV dock on DAs/TFs is unknown. After PCV formation, the physical association between the CEP164-Cby complex and the Rabin8/Rab8 trafficking machinery likely recruits more Rab8-positive vesicles to TFs to ensure PCV extension and support axoneme elongation [24, 33, 35, 36] (Fig. 3).
Figure 3. The roles of transition fibers in the context of cilia.
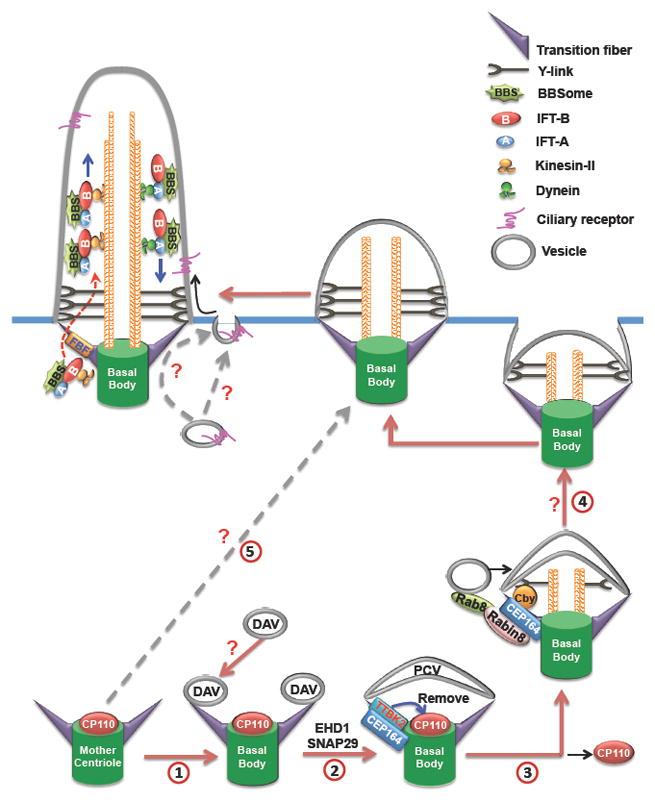
In some mammalian cell types, during the very early stage of ciliogenesis, small DAVs first dock to the DAs of the mother centriole through an unknown mechanism (1). EHD1 and SNAP29 then regulate the fusion of DAVs into the large PCV (2). During the formation of PCV, TTBK2 is recruited to TFs by CEP164 to remove microtubule cap protein CP110 to initiate axoneme elongation. Then the interaction between Rabin8/Rab8 complex and CEP164/Cby complex mediates the recruitment of more Rab8 positive vesicles to support membrane extension of the PCV (3). Meantime, the TZ starts to form, the basal body-PCV migrates to the plasma membrane, and then the PCV fuses with the cell membrane through an unknown mechanism (4). Lastly, IFT regulates the extension of the axoneme. FBF1 acts as the functional component on TFs to facilitate the ciliary import of assembled IFT complex. Polarized vesicle trafficking and exocytosis have been implicated in mediating ciliary cargos targeting to periciliary membrane, and then these cargos enter into cilia through lateral diffusion. But, whether TFs play a role in this process is not clear. In other cell types, an alternative ciliogenesis pathway (5) may be employed: the basal body directly docks to the plasma membrane independent of DAV/PCV route. TFs, transition fibers; TZ, transition zone; DAs, distal appendages; DAVs, distal appendage vesicles; PCV, primary ciliary vesicle.
In addition to their indispensable role in membrane docking, TFs are required to initiate axoneme elongation. When cells exit the cell cycle to form cilia, CEP164 recruits TTBK2 to TFs [25]. Once associated with TFs, TTBK2 regulates the removal of microtubule cap protein CP110 and the recruitment of IFT components [37]. In TF-defective cells, CP110 is not removed from the distal end of the mother centriole, and ciliogenesis is blocked [20, 25, 29]. Although both are regulated by CEP164, TTBK2 recruitment and CP110 removal appear to occur before Rab8-dependent membrane extension in the early steps of ciliogenesis [35] (Fig. 3).
TFs, diffusion barrier, and the ciliary gate
Mounting evidence suggests that diffusion barriers exist for either membrane [38] or soluble proteins [39–42] at the ciliary base. For soluble proteins, different approaches gave different estimations for the physical size or the diffusion rate of the barrier. Proteins larger than 100 kDa are commonly assumed not to efficiently pass through the ciliary base without the assistance of active importing mechanisms. A group of proteins mutated in different ciliopathies, including NPHP, MKS, and JBTS syndromes, form multimeric complexes to regulate the integrity of the TZ and the function of the diffusion barrier [43–48]. In C. elegans, the TRAM protein abnormally enters cilia when the integrity of the TZ is compromised [46]. In mammalian cells, the plasma membrane proteins GFP-CEACAM1 and GFP-GPI accumulate abnormally inside cilia when the TZ was disrupted [44]. However, the role of TFs as part of the diffusion barrier for the ciliary proteins has not been directly confirmed. Unlike with TZ, disruption of TFs completely abolishes ciliogenesis initiation and would thus make it impossible to study whether TFs act as part of diffusion barrier or not. To this end, a hypomorphic mutant that only partially disrupts TF integrity or the identification of structural component(s) of the barrier on or near TFs would answer the question.
The existence of a diffusion barrier at the ciliary base indicates that active transport is required for the import of large proteins. Remarkably, the IFT complex is a large multimeric complex comprising >20 proteins [11–13], with many components larger than 100 kDa. Some IFT cargoes, such as radial spokes and dynein arms, are also large protein complexes assembled before they enter cilia [49, 50]. Thus, certain mechanism must facilitate the ciliary import of the IFT machinery. The discovery of the pore-like structures formed by the TFs and the TZ (Fig. 1) led to the hypothesis that they are analogous to the nuclear pore complex (NPC) and may act as a passive diffusion barrier and an active cilia gate to actively transport soluble proteins into cilia [13, 17]. Immuno-EM studies revealed that the IFT-B component IFT52 clusters along the TFs and especially where the TFs connect to the flagellar membrane in Chlamydomonas [16]. Recruitment of IFT proteins is abnormal in CEP164- and CEP83-deficient cells [20, 23, 25, 33]. All these data lead to the assertion that TFs might be the site for actively regulating the ciliary entry of IFT particles.
Recently, a study in C. elegans revealed how TFs facilitate the ciliary entry of assembled IFT particles. DYF-19 (the homologue of human FBF1) facilitates the ciliary import of assembled IFT particles through direct interaction with IFT component DYF-11 (the homologue of human IFT54) (Fig. 3) [21]. FBF1 (DYF-19) might provide a docking site on TFs for the assembled IFT particles. After docking, the proposed ciliary pore complex (CPC), possibly positioned at inter-fiber space of TFs, facilitates the import of assembled IFT particles. The molecular identities of the CPC remain mysterious, and molecular evidence supports [40, 51, 52] or challenges [42] the similarity of the CPC and NPC. It is worth determining if this active transport mechanism applies to other non-IFT ciliary proteins, how the CPC recognizes the cilia-targeting signal of ciliary proteins, and what is the functional and structural relationship among the CPC, TFs, and the TZ.
TFs and cilia-targeted vesicular trafficking
Apart from the roles in guiding ciliogenesis initiation and membrane extension, cilia-related vesicles presumptively regulate the transport of ciliary proteins from post-Golgi network or recycling endosomes to the periciliary membrane [13, 39, 53]. Small GTPases along with their regulators have been implicated in regulating the sorting, docking, or fusion of vesicles at the ciliary base [54, 55]. In particular, Rab8 has been implicated in facilitating cilia trafficking by several studies [36, 56–61]. Notably, ultrastructure studies also suggest that polarized exocytosis takes place at the base of either Ochromonas flagella or photoreceptor cell connecting cilia [62, 63]. Polarized exocytosis adjacent to the basal body has been widely accepted as an early step for the ciliary import of membrane proteins [13, 39, 53]. Multiple components of the exocyst, a multiprotein complex that mediates the polarized exocytosis, localize at the ciliary base or on the ciliary membrane, interact with either Rab11 or Rab10, and are required for ciliogenesis [64–67]. Since the Rab11-Rab8 small GTPase cascade and the TFs are involved in ciliogenesis initiation, it would be interesting to determine if Rabs and the exocyst also regulate the docking of ciliary vesicles on TFs and the polarized exocytosis at the base of mature cilia.
Tight connection between TFs and ciliopathies
Consistent with the important roles of TFs in the context of cilia discussed here, many identified TF components were recently identified as causal loci for specific human ciliopathies (Table 1). Mutations in CEP164 (also called NPHP15) and CEP83 (also called NPHP18) cause nephronophthisis-related ciliopathies (NPHP-RC) [68, 69]. SCLT1 is mutated in patients with orofaciodigital syndrome (OFD) type IX [70], and mutations in TTBK2 cause neurodegenerative disease spinocerebellar ataxia type 11 [37]. In addition, mutations in OFD1 and C2CD3, two centriole proteins required for TF formation, are associated with OFD syndrome [28]. It will not be surprising to see that other identified TF components are identified as causal loci for certain ciliopathies in the near future. Exploring how TF components and their modifiers/effectors function in vivo, as well as their functional crosstalk with other characterized ciliary proteins, especially ciliopathy proteins, will greatly enhance our understanding of the role of TFs in the pathogenesis of various ciliopathies.
Perspectives
In recent years, our understanding of TFs in the context of cilia has deepened significantly. However, many questions remain unanswered and will likely be the focus of future research. First, what are the structural components of DAs/TFs? Given the highly conserved structure of cilia and flagella during evolution, TFs are not likely built from different structural parts in different ciliated species. Thus, dissecting the interaction network of highly conserved TF components, such as FBF1 and TTBK2, will likely have a greater chance of identifying the bona fide structural components of TFs. Second, what signals trigger the initial docking of DAVs at the tip of the mother centriole? When cells exit the cell cycle and form cilia, a checkpoint must initiate recruitment of DAVs to the DAs of the mother centriole. Only after this checkpoint can the mother centriole mature into the basal body and the DAs transform into TFs, which ultimately support the formation of the sensory antenna for the cells. Third, while we have amassed a reasonable candidate list for the roles of TFs in basal body docking, ciliary gate formation, vesicle trafficking, and IFT docking and sorting, how are these players are integrated to execute their function? Different protein modules are likely responsible for different aspects of TF function. For example, CEP164 and its interacting partners regulate basal body docking and the initiation of ciliogenesis; while the FBF1 protein network regulates active import through the ciliary gate. Finally, how do TFs molecularly regulate the direct docking of the mother centriole to the cell membrane in those cell types that do not use DAV-PCV vesicle route in ciliogenesis initiation? To this end, identification of structural and other functional components of TFs and a better understanding of how TF dysfunctions lead to human ciliopathies will provide seminal insights into our understanding of cilia development and function in normal and pathological states, and represent an interesting challenge for cell biology.
Acknowledgments
We thank our many colleagues who have contributed to the work described herein, and apologize to those whose work we could not cite due to the space issue. We are grateful for funding from the National Institutes of Health research grant 1R01DK090038, 1R01DK099160-01A1, and P30 center grant P30DK90728. K.L. was supported by the National Cancer Institute (NCI; 1R01CA149039-01A1), Susan G. Komen for the Cure (KG100902).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
Papers of particular interest, published within the last two years of review, have been highlighted as:
* of special interest
** of outstanding interest
- 1.Gerdes JM, Davis EE, Katsanis N. The vertebrate primary cilium in development, homeostasis, and disease. Cell. 2009;137:32–45. doi: 10.1016/j.cell.2009.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. 2010;11:331–344. doi: 10.1038/nrg2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med. 2011;364:1533–1543. doi: 10.1056/NEJMra1010172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet. 2006;7:125–148. doi: 10.1146/annurev.genom.7.080505.115610. [DOI] [PubMed] [Google Scholar]
- 5.Baker K, Beales PL. Making sense of cilia in disease: the human ciliopathies. American journal of medical genetics Part C, Seminars in medical genetics. 2009;151C:281–295. doi: 10.1002/ajmg.c.30231. [DOI] [PubMed] [Google Scholar]
- 6.Davis EE, Katsanis N. The ciliopathies: a transitional model into systems biology of human genetic disease. Current opinion in genetics & development. 2012;22:290–303. doi: 10.1016/j.gde.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7**.Sorokin S. Centrioles and the formation of rudimentary cilia by fibroblasts and smooth muscle cells. The Journal of cell biology. 1962;15:363–377. doi: 10.1083/jcb.15.2.363. This ultrastructural study demonstrated that the centriole-vesicle-docking happens in the early stage of ciliogenesis in fibroblasts and smooth muscle cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8**.Sotelo JR, Trujillo-Cenoz O. Electron microscope study on the development of ciliary components of the neural epithelium of the chick embryo. Zeitschrift fur Zellforschung und mikroskopische Anatomie. 1958;49:1–12. doi: 10.1007/BF00335059. This ultrastructural study demonstrated that the the basal body directly docks to the plasma membrane but not vesicles in chicken neuroepithelial cells. [DOI] [PubMed] [Google Scholar]
- 9.Sorokin SP. Reconstructions of centriole formation and ciliogenesis in mammalian lungs. J Cell Sci. 1968;3:207–230. doi: 10.1242/jcs.3.2.207. [DOI] [PubMed] [Google Scholar]
- 10.Anderson RG. The three-dimensional structure of the basal body from the rhesus monkey oviduct. The Journal of cell biology. 1972;54:246–265. doi: 10.1083/jcb.54.2.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pedersen LB, Rosenbaum JL. Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr Top Dev Biol. 2008;85:23–61. doi: 10.1016/S0070-2153(08)00802-8. [DOI] [PubMed] [Google Scholar]
- 12.Hao L, Scholey JM. Intraflagellar transport at a glance. J Cell Sci. 2009;122:889–892. doi: 10.1242/jcs.023861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosenbaum JL, Witman GB. Intraflagellar transport. Nat Rev Mol Cell Biol. 2002;3:813–825. doi: 10.1038/nrm952. [DOI] [PubMed] [Google Scholar]
- 14.Reiter JF, Blacque OE, Leroux MR. The base of the cilium: roles for transition fibres and the transition zone in ciliary formation, maintenance and compartmentalization. EMBO Rep. 2012;13:608–618. doi: 10.1038/embor.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gilula NB, Satir P. The ciliary necklace. A ciliary membrane specialization. The Journal of cell biology. 1972;53:494–509. doi: 10.1083/jcb.53.2.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deane JA, Cole DG, Seeley ES, Diener DR, Rosenbaum JL. Localization of intraflagellar transport protein IFT52 identifies basal body transitional fibers as the docking site for IFT particles. Curr Biol. 2001;11:1586–1590. doi: 10.1016/s0960-9822(01)00484-5. [DOI] [PubMed] [Google Scholar]
- 17.Satir P, Christensen ST. Overview of structure and function of mammalian cilia. Annu Rev Physiol. 2007;69:377–400. doi: 10.1146/annurev.physiol.69.040705.141236. [DOI] [PubMed] [Google Scholar]
- 18.Graser S, Stierhof YD, Lavoie SB, Gassner OS, Lamla S, Le Clech M, Nigg EA. Cep164, a novel centriole appendage protein required for primary cilium formation. The Journal of cell biology. 2007;179:321–330. doi: 10.1083/jcb.200707181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jakobsen L, Vanselow K, Skogs M, Toyoda Y, Lundberg E, Poser I, Falkenby LG, Bennetzen M, Westendorf J, Nigg EA, et al. Novel asymmetrically localizing components of human centrosomes identified by complementary proteomics methods. Embo J. 2011;30:1520–1535. doi: 10.1038/emboj.2011.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20**.Tanos BE, Yang HJ, Soni R, Wang WJ, Macaluso FP, Asara JM, Tsou MF. Centriole distal appendages promote membrane docking, leading to cilia initiation. Genes Dev. 2013;27:163–168. doi: 10.1101/gad.207043.112. This study identified five transition fiber proteins from a systematically proteomic study, and demonstrated that CEP83 is required for centriole-to-membrane docking. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21**.Wei Q, Xu Q, Zhang Y, Li Y, Zhang Q, Hu Z, Harris PC, Torres VE, Ling K, Hu J. Transition fibre protein FBF1 is required for the ciliary entry of assembled intraflagellar transport complexes. Nat Commun. 2013;4:2750. doi: 10.1038/ncomms3750. This study supports that FBF1 acts as the functional component of transition fibers to regulate the ciliary entry of aeeembled intraflagellar transport complexes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sillibourne JE, Specht CG, Izeddin I, Hurbain I, Tran P, Triller A, Darzacq X, Dahan M, Bornens M. Assessing the localization of centrosomal proteins by PALM/STORM nanoscopy. Cytoskeleton (Hoboken) 2011;68:619–627. doi: 10.1002/cm.20536. [DOI] [PubMed] [Google Scholar]
- 23*.Joo K, Kim CG, Lee MS, Moon HY, Lee SH, Kim MJ, Kweon HS, Park WY, Kim CH, Gleeson JG, et al. CCDC41 is required for ciliary vesicle docking to the mother centriole. Proc Natl Acad Sci U S A. 2013;110:5987–5992. doi: 10.1073/pnas.1220927110. An independent study revealed that CEP83 is required for centriole docking to the cilairy vesicle, indicating the role of transition fibers in basal body docking. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24**.Burke MC, Li FQ, Cyge B, Arashiro T, Brechbuhl HM, Chen X, Siller SS, Weiss MA, O’Connell CB, Love D, et al. Chibby promotes ciliary vesicle formation and basal body docking during airway cell differentiation. The Journal of cell biology. 2014;207:123–137. doi: 10.1083/jcb.201406140. This study demonstrated that Chibby interacts with CEP164, and coorprates with Rab8-Rabin8 to regulate ciliary vesicle fromation and basal body docking. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25**.Cajanek L, Nigg EA. Cep164 triggers ciliogenesis by recruiting Tau tubulin kinase 2 to the mother centriole. Proc Natl Acad Sci U S A. 2014;111:E2841–2850. doi: 10.1073/pnas.1401777111. TTBK2 was identifed as a CEP164 interactor on transtion fibers, and acts to remove microtube cap protein CP110 to initiate ciliogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26*.Ishikawa H, Kubo A, Tsukita S. Odf2-deficient mother centrioles lack distal/subdistal appendages and the ability to generate primary cilia. Nat Cell Biol. 2005;7:517–524. doi: 10.1038/ncb1251. This paper reported that both distal appendages and sub-distal appendages dispear from the mother centriole in Odf2 KO mouse F9 cells, indicating that ODF2 is indispensable for DA assembly. [DOI] [PubMed] [Google Scholar]
- 27*.Singla V, Romaguera-Ros M, Garcia-Verdugo JM, Reiter JF. Ofd1, a human disease gene, regulates the length and distal structure of centrioles. Dev Cell. 2010;18:410–424. doi: 10.1016/j.devcel.2009.12.022. This study revealed that mutated Ofd1 in mouse ES cells results in abnormally long centriole, lacking of DAs and defects in recruiting IFT component IFT88 to the centrosome. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28*.Thauvin-Robinet C, Lee JS, Lopez E, Herranz-Perez V, Shida T, Franco B, Jego L, Ye F, Pasquier L, Loget P, et al. The oral-facial-digital syndrome gene C2CD3 encodes a positive regulator of centriole elongation. Nat Genet. 2014;46:905–911. doi: 10.1038/ng.3031. In this paper, C2CD3 was identified as an OFD syndrome protein, promotes centriole elongation, and regulates the formation of both DAs and sub-distal appendages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29*.Ye X, Zeng H, Ning G, Reiter JF, Liu A. C2cd3 is critical for centriolar distal appendage assembly and ciliary vesicle docking in mammals. Proc Natl Acad Sci U S A. 2014;111:2164–2169. doi: 10.1073/pnas.1318737111. This study demonstrated that C2CD3 is required for DAs assebmly, ciliary vesicle docking, recruitment of TTBK2, removal of CP110 and recruitment of IFT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang C, Low WC, Liu A, Wang B. Centrosomal protein DZIP1 regulates Hedgehog signaling by promoting cytoplasmic retention of transcription factor GLI3 and affecting ciliogenesis. J Biol Chem. 2013;288:29518–29529. doi: 10.1074/jbc.M113.492066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tateishi K, Yamazaki Y, Nishida T, Watanabe S, Kunimoto K, Ishikawa H, Tsukita S. Two appendages homologous between basal bodies and centrioles are formed using distinct Odf2 domains. The Journal of cell biology. 2013;203:417–425. doi: 10.1083/jcb.201303071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang Z, Lin MG, Stowe TR, Chen S, Zhu M, Stearns T, Franco B, Zhong Q. Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature. 2013;502:254–257. doi: 10.1038/nature12606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33*.Schmidt KN, Kuhns S, Neuner A, Hub B, Zentgraf H, Pereira G. Cep164 mediates vesicular docking to the mother centriole during early steps of ciliogenesis. The Journal of cell biology. 2012;199:1083–1101. doi: 10.1083/jcb.201202126. This paper demonstrated that CEP164 mediates ciliary vesiclar docking to the mother centriole by interacting with Rab8-Rabin8 vesicle trafficking machinery. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sillibourne JE, Hurbain I, Grand-Perret T, Goud B, Tran P, Bornens M. Primary ciliogenesis requires the distal appendage component Cep123. Biol Open. 2013;2:535–545. doi: 10.1242/bio.20134457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35**.Lu Q, Insinna C, Ott C, Stauffer J, Pintado PA, Rahajeng J, Baxa U, Walia V, Cuenca A, Hwang YS, et al. Early steps in primary cilium assembly require EHD1/EHD3-dependent ciliary vesicle formation. Nat Cell Biol. 2015;17:228–240. doi: 10.1038/ncb3109. This work revealed that EHD proteins and SNAP29 are required for the formation of the primary ciliary vesicle from smaller distal appendages vesicles. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Westlake CJ, Baye LM, Nachury MV, Wright KJ, Ervin KE, Phu L, Chalouni C, Beck JS, Kirkpatrick DS, Slusarski DC, et al. Primary cilia membrane assembly is initiated by Rab11 and transport protein particle II (TRAPPII) complex-dependent trafficking of Rabin8 to the centrosome. Proc Natl Acad Sci U S A. 2011;108:2759–2764. doi: 10.1073/pnas.1018823108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goetz SC, Liem KF, Jr, Anderson KV. The spinocerebellar ataxia-associated gene Tau tubulin kinase 2 controls the initiation of ciliogenesis. Cell. 2012;151:847–858. doi: 10.1016/j.cell.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vieira OV, Gaus K, Verkade P, Fullekrug J, Vaz WL, Simons K. FAPP2, cilium formation, and compartmentalization of the apical membrane in polarized Madin-Darby canine kidney (MDCK) cells. Proc Natl Acad Sci U S A. 2006;103:18556–18561. doi: 10.1073/pnas.0608291103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nachury MV, Seeley ES, Jin H. Trafficking to the ciliary membrane: how to get across the periciliary diffusion barrier? Annu Rev Cell Dev Biol. 2010;26:59–87. doi: 10.1146/annurev.cellbio.042308.113337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40*.Kee HL, Dishinger JF, Blasius TL, Liu CJ, Margolis B, Verhey KJ. A size-exclusion permeability barrier and nucleoporins characterize a ciliary pore complex that regulates transport into cilia. Nat Cell Biol. 2012;14:431–437. doi: 10.1038/ncb2450. This study first demonstrated the existence of a size-dependent diffusion barrier at the ciliary base and suggested its correlation with the nuclear pore complex. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin YC, Niewiadomski P, Lin B, Nakamura H, Phua SC, Jiao J, Levchenko A, Inoue T, Rohatgi R. Chemically inducible diffusion trap at cilia reveals molecular sieve-like barrier. Nat Chem Biol. 2013;9:437–443. doi: 10.1038/nchembio.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42*.Breslow DK, Koslover EF, Seydel F, Spakowitz AJ, Nachury MV. An in vitro assay for entry into cilia reveals unique properties of the soluble diffusion barrier. The Journal of cell biology. 2013;203:129–147. doi: 10.1083/jcb.201212024. This study confirmed the existence of a soluble diffusion barrier, but questioned that the barrier is mechanistically distinct from the nuclear pore complex. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sang L, Miller JJ, Corbit KC, Giles RH, Brauer MJ, Otto EA, Baye LM, Wen X, Scales SJ, Kwong M, et al. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell. 2011;145:513–528. doi: 10.1016/j.cell.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chih B, Liu P, Chinn Y, Chalouni C, Komuves LG, Hass PE, Sandoval W, Peterson AS. A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat Cell Biol. 2012;14:61–72. doi: 10.1038/ncb2410. [DOI] [PubMed] [Google Scholar]
- 45.Garcia-Gonzalo FR, Corbit KC, Sirerol-Piquer MS, Ramaswami G, Otto EA, Noriega TR, Seol AD, Robinson JF, Bennett CL, Josifova DJ, et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet. 2011;43:776–784. doi: 10.1038/ng.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Williams CL, Li C, Kida K, Inglis PN, Mohan S, Semenec L, Bialas NJ, Stupay RM, Chen N, Blacque OE, et al. MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. The Journal of cell biology. 2011;192:1023–1041. doi: 10.1083/jcb.201012116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Craige B, Tsao CC, Diener DR, Hou Y, Lechtreck KF, Rosenbaum JL, Witman GB. CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. The Journal of cell biology. 2010;190:927–940. doi: 10.1083/jcb.201006105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Czarnecki PG, Shah JV. The ciliary transition zone: from morphology and molecules to medicine. Trends Cell Biol. 2012;22:201–210. doi: 10.1016/j.tcb.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Qin H, Diener DR, Geimer S, Cole DG, Rosenbaum JL. Intraflagellar transport (IFT) cargo: IFT transports flagellar precursors to the tip and turnover products to the cell body. The Journal of cell biology. 2004;164:255–266. doi: 10.1083/jcb.200308132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fowkes ME, Mitchell DR. The role of preassembled cytoplasmic complexes in assembly of flagellar dynein subunits. Mol Biol Cell. 1998;9:2337–2347. doi: 10.1091/mbc.9.9.2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ounjai P, Kim KD, Liu H, Dong M, Tauscher AN, Witkowska HE, Downing KH. Architectural insights into a ciliary partition. Curr Biol. 2013;23:339–344. doi: 10.1016/j.cub.2013.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Takao D, Dishinger JF, Kee HL, Pinskey JM, Allen BL, Verhey KJ. An assay for clogging the ciliary pore complex distinguishes mechanisms of cytosolic and membrane protein entry. Curr Biol. 2014;24:2288–2294. doi: 10.1016/j.cub.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pazour GJ, Bloodgood RA. Targeting proteins to the ciliary membrane. Curr Top Dev Biol. 2008;85:115–149. doi: 10.1016/S0070-2153(08)00805-3. [DOI] [PubMed] [Google Scholar]
- 54.Lim YS, Chua CE, Tang BL. Rabs and other small GTPases in ciliary transport. Biology of the cell / under the auspices of the European Cell Biology Organization. 2011;103:209–221. doi: 10.1042/BC20100150. [DOI] [PubMed] [Google Scholar]
- 55.Li Y, Hu J. Small GTPases and cilia. Protein & cell. 2011;2:13–25. doi: 10.1007/s13238-011-1004-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56*.Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peranen J, Merdes A, Slusarski DC, Scheller RH, Bazan JF, Sheffield VC, et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129:1201–1213. doi: 10.1016/j.cell.2007.03.053. This study confirmed the role of Rab8 in promoting membrane biogenesis in cilia after centriole-membrane docking. [DOI] [PubMed] [Google Scholar]
- 57.Yoshimura S, Egerer J, Fuchs E, Haas AK, Barr FA. Functional dissection of Rab GTPases involved in primary cilium formation. The Journal of cell biology. 2007;178:363–369. doi: 10.1083/jcb.200703047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58**.Knodler A, Feng S, Zhang J, Zhang X, Das A, Peranen J, Guo W. Coordination of Rab8 and Rab11 in primary ciliogenesis. Proc Natl Acad Sci U S A. 2010;107:6346–6351. doi: 10.1073/pnas.1002401107. This study suggested the role of Rab11-Rabin8-Rab8 small GTPase cascade in coordinating vesicular trafficking during primary ciliogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moritz OL, Tam BM, Hurd LL, Peranen J, Deretic D, Papermaster DS. Mutant rab8 Impairs docking and fusion of rhodopsin-bearing post-Golgi membranes and causes cell death of transgenic Xenopus rods. Mol Biol Cell. 2001;12:2341–2351. doi: 10.1091/mbc.12.8.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang J, Deretic D. The Arf and Rab11 effector FIP3 acts synergistically with ASAP1 to direct Rabin8 in ciliary receptor targeting. J Cell Sci. 2015;128:1375–1385. doi: 10.1242/jcs.162925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang J, Morita Y, Mazelova J, Deretic D. The Arf GAP ASAP1 provides a platform to regulate Arf4- and Rab11-Rab8-mediated ciliary receptor targeting. Embo J. 2012;31:4057–4071. doi: 10.1038/emboj.2012.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bouck GB. The structure, origin, isolation, and composition of the tubular mastigonemes of the Ochromas flagellum. The Journal of cell biology. 1971;50:362–384. doi: 10.1083/jcb.50.2.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Papermaster DS, Schneider BG, Besharse JC. Vesicular transport of newly synthesized opsin from the Golgi apparatus toward the rod outer segment. Ultrastructural immunocytochemical and autoradiographic evidence in Xenopus retinas. Investigative ophthalmology & visual science. 1985;26:1386–1404. [PubMed] [Google Scholar]
- 64*.Rogers KK, Wilson PD, Snyder RW, Zhang X, Guo W, Burrow CR, Lipschutz JH. The exocyst localizes to the primary cilium in MDCK cells. Biochem Biophys Res Commun. 2004;319:138–143. doi: 10.1016/j.bbrc.2004.04.165. [DOI] [PubMed] [Google Scholar]
- 65*.Zuo X, Guo W, Lipschutz JH. The exocyst protein Sec10 is necessary for primary ciliogenesis and cystogenesis in vitro. Mol Biol Cell. 2009;20:2522–2529. doi: 10.1091/mbc.E08-07-0772. This study and the above one provide molecular evidences that the exocyst localizes at the cilairy base and play essential role in the context of cilia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Babbey CM, Bacallao RL, Dunn KW. Rab10 associates with primary cilia and the exocyst complex in renal epithelial cells. Am J Physiol Renal Physiol. 2010;299:F495–506. doi: 10.1152/ajprenal.00198.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Oztan A, Silvis M, Weisz OA, Bradbury NA, Hsu SC, Goldenring JR, Yeaman C, Apodaca G. Exocyst requirement for endocytic traffic directed toward the apical and basolateral poles of polarized MDCK cells. Mol Biol Cell. 2007;18:3978–3992. doi: 10.1091/mbc.E07-02-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chaki M, Airik R, Ghosh AK, Giles RH, Chen R, Slaats GG, Wang H, Hurd TW, Zhou W, Cluckey A, et al. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell. 2012;150:533–548. doi: 10.1016/j.cell.2012.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Failler M, Gee HY, Krug P, Joo K, Halbritter J, Belkacem L, Filhol E, Porath JD, Braun DA, Schueler M, et al. Mutations of CEP83 cause infantile nephronophthisis and intellectual disability. Am J Hum Genet. 2014;94:905–914. doi: 10.1016/j.ajhg.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Adly N, Alhashem A, Ammari A, Alkuraya FS. Ciliary genes TBC1D32/C6orf170 and SCLT1 are mutated in patients with OFD type IX. Hum Mutat. 2014;35:36–40. doi: 10.1002/humu.22477. [DOI] [PubMed] [Google Scholar]