

Abstract
Background
Cardiovascular disease (CVD) remains the major cause of excess mortality in patients with non-alcoholic fatty liver disease (NAFLD). The aim of this study was to investigate the individual contribution of NAFLD to CVD risk factors in the absence of pathogenic influences from other comorbidities often found in NAFLD patients, by using an established in-vitro model of hepatic steatosis.
Methods
Histopathological events in non-alcoholic fatty liver disease were recapitulated by focused metabolic nutrient overload of hepatoblastoma C3A cells, using oleate-treated-cells and untreated controls for comparison. Microarray and proteomic data from cell culture experiments were integrated into a custom-built systems biology database and proteogenomics analysis performed. Candidate genes with significant dysregulation and concomitant changes in protein abundance were identified and STRING association and enrichment analysis performed to identify putative pathogenic pathways.
Results
The search strategy yielded 3 candidate genes that were specifically and significantly up-regulated in nutrient-overloaded cells compared to untreated controls: fibrinogen alpha chain (2.2 fold), fibrinogen beta chain (2.3 fold) and fibrinogen gamma chain (2.1 fold) (all rank products pfp <0.05). Fibrinogen alpha and gamma chain also demonstrated significant concomitant increases in protein abundance (3.8-fold and 2.0-fold, respectively, p <0.05).
Conclusions
In-vitro modelling of NAFLD and reactive oxygen species formation in nutrient overloaded C3A cells, in the absence of pathogenic influences from other comorbidities, suggests that NAFLD is an isolated determinant of CVD. Nutrient overload-induced up-regulation of all three fibrinogen component subunits of the coagulation cascade provides a possible mechanism to explain the excess CVD mortality observed in NAFLD patients.
Electronic supplementary material
The online version of this article (doi:10.1186/s12944-015-0069-3) contains supplementary material, which is available to authorized users.
Keywords: Systems biology, Proteomics, Microarray, STRING, Non-alcoholic fatty liver disease, Cardiovascular disease, Metabolic syndrome, C3A cells
Background
Non-Alcoholic Fatty Liver Disease (NAFLD) has been traditionally regarded as the consequence of a high-fat western diet and sedentary lifestyle [1, 2]. Its increasing worldwide prevalence, however, suggests that NAFLD is more than a lifestyle disease. Studies have identified susceptibility genes and genetic polymorphisms that associate with development and severity of NAFLD [3]. This may explain the differences in demographic data observed in Asia compared to elsewhere in the world, including a younger age distribution and a higher proportion of patients who are judged lean by Body Mass Index, but show an altered metabolic profile associated with obesity [4, 5].
Cardiovascular disease (CVD) remains one of the major causes of excess mortality in NAFLD patients [6–8]. The term “Metabolic Syndrome” (MetS) describes a clinical cluster of diseases that tend to aggregate in individuals over time, including central obesity, hypertension, impaired fasting glucose and dyslipidemia [9–11]. Together these components create a pro-atherogenic environment that is postulated to accelerate atherosclerosis and increase the risk of cardiovascular diseases. Whilst the definition of MetS does not presently include NAFLD, the association of atherosclerotic markers, such as carotid artery wall thickness, with NAFLD has been reported previously [12, 13]. Further attempts to understand the possible causative role of NAFLD in CVD have used statistical models to exclude atherogenic contributions from traditional CVD risk factors and other components of MetS [14, 15]. However, to date, there are no experimental genomic or proteomic studies that examine specifically whether NAFLD is an isolated risk factor for CVD in absence of other components of MetS, or if these conditions have a common cause.
Previously, we developed an in vitro model of cellular steatosis by exposing hepatoblastoma C3A cells to nutrient overload (treatment with lactate, pyruvate, octanoate and ammonia), which reproduces the characteristic pathophysiological changes found in NAFLD, including intracellular triglyceride accumulation and reactive oxygen species (ROS) formation [16]. This model allows the opportunity to assess the individual contribution of NAFLD to CVD risk factors in the absence of pathogenic influences from other comorbidities often found in NAFLD patients.
In the present study, changes in hepatoblastoma C3A gene transcription and protein expression in response to cellular steatosis and ROS formation induced specifically by nutrient overload were assembled into a custom-built data portal allowing evaluation of integrated transcriptomic and proteomic data to identify gene products potentially involved in pathogenic pathways. Candidates showing consistently greater than two fold alterations in specific nutrient overload-mediated gene transcription and protein expression were subjected to further analysis by the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) v9.1 database (http://string-db.org) and enrichment analysis to identify predicted functional partners and pathogenic pathways contributing potentially to a pro-atherogenic environment.
Methods
Cell culture, treatment and sample collection
Hepatoblastoma C3A cells (ATCC® CRL-10741TM, LGC Standards, Teddington, UK) were cultured as previously described [16]. Briefly, cell cultures were treated either with the combination of lactate, pyruvate, octanoate and ammonia (LPON), oleate (OLE), or untreated controls. Both octanoate and oleate readily diffuse into mitochondria to promote efficient ß-oxidation and lipid accumulation, but while OLE treatment causes simple cellular steatosis, LPON treatment induces both ROS formation and mitochondrial dysfunction, in addition to steatosis, typically seen in NAFLD [16]. C3A cells were treated in three separate experiments either with LPON, OLE, or untreated controls and processed for transcriptomic or proteomic analysis as described in the following sections.
Sample preparation and transcriptomics
Cells were washed twice in cold PBS and transferred to cold RNALater® (Life Technologies, Paisley, UK) for overnight incubation at 4 °C. Afterwards, RNA was isolated with an RNAqueous®-4PCR kit (Life Technologies) and subsequently amplified and biotinylated with an Illumina® TotalPrep RNA Amplification kit (Life Technologies), following the manufacturer’s instructions. RNA expression was measured by hybridization to the Illumina® Whole Human Genome BeadChip H12 Microarray (Illumina United Kingdom, Essex, UK). Data were extracted through the GCOS software (Affymetrix UK Ltd., High Wycombe, UK). CELfiles were used for additional data processing and imported into Bioconductor [17] to examine differences between LPON- and OLE-treated groups and untreated controls. Data were normalized by robust multi-array average (RMA) in the Oligo module (http://www.bioconductor.org/packages/2.0/bioc/html/oligo.html). Gene ontology and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was performed with the DAVID tool [18, 19] on genes that were significantly differentially expressed. Data was statistically analyzed with the bioconductor Limma package [20].
Sample preparation and proteomics
Protein extraction was performed as previously described [21]. Briefly, samples were denatured in 8 M urea, reduced by incubating with dithiothreithol prior to cysteine alkylation with iodoacetamide and overnight digestion with 8 μg trypsin. Protein concentrations were estimated by Bradford protein assay (Thermo Scientific, Rockford, IL, USA) on a 10 μl sample, diluted to 2 M Urea and quantified against a BSA standard curve. 4 μg peptide samples were acidified (1 % formic acid), centrifuged and cleaned using Stagetips [22], dried by SpeedVac, and stored at −20 °C.
2 μg peptide samples were analysed in a randomised sequence by capillary-HPLC- MSMS as described previously [23], using an on-line system consisting of a micro-pump (1200 binary HPLC system, Agilent, UK) coupled to a hybrid LTQ-Orbitrap XL instrument (Thermo-Fisher, Leicestershire, UK). Acetonitrile and water were HPLC quality (Fisher). Formic acid was Suprapure 98-100 % (Merck, Darmstadt, Germany) and trifluoroacetic acid was 99 % purity sequencing grade. LC-MSMS label-free quantification was performed using Progenesis 4.0 (Nonlinear Dynamics, Newcastle upon Tyne, UK) as described previously [24]. Multicharged (2+,3+,4+) ion intensities were extracted from LC-MS files and MSMSdata were searched using Mascot Version 2.4 (Matrix Science Ltd, London, UK) against the Homo Sapiens subset of the NCBI protein database (12/01/2011; 34,281 sequences) using a maximum missed-cut value of 2, variable oxidation (M), N-terminal protein acetylation and fixed carbamidomethylation (C); precursor mass tolerance was 7 ppm and MSMS tolerance 0.4 Da. The significance threshold (p) was < 0.05 (MudPIT scoring). A minimum peptide cut off score of 20 was set, corresponding to <3 % global false discovery rate (FDR) using a decoy database search. Proteins identified and quantified with two or more peptide sequences were retained. A two-tailed t-test for independent samples or biological triplicates was performed on arcsinh transformed, normally distributed intensity data.
Data mining and candidate gene identification
Changes in hepatoblastoma C3A gene transcription and protein expression in response to cellular steatosis induced by nutrient overload, compared with OLE-treatment and untreated controls, were analysed in a custom built bioinformatics data mining tool established by the BHF Centre of Research Excellence Bioinformatics Team at the Queen’s Medical Research Institute at the University of Edinburgh. This allows evaluation of integrated transcriptomic and proteomic data, by matching differential gene transcription with altered protein abundance, to identify gene products potentially involved in pathogenic pathways. Genes of interest were analysed in a stepwise approach outlined in Fig. 1 to identify genes showing a consistently greater than 2.0 fold increase in both gene transcription and protein abundance. To focus on the specific effects of steatosis with ROS formation and mitochondrial dysfunction and to eliminate candidates up-regulated by simple steatosis, genes showing similar > 2.0 fold increases in expression in both OLE- and LPON-treated cells compared to untreated controls were excluded from the analysis.
Fig. 1.

Strategy for data mining and candidate gene identification. Step 1: C3A cell cultures treated with LPON, OLE or untreated controls, as described under ‘Methods’, were subject to transcriptomic or proteomic analysis and data screened for candidate genes specifically up-regulated by nutrient overload using the screening criteria outlined in Box 1. Step 2: Predicted functional partners of primary candidates showing >2.0 fold nutrient overload-induced increases in gene transcripts and protein abundance were identified by STRING network associations. Step 3: Primary gene candidates, identified in step 1, and their predicted functional partners, identified in step 2, were grouped by enrichment analysis according to their functional annotations to identify representation of common biological processes among the candidate genes. Further details of these bioinformatics procedures are described under ‘Methods’.
STRING Network Associations
Predicted functional partners of candidate genes induced specifically in response to nutrient overload were identified using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) v9.1 database (http://string-db.org). Only interactions in Homo sapiens with a probabilistic confidence score ≥0.900, corresponding to a "highest-confidence" network, were considered in this study. STRING PFPs were cross-validated against the original transcriptomic and proteomic data for differential expression and protein abundance, using similar criteria (>2.0 fold increase in LPON-treated cells, excluding PFPs with >2.0 fold increase in OLE-treated cells: see Fig. 1) in order to identify candidate genes that had otherwise been excluded by the more stringent primary search strategy.
Enrichment analysis
To identify dysregulated pathways and biological processes contributing potentially to a pathogenic phenotype, candidate genes differentially expressed in response to nutrient overload and their PFPs were grouped according to their functional annotations with data from the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) database via the Gene Ontology enRIchment anaLysis and visuaLizAtion (GOrilla) tool (http://cbl-gorilla.cs.technion.ac.il) [25, 26]. Only dysregulations with p < 0.05 were considered in this study, with p-values being corrected for multiple testing using the Benjamini and Hochberg method [27]. Enrichment was based on gene ranking, which was indicated by the STRING analysis confidence score.
Results
Bioinformatics, using the screening criteria defined in step 1 of Fig. 1, identified 2 genes that showed a >2-fold increase in both gene transcription and protein abundance specifically in response to nutrient overload, namely fibrinogen alpha chain and farnesyl-diphosphate farnesyltransferase (Tables 1 and 2). These were selected for STRING association and enrichment analysis to identify predicted functional partners and pathogenic pathways as described below. Several other genes demonstrated a >2-fold increase in specific nutrient overload- induced gene transcription, but lacked corroborative proteomics data and consequently were not used for primary STRING association and enrichment analysis. No examples were found of any gene showing a >2-fold reduction in gene transcription and protein abundance, supporting either unchanged or increased gene expression specifically in response to nutrient overload.
Table 1.
Microarray data for candidate genes specifically up-regulated by nutrient overload
| Entrez gene ID | Accession | Gene Symbol | Description | Control | LPON | Control-LPON | OLE | Control-OLE | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Max Mean value | Max Mean value | Direction of change wrt control | Maximum FC | RP pfp | Max Mean value | Direction of change wrt control | Max Mean value | RP pfp | ||||
| 2222 | FDFT | farnesyl-diphosphate farnesyltransferase 1 | 1055.01 | 2501.65 | ↑ | 2.37121 | 0 | 1558.93 | ↑ | 1.47764 | 0.0194 | |
| 2243 | NP_000499 | FGA | Fibrinogen alpha chain | 3686.45 | 7280.47 | ↑ | 2.24956 | 0.0002 | 6716.9 | ↑ | 1.97551 | 0.0009 |
FGA and FDFT were identified as candidate genes showing > 2.0 fold specific up-regulation of gene expression and protein abundance (see Table 2) in LPON-treated cells compared to untreated controls.
LPON Lactate, pyruvate, octanoate and ammonia, FC fold change, RP pfp Rank products estimated percentage of false positive predictions; FGA Fibrinogen alpha chain, FDFT Farnesyl-diphosphate farnesyltransferase 1.
Table 2.
Proteomics data for candidate genes specifically up-regulated by nutrient overload
Fibrinogen alpha chain (FGA)
FGA gene expression was significantly higher in LPON-treated cells compared to untreated controls by 2.3 fold (rank products pfp = 0.0002). The FGA protein product was also more abundant in LPON-treated cells (3.8 fold change, p = 0.009).
STRING network analysis (step 2, Fig. 1) identified 32 STRING PFPs of FGA in Homo sapiens with a probabilistic confidence score ≥ 0.900 (Supplementary material; Additional File 1). Expression of four out of 32 PFPs were up-regulated by >2 fold in LPON-treated cells compared to untreated controls (rank products pfp <0.05). This included the remaining two fibrinogen beta (FGB) and fibrinogen gamma (FGG) chains, fibrinogen-like 1 (FGL1), and cystatin C (CST3) (Table 3). These 4 genes were not identified in the primary analytical strategy either because no proteomics data was available (FGB, FLG1, CST3; Table 4) or the differential protein abundance did not quite reach the primary fold change search criteria and/or statistical significance (FGG, fold change 1.97462, p = 0.07, Table 4).Of these up-regulated PFPs, FGL1 and CST3 were subsequently excluded from further analysis, as transcription of both genes was also up-regulated by more than 2-fold in OLE-treated cells compared to untreated controls (Table 3). In contrast, FGG and FGB, along with FGA identified in the primary screen, were up-regulated to a much lesser extent in OLE-treated compared to LPON-treated cells (all less than 2 fold-change, see Tables 1 and 4). In total, three genes of the coagulant cascade (FGA, FGG, and FGB) met the final inclusion criteria as being specifically up-regulated in response to nutrient overload.
Table 3.
Microarray data for predicted functional partners of FGA up-regulated by nutrient overload
| Entrez gene ID | Accession | Gene symbol | Description | Control | LPON | Control-LPON | OLE | Control-OLE | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Maximum mean value | Maximum mean value | Direction of change wrt control | Maximum FC | RP pfp | Maximum mean value | Direction of change wrt control | Maximum FC | RP pfp | ||||
| 2266 | NP_000500 | FGG | Fibrinogen gamma chain | 4612.12 | 1227.85 | ↑ | 2.10864 | 0.003 | 7458.56 | ↑ | 1.61716 | 0 |
| 2244 | NP_001171670 | FGB | Fibrinogen beta chain | 644.648 | 1505.62 | ↑ | 2.33556 | 0.0018 | 1239.23 | ↑ | 1.92234 | 0.02 |
| 2267 | NP_004458 | FGL1 | Fibrinogen-like 1 | 400.579 | 1086.52 | ↑ | 2.43005 | 0.003 | 884.484 | ↑ | 2.20802 | 0.0178 |
| 1471 | NP_000090 | CST3 | Cystatin C | 2153.68 | 5232.34 | ↑ | 2.42948 | 0 | 4730.95 | ↑ | 2.19668 | 0 |
>2 fold up-regulation in gene expression of FGG, FGB, FGL1 and CST3 were observed in LPON-treated cells compared to untreated controls.
For abbreviations: see Table 1.
Table 4.
Proteomics data for predicted functional partners of FGA up-regulated by nutrient overload
| Gene | Unique | Ratio | |||
|---|---|---|---|---|---|
| Accession | Symbol | Peptides | Description | P value | LPON/ctrl |
| NP_000500 | FGG | 3 | Fibrinogen gamma chain | 0.072855965 | 1.97 |
For abbreviations: see Table 1.
Enrichment analysis of FGA and its STRING-predicted functional partners by gene ontology revealed platelet activation, coagulation, and hemostasis as significantly over-represented biological processes (Table 5 ).
Table 5.
Enrichment analysis based on the number of predicted functional partners of FGA indicated by STRING analysis
| GO term | Description | P-value | FDR q-value | Enrichment (N, B, n, b) |
|---|---|---|---|---|
| GO:0030168 | Platelet activation | 1.43E-05 | 1.96E-02 | 3.00 (33,11,8,8) |
| GO:0051592 | Response to calcium ion | 1.83E-04 | 1.26E-01 | 11.00 (33,3,3,3) |
| GO:0050817 | Coagulation | 3.02E-04 | 1.38E-01 | 1.55 (33,16,20,15) |
| GO:0007599 | Chemostasis | 3.02E-04 | 1.04E-01 | 1.55 (33,16,20,15) |
| GO:0007596 | Blood coagulation | 3.02E-04 | 8.30E-02 | 1.55 (33,16,20,15) |
| GO:0001775 | Cell activation | 4.88E-04 | 1.12E-01 | 2.36 (33,14,8,8) |
| GO:0006887 | Exocytosis | 5.88E-04 | 1.15E-01 | 2.89 (33,10,8,7) |
| GO:0002576 | Platelet degranulation | 5.88E-04 | 1.01E-01 | 2.89 (33,10,8,7) |
| GO:0032940 | Secretion by cell | 5.88E-04 | 8.96E-02 | 2.89 (33,10,8,7) |
Enrichment was calculated as (b/n) / (B/N), where N is the total number of partners including FGA; B is the total number of genes associated with a specific GO term; n is the number of genes in the STRING partner list, as ranked by confidence score or in the target set when appropriate b is the number of genes in the intersection.
Farnesyl-diphosphate farnesyltransferase 1 (FDFT1)
Gene expression of FDFT1, which catalyses the first committed step in sterol synthesis on the pathway to cholesterol, was up-regulated by 2.4 fold (rank products pfp = 0) in LPON-treated cells compared to untreated controls. A concomitant increase in protein abundance by 3.2 fold (p = 0.02) in LPON-treated cells was also recorded. 10 STRING PFPs of FDFT1 in Homo sapiens with a probabilistic confidence score ≥0.900 were identified (Supplementary material; Additional File 2); however none of these genes satisfied the criteria of >2.0 fold change in gene expression (Rank products pfp <0.05) when comparing LPON-treated cells to untreated controls.
Discussion
CVD remains a major cause of excess mortality in NAFLD patients [6–8]. In the present study, we sought to clarify the possible causative role of NAFLD in CVD by identifying gene and protein dysregulation using an in vivo model of cellular steatosis. Use of an in vitro model is particularly relevant as it enables experimental strategies to be designed to exclude pathogenic influences from conditions, such as diabetes mellitus and hypertension, which commonly co-exist with NAFLD [28].
Fibrinogen is produced in the liver and is made up of alpha, beta, and gamma chains in a 1:1:1 ratio. We found similar up-regulation in gene expression of all three fibrinogen subunits (see Tables 1 and 3), which provides a potential mechanism to explain the excess CVD mortality observed in NAFLD patients. In the common pathway of the coagulation cascade, fibrinogen (factor I) is a precursor of fibrin (Ia), which contributes to the subsequent formation of a stable fibrin clot (see Fig. 2). An increased plasma fibrinogen level is therefore prothrombotic and its potential clinical application as a surrogate biomarker for predicting future CVD events is currently being explored [29, 30]. Two large-scale studies, which together included 400,880 patients, concluded that fibrinogen is a reliable predictor of cardiovascular mortality [31, 32]. Such a correlation persists even after adjustment for other established risk factors of CVD known to associate with fibrinogen levels, including total cholesterol and blood pressure. This suggests that there is an independent association between plasma fibrinogen levels and CVD. Therefore NAFLD, through up-regulation of fibrinogen coagulation factor, is an independent risk factor of CVD, potentially by enhancing clot strength [33]. This is logical, because liver is the site of synthesis of most coagulation factors, and therefore pathologies of the liver would most probably contribute to systemic cardiometabolic dysregulation. Indeed, elevated plasma fibrinogen is frequently reported as an additional and independent cardiovascular risk factor in a clinical context of MetS, with a significant association with the severity of hyperinsulinaemia [34, 35]. To our knowledge, our in vitro approach is unique in providing a controlled environment for isolating events that occur specifically in the hepatocytes, independent of the influence exerted by other disease variables forming the clinical cluster of MetS. This allows us to identify hepatocyte dysfunction as the pathophysiological culprit of increased cardiovascular risk observed in these individuals.
Fig. 2.
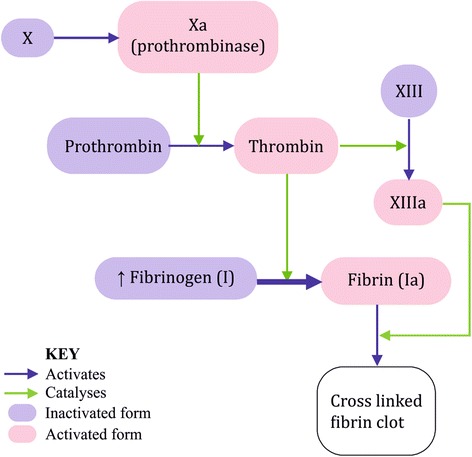
Common pathway of the coagulation cascade. Common pathway of the coagulation cascade, showing how fibrinogen (factor I) as a precursor of fibrin (Ia), contributes to the subsequent formation of a stable fibrin clot.
Difference between OLE and LPON-treated cells
Up-regulation in the genetic expressions of all 3 fibrinogen chains was consistently more dramatic in LPON-treated compared OLE-treated cells, the latter of which models simple cellular steatosis. In contrast, LPON treatment additionally induced ROS formation and mitochondrial dysfunction [16] Thus, we hypothesize that dysregulation of gene expression in LPON-treated cells was made more dramatic by oxidative stress due to free radical formation.
Previous cross-sectional studies have demonstrated increased blood fibrinogen concentrations in response to mild pulmonary inflammation in healthy humans caused by diesel exhaust particulates, a major constituent of city air pollution [36, 37]. In a previous double-blinded randomised crossover study, diesel exhaust inhalation has also been shown to mediate excess CVD risk caused by air pollution by increasing thrombus formation following platelet activation secondary to ROS formation and oxidative stress [38]. This supports the notion that multiple mechanisms intersecting at the level of ROS formation may contribute to excess CVD risk. However, while ROS-formation in C3A cells after exposure to nanoparticles has been confirmed in our laboratory, such observations should be taken with caution as the mechanism of free radical generation may be different in particulate-induced and steatosis-induced conditions [39]. Thus, while ROS formation observed after inhalation of diesel exhaust arises from the alteration of redox potential by particulates and inflammatory cells [40], in our model of NAFLD, ROS are formed endogenously due to nutrient excess [16].
In conclusion, this study presents evidence supporting the hypothesis of NAFLD being an isolated determinant of CVD, as use of a proven in vitro model of NAFLD eliminates the confounding influences of other comorbidities found in NAFLD patients. This supports the notion that the liver is a source of pro-coagulant molecules which contributes to the risk of thrombosis and enhanced CVD risk in the NAFLD population. However, in using an in vitro model examines only one aspect of the concomitant pathophysiological mechanisms operating in vivo. Other factors operating in concert may play significant roles in modifying the systemic response to the observed cellular effects, for example the role of visceral fat as a systemic pro-inflammatory signal in CVD risk in NAFLD patients. Further work is required to elucidate the pathophysiological mechanisms explaining the association between free radical-induced oxidative stress and CVD in a NAFLD setting. Clinical evidence could be sought by measuring the plasma levels of FGA, FGB and FGG in NAFLD patients, to investigate potential relationships between plasma fibrinogen levels and severity of CVD determined by conventional atherosclerotic markers, such as carotid artery wall thickness. Indeed, verification of FGA, FGB and FGG as early plasma predictors of CVD development would provide a rationale to explore the possibility of prophylactic fibrinolytic therapy in a NAFLD context, similar to that already used in myocardial infarction [41].
Acknowledgements
This work was funded internally, from departmental funds of the Hepatology Laboratory, Division of Health Sciences, University of Edinburgh. The decision to submit the article for publication was not influenced by the funding body.
Additional files
Functional partners of FGA as predicted by STRING analysis.
Functional partners of FDFT1 as predicted by STRING analysis.
Footnotes
Emily N. W. Yeung and Philipp Treskes contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Conceived & designed the study: LJN, JNP. Performed the research: ENWY, PT, SFM, TLB, LJN. Contributed reagents/materials/analysis tools: JRM, DRD, LJN. Analysed the data: ENWY, PT, SFM, JRM, SMR, TLB, KLA, SDM, PCH, LJN, JNP. Wrote the manuscript: ENWY, PT, SDM, LJN, JNP. All authors critically revised and approved the final manuscript for intellectual content.
Authors’ information
Leonard J. Nelson and John N. Plevris are joint senior authors.
Contributor Information
Emily N. W. Yeung, Email: N.W.Yeung@sms.ed.ac.uk
Philipp Treskes, Email: p.treskes@ed.ac.uk.
Sarah F. Martin, Email: smartoak@gmail.com
Jonathan R. Manning, Email: jonathan.manning@ed.ac.uk
Donald R. Dunbar, Email: donald.dunbar@sistemic.co.uk
Sophie M. Rogers, Email: sophie_rogers@hotmail.co.uk
Thierry Le Bihan, Email: thierry.lebihan@ed.ac.uk.
K. Ann Lockman, Email: Ann.Lockman@nhslothian.scot.nhs.uk.
Steven D. Morley, Email: steve.morley@ed.ac.uk
Peter C. Hayes, Email: P.Hayes@ed.ac.uk
Leonard J. Nelson, Email: L.Nelson@ed.ac.uk
John N. Plevris, Email: j.plevris@ed.ac.uk
References
- 1.Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, et al. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40:6. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 2.Feldstein AE. Novel insights into the pathophysiology of nonalcoholic fatty liver disease. Semin Liver Dis. 2010;30:4. doi: 10.1055/s-0030-1267539. [DOI] [PubMed] [Google Scholar]
- 3.Farrell GC, Wong VWS, Chitturi S. NAFLD in Asia – as common and important as in the West. Nat Rev Gastrolenterol Hepatol. 2013;10:5. doi: 10.1038/nrgastro.2013.34. [DOI] [PubMed] [Google Scholar]
- 4.Denis GV, Hamilton JA. Healthy obese persons: How can they be identified and do metabolic profiles stratify risk? Curr Opin Endocrinol Diabetes Obes. 2013;20:5. doi: 10.1097/01.med.0000433058.78485.b3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu CJ. Prevalence and risk factors for NAFLD in Asian people who are not obese. J Gastroenterol Hepatol. 2012;27:10. doi: 10.1111/j.1440-1746.2012.07222.x. [DOI] [PubMed] [Google Scholar]
- 6.Adams LA, Lymp JF, St Sauver J, Sanderson SO, Lindor KD, Feldstein A, et al. The natural history of nonalcoholic fatty liver disease: a population- based cohort study. Gastroenterology. 2005;129:1. doi: 10.1053/j.gastro.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 7.Harrison SA. Day CP Benefits of lifestyle modification in NAFLD. Gut. 2007;56:12. doi: 10.1136/gut.2006.112094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Preiss D, Sattar N. Non-alcoholic fatty liver disease: an overview of prevalence, diagnosis, pathogenesis and treatment considerations. Clin Sci (Lond) 2008;115:5. doi: 10.1042/CS20070402. [DOI] [PubMed] [Google Scholar]
- 9.Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2005;365:9468. doi: 10.1016/S0140-6736(05)66378-7. [DOI] [PubMed] [Google Scholar]
- 10.Ekstedt M, Franzen LE, Mathiesen UL, Thorelius L, Holmqvist M, Bodemar G, et al. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology. 2006;44:4. doi: 10.1002/hep.21327. [DOI] [PubMed] [Google Scholar]
- 11.Soderberg C, Stal P, Askling J, Glaumann H, Lindberg G, Marmur J, et al. Decreased survival of subjects with elevated liver function tests during a 28- year follow-up. Hepatology. 2010;51:2. doi: 10.1002/hep.23314. [DOI] [PubMed] [Google Scholar]
- 12.Anstee QM, Targher G, Day CP. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat Rev Gastroenterol Hepatol. 2013;10:6. doi: 10.1038/nrgastro.2013.41. [DOI] [PubMed] [Google Scholar]
- 13.Targher G, Bertolini L, Padovani R, Rodella S, Zoppini R, Zenari L, et al. Relations between carotid artery wall thickness and liver histology in subjects with nonalcoholic fatty liver disease. Diabetes Care. 2006;29:6. doi: 10.2337/dc06-0135. [DOI] [PubMed] [Google Scholar]
- 14.Choi SY, Kim D, Kang JH, Park MJ, Kim YS, Lim SH, et al. Nonalcoholic fatty liver disease as a risk factor of cardiovascular disease: relation of non-alcoholic fatty liver disease to carotid atherosclerosis] Korean J Hepatol. 2008;14:1. doi: 10.3350/kjhep.2008.14.1.1. [DOI] [PubMed] [Google Scholar]
- 15.Hamaguchi M, Kojima T, Takeda N, Nagata C, Takeda J, Sarui H, et al. Nonalcoholic fatty liver disease is a novel predictor of cardiovascular disease. World J Gastroenterol. 2007;13:10. doi: 10.3748/wjg.v13.i10.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lockman KA, Baren JP, Pemberton CJ, Baghdadi H, Burgess KE, Plevris-Papaioannou N, et al. Oxidative stress rather than triglyceride accumulation is a determinant of mitochondrial dysfunction in in vitro models of hepatic cellular steatosis. Liver Int. 2012;32:7. doi: 10.1111/j.1478-3231.2012.02775.x. [DOI] [PubMed] [Google Scholar]
- 17.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:10. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:1. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 19.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:5. [PubMed] [Google Scholar]
- 20.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 21.Le Bihan T, Martin SF, Chirnside ES, van Ooijen G, Barrios-Llerena ME, O'Neill JS, et al. Shotgun proteomic analysis of the unicellular alga Ostreococcus tauri. J Proteomics. 2011;74:10. doi: 10.1016/j.jprot.2011.05.028. [DOI] [PubMed] [Google Scholar]
- 22.Rappsilber J, Mann M, Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc. 2007;2:8. doi: 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- 23.Martin SF, Munagapati VS, Salvo-Chirnside E, Kerr LE, Le Bihan T. Proteome turnover in the green alga Ostreococcus tauri by time course 15 N metabolic labeling mass spectrometry. J Proteome Res. 2012;11:1. doi: 10.1021/pr201195p. [DOI] [PubMed] [Google Scholar]
- 24.Le Bihan T, Grima R, Martin S, Forster T, Le Bihan Y. Quantitative analysis of low-abundance peptides in HeLa cell cytoplasm by targeted liquid chromatography/mass spectrometry and stable isotope dilution: emphasising the distinction between peptide detection and peptide identification. Rapid Commun Mass Spectrom. 2010;24:7. doi: 10.1002/rcm.4487. [DOI] [PubMed] [Google Scholar]
- 25.Eden E, Lipson D, Yogev S, Yakhini Z. Discovering motifs in ranked lists of DNA sequences. PLoS computational biology. 2007;3:3. doi: 10.1371/journal.pcbi.0030039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC bioinformatics. 2009;10:1. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. 1995;Series B:Methodological.
- 28.Adams LA, Waters OR, Knuiman MW, Elliott RR, Olynyk JK. NAFLD as a risk factor for the development of diabetes and the metabolic syndrome: an eleven-year follow-up study. The American journal of gastroenterology. 2009;104:4. doi: 10.1038/ajg.2008.146. [DOI] [PubMed] [Google Scholar]
- 29.Kaptoge S, Di Angelantonio E, Pennells L, Wood AM, White IR, Gao P, et al. C-reactive protein, fibrinogen, and cardiovascular disease prediction. N Engl J Med. 2012;367:14. doi: 10.1056/NEJMicm1114392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lisman T, Weeterings C, de Groot PG. Platelet aggregation: involvement of thrombin and fibrin(ogen) Front Biosci. 2005;10:2504. doi: 10.2741/1715. [DOI] [PubMed] [Google Scholar]
- 31.Danesh J, Lewington S, Thompson SG, Lowe GD, Collins R, Kostis JB, et al. Plasma fibrinogen level and the risk of major cardiovascular diseases and nonvascular mortality: an individual participant meta-analysis. Jama. 2005;294:14. doi: 10.1001/jama.294.14.1799. [DOI] [PubMed] [Google Scholar]
- 32.Ndrepepa G, Braun S, King L, Fusaro M, Keta D, Cassese S, et al. Relation of fibrinogen level with cardiovascular events in patients with coronary artery disease. Am J Cardiol. 2013;111:6. doi: 10.1016/j.amjcard.2012.11.060. [DOI] [PubMed] [Google Scholar]
- 33.Hickman IJ, Sullivan CM, Flight S, Campbell C, Crawford DH, Masci PP, et al. Altered clot kinetics in patients with non-alcoholic fatty liver disease. Ann Hepatol. 2009;8:4. [PubMed] [Google Scholar]
- 34.Mertens I, Van Gaal LF. Obesity, haemostasis and the fibrinolytic system. Obes Rev. 2002;3:2. doi: 10.1046/j.1467-789X.2002.00056.x. [DOI] [PubMed] [Google Scholar]
- 35.Russo I. The Prothrombotic Tendency in Metabolic Syndrome: Focus on the Potential Mechanisms Involved in Impaired Haemostasis and Fibrinolytic Balance. Scientifica (Cairo) 2012;2012:525374. doi: 10.6064/2012/525374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ghio AJ, Kim C, Devlin RB. Concentrated ambient air particles induce mild pulmonary inflammation in healthy human volunteers. Am J Respir Crit Care Med. 2000;162:3. doi: 10.1164/ajrccm.162.3.9911115. [DOI] [PubMed] [Google Scholar]
- 37.Pekkanen J, Brunner EJ, Anderson HR, Tiittanen P, Atkinson RW. Daily concentrations of air pollution and plasma fibrinogen in London. Occup Environ Med. 2000;57:12. doi: 10.1136/oem.57.12.818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lucking AJ, Lundback M, Mills NL, Faratian D, Barath SL, Pourazar J, et al. Diesel exhaust inhalation increases thrombus formation in man. Eur Heart J. 2008;29:24. doi: 10.1093/eurheartj/ehn464. [DOI] [PubMed] [Google Scholar]
- 39.Filippi C, Pryde A, Cowan P, Lee T, Hayes P, Donaldson K, et al. Toxicology of ZnO and TiO2 nanoparticles on hepatocytes: Impact on metabolism and bioenergetics. Nanotoxicoloty. 2015;9:1. doi: 10.3109/17435390.2014.895437. [DOI] [PubMed] [Google Scholar]
- 40.Tornqvist H, Mills NL, Gonzalez M, Miller MR, Robinson SD, Megson IL, et al. Persistent endothelial dysfunction in humans after diesel exhaust inhalation. Am J Respir Crit Care Med. 2007;176:4. doi: 10.1164/rccm.200606-872OC. [DOI] [PubMed] [Google Scholar]
- 41.Buczko W, Mogielnicki A, Kramkowski K, Chabielska E. Aspirin and the fibrinolytic response. Thrombosis Research. 2003;110:5–6. doi: 10.1016/j.thromres.2003.08.006. [DOI] [PubMed] [Google Scholar]