

Abstract
The merging of knowledge from genomics, cellular signal transduction and molecular evolution is producing new paradigms of cancer analysis. Protein kinases have long been understood to initiate and promote malignant cell growth and targeting kinases to fight cancer has been a major strategy within the pharmaceutical industry for over two decades. Despite the initial success of kinase inhibitors (KIs), the ability of cancer to evolve resistance and reprogram oncogenic signaling networks has reduced the efficacy of kinase targeting. The molecular chaperone HSP90 physically supports global kinase function while also acting as an evolutionary capacitor. The Cancer Genome Atlas (TCGA) has compiled a trove of data indicating that a large percentage of tumors overexpress or possess mutant kinases that depend on the HSP90 molecular chaperone complex. Moreover, the overexpression or mutation of parallel activators of kinase activity (PAKA) increases the number of components that promote malignancy and indirectly associate with HSP90. Therefore, targeting HSP90 is predicted to complement kinase inhibitors by inhibiting oncogenic reprogramming and cancer evolution. Based on this hypothesis, consideration should be given by both the research and clinical communities towards combining kinase inhibitors and HSP90 inhibitors (H90Ins) in combating cancer. The purpose of this perspective is to reflect on the current understanding of HSP90 and kinase biology as well as promote the exploration of potential synergistic molecular therapy combinations through the utilization of The Cancer Genome Atlas.
Electronic supplementary material
The online version of this article (doi:10.1007/s12192-015-0604-1) contains supplementary material, which is available to authorized users.
Keywords: Cancer, Drug resistance, HSP90, Kinase, Evolution, TCGA
Background
Cancer is a disease of deregulated cell growth. The presence of continuous pro-growth signals and overriding of cell cycle checkpoints allows for the initiation of neoplastic transformation and eventual cancer. Kinases, along with the phosphoinositide 3-kinase and RAS signaling pathways often perpetuate pro-growth signals that can lead to malignancy (Blume-Jensen and Hunter 2001); (Yuan and Cantley 2008); (Chang et al. 1982). The human genome encodes over 500 protein kinases, 90 of which are tyrosine kinases, and of these, 58 are receptor tyrosine kinases (Manning et al. 2002). Together, these kinases form cascading networks that signal for normal cell growth and differentiation. However, when overexpressed, mutated, or otherwise deregulated, kinases can drive a mass of cells toward malignancy (Levinson et al. 1978; Di Fiore et al. 1987); (Hudziak et al. 1987); (Davies et al. 2002); (Wong et al. 1987) (Fig. 1). Profiling these malignancy-driving alterations in distinct cancers is now possible with the establishment of The Cancer Genome Atlas (TCGA). Equally interesting is the understanding that the majority of kinases in a cancer cell associate with and depend on the HSP90 molecular chaperone complex along with CDC37 and HSP70 to bind, hold, and fold newly synthesized kinases into their proper three-dimensional arrangement—maturing them into functional signaling components (Pratt and Toft 2003); (Prince and Matts 2004); (Shao et al. 2001). Moreover, when kinases become structurally destabilized as a result of over-activation, mutation and/or proteotoxic stress, HSP90 and CDC37 reassociate, refold them, and restore their kinase function (Fig. 2) (Gray et al. 2008); (Xu et al. 2005); (Citri et al. 2006); (Miyajima et al. 2013). Inhibiting HSP90 destabilizes the kinase, resulting in its subsequent degradation and in a reduction in overall pro-growth signaling (Xu et al. 2002); (Trepel et al. 2010); (Citri et al. 2002); (Lerdrup et al. 2006). Based on the premise that structure dictates function, this relationship suggests that kinase activity is at least partially dependent on HSP90. Due to this relationship and the fact that a number of clinically relevant HSP90 inhibitors (H90Ins) currently exist (Alarcon et al. 2012), the concept of targeting HSP90 as a way to broadly inhibit kinase activity in cancer deserves continued consideration (Whitesell and Lindquist 2005); (Trepel et al. 2010); (Lu et al. 2012a); (Barrott and Haystead 2013).
Fig. 1.

Simplified model of kinase driven signaling cascades that promote pro-growth gene expression and their dependency on HSP90
Fig. 2.

Cartoon of molecular chaperone-dependent kinase folding, maturation, and maintenance along with the possible effect of H90Ins on distinct kinase populations
While the success of the small molecule kinase inhibitor (KI) imatinib, which targets the BCR-ABL fusion protein in treating chronic myelogenous leukemia (CML), and that of the ALK inhibitor crizotinib in treating certain forms of non-small-cell lung cancer (NSCLC) is certainly promising (Druker et al. 1996); (Ou et al. 2011); the clinical benefit tends to be short lived, as most cancers evolve resistance to such targeted KIs (Carroll 2006); (Vaidya et al. 2015). This evolved resistance often is a consequence of a number of cellular events that allow the reprogramming of oncogenic signals in order to compensate for the loss of activity of the targeted kinase (Garraway and Janne 2012). Some of these cellular events include the following: increased rates of mutagenesis resulting in alteration of the drug-binding site (Vaidya et al. 2015); (Ma et al. 2002); (Pao et al. 2005); (Yu et al. 2014), chromosomal deletions or rearrangements creating chimeric transcripts that provide deregulated growth signals (Grammatikakis et al. 2002); (Duesberg et al. 2001); (Lee et al. 2011); (Hingorani et al. 2005); (Hashida et al. 2015), epigenetic rewiring of gene expression (Ricketts et al. 2014); (Hill et al. 2011); (Abdel-Hafiz and Horwitz 2015), hyper-activation of alternative but overlapping kinase signaling cascades (Drake et al. 2014); (Shattuck et al. 2008); (Maroun and Rowlands 2014); (Chen et al. 2012), relaxation of protein translational control to favor increased production of oncogenes (Pelletier et al. 2015); (Boussemart et al. 2014); (Konicek et al. 2008), and additional mechanisms as yet not fully appreciated such as altered non-coding RNA expression and cellular metabolic reprogramming (Klinge 2015); (Ward et al. 2014); (Ward and Thompson 2012); (DeBerardinis et al. 2008); (Linehan and Rouault 2013). The relationship of oncogenic reprogramming to evolved resistance is supported by the clonal diversity and genetic heterogeneity of most tumors along with the ability of most cancers to relapse after several rounds of therapy (Hiley and Swanton 2014); (Calderwood 2013). Therefore, targeting this phenomenon is critical to preventing resistance to KIs. HSP90 is associated with each cellular event required for oncogenic reprogramming, consistent with its ability to act as an evolutionary capacitor, and suggesting that inhibition of HSP90 may represent a viable strategy to combat resistance (Trepel et al. 2010); (Hanahan and Weinberg 2011) (Taipale et al. 2010); (Rutherford and Lindquist 1998); (Zhao et al. 2005); (Lu et al. 2012b); (Barrott and Haystead 2013); (Methot et al. 2015); (Fig. 3).
Fig. 3.

Model of the oncogenic reprogramming concept that allows for evolved drug resistance in cancer
KIs and H90Ins
Kinases and HSP90 both utilize ATP in their molecular function. Kinases employ ATP by adding the gamma phosphate group onto substrates in order to transmit a signal (Burnett and Kennedy 1954; Fischer et al. 1959), while HSP90 uses ATP hydrolysis to fuel the conformational dynamics that drive its chaperone activity (Grenert et al. 1997; Prodromou et al. 1997). Targeting the structural pocket that binds ATP in kinases has been a major focus of the pharmaceutical industry for over two decades and has yielded an arsenal of kinase inhibitors. These KIs vary as the ATP-binding pocket varies from kinase to kinase allowing for a degree of specificity and the ability to target key signaling pathways that promote malignant growth (Knight and Shokat 2005). However, in response to such inhibition, tumors often mutate the ATP-binding pocket and other structural features of the kinase domain in order to greatly reduce the affinity of the KI, and thus reinitiating pro-growth signaling (Pao et al. 2005; Miyajima et al. 2013); (Katayama et al. 2011). Importantly, many of these mutations make the kinase structurally unstable and more dependent on HSP90 to maintain function (Miyajima et al. 2013); (Shimamura et al. 2008); (Barrott and Haystead 2013); (Shimamura et al. 2005; Sang et al. 2013; Lachowiec et al. 2015). Conversely, a kinase may also evolve HSP90 independence through mutations that stabilize its structure as observed throughout evolution (Nony et al. 2003; Taipale et al. 2012). However, combined administration of KIs and H90Ins would place opposing pressures upon the kinase to alter the KI-binding site in order to regain kinase activity, while at the same time, altering its structure to attain HSP90 independence, thus making the evolutionary walk of such a kinase improbable. Indeed, in a random mutagenesis screen of BCR-ABL, while numerous KI resistance mutants were identified, no H90Ins resistance mutants were identified (Tauchi et al. 2011). Alternatively, a tumor may also compensate for the inhibition of one growth promoting kinase by activating another kinase signaling pathway (Chen et al. 2012); (Lee et al. 2013). Such phenomena demonstrate the need to simultaneously inhibit multiple kinases, which may be accomplished by targeting HSP90. Clinically evaluated H90Ins target the ATP-binding pocket of HSP90, which is distinct from kinase ATP-binding pockets (Whitesell et al. 1994; Neckers and Trepel 2014). H90Ins operate by locking HSP90 into a static structural conformation that prevents it from chaperoning its client kinases. This leads to destabilization of the kinases and their eventual degradation by the proteasome (Trepel et al. 2010); (Barrott and Haystead 2013); (Miyata et al. 2013); (Fig. 4).
Fig. 4.

Model of oncogenic growth signal output reduced by combined treatment
Despite their theoretical efficacy and initial promise (Kamal et al. 2003); (Neckers and Workman 2012), treating cancer patients with H90Ins alone has not proven to be particularly effective (Barrott and Haystead 2013). As single agents, H90Ins require higher effective dosages for inhibiting tumor growth, often resulting in increased toxicity (Barrott and Haystead 2013); (Johnson et al. 2015); (Jhaveri et al. 2012); (Sequist et al. 2010); (Sessa et al. 2013). Some of these side effects may be the result of off-target inhibition of the HSP90 homologues, which reside in the endoplasmic reticulum (GRP94) and mitochondria (TRAP1), although this has not been determined. Moreover, these side effects and the need for increased dosing may be related in part to the phenomenon that HSP90 inhibition leads to activation of a feedback loop that involves HSF1, the master stress response transcription factor (Bagatell et al. 2000); (Ciocca et al. 2013). HSF1 is responsible for initiating an adaptive, pro-survival, and anti-apoptotic gene expression program, which includes a number of molecular chaperones. Consequently, HSF1 activation likely reduces the effectiveness of HSP90 inhibition in cancer and may even enhance transcription of certain cancer-promoting genes that comprise a unique cancer-specific HSF1 transcriptional signature (Dai et al. 2007); (Santagata et al. 2013); (Mendillo et al. 2012). Thus, identifying approaches to uncouple this feedback loop are a major focus in the HSP90 research field. Combining H90Ins with specific KIs is one possible strategy since HSF1 activity is regulated by kinase cascades to some degree (Guettouche et al. 2005); (Holmberg et al. 2002); (Calderwood et al. 2010). For example, mTORC and MEK1 have been shown to phosphorylate HSF1 on S326, a post-translational modification important for inducing the heat shock response. Moreover, inhibition of these kinases reduces HSF1 overall transcriptional activity, suggesting a possible combination of inhibitors that may prove effective in treating specific tumor types (Chou et al. 2012); (Tang et al. 2015); (Acquaviva et al. 2014a, b).
TCGA
The Cancer Genome Atlas is a national initiative to characterize over 80 forms of cancer at the molecular level. Its goal is to sequence the entire genome and quantitatively characterize a representative number of cases of a defined tumor type, providing information on gene copy number, promoter methylation patterns, RNA expression levels, global mutation analysis, and eventually proteomic profiling (http://cancergenome.nih.gov/). These data are made publicly available as they are processed and published, allowing for further analysis by the cancer research community. Several sites and institutes host and distribute TCGA data including cBioPortal at Memorial Sloan-Kettering Cancer Center, which was utilized here (Gao et al. 2013); (Cerami et al. 2012).
In our analysis of the mRNA expression levels and open reading frame mutations of 31 representative kinases that associate with HSP90, we found that a large percentage of samples from each of 23 tumor types either overexpress or possess a mutated HSP90-dependent kinase or parallel activator of kinase activity (PAKA) such as PIK3CA or KRAS (Table 1, see Supplemental Tables 2 and 3 for sources). When the number of overexpressed (blue column) HSP90-dependent kinases is combined, this percentage exceeds 50 % in all tumor types compared to normal corresponding tissue. This frequency reaches 80 % or greater in seven tumor types: cervical cancer (CESC), chromophobe renal cell carcinoma (KICH), glioblastoma multiforme (GBM), head and neck squamous cell carcinoma (HNSC), stomach adenocarcinoma (STAD), bladder cancer (BLCA), and uterine carcinoma (UCS). Further, when combined with PAKA overexpression, this frequency increased by an additional 12 % in lung squamous cell carcinoma (LUSC) and by 15 % in ovarian cancer (OV). When the sum total (orange row) of all overexpressed kinases and PAKAs within the table is calculated, the value exceeds 100 and is often more than twice the actual grand total (red row) provided by TCGA, indicating that more than one HSP90-dependent kinase is overexpressed in a single tumor. Indeed, this has been observed in other studies where one third of stomach cancers were found to overexpress multiple HSP90-dependent receptor tyrosine kinases (Nagatsuma et al. 2014); (Sjoblom et al. 2006); (Greenman et al. 2007). In contrast, the combined number of mutated (green column) HSP90 dependent kinases and PAKAs does not exceed a frequency of 50 % in most tumor types. However, in certain tumor types, including colon and rectal adenocarcinoma (COAD), cutaneous melanoma (SKCM), lung adenocarcinoma (LUAD), papillary thyroid carcinoma (THCA), and uterine corpus endometrial carcinoma (UCEC), mutation frequencies are above 70 %.
Table 1.
Profile of the percentage of tumors upregulating mRNA expression by two standard deviations or possessing a mutant form of an HSP90-dependent kinase or parallel activator of kinase activity (PAKA). Blue columns, list of the percentage of tumors with mRNA expression; green columns, list of the percentage of tumors with mutants; gold rows, list of the TCGA totals for each tumor type; red row, list of the TCGA grand totals of both kinases and PAKAs; and orange row, list of the sum totals of kinases and PAKAs, which was calculated by adding up all the observed percentages within the column. Inhibitors are listed on the right. Data was collected from TCGA cBioPortal (Gao et al. 2013); (Cerami et al. 2012) and Blue Ridge Institute for Medical Research (Roskoski 2015)
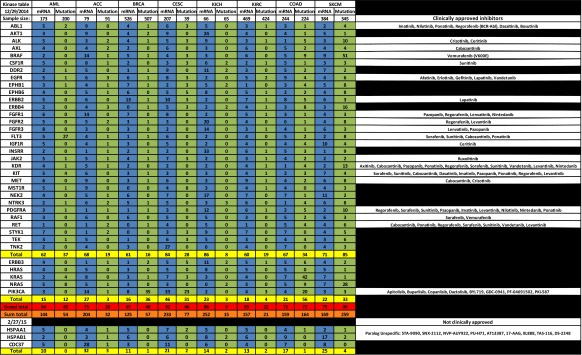
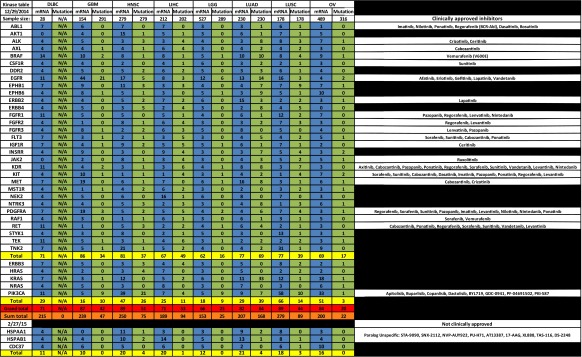
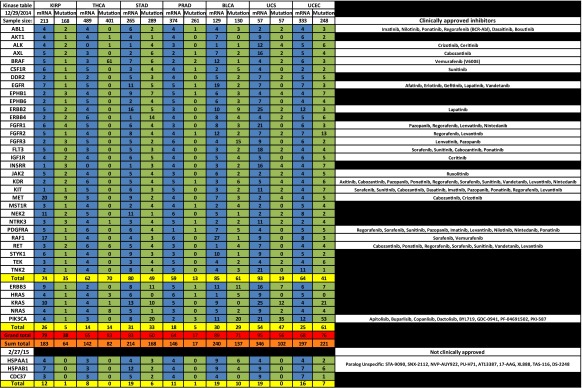
Based on these TCGA data, we suggest that individual KI and H90In combinations should be explored in a variety of tumor types. For example, in breast cancer (BRCA), the kinase ERBB2 is overexpressed in 13 % of 526 tumor samples while the signaling component PIK3CA is mutated in 35 % of 507 tumor samples. Combining an H90In with either an ERBB2 inhibitor or a number of PIK3CA inhibitors may prove effective in treating certain populations of BRCA. Indeed, combination of the monoclonal antibody herceptin and H90In has shown clinical benefit (O’Connell et al. 2014). In bladder cancer (BLCA), the most cost intensive cancer to treat (Kaplan et al. 2014) RAF1 is overexpressed in 27 % while EGFR is overexpressed in 19 % of the sampled tumors. In specific cases, a combination of the RAF1 inhibitors sorafenib or vemurafenib, or a number of EGFR inhibitors, along with an H90In may prove to be effective (Acquaviva et al. 2014a, b; Huang et al. 2015). Glioblastoma multiforme (GBM) overexpresses one or more HSP90-dependent kinases in 86 % of tumors sampled. Consequently, a combination of at least 11 different KIs with an H90In is a treatment option worth exploring (Fu et al. 2013) (Wachsberger et al. 2014). Lung adenocarcinoma (LUAD), which is responsible for the largest number of cancer deaths, overexpresses EGFR in 13 %, ERBB2 in 15 %, and MET in 16 % of tumor samples. Combining a number of kinase inhibitors with an H90In may be beneficial in treating up to 77 % of patients with lung adenocarcinoma (Chen et al. 2014); (Ohkubo et al. 2015). A similar approach may be taken for uterine carcinosarcomas (UCS), which overexpress HSP90-dependent kinases in 93 % of tumors sampled.
Conclusion
Cancer is resilient. Its only function is to proliferate, and to this end, it utilizes every biological mechanism available to it to gain a proliferative advantage (Wachsberger et al. 2014); (Holmberg et al. 2002). Consequently, efforts must be focused on anticipating the routes available to cancer cells for this purpose and to implement therapies able to counter them. Indeed, evidence from preclinical models suggests that early simultaneous targeting of the HSP90 chaperone complex and specific tumor-driving kinases prolongs efficacy while possibly reducing toxicity by lowering effective drug dose (Fiskus et al. 2011); (Lu et al. 2012a, b), (Barrott and Haystead 2013); (Tonini 2015); (Miyajima et al. 2013); (Solarova et al. 2015); (Xiao et al. 2007); (Fig. 5). As with all novel treatments, determining the optimal dose combination and schedule for each tumor type will require further study.
Fig. 5.

Hypothesized timeline for cancer growth inhibition and overall toxicity comparing single agent KI therapy vs. combined H90In therapy
The synergy provided by combining H90Ins with KIs is predicted to reduce the evolutionary space available to cancer by simultaneously targeting cellular proteostasis and multiple pro-growth and metastatic signaling pathways (Rutherford and Lindquist 1998); (Workman et al. 2007a; Gerlinger et al. 2014); (Whitesell et al. 2014). This hypothesis is further supported by recent observations that HSP90 influences the function of a number of other signaling components that are overexpressed, or otherwise deregulated in cancer, including transcription factors, E3-ligases, metabolic enzymes, and protein translational machinery (Taipale et al. 2014); (Liu et al. 2015); (Solier et al. 2012); (Supplemental Table 2). While development of specific inhibitors of these various signaling components lags behind the development of KIs, preliminary findings provide evidence of synergy (Brady et al. 2015).
Equally there is always the possibility that administration of H90Ins along with KIs or any other molecular therapy may result in unintended outcomes. H90Ins have the possibility of being weapons that cut both ways, and therefore, their use will require great forethought and care in wielding them. In both mouse and drosophila model systems, HSP90 inhibition has been shown to increase transposon activity in germ line cells (Specchia et al. 2010); (Ichiyanagi et al. 2014). This is understood to be related to the ability of HSP90 to maintain Piwi protein function and piRNA loading (Gangaraju et al. 2011; Izumi et al. 2013), which together function to repress transposon mobility. Uncontrolled transposon activity has been shown to result in sterility and alteration of the germ line (Fu and Wang 2014; Hadziselimovic et al. 2015). Therefore, administration of H90Ins to only the non-breeding population may be warranted, as is careful monitoring of metabolized and unmetabolized H90Ins in the water table and environment.
There is also the concern that H90Ins could impact the effectiveness of tumor suppressor pathways (Fierro-Monti et al. 2013); (Manjarrez et al. 2014). HSP90 interacts with a large portion of the proteome and is involved in maintaining tumor promoting as well as tumor suppressing cellular components (Taipale et al. 2014); (Nony et al. 2003). The tumor suppressor TP53 is mutated in a vast number of tumors, and HSP90 has been shown to associate with both mutant and WT versions of TP53 (Blagosklonny et al. 1996); (Nagata et al. 1999); (Walerych et al. 2004). However, if TP53 mutants do not function as tumor suppressors, and may even serve as tumor promoters (Walerych et al. 2012); (Shetzer et al. 2014), then compromising their stabilization by HSP90 inhibition is likely to promote anti-tumor activity (Powell et al. 2014).
HSP90 dependence of the tumor suppressor kinase STK11 (or LKB1) may be more significant, as this kinase plays a major role in regulating cellular metabolism (Nony et al. 2003); (Taipale et al. 2012); (Zhao and Xu 2014). HSP90 also participates in regulation of gene expression and genome maintenance (Fang et al. 2014); (Sollars et al. 2003; Lu et al. 2012a, b). Despite these uncertainties, the observation that H90Ins as a class concentrate in tumor cells to a greater degree than in normal tissue is of importance and a point of hope (Kamal et al. 2003); (Moulick et al. 2011); (Taldone et al. 2014); (Suzuki et al. 2015); (Moses et al. 2015). The notion that rapidly proliferating malignant cells in a toxic microenvironment have a larger population of targetable molecular chaperones such as HSP90, and depend more on these proteostasis components for survival, fits well with the concept of chaperone addiction put forth by many others in the field (Miyata et al. 2013); (Prodromou 2009); (Workman et al. 2007a); (Calderwood et al. 2006); (Xiao et al. 2007); (Barrott and Haystead 2013).
The pioneers of combinational drug therapy, Emil Freireich, James Holland, and Emil Frei, laid the groundwork for synergistic drug combinations with their development of successful treatments for children suffering from acute lymphoblastic leukemia (Frei et al. 1958). A more recent example of this concept is the development of multi-drug cocktails for controlling HIV (Fauci et al. 2013). Unfortunately, for individuals with cancer, development of KI resistance is all too common. As a consequence, there are currently 69 clinical trials focused on targeting HSP90 (Supplemental Table 4), with 8 trials testing combinations of KIs and H90Ins (NCT01613950, NCT02192541, NCT02008877, NCT01712217, NCT02097225, NCT01657591, and NCT01236144) (Clinicaltrials.gov 2015) (Supplemental Table 5). Early reporting of the NCT01259089 phase I/II trial that combined erlotinib with the HSP90In, AUY922, for treating erlotinib-resistant non-small-cell lung cancer (NSCLC), however demonstrated only partial efficacy with an elevated toxicity profile (Johnson et al. 2015). These unfortunate findings indicate the need for improved understanding of HSP90 biology and inhibitor development but should not discourage further clinical evaluation of HS90Ins (Ohkubo et al. 2015); (Besse et al. 2014); (Hubbard 2014). Early, simultaneous administration of combined molecular therapies based on insights gathered from TCGA data should improve upon current outcomes that rely solely on single agents. Indeed, this strategy aligns with the Precision Medicine Initiative to provide quality healthcare based on individual variations in genes, environment, and lifestyle (Collins and Varmus 2015); (Zhao et al. 2015).
Cancer and its evolving genome is a complex operation given only the simple task to proliferate. An immense amount of information remains to be discovered and understood concerning the origins and driving forces of cancer. The results of ongoing clinical trials along with increased utilization of TCGA data will help not only in testing the hypothesis put forth here, but also in designing future clinical trials that incorporate HSP90 inhibition as a mechanism to combat cancer robustness and prevent oncogenic reprogramming.
Electronic supplementary material
(XLSX 92 kb)
Acknowledgments
We thank Jane Trepel, Young Lee, and Chris Ricketts for thoughtful scientific discussion. This work was supported by funds from the Intramural Research Program, National Cancer Institute. We sincerely regret that we were not able to include all the references and sources that influenced or provided the scientific foundation for this manuscript.
Supplemental Tables 2 and 3 provide the sources for making Table 1 and Supplemental Table 1. Supplemental Tables 4 and 5 list the current information on clinical trials using H90Ins.
References
- Abdel-Hafiz HA, Horwitz KB. Role of epigenetic modifications in luminal breast cancer. Epigenomics. 2015;17:1–16. doi: 10.2217/epi.15.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acquaviva J, He S, et al. mTOR inhibition potentiates HSP90 inhibitor activity via cessation of HSP synthesis. Mol Cancer Res. 2014;12(5):703–713. doi: 10.1158/1541-7786.MCR-13-0605. [DOI] [PubMed] [Google Scholar]
- Acquaviva J, Smith DL, et al. Overcoming acquired BRAF inhibitor resistance in melanoma via targeted inhibition of HSP90 with ganetespib. Mol Cancer Ther. 2014;13(2):353–363. doi: 10.1158/1535-7163.MCT-13-0481. [DOI] [PubMed] [Google Scholar]
- Alarcon SV, Mollapour M, et al. Tumor-intrinsic and tumor-extrinsic factors impacting HSP90-targeted therapy. Curr Mol Med. 2012;12(9):1125–1141. doi: 10.2174/156652412803306729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagatell R, Paine-Murrieta GD, et al. Induction of a heat shock factor 1-dependent stress response alters the cytotoxic activity of HSP90-binding agents. Clin Cancer Res. 2000;6(8):3312–3318. [PubMed] [Google Scholar]
- Barrott JJ, Haystead TA. HSP90, an unlikely ally in the war on cancer. Febs J. 2013;280(6):1381–1396. doi: 10.1111/febs.12147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- B Besse, E. B., N.A. Pennell, A. Wozniak, D. Mahadevan, A. Spira, A. Oganesian, L. Manlapaz-Espiritu, H. Keer, J. Soria, D.R. Camidge. (2014). A study of Hsp90 inhibitor AT13387 alone and in combination with crizotinib (CZT) in the treatment of non-small cell lung cancer (NSCLC). Annals of Oncology (2014) 25 (suppl_4): iv426-iv470. 10.1093/annonc/mdu34. from http://oncologypro.esmo.org/Meeting-Resources/ESMO-2014/NSCLC-Metastatic/A-study-of-Hsp90-inhibitor-AT13387-alone-and-in-combination-with-crizotinib-CZT-in-the-treatment-of-non-small-cell-lung-cancer-NSCLC
- Blagosklonny MV, Toretsky J, et al. Mutant conformation of p53 translated in vitro or in vivo requires functional HSP90. Proc Natl Acad Sci U S A. 1996;93(16):8379–8383. doi: 10.1073/pnas.93.16.8379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411(6835):355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- Boussemart L, Malka-Mahieu H, et al. eIF4F is a nexus of resistance to anti-BRAF and anti-MEK cancer therapies. Nature. 2014;513(7516):105–109. doi: 10.1038/nature13572. [DOI] [PubMed] [Google Scholar]
- Brady SW, Zhang J, et al. PI3K-independent mTOR activation promotes lapatinib resistance and IAP expression that can be effectively reversed by mTOR and HSP90 inhibition. Cancer Biol Ther. 2015;16(3):402–411. doi: 10.1080/15384047.2014.1002693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett G, Kennedy EP. The enzymatic phosphorylation of proteins. J Biol Chem. 1954;211(2):969–980. [PubMed] [Google Scholar]
- Calderwood SK. Tumor heterogeneity, clonal evolution, and therapy resistance: an opportunity for multitargeting therapy. Discov Med. 2013;15(82):188–194. [PMC free article] [PubMed] [Google Scholar]
- Calderwood SK, Khaleque MA, et al. Heat shock proteins in cancer: chaperones of tumorigenesis. Trends Biochem Sci. 2006;31(3):164–172. doi: 10.1016/j.tibs.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Calderwood SK, Xie Y, et al. Signal transduction pathways leading to heat shock transcription. Sign Transduct Insights. 2010;2:13–24. doi: 10.4137/STI.S3994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll SB. The making of the fittest: DNA and the ultimate forensic record of evolution. New York: W.W. Norton & Co.; 2006. [Google Scholar]
- Cerami E, Gao J, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang EH, Furth ME, et al. Tumorigenic transformation of mammalian cells induced by a normal human gene homologous to the oncogene of Harvey murine sarcoma virus. Nature. 1982;297(5866):479–483. doi: 10.1038/297479a0. [DOI] [PubMed] [Google Scholar]
- Chen CT, Kim H, et al. MET activation mediates resistance to lapatinib inhibition of HER2-amplified gastric cancer cells. Mol Cancer Ther. 2012;11(3):660–669. doi: 10.1158/1535-7163.MCT-11-0754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Akbay E, et al. Co-clinical trials demonstrate superiority of crizotinib to chemotherapy in ALK-rearranged non-small cell lung cancer and predict strategies to overcome resistance. Clin Cancer Res. 2014;20(5):1204–1211. doi: 10.1158/1078-0432.CCR-13-1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou SD, Prince T, et al. mTOR is essential for the proteotoxic stress response, HSF1 activation and heat shock protein synthesis. PLoS One. 2012;7(6):29. doi: 10.1371/journal.pone.0039679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciocca DR, Arrigo AP, et al. Heat shock proteins and heat shock factor 1 in carcinogenesis and tumor development: an update. Arch Toxicol. 2013;87(1):19–48. doi: 10.1007/s00204-012-0918-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citri A, Alroy I, et al. Drug-induced ubiquitylation and degradation of ErbB receptor tyrosine kinases: implications for cancer therapy. Embo J. 2002;21(10):2407–2417. doi: 10.1093/emboj/21.10.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citri A, Harari D, et al. HSP90 recognizes a common surface on client kinases. J Biol Chem. 2006;281(20):14361–14369. doi: 10.1074/jbc.M512613200. [DOI] [PubMed] [Google Scholar]
- clinicaltrials.gov (2015). https://clinicaltrials.gov/ct2/results?term=hsp90+kinase+inhibitor&Search=Search
- Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372(9):793–795. doi: 10.1056/NEJMp1500523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C, Whitesell L, et al. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell. 2007;130(6):1005–1018. doi: 10.1016/j.cell.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Bignell GR, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- DeBerardinis RJ, Lum JJ, et al. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7(1):11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Di Fiore PP, Pierce JH, et al. erbB-2 is a potent oncogene when overexpressed in NIH/3T3 cells. Science. 1987;237(4811):178–182. doi: 10.1126/science.2885917. [DOI] [PubMed] [Google Scholar]
- Drake JM, Lee JK, et al. Clinical targeting of mutated and wild-type protein tyrosine kinases in cancer. Mol Cell Biol. 2014;34(10):1722–1732. doi: 10.1128/MCB.01592-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druker BJ, Tamura S, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2(5):561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- Duesberg P, Stindl R, et al. Origin of multidrug resistance in cells with and without multidrug resistance genes: chromosome reassortments catalyzed by aneuploidy. Proc Natl Acad Sci U S A. 2001;98(20):11283–11288. doi: 10.1073/pnas.201398998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Q, Inanc B et al (2014) HSP90 regulates DNA repair via the interaction between XRCC1 and DNA polymerase beta. Nat Commun 5(5513) [DOI] [PMC free article] [PubMed]
- Fauci AS, Folkers GK, et al. HIV-AIDS: much accomplished, much to do. Nat Immunol. 2013;14(11):1104–1107. doi: 10.1038/ni.2735. [DOI] [PubMed] [Google Scholar]
- Fierro-Monti I, Echeverria P et al (2013) Dynamic impacts of the inhibition of the molecular chaperone HSP90 on the T cell proteome have implications for anti-cancer therapy. PLoS One 8(11) [DOI] [PMC free article] [PubMed]
- Fischer EH, Graves DJ, et al. Structure of the site phosphorylated in the phosphorylase b to a reaction. J Biol Chem. 1959;234(7):1698–1704. [PubMed] [Google Scholar]
- Fiskus W, Verstovsek S, et al. Heat shock protein 90 inhibitor is synergistic with JAK2 inhibitor and overcomes resistance to JAK2-TKI in human myeloproliferative neoplasm cells. Clin Cancer Res. 2011;17(23):7347–7358. doi: 10.1158/1078-0432.CCR-11-1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frei E, 3rd, Holland JF, et al. A comparative study of two regimens of combination chemotherapy in acute leukemia. Blood. 1958;13(12):1126–1148. [PubMed] [Google Scholar]
- Fu Q, Wang PJ. Mammalian piRNAs: biogenesis, function, and mysteries. Spermatogenesis. 2014;4:e27889. doi: 10.4161/spmg.27889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Koul D, et al. Novel HSP90 inhibitor NVP-HSP990 targets cell-cycle regulators to ablate Olig2-positive glioma tumor-initiating cells. Cancer Res. 2013;73(10):3062–3074. doi: 10.1158/0008-5472.CAN-12-2033. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Gangaraju VK, Yin H, et al. Drosophila Piwi functions in HSP90-mediated suppression of phenotypic variation. Nat Genet. 2011;43(2):153–158. doi: 10.1038/ng.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):2004088. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway LA, Janne PA. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012;2(3):214–226. doi: 10.1158/2159-8290.CD-12-0012. [DOI] [PubMed] [Google Scholar]
- Gerlinger M, McGranahan N, et al. Cancer: evolution within a lifetime. Annu Rev Genet. 2014;48:215–236. doi: 10.1146/annurev-genet-120213-092314. [DOI] [PubMed] [Google Scholar]
- Grammatikakis N, Vultur A, et al. The role of HSP90N, a new member of the HSP90 family, in signal transduction and neoplastic transformation. J Biol Chem. 2002;277(10):8312–8320. doi: 10.1074/jbc.M109200200. [DOI] [PubMed] [Google Scholar]
- Gray PJ, Jr, Prince T, et al. Targeting the oncogene and kinome chaperone CDC37. Nat Rev Cancer. 2008;8(7):491–495. doi: 10.1038/nrc2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenman C, Stephens P, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446(7132):153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenert JP, Sullivan WP, et al. The amino-terminal domain of heat shock protein 90 (HSP90) that binds geldanamycin is an ATP/ADP switch domain that regulates HSP90 conformation. J Biol Chem. 1997;272(38):23843–23850. doi: 10.1074/jbc.272.38.23843. [DOI] [PubMed] [Google Scholar]
- Guettouche T, Boellmann F, et al. Analysis of phosphorylation of human heat shock factor 1 in cells experiencing a stress. BMC Biochem. 2005;6:4. doi: 10.1186/1471-2091-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadziselimovic F, Hadziselimovic NO, et al. Piwi-pathway alteration induces LINE-1 transposon derepression and infertility development in cryptorchidism. Sex Dev. 2015;9(2):98–104. doi: 10.1159/000375351. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hashida S, Yamamoto H, et al. HSP90 inhibitor NVP-AUY922 enhances the radiation sensitivity of lung cancer cell lines with acquired resistance to EGFR-tyrosine kinase inhibitors. Oncol Rep. 2015;33(3):1499–1504. doi: 10.3892/or.2015.3735. [DOI] [PubMed] [Google Scholar]
- Hiley CT, Swanton C. Spatial and temporal cancer evolution: causes and consequences of tumour diversity. Clin Med. 2014;14(6):14–16. doi: 10.7861/clinmedicine.14-6-s33. [DOI] [PubMed] [Google Scholar]
- Hill VK, Ricketts C, et al. Genome-wide DNA methylation profiling of CpG islands in breast cancer identifies novel genes associated with tumorigenicity. Cancer Res. 2011;71(8):2988–2999. doi: 10.1158/0008-5472.CAN-10-4026. [DOI] [PubMed] [Google Scholar]
- Hingorani SR, Wang L, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7(5):469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- Holmberg CI, Tran SE, et al. Multisite phosphorylation provides sophisticated regulation of transcription factors. Trends Biochem Sci. 2002;27(12):619–627. doi: 10.1016/S0968-0004(02)02207-7. [DOI] [PubMed] [Google Scholar]
- Huang W, Wu QD, et al. Novel HSP90 inhibitor FW-04-806 displays potent antitumor effects in HER2-positive breast cancer cells as a single agent or in combination with lapatinib. Cancer Lett. 2015;356(2 Pt B):862–871. doi: 10.1016/j.canlet.2014.10.040. [DOI] [PubMed] [Google Scholar]
- Hubbard, S. (2014). Exelixis announces positive preliminary data from an investigator-sponsored phase 1 trial of XL888 and vemurafenib. from http://finance.yahoo.com/news/exelixis-announces-positive-preliminary-data-092000599.html
- Hudziak RM, Schlessinger J, et al. Increased expression of the putative growth factor receptor p185HER2 causes transformation and tumorigenesis of NIH 3T3 cells. Proc Natl Acad Sci U S A. 1987;84(20):7159–7163. doi: 10.1073/pnas.84.20.7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichiyanagi T, Ichiyanagi K, et al. HSP90 alpha plays an important role in piRNA biogenesis and retrotransposon repression in mouse. Nucleic Acids Res. 2014;42(19):11903–11911. doi: 10.1093/nar/gku881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi N, Kawaoka S, et al. HSP90 facilitates accurate loading of precursor piRNAs into Piwi proteins. RNA. 2013;19(7):896–901. doi: 10.1261/rna.037200.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhaveri K, Taldone T, et al. Advances in the clinical development of heat shock protein 90 (HSP90) inhibitors in cancers. Biochim Biophys Acta. 2012;3:742–755. doi: 10.1016/j.bbamcr.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ML, Yu HA et al (2015) Phase I/II study of HSP90 Inhibitor AUY922 and erlotinib for EGFR-mutant lung cancer with acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors. J Clin Oncol 13(59) [DOI] [PMC free article] [PubMed]
- Kamal A, Thao L, et al. A high-affinity conformation of HSP90 confers tumour selectivity on HSP90 inhibitors. Nature. 2003;425(6956):407–410. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- Kaplan AL, Litwin MS, et al. The future of bladder cancer care in the USA. Nat Rev Urol. 2014;11(1):59–62. doi: 10.1038/nrurol.2013.180. [DOI] [PubMed] [Google Scholar]
- Katayama R, Khan TM, et al. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci U S A. 2011;108(18):7535–7540. doi: 10.1073/pnas.1019559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinge CM (2015) miRNAs regulated by estrogens, tamoxifen, and endocrine disruptors and their downstream gene targets. Mol Cell Endocrinol [DOI] [PMC free article] [PubMed]
- Knight ZA, Shokat KM. Features of selective kinase inhibitors. Chem Biol. 2005;12(6):621–637. doi: 10.1016/j.chembiol.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Konicek BW, Dumstorf CA, et al. Targeting the eIF4F translation initiation complex for cancer therapy. Cell Cycle. 2008;7(16):2466–2471. doi: 10.4161/cc.7.16.6464. [DOI] [PubMed] [Google Scholar]
- Lachowiec J, Lemus T, et al. HSP90 promotes kinase evolution. Mol Biol Evol. 2015;32(1):91–99. doi: 10.1093/molbev/msu270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AJ, Endesfelder D, et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 2011;71(5):1858–1870. doi: 10.1158/0008-5472.CAN-10-3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YY, Kim HP, et al. Phosphoproteomic analysis identifies activated MET-axis PI3K/AKT and MAPK/ERK in lapatinib-resistant cancer cell line. Exp Mol Med. 2013;22(45):115. doi: 10.1038/emm.2013.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerdrup M, Hommelgaard AM, et al. Geldanamycin stimulates internalization of ErbB2 in a proteasome-dependent way. J Cell Sci. 2006;119(Pt 1):85–95. doi: 10.1242/jcs.02707. [DOI] [PubMed] [Google Scholar]
- Levinson AD, Oppermann H, et al. Evidence that the transforming gene of avian sarcoma virus encodes a protein kinase associated with a phosphoprotein. Cell. 1978;15(2):561–572. doi: 10.1016/0092-8674(78)90024-7. [DOI] [PubMed] [Google Scholar]
- Linehan WM, Rouault TA. Molecular pathways: fumarate hydratase-deficient kidney cancer—targeting the Warburg effect in cancer. Clin Cancer Res. 2013;19(13):3345–3352. doi: 10.1158/1078-0432.CCR-13-0304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Lu J, et al. Targeting heat-shock protein 90 with ganetespib for molecularly targeted therapy of gastric cancer. Cell Death Dis. 2015;15(6):555. doi: 10.1038/cddis.2014.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Wang L, et al. HSP90 inhibitors and the reduction of anti-cancer drug resistance by non-genetic and genetic mechanisms. Pharmaceuticals (Basel) 2012;5(9):890–898. doi: 10.3390/ph5090890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Xiao L, et al. HSP90 inhibitors and drug resistance in cancer: the potential benefits of combination therapies of HSP90 inhibitors and other anti-cancer drugs. Biochem Pharmacol. 2012;83(8):995–1004. doi: 10.1016/j.bcp.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Zeng S, et al. The c-KIT mutation causing human mastocytosis is resistant to STI571 and other KIT kinase inhibitors; kinases with enzymatic site mutations show different inhibitor sensitivity profiles than wild-type kinases and those with regulatory-type mutations. Blood. 2002;99(5):1741–1744. doi: 10.1182/blood.V99.5.1741. [DOI] [PubMed] [Google Scholar]
- Manjarrez JR, Sun L et al (2014) HSP90-dependent assembly of the DBC2/RhoBTB2-Cullin3 E3-ligase complex. PLoS One 9(3) [DOI] [PMC free article] [PubMed]
- Manning G, Whyte DB, et al. The protein kinase complement of the human genome. Science. 2002;298(5600):1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Maroun CR, Rowlands T. The MET receptor tyrosine kinase: a key player in oncogenesis and drug resistance. Pharmacol Ther. 2014;142(3):316–338. doi: 10.1016/j.pharmthera.2013.12.014. [DOI] [PubMed] [Google Scholar]
- Mendillo ML, Santagata S, et al. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell. 2012;150(3):549–562. doi: 10.1016/j.cell.2012.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methot SP, Litzler LC, et al. Consecutive interactions with HSP90 and eEF1A underlie a functional maturation and storage pathway of AID in the cytoplasm. J Exp Med. 2015;212(4):581–596. doi: 10.1084/jem.20141157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyajima N, Tsutsumi S, et al. The HSP90 inhibitor ganetespib synergizes with the MET kinase inhibitor crizotinib in both crizotinib-sensitive and -resistant MET-driven tumor models. Cancer Res. 2013;73(23):7022–7033. doi: 10.1158/0008-5472.CAN-13-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata Y, Nakamoto H, et al. The therapeutic target HSP90 and cancer hallmarks. Curr Pharm Des. 2013;19(3):347–365. doi: 10.2174/138161213804143725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moses MA, Henry EC, et al. The heat shock protein 90 inhibitor, (-)-epigallocatechin gallate, has anticancer activity in a novel human prostate cancer progression model. Cancer Prev Res. 2015;8(3):249–257. doi: 10.1158/1940-6207.CAPR-14-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulick K, Ahn JH, et al. Affinity-based proteomics reveal cancer-specific networks coordinated by HSP90. Nat Chem Biol. 2011;7(11):818–826. doi: 10.1038/nchembio.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata Y, Anan T, et al. The stabilization mechanism of mutant-type p53 by impaired ubiquitination: the loss of wild-type p53 function and the HSP90 association. Oncogene. 1999;18(44):6037–6049. doi: 10.1038/sj.onc.1202978. [DOI] [PubMed] [Google Scholar]
- Nagatsuma AK, Aizawa M et al (2014) Expression profiles of HER2, EGFR, MET and FGFR2 in a large cohort of patients with gastric adenocarcinoma. Gastric Cancer [DOI] [PubMed]
- Neckers L, Trepel JB. Stressing the development of small molecules targeting HSP90. Clin Cancer Res. 2014;20(2):275–277. doi: 10.1158/1078-0432.CCR-13-2571. [DOI] [PubMed] [Google Scholar]
- Neckers L, Workman P. HSP90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res. 2012;18(1):64–76. doi: 10.1158/1078-0432.CCR-11-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nony P, Gaude H, et al. Stability of the Peutz-Jeghers syndrome kinase LKB1 requires its binding to the molecular chaperones HSP90/Cdc37. Oncogene. 2003;22(57):9165–9175. doi: 10.1038/sj.onc.1207179. [DOI] [PubMed] [Google Scholar]
- O’Connell BC, O’Callaghan K et al (2014) HSP90 inhibition enhances antimitotic drug-induced mitotic arrest and cell death in preclinical models of non-small cell lung cancer. PLoS One 9(12) [DOI] [PMC free article] [PubMed]
- Ohkubo S, Kodama Y, et al. TAS-116, a highly selective inhibitor of heat shock protein 90 alpha and beta, demonstrates potent antitumor activity and minimal ocular toxicity in preclinical models. Mol Cancer Ther. 2015;14(1):14–22. doi: 10.1158/1535-7163.MCT-14-0219. [DOI] [PubMed] [Google Scholar]
- Ou SH, Kwak EL, et al. Activity of crizotinib (PF02341066), a dual mesenchymal-epithelial transition (MET) and anaplastic lymphoma kinase (ALK) inhibitor, in a non-small cell lung cancer patient with de novo MET amplification. J Thorac Oncol. 2011;6(5):942–946. doi: 10.1097/JTO.0b013e31821528d3. [DOI] [PubMed] [Google Scholar]
- Pao W, Miller VA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2(3):22. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier J, Graff J, et al. Targeting the eIF4F translation initiation complex: a critical nexus for cancer development. Cancer Res. 2015;75(2):250–263. doi: 10.1158/0008-5472.CAN-14-2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell E, Piwnica-Worms D, et al. Contribution of p53 to metastasis. Cancer Discov. 2014;4(4):405–414. doi: 10.1158/2159-8290.CD-13-0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the HSP90/HSP70-based chaperone machinery. Exp Biol Med. 2003;228(2):111–133. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- Prince T, Matts RL. Definition of protein kinase sequence motifs that trigger high affinity binding of HSP90 and Cdc37. J Biol Chem. 2004;279(38):39975–39981. doi: 10.1074/jbc.M406882200. [DOI] [PubMed] [Google Scholar]
- Prodromou C. Strategies for stalling malignancy: targeting cancer’s addiction to HSP90. Curr Top Med Chem. 2009;9(15):1352–1368. doi: 10.2174/156802609789895656. [DOI] [PubMed] [Google Scholar]
- Prodromou C, Roe SM, et al. Identification and structural characterization of the ATP/ADP-binding site in the HSP90 molecular chaperone. Cell. 1997;90(1):65–75. doi: 10.1016/S0092-8674(00)80314-1. [DOI] [PubMed] [Google Scholar]
- Ricketts CJ, Hill VK et al (2014) Tumor-specific hypermethylation of epigenetic biomarkers, including SFRP1, predicts for poorer survival in patients from the TCGA Kidney Renal Clear Cell Carcinoma (KIRC) project. PLoS One 9(1) [DOI] [PMC free article] [PubMed]
- Roskoski, R. (2015). “http://www.brimr.org/.” from http://www.brimr.org/PKI/PKIs.htm
- Rutherford SL, Lindquist S. HSP90 as a capacitor for morphological evolution. Nature. 1998;396(6709):336–342. doi: 10.1038/24550. [DOI] [PubMed] [Google Scholar]
- Sang J, Acquaviva J, et al. Targeted inhibition of the molecular chaperone HSP90 overcomes ALK inhibitor resistance in non-small cell lung cancer. Cancer Discov. 2013;3(4):430–443. doi: 10.1158/2159-8290.CD-12-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santagata S, Mendillo ML, et al. Tight coordination of protein translation and HSF1 activation supports the anabolic malignant state. Science. 2013;341(6143):1238303. doi: 10.1126/science.1238303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequist LV, Gettinger S, et al. Activity of IPI-504, a novel heat-shock protein 90 inhibitor, in patients with molecularly defined non-small-cell lung cancer. J Clin Oncol. 2010;28(33):4953–4960. doi: 10.1200/JCO.2010.30.8338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessa C, Shapiro GI, et al. First-in-human phase I dose-escalation study of the HSP90 inhibitor AUY922 in patients with advanced solid tumors. Clin Cancer Res. 2013;19(13):3671–3680. doi: 10.1158/1078-0432.CCR-12-3404. [DOI] [PubMed] [Google Scholar]
- Shao J, Grammatikakis N, et al. HSP90 regulates p50(cdc37) function during the biogenesis of the active conformation of the heme-regulated eIF2 alpha kinase. J Biol Chem. 2001;276(1):206–214. doi: 10.1074/jbc.M007583200. [DOI] [PubMed] [Google Scholar]
- Shattuck DL, Miller JK, et al. Met receptor contributes to trastuzumab resistance of Her2-overexpressing breast cancer cells. Cancer Res. 2008;68(5):1471–1477. doi: 10.1158/0008-5472.CAN-07-5962. [DOI] [PubMed] [Google Scholar]
- Shetzer Y, Solomon H, et al. The paradigm of mutant p53-expressing cancer stem cells and drug resistance. Carcinogenesis. 2014;35(6):1196–1208. doi: 10.1093/carcin/bgu073. [DOI] [PubMed] [Google Scholar]
- Shimamura T, Lowell AM, et al. Epidermal growth factor receptors harboring kinase domain mutations associate with the heat shock protein 90 chaperone and are destabilized following exposure to geldanamycins. Cancer Res. 2005;65(14):6401–6408. doi: 10.1158/0008-5472.CAN-05-0933. [DOI] [PubMed] [Google Scholar]
- Shimamura T, Li D, et al. HSP90 inhibition suppresses mutant EGFR-T790M signaling and overcomes kinase inhibitor resistance. Cancer Res. 2008;68(14):5827–5838. doi: 10.1158/0008-5472.CAN-07-5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjoblom T, Jones S, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314(5797):268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- Solarova Z, Mojzis J, et al. HSP90 inhibitor as a sensitizer of cancer cells to different therapies (review) Int J Oncol. 2015;46(3):907–926. doi: 10.3892/ijo.2014.2791. [DOI] [PubMed] [Google Scholar]
- Solier S, Kohn KW, et al. Heat shock protein 90 alpha (HSP90 alpha), a substrate and chaperone of DNA-PK necessary for the apoptotic response. Proc Natl Acad Sci U S A. 2012;109(32):12866–12872. doi: 10.1073/pnas.1203617109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollars V, Lu X, et al. Evidence for an epigenetic mechanism by which HSP90 acts as a capacitor for morphological evolution. Nat Genet. 2003;33(1):70–74. doi: 10.1038/ng1067. [DOI] [PubMed] [Google Scholar]
- Specchia V, Piacentini L, et al. HSP90 prevents phenotypic variation by suppressing the mutagenic activity of transposons. Nature. 2010;463(7281):662–665. doi: 10.1038/nature08739. [DOI] [PubMed] [Google Scholar]
- Suzuki R, Hideshima T, et al. Anti-tumor activities of selective HSP90 alpha/beta inhibitor, TAS-116, in combination with bortezomib in multiple myeloma. Leukemia. 2015;29(2):510–514. doi: 10.1038/leu.2014.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale M, Jarosz DF, et al. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010;11(7):515–528. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- Taipale M, Krykbaeva I, et al. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell. 2012;150(5):987–1001. doi: 10.1016/j.cell.2012.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale M, Tucker G, et al. A quantitative chaperone interaction network reveals the architecture of cellular protein homeostasis pathways. Cell. 2014;158(2):434–448. doi: 10.1016/j.cell.2014.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taldone T, Ochiana SO, et al. Selective targeting of the stress chaperome as a therapeutic strategy. Trends Pharmacol Sci. 2014;35(11):592–603. doi: 10.1016/j.tips.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Dai S, et al. MEK guards proteome stability and inhibits tumor-suppressive amyloidogenesis via HSF1. Cell. 2015;160(4):729–744. doi: 10.1016/j.cell.2015.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauchi T, Okabe S, et al. Combined effects of novel heat shock protein 90 inhibitor NVP-AUY922 and nilotinib in a random mutagenesis screen. Oncogene. 2011;30(24):2789–2797. doi: 10.1038/onc.2011.3. [DOI] [PubMed] [Google Scholar]
- Tonini G. Trends in the early investigational drug development and areas for improvement. Expert Opin Investig Drugs. 2015;20:1–6. doi: 10.1517/13543784.2015.1037881. [DOI] [PubMed] [Google Scholar]
- Trepel J, Mollapour M, et al. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010;10(8):537–549. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidya S, Vundinti BR et al (2015) Evolution of BCR/ABL gene mutation in CML is time dependent and dependent on the pressure exerted by tyrosine kinase inhibitor. PLoS One 10(1) [DOI] [PMC free article] [PubMed]
- Wachsberger PR, Lawrence YR, et al. HSP90 inhibition enhances PI-3 kinase inhibition and radiosensitivity in glioblastoma. J Cancer Res Clin Oncol. 2014;140(4):573–582. doi: 10.1007/s00432-014-1594-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walerych D, Kudla G, et al. HSP90 chaperones wild-type p53 tumor suppressor protein. J Biol Chem. 2004;279(47):48836–48845. doi: 10.1074/jbc.M407601200. [DOI] [PubMed] [Google Scholar]
- Walerych D, Napoli M, et al. The rebel angel: mutant p53 as the driving oncogene in breast cancer. Carcinogenesis. 2012;33(11):2007–2017. doi: 10.1093/carcin/bgs232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell. 2012;21(3):297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward A, Shukla K, et al. MicroRNA-519a is a novel oncomir conferring tamoxifen resistance by targeting a network of tumour-suppressor genes in ER+ breast cancer. J Pathol. 2014;233(4):368–379. doi: 10.1002/path.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5(10):761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- Whitesell L, Mimnaugh EG, et al. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci U S A. 1994;91(18):8324–8328. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitesell L, Santagata S, et al. HSP90 empowers evolution of resistance to hormonal therapy in human breast cancer models. Proc Natl Acad Sci U S A. 2014;111(51):18297–18302. doi: 10.1073/pnas.1421323111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AJ, Bigner SH, et al. Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplification. Proc Natl Acad Sci U S A. 1987;84(19):6899–6903. doi: 10.1073/pnas.84.19.6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman P, Burrows F, et al. Drugging the cancer chaperone HSP90: combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann N Y Acad Sci. 2007;1113:202–216. doi: 10.1196/annals.1391.012. [DOI] [PubMed] [Google Scholar]
- Xiao L, Rasouli P, et al. Possible effects of early treatments of HSP90 inhibitors on preventing the evolution of drug resistance to other anti-cancer drugs. Curr Med Chem. 2007;14(2):223–232. doi: 10.2174/092986707779313372. [DOI] [PubMed] [Google Scholar]
- Xu W, Marcu M, et al. Chaperone-dependent E3 ubiquitin ligase CHIP mediates a degradative pathway for c-ErbB2/Neu. Proc Natl Acad Sci U S A. 2002;99(20):12847–12852. doi: 10.1073/pnas.202365899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Yuan X, et al. Surface charge and hydrophobicity determine ErbB2 binding to the HSP90 chaperone complex. Nat Struct Mol Biol. 2005;12(2):120–126. doi: 10.1038/nsmb885. [DOI] [PubMed] [Google Scholar]
- Yu HA, Riely GJ, et al. Therapeutic strategies utilized in the setting of acquired resistance to EGFR tyrosine kinase inhibitors. Clin Cancer Res. 2014;20(23):5898–5907. doi: 10.1158/1078-0432.CCR-13-2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27(41):5497–5510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao RX, Xu ZX. Targeting the LKB1 tumor suppressor. Curr Drug Targets. 2014;15(1):32–52. doi: 10.2174/1389450114666140106095811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R, Davey M, et al. Navigating the chaperone network: an integrative map of physical and genetic interactions mediated by the HSP90 chaperone. Cell. 2005;120(5):715–727. doi: 10.1016/j.cell.2004.12.024. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Polley EC, et al. GeneMed: an informatics hub for the coordination of next-generation sequencing studies that support precision oncology clinical trials. Cancer Inform. 2015;14(Suppl 2):45–55. doi: 10.4137/CIN.S17282. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLSX 92 kb)