

Abstract
Numerous studies conducted on obese humans and various rodent models of obesity have identified a correlation between hepatic lipid content and the development of insulin resistance in liver and other tissues. Despite a large body of the literature on this topic, the cause and effect relationship between hepatic steatosis and insulin resistance remains controversial. If, as many believe, lipid aggregation in liver drives insulin resistance and other metabolic abnormalities, there are significant unanswered questions as to which lipid mediators are causative in this cascade. Several published papers have now correlated levels of diacylglycerol (DAG), the penultimate intermediate in triglyceride synthesis, with development of insulin resistance and have postulated that this occurs via activation of protein kinase C signaling. Although many studies have confirmed this relationship, many others have reported a disconnect between DAG content and insulin resistance. It has been postulated that differences in methods for DAG measurement, DAG compartmentalization within the cell, or fatty acid composition of the DAG may explain these discrepancies. The purpose of this review is to compare and contrast some of the relevant findings in this area and to discuss a number of unanswered questions regarding the relationship between DAG and insulin resistance.
1. Introduction
Hepatic insulin resistance, lipid accumulation, and inflammation seem to be tightly interconnected. Indeed, strong correlations among these variables have been detected in obese human subjects and in studies conducted on a variety of mouse and rat models. However, the cause and effect relationship among these factors is not always clear and usually difficult to discern [1]. Furthermore, the largest genetic predictors of NAFLD are not always associated with hepatic insulin resistance [2, 3]. Experimental approaches to modulate the abundance of a given lipid unavoidably lead to changes in the abundance of other interconverted lipids such that manipulating the concentrations of one lipid in isolation seems impossible. Furthermore, difficulties in measuring the abundance of lipids present at very low levels and reproducibility across different model systems have raised questions regarding whether a specific lipid can be causally linked to development of insulin resistance. Whereas several lipids accumulate in steatotic liver, this review will focus on diacylglycerol (DAG), the evidence linking it to insulin resistance, and the controversy surrounding this linkage.
On the surface, DAG is a simple hydrophobic lipid that is normally a component of cellular membranes or is stored in lipid droplets. DAG is composed of a glycerol backbone and two fatty acyl groups. However, the biophysical properties and the physiological effects of DAG can be strongly influenced by the composition of the fatty acyl groups and its physical location within the cell. For example, acyl moieties can be esterified at either the sn-1,2 or the sn-1,3 positions of glycerol depending upon the pathway used to generate the DAG molecule. These two stereoisomers have different biophysical properties in membranes and previous work has shown that the sn-1,2 stereoisomer is much more potent, compared to sn-1,3-DAG, at activating certain signaling cascades linked to insulin resistance [4]. Accumulation of DAG containing saturated fatty acids has also been linked to development of insulin resistance. There is also correlative evidence that the abundance of DAG in the various intracellular compartments (membrane versus lipid droplet) can be more strongly associated with insulin resistance [5, 6].
2. Connections between DAG and Insulin Resistance
Some of the original work correlating tissue DAG concentrations to insulin resistance was conducted in obese rats almost 25 years ago [7]. Based on previous work showing that phorbol ester, a DAG analog, could impair insulin action, Turinsky and colleagues hypothesized that endogenous DAG might be increased in insulin-resistant rodents. Measurement of DAG in obese Zucker rat tissues revealed that 1,2-DAG was elevated in multiple tissues in this model of type 2 diabetes. Since then, elevated DAG has been correlated to impaired insulin action in a variety of studies [8–13]. Ectopic accumulation of DAG in liver may be due to a variety of factors including consumption of a high fat or high sugar diet, inability of adipose tissue to appropriately store lipids leading to elevated circulating free fatty acids [14–16], or effects of oxidative stress in the liver causing DAG formation [17–19]. Because many lipids accumulate ectopically in obesity, which is correlated with insulin resistance, a variety of lipid species can be correlated with insulin resistance in obese animal models or humans. However, much of the recent attention in this area has focused on DAG due to the identification of clear mechanisms linking DAG to impaired insulin signaling.
Specifically, DAG has been shown in a variety of model systems to activate protein kinase C (PKC) family kinases, which physically interact with membrane-embedded DAG [20]. In hepatocytes or intact liver, links among DAG accumulation, PKC activation, and impaired insulin action have been made for PKCε [21] and PKCδ [22]. The mechanism of PKCε inhibition of insulin action was mediated via a direct interaction of PKCε with the insulin receptor to inhibit its intrinsic kinase activity [21] (Figure 1). The link between PKCε activation and hepatic insulin resistance is supported in correlative fashion by several papers [12, 13, 16, 23, 24] and in more convincing fashion by “knocking down” PKCε in liver by RNAi [21]. PKCε knockout mice exhibit improved glycemic control on a high fat diet, but this is likely mediated via enhanced insulin secretion [25]. PKCδ is also activated in steatotic liver [22, 25] and PKCδ knockout mice are protected from high fat diet induced hepatic steatosis while PKCδ overexpression was sufficient to drive insulin resistance [22].
Figure 1.
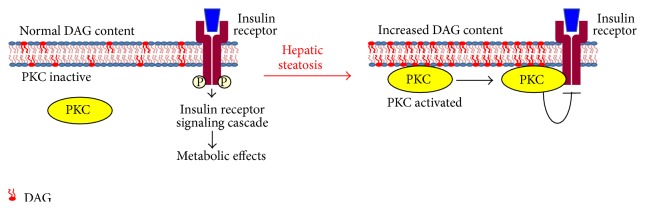
Proposed mechanism for DAG-mediated insulin resistance through activation of PKC is shown.
As delineated above, numerous studies have correlated altered DAG concentrations to PKC activation and insulin resistance. However, it should be noted that several notable exceptions to these correlations have been detected where DAG accumulation in liver was not associated with development of insulin resistance [26–33]. One interpretation of these data is that DAG elevation is not, per se, sufficient to cause insulin resistance. The caveat to this conclusion is that it is not always clear whether all species, stereoisomers, or subcellular compartments are affected similarly. Could a change in the ratio or absolute amounts of sn-1,2 and sn-1,3 affect downstream signaling cascade activity? Similarly, could the chain length and degree of saturation also impact interpretation of these findings? Lastly, the subcellular compartmentalization of DAG has been reported to impact whether DAG accumulation drives insulin resistance or not [5, 6].
Though a large number of studies have examined the correlation between DAG and insulin resistance, this review is going to focus primarily on data generated by targeting enzymes that directly synthesize or degrade DAG. These studies were mostly conducted in animal models with gene deletion, gene expression knockdown, or overexpression. As discussed below, there are multiple enzymatic reactions involving glycerolipid and phosphoglycerolipid substrates that can result in DAG synthesis. The compartmentalization of many of these pathways, the substrate used, and the subcellular location where the reaction occurs could influence the resulting effect on metabolism and signaling. Moreover, despite the focus on enzymes that directly regulate DAG synthesis or turnover, this is not to say that other lipids derived from DAG or substrates for DAG synthesis are not affected. Indeed, as noted above, it is practically impossible to affect the concentration of one lipid in isolation.
3. Mechanisms for DAG Synthesis
In the liver, one of the primary pathways for synthesizing DAG is from the dephosphorylation of ER membrane-embedded phosphatidic acid (PA) by the lipin family of proteins (lipin 1, lipin 2, and lipin 3) (Figure 2) [34, 35]. This pathway can only produce 1,2-DAG since PA is phosphorylated at the sn-3 position. Evidence exists that both lipin 1 and lipin 2 encode significant hepatic PAP activity [36] and may play a role in development of NAFLD and related metabolic abnormalities. Acute adenoviral-mediated knockdown of lipin 1 or lipin 2 was shown to reduce hepatic DAG, PKCε activation, and associated insulin resistance [37, 38]. However, when hepatic steatosis was examined in liver-specific lipin 1 knockout mice fed a diet containing high amounts of ethanol, alcoholic hepatic steatosis and liver diseases were exacerbated by lipin 1 deficiency [39]. Similarly, we have recently found that liver-specific lipin 1 knockout mice are not protected from hepatic steatosis and insulin resistance after high fat diet (our unpublished results). It is not clear whether these discordant results between knockout mouse studies and RNAi approaches are due to duration of lipin inhibition, chronic compensatory mechanisms, or some other experimental differences.
Figure 2.
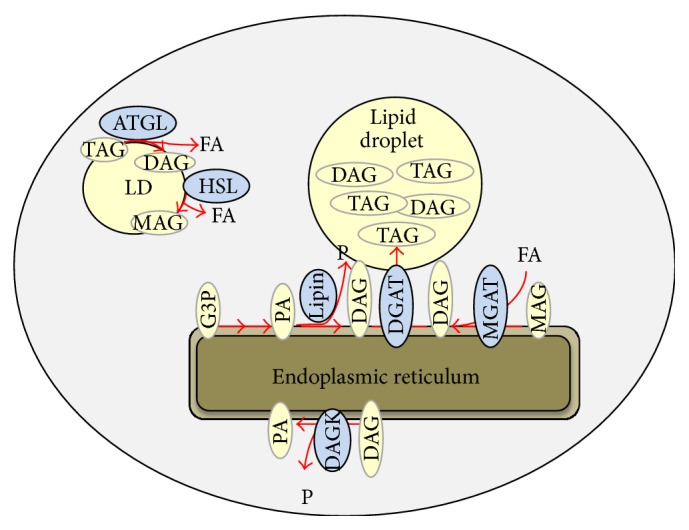
The pathways for DAG synthesis and hydrolysis are shown. FA: fatty acid, P: phosphate, G-3-P: glycerol-3-phosphate, PA: phosphatidic acid (PA), MAG: monoacylglycerol, MGAT: MAG acyltransferase, DAG: diacylglycerol, DGAT: DAG acyltransferase, TAG: triacylglycerol, ATGL: adipose tissue triglyceride lipase, HSL: hormone sensitive lipase, and DAGK: DAG kinase.
Monoacylglycerol acyltransferase enzymes (MGAT1, MGAT2, and MGAT3) also generate DAG by acylating monoacylglycerol (Figure 1), and both sn-1,2 and sn-1,3 DAG can be synthesized by MGAT enzymes. Recent work has suggested that the expression of genes encoding MGATs (Mogats) is markedly induced in human patients with NAFLD [40] as well as rodent models of obesity [29, 41]. Mogat1 knockdown or Mogat2 KO in mice led to a reversal or prevention of insulin resistance in high fat diet fed mice [29, 41–43]. Interestingly, Mogat1 knockdown in diet-induced obese mice, which caused a marked insulin sensitization, did not affect hepatic DAG content or compartmentalization [29, 42]. Despite this, membrane-associated PKC activity was reduced by Mogat1 knockdown, with the caveat that the PKC activity was not increased by the high fat diet compared to low fat controls [29].
Adipose tissue triglyceride lipase (ATGL) is a major hepatic triglyceride lipase [44]. Genetic deficiency in ATGL leads to ectopic lipid accumulation, due to the inability to mobilize stored triglycerides, in a number of tissues including the liver [28, 31]. ATGL deficiency led to hepatic steatosis, but this was not associated with development of hepatic insulin resistance, inflammation, or fibrosis [28, 31, 32, 45], despite the accumulation of DAG [31]. ATGL activity is also controlled by an enhancer protein called CGI-58 [46]. Knockdown of CGI-58 also resulted in accumulation of hepatic lipids, including DAG, but this did not cause insulin resistance in high fat diet fed mice [26]. A follow-up study concluded that loss of CGI-58 caused accumulation of DAG specifically in lipid droplets rather than ectopically in cell membranes, which prevented activation of PKCε signaling [6]. However, this contradicted another previous study by the same group showing a strong correlation between lipid droplet DAG content and insulin resistance in human liver [5]. These contradictory findings have not been reconciled.
4. Mechanisms for DAG Degradation
There are multiple enzymes that convert DAG to other chemical forms. This can be accomplished by addition or removal of a fatty acyl molecule or addition of a phosphate group. The effects of some of these pathways have now been examined by using transgenic mouse systems or RNAi methodology.
The terminal step in triglyceride synthesizes the diacylglycerol acyltransferases (DGAT1 and DGAT2). DGATs are well expressed in liver and have been targeted for gene deletion or knockdown by a number of studies. DGAT1 inhibition did not affect insulin sensitivity in high fat diet fed rats, while DGAT2 knockdown reduced hepatic lipid accumulation and improved hepatic and whole body insulin sensitivity [47]. The improvement in insulin sensitivity was correlated with a reduction in hepatic content of DAG and a corresponding reduction in PKCε activity [47]. Liver-specific overexpression of DGAT2 in transgenic mice somewhat surprisingly led to an accumulation of DAG and TAG but, interestingly, did not affect insulin sensitivity [27]. Subsequent analyses of these mice contradicted this and suggested that hepatic insulin sensitivity was impaired [11]. The discrepant results between the two studies have not yet been explained. It is also unclear why DGAT deficiency and overexpression had paradoxical effects on DAG content, though an unexpected increase in DAG was also observed with Mogat1 inhibition [29].
Hydrolysis of a fatty acyl group from DAG by fatty acid lipases is another way to degrade DAG to other chemical forms. Two genes encoding DAG lipases (Dagla and Daglb) have been cloned, but their role in the liver and in hepatic lipid homeostasis seems to be unknown. Hormone sensitive lipase (HSL) was once considered the primary triglyceride hydrolase but is now considered to be primarily a DAG lipase. HSL deficient mice exhibit increased hepatic insulin sensitivity with reduced hepatic triglyceride content [30, 48], while adenoviral-mediated overexpression of HSL also reduced hepatic steatosis [49]. It is not clear whether hepatic DAG content was affected by HSL loss or gain of function, and thus the evidence provided by these studies may not inform us about the linkage between DAG and insulin resistance.
DAG phosphorylation by DAG kinase to produce PA is another mechanism by which DAG concentrations could be affected. Whereas DAG kinase δ (DAGKδ) activity in skeletal muscle has been linked to obesity-related insulin resistance, no effect of diminished DAGKδ activity in liver was detected [50]. Could this mean that DAGKδ is not well expressed in liver or that another isoform of this family, of which there are many, could be the predominant form in liver? This has not been explored to our knowledge and it is not clear which, if any, DAGK family members are highly expressed in liver. Future studies may address this question.
5. Conclusions
The review of the relevant literature focused on enzymes that directly synthesize or metabolize DAG reveals a pattern of findings that is extremely mixed. Whereas some of the studies support a link between altered DAG content and insulin resistance through PKCs, other works fail to find a relationship. Again, there is an important limitation to interpreting data from this area; all of the generated data are essentially correlative. It is also unclear how the proposed mechanism for PKCε-mediated impairment in insulin action through inhibiting insulin receptor phosphorylation fits with the concept of selective insulin resistance [51]. Selective insulin resistance refers to the observation that although insulin-mediated suppression of gluconeogenic pathways is impaired in insulin-resistant liver, another pathway that stimulates de novo lipogenesis through the sterol response element binding protein (SREBP1) remains intact [51–54]. Though there are somewhat contradictory findings regarding at which step in the bifurcating insulin signaling cascades the selective insulin resistance occurs [52–54], it is generally believed to be downstream of the insulin receptor. Therefore, it is unclear how the PKCε-mediated impingement on insulin receptor activity fits with this widely observed concept and meshes into the broader model of hepatic insulin resistance. Future work will be needed to address these discrepancies and reconcile existing inconsistencies.
Acknowledgments
This work was supported by NIH grants K01-DK087821 to Angela M. Hall and R01-DK078187 and R42-AA021228 to Brian N. Finck. The authors would like to thank all members of the lab, past and present, that contributed to this work with helpful discussions.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Farese R. V., Jr., Zechner R., Newgard C. B., Walther T. C. The problem of establishing relationships between hepatic steatosis and hepatic insulin resistance. Cell Metabolism. 2012;15(5):570–573. doi: 10.1016/j.cmet.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou Y., Llauradó G., Orešič M., Hyötyläinen T., Orho-Melander M., Yki-Järvinen H. Circulating triacylglycerol signatures and insulin sensitivity in NAFLD associated with the E167K variant in TM6SF2. Journal of Hepatology. 2014 doi: 10.1016/j.jhep.2014.10.010. [DOI] [PubMed] [Google Scholar]
- 3.Sookoian S., Pirola C. J. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology. 2011;53(6):1883–1894. doi: 10.1002/hep.24283. [DOI] [PubMed] [Google Scholar]
- 4.Rando R. R., Young N. The stereospecific activation of protein kinase C. Biochemical and Biophysical Research Communications. 1984;122(2):818–823. doi: 10.1016/S0006-291X(84)80107-2. [DOI] [PubMed] [Google Scholar]
- 5.Kumashiro N., Erion D. M., Zhang D., et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(39):16381–16385. doi: 10.1073/pnas.1113359108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cantley J. L., Yoshimura T., Camporez J. P. G., et al. CGI-58 knockdown sequesters diacylglycerols in lipid droplets/ER-preventing diacylglycerol-mediated hepatic insulin resistance. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(5):1869–1874. doi: 10.1073/pnas.1219456110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turinsky J., O'Sullivan D. M., Bayly B. P. 1,2-diacylglycerol and ceramide levels in insulin-resistant tissues of the rat in vivo. Journal of Biological Chemistry. 1990;265(28):16880–16885. [PubMed] [Google Scholar]
- 8.Birkenfeld A. L., Lee H. Y., Guebre-Egziabher F., et al. Deletion of the mammalian INDY homolog mimics aspects of dietary restriction and protects against adiposity and insulin resistance in mice. Cell Metabolism. 2011;14(2):184–195. doi: 10.1016/j.cmet.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi S. C., Savage D. B., Abu-Elheiga L., et al. Continuous fat oxidation in acetyl-CoA carboxylase 2 knockout mice increases total energy expenditure, reduces fat mass, and improves insulin sensitivity. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(42):16480–16485. doi: 10.1073/pnas.0706794104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erion D. M., Ignatova I. D., Yonemitsu S., et al. Prevention of hepatic steatosis and hepatic insulin resistance by knockdown of cAMP response element-binding protein. Cell Metabolism. 2009;10(6):499–506. doi: 10.1016/j.cmet.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jornayvaz F. R., Birkenfeld A. L., Jurczak M. J., et al. Hepatic insulin resistance in mice with hepatic overexpression of diacylglycerol acyltransferase 2. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(14):5748–5752. doi: 10.1073/pnas.1103451108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neschen S., Morino K., Hammond L. E., et al. Prevention of hepatic steatosis and hepatic insulin resistance in mitochondrial acyl-CoA:glycerol-sn-3-phosphate acyltransferase 1 knockout mice. Cell Metabolism. 2005;2(1):55–65. doi: 10.1016/j.cmet.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 13.Savage D. B., Cheol S. C., Samuel V. T., et al. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. Journal of Clinical Investigation. 2006;116(3):817–824. doi: 10.1172/JCI27300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Samuel V. T., Liu Z.-X., Qu X., et al. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. Journal of Biological Chemistry. 2004;279(31):32345–32353. doi: 10.1074/jbc.M313478200. [DOI] [PubMed] [Google Scholar]
- 15.Donnelly K. L., Smith C. I., Schwarzenberg S. J., Jessurun J., Boldt M. D., Parks E. J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. Journal of Clinical Investigation. 2005;115(5):1343–1351. doi: 10.1172/JCI200523621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagai Y., Yonemitsu S., Erion D. M., et al. The role of peroxisome proliferator-activated receptor γ coactivator-1 β in the pathogenesis of fructose-induced insulin resistance. Cell Metabolism. 2009;9(3):252–264. doi: 10.1016/j.cmet.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakajima T., Yukawa O. Mechanism of radiation-induced diacylglycerol production in primary cultured rat hepatocytes. Journal of Radiation Research. 1999;40(2):135–144. doi: 10.1269/jrr.40.135. [DOI] [PubMed] [Google Scholar]
- 18.Toriumi K., Horikoshi Y., Yoshiyuki Osamura R., Yamamoto Y., Nakamura N., Takekoshi S. Carbon tetrachloride-induced hepatic injury through formation of oxidized diacylglycerol and activation of the PKC/NF-κB pathway. Laboratory Investigation. 2013;93(2):218–229. doi: 10.1038/labinvest.2012.145. [DOI] [PubMed] [Google Scholar]
- 19.Yoon S., Maruyama Y., Kazusaka A., Fujita S. Accumulation of diacylglycerol induced by CCl4-derived radicals in rat liver membrane and its inhibition with radical trapping reagent—FT-IR spectroscopic and HPLC chromatographic observations. Japanese Journal of Veterinary Research. 2000;47(3-4):135–144. [PubMed] [Google Scholar]
- 20.Takai Y., Kishimoto A., Kikkawa U., Mori T., Nishizuka Y. Unsaturated diacylglycerol as a possible messenger for the activation of calcium-activated, phospholipid-dependent protein kinase system. Biochemical and Biophysical Research Communications. 1979;91(4):1218–1224. doi: 10.1016/0006-291X(79)91197-5. [DOI] [PubMed] [Google Scholar]
- 21.Samuel V. T., Liu Z.-X., Wang A., et al. Inhibition of protein kinase Cε prevents hepatic insulin resistance in nonalcoholic fatty liver disease. The Journal of Clinical Investigation. 2007;117(3):739–745. doi: 10.1172/jci30400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bezy O., Tran T. T., Pihlajamäki J., et al. PKCδ regulates hepatic insulin sensitivity and hepatosteatosis in mice and humans. Journal of Clinical Investigation. 2011;121(6):2504–2517. doi: 10.1172/JCI46045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Erion D. M., Yonemitsu S., Nie Y., et al. SirT1 knockdown in liver decreases basal hepatic glucose production and increases hepatic insulin responsiveness in diabetic rats. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(27):11288–11293. doi: 10.1073/pnas.0812931106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang D., Liu Z.-X., Cheol S. C., et al. Mitochondrial dysfunction due to long-chain Acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(43):17075–17080. doi: 10.1073/pnas.0707060104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frangioudakis G., Burchfield J. G., Narasimhan S., et al. Diverse roles for protein kinase C δ and protein kinase C ε in the generation of high-fat-diet-induced glucose intolerance in mice: regulation of lipogenesis by protein kinase C δ . Diabetologia. 2009;52(12):2616–2620. doi: 10.1007/s00125-009-1543-0. [DOI] [PubMed] [Google Scholar]
- 26.Brown J. M., Betters J. L., Lord C., et al. CGI-58 knockdown in mice causes hepatic steatosis but prevents diet-induced obesity and glucose intolerance. Journal of Lipid Research. 2010;51(11):3306–3315. doi: 10.1194/jlr.M010256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Monetti M., Levin M. C., Watt M. J., et al. Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metabolism. 2007;6(1):69–78. doi: 10.1016/j.cmet.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 28.Hoy A. J., Bruce C. R., Turpin S. M., Morris A. J., Febbraio M. A., Watt M. J. Adipose triglyceride lipase-null mice are resistant to high-fat diet-induced insulin resistance despite reduced energy expenditure and ectopic lipid accumulation. Endocrinology. 2011;152(1):48–58. doi: 10.1210/en.2010-0661. [DOI] [PubMed] [Google Scholar]
- 29.Hall A. M., Soufi N., Chambers K. T., et al. Abrogating monoacylglycerol acyltransferase activity in liver improves glucose tolerance and hepatic insulin signaling in obese mice. Diabetes. 2014;63(7):2284–2296. doi: 10.2337/db13-1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Voshol P. J., Haemmerle G., Ouwens D. M., et al. Increased hepatic insulin sensitivity together with decreased hepatic triglyceride stores in hormone-sensitive lipase-deficient mice. Endocrinology. 2003;144(8):3456–3462. doi: 10.1210/en.2002-0036. [DOI] [PubMed] [Google Scholar]
- 31.Turpin S. M., Hoy A. J., Brown R. D., et al. Adipose triacylglycerol lipase is a major regulator of hepatic lipid metabolism but not insulin sensitivity in mice. Diabetologia. 2011;54(1):146–156. doi: 10.1007/s00125-010-1895-5. [DOI] [PubMed] [Google Scholar]
- 32.Ong K. T., Mashek M. T., Bu S. Y., Mashek D. G. Hepatic ATGL knockdown uncouples glucose intolerance from liver TAG accumulation. The FASEB Journal. 2013;27(1):313–321. doi: 10.1096/fj.12-213454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun Z., Miller R. A., Patel R. T., et al. Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nature Medicine. 2012;18(6):934–942. doi: 10.1038/nm.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Donkor J., Sariahmetoglu M., Dewald J., Brindley D. N., Reue K. Three mammalian lipins act as phosphatidate phosphatases with distinct tissue expression patterns. Journal of Biological Chemistry. 2007;282(6):3450–3457. doi: 10.1074/jbc.M610745200. [DOI] [PubMed] [Google Scholar]
- 35.Han G.-S., Wu W.-I., Carman G. M. The Saccharomyces cerevisiae lipin homolog is a Mg2+-dependent phosphatidate phosphatase enzyme. Journal of Biological Chemistry. 2006;281(14):9210–9218. doi: 10.1074/jbc.m600425200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gropler M. C., Harris T. E., Hall A. M., et al. Lipin 2 is a liver-enriched phosphatidate phosphohydrolase enzyme that is dynamically regulated by fasting andobesity in mice. The Journal of Biological Chemistry. 2009;284(11):6763–6772. doi: 10.1074/jbc.m807882200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ryu D., Oh K.-J., Jo H.-Y., et al. TORC2 regulates hepatic insulin signaling via a mammalian phosphatidic acid phosphatase, LIPIN1. Cell Metabolism. 2009;9(3):240–251. doi: 10.1016/j.cmet.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 38.Ryu D., Seo W.-Y., Yoon Y.-S., et al. Endoplasmic reticulum stress promotes LIPIN2-dependent hepatic insulin resistance. Diabetes. 2011;60(4):1072–1081. doi: 10.2337/db10-1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu M., Yin H., Mitra M. S., et al. Hepatic-specific lipin-1 deficiency exacerbates experimental alcohol-induced steatohepatitis in mice. Hepatology. 2013;58(6):1953–1963. doi: 10.1002/hep.26589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hall A. M., Kou K., Chen Z., et al. Evidence for regulated monoacylglycerol acyltransferase expression and activity in human liver. Journal of Lipid Research. 2012;53(5):990–999. doi: 10.1194/jlr.P025536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee Y. J., Ko E. H., Kim J. E., et al. Nuclear receptor PPARγ-regulated monoacylglycerol O-acyltransferase 1 (MGAT1) expression is responsible for the lipid accumulation in diet-induced hepatic steatosis. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(34):13656–13661. doi: 10.1073/pnas.1203218109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Soufi N., Hall A. M., Chen Z., et al. Inhibiting monoacylglycerol acyltransferase 1 ameliorates hepatic metabolic abnormalities but not inflammation and injury in mice. The Journal of Biological Chemistry. 2014;289(43):30177–30188. doi: 10.1074/jbc.m114.595850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yen C.-L. E., Cheong M.-L., Grueter C., et al. Deficiency of the intestinal enzyme acyl CoA:monoacylglycerol acyltransferase-2 protects mice from metabolic disorders induced by high-fat feeding. Nature Medicine. 2009;15(4):442–446. doi: 10.1038/nm.1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zimmermann R., Strauss J. G., Haemmerle G., et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science. 2004;306(5700):1383–1386. doi: 10.1126/science.1100747. [DOI] [PubMed] [Google Scholar]
- 45.Wu J. W., Wang S. P., Alvarez F., et al. Deficiency of liver adipose triglyceride lipase in mice causes progressive hepatic steatosis. Hepatology. 2011;54(1):122–132. doi: 10.1002/hep.24338. [DOI] [PubMed] [Google Scholar]
- 46.Lass A., Zimmermann R., Haemmerle G., et al. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome. Cell Metabolism. 2006;3(5):309–319. doi: 10.1016/j.cmet.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 47.Choi C. S., Savage D. B., Kulkarni A., et al. Suppression of diacylglycerol acyltransferase-2 (DGAT2), but not DGAT1, with antisense oligonucleotides reverses diet-induced hepatic steatosis and insulin resistance. The Journal of Biological Chemistry. 2007;282(31):22678–22688. doi: 10.1074/jbc.m704213200. [DOI] [PubMed] [Google Scholar]
- 48.Park S. Y., Kim H. J., Wang S., et al. Hormone-sensitive lipase knockout mice have increased hepatic insulin sensitivity and are protected from short-term diet-induced insulin resistance in skeletal muscle and heart. The American Journal of Physiology—Endocrinology and Metabolism. 2005;289(1):E30–E39. doi: 10.1152/ajpendo.00251.2004. [DOI] [PubMed] [Google Scholar]
- 49.Reid B. N., Ables G. P., Otlivanchik O. A., et al. Hepatic overexpression of hormone-sensitive lipase and adipose triglyceride lipase promotes fatty acid oxidation, stimulates direct release of free fatty acids, and ameliorates steatosis. The Journal of Biological Chemistry. 2008;283(19):13087–13099. doi: 10.1074/jbc.m800533200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chibalin A. V., Leng Y., Vieira E., et al. Downregulation of diacylglycerol kinase delta contributes to hyperglycemia-induced insulin resistance. Cell. 2008;132(3):375–386. doi: 10.1016/j.cell.2007.12.035. [DOI] [PubMed] [Google Scholar]
- 51.Brown M. S., Goldstein J. L. Selective versus total insulin resistance: a pathogenic paradox. Cell Metabolism. 2008;7(2):95–96. doi: 10.1016/j.cmet.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 52.Semple R. K., Sleigh A., Murgatroyd P. R., et al. Postreceptor insulin resistance contributes to human dyslipidemia and hepatic steatosis. The Journal of Clinical Investigation. 2009;119(2):315–322. doi: 10.1172/jci37432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bae E. J., Xu J., Oh D. Y., et al. Liver-specific p70 S6 kinase depletion protects against hepatic steatosis and systemic insulin resistance. Journal of Biological Chemistry. 2012;287(22):18769–18780. doi: 10.1074/jbc.m112.365544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li S., Brown M. S., Goldstein J. L. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(8):3441–3446. doi: 10.1073/pnas.0914798107. [DOI] [PMC free article] [PubMed] [Google Scholar]