

Abstract
Correlational data suggest that learned associations are encoded within neuronal ensembles. However, it has been difficult to prove that neuronal ensembles mediate learned behaviours because traditional pharmacological and lesion methods, and even newer cell type-specific methods, affect both activated and non-activated neurons. Additionally, previous studies on synaptic and molecular alterations induced by learning did not distinguish between behaviourally activated and non-activated neurons. Here, we describe three new approaches—Daun02 inactivation, FACS sorting of activated neurons and c-fos-GFP transgenic rats — that have been used to selectively target and study activated neuronal ensembles in models of conditioned drug effects and relapse. We also describe two new tools — c-fos-tTA mice and inactivation of CREB-overexpressing neurons — that have been used to study the role of neuronal ensembles in conditioned fear.
Introduction
In 1949, Hebb proposed that learned associations are encoded within specific patterns of neurons called cell assemblies (now called neuronal ensembles) that were selectively activated by environmental cues1. Since then, many electrophysiology and cellular imaging studies have found correlational evidence that supports the idea that learned associations between environmental cues and unconditioned rewards are encoded by neuronal ensembles that are activated by these same cues and rewards 2 (Figure 1). The neuronal ensemble hypothesis has had a transforming and long-lasting impact on modern neuroscience research, and has been the conceptual framework for numerous learning and memory studies 2–8. Since the 1950s 9, investigators have primarily used in vivo electrophysiology to characterize temporal activity patterns of putative neuronal ensembles in different brain areas in learned behaviours 5, 10–13. Since the late 1990s, investigators have also used double-labelling methods with immediate-early genes (IEGs) as markers of neural activity to characterize the spatial pattern of activated neuronal ensembles in the brain 14–19 (Box 1). More recently, in vivo two-photon calcium imaging methods were developed to simultaneously record from hundreds of activated neurons 20. These methods, which use calcium-sensitive synthetic dyes and genetically encoded calcium indicator proteins (GCaMPs), have been used to record learning-related alterations in the activity of neuronal ensembles in head-fixed 21 or freely moving awake behaving mice 22.
Figure 1. Neuronal ensembles in the mesocorticolimbic dopamine reward system.

Hypothetical schematic of how drug-associated stimuli activate specific patterns of neurons or neuronal ensembles in this brain system. Environmental stimuli (e.g., tones, lights, odours) and drug-induced interoceptive stimuli (e.g., heart rate, blood vessel tone) during drug self-administration activate specific neuronal ensembles in sensory regions of the neocortex and olfactory bulb that in turn activate specific neuronal ensembles in the prefrontal cortex, hippocampus, basolateral amygdala (BLA), and thalamus. Activated principal (glutamatergic) neurons in each brain area are indicated with red circles and non-activated principal neurons are indicated by light blue circles. Neurons in the nucleus accumbens that receive the highest levels of convergent excitatory glutamatergic input (blue lines) from these latter brain areas are selectively activated to form a neuronal ensemble that corresponds to or encodes the specific combination of stimuli and their relationships on the basis of past experience. Depending on the salience and reward value of these stimuli, the ventral tegmental area (VTA) sends dopaminergic input (red lines) to the prefrontal cortex and nucleus accumbens that further enhances ongoing activity of the more highly activated neuronal ensembles while attenuating activity in the less activated majority of neurons in the prefrontal cortex and nucleus accumbens. Yellow lines indicate GABAergic outputs from the nucleus accumbens to the ventral pallidum (VP) and VTA. CeA, central nucleus of the amygdala.
Box 1. Immediate early gene-based methods.
Over the years, several immediate early gene (IEG)-based methods have been used to identify putative neuronal ensembles in the brain34, 130, 131. The general principle has been to use one neuronal activity marker to label neurons activated during the initial learning session or sessions and a different neuronal marker to label neurons that are activated during a subsequent session (which is typically used to assess the expression of the learned behaviour). A high level of double-labelling of the two activity markers would suggest the recruitment of neuronal ensembles that encode the learned behaviours.
In the late 1990s, Guzowski and colleagues introduced the ‘cellular compartment analysis of temporal activity by fluorescence in situ hybridization’ (catFISH) method 14. This procedure was based on the temporal characteristics of the IEG arc after neuronal activation: a nuclear arc RNA signal emerges 2 min after neuronal activation and persists for up to 16 min, whereas a cytoplasmic RNA signal emerges 20–45 min after activation 17. Accordingly, in situ hybridization can reveal neurons with cytoplasmic arc mRNA, which are neurons that were active earlier (e.g., during the first learning session), and neurons with nuclear arc mRNA, which are neurons that were active more recently (e.g. during the second learning session)17. Along with a variation on the procedure in which the IEG Homer1 is used to label initial neuronal activation and nuclear arc is used to label subsequent neuronal activation 112, 132, this method has been used to identify putative neuronal ensembles that encode specific cues or contexts. The method is useful in identifying neuronal ensembles that are activated during short (about 30 min) learning tasks or time intervals between presentations of the same or different stimuli 14, 17. The main limitation of the catFISH method is that it cannot be used in learning tasks in which the learning and the expression of the learned behaviour are separated by hours or days.
Another IEG-based method is double-labelling of the IEGs c-fos (using in situ hybridization) and FosB (using immunohistochemistry). FosB immunoreactivity labels neurons that were repeatedly activated during the first training or learning sessions, whereas c-fos in situ hybridization labels neurons that were activated during the second session. This method is based on the accumulation of long-lasting protein isoforms from a truncated fosB splice variant called deltaFosB in repeatedly activated neurons 133. This method has been used to identify putative neuronal ensembles in the nucleus accumbens in context-specific cocaine locomotor sensitization 15.
In a recently developed approach, a transgenic c-fos-tTA mouse is used to identify neuronal ensembles 16, 114. The c-fos-tTA transgene uses the c-fos promoter to induce the expression of a tetracycline (tet)-off transcriptional activator (tTA) protein in neurons that are activated during the first learning session. The tTA protein can then bind to a tet operator in the promoter of a second transgene to induce expression of a marker gene. tTA can be induced in activated neurons before and after training, but doxycycline, which binds to and represses tTA, can be added to the mouse diet to block expression of the marker gene. If doxycycline is removed from the diet before the mouse undergoes the first learning session, the marker gene (e.g., lacZ, histone2B-green fluorescent protein (GFP)) can be expressed in neurons that were activated during a selected time window. Immunohistochemical labelling of the protein products of zif268 16 or c-fos 116 can be used to label neurons that were activated during the second session. The c-fos-tTA tool has been used to identify neuronal ensembles that control fear conditioning 16, 19, 33, 116.
An extensive literature, reviewed in the citations above and many other reviews, supports the notion that neuronal ensemble activity encodes diverse forms of learned associations that mediate learned behaviours. However, until recently this evidence was based only on correlations between behaviour and in vivo electrophysiological firing or two-photon calcium-imaging patterns during learning and memory tasks or postmortem activity patterns of different IEGs. Therefore, a causal involvement of the putative neuronal ensembles in learned behaviours had not been established. Until recently, methods aimed at assessing such causal roles in behaviour manipulated neuronal activity within specific brain areas — such as permanent excitotoxic lesions, reversible inactivation using GABAergic agonists or the sodium channel blocker tetrodotoxin, intracranial injections of selective receptor antagonists (e.g., dopamine or glutamate receptor antagonists), activation or inhibition using optogenetics or DREADD (designer receptors exclusively activated by designer drugs) 23 — affect neuronal activity of either all neurons or all neurons of a particular cell type, regardless of their activation state during the learned behaviours. Additionally, it had not been possible to characterize molecular and electrophysiological alterations within the activated neuronal ensembles that presumably mediate memory formation and learned behaviours. Indeed, the published literature on synaptic plasticity (as assessed by ex vivo slice electrophysiology techniques) and molecular mechanisms of learning and memory are based on results obtained from either randomly selected neurons or neurons of a particular cell type, independently of their activation state during learned behaviours 24–27.
The goal of this review is to describe several recent technical developments that make it possible to determine causal roles of putative activated neuronal ensembles in learned behaviours, and to characterize molecular and electrophysiological alterations within the activated neurons. We first describe three recently developed tools to study the role of neuronal ensembles in conditioned drug effects and relapse in rats. These include the Daun02 inactivation method 28, flow cytometry and fluorescence-activated cell sorting (FACS) of activated Fos-expressing neurons 29 and c-fos-GFP transgenic rats 30, which can be used to selectively inactivate neuronal ensembles and assess molecular and electrophysiological alterations within activated neuronal ensembles. We also describe two other methods — inactivation of CREB-overexpressing neurons 31, 32 and c-fos-tTA mice 16, 19, 33 — that have been used to study the role of neuronal ensembles in conditioned fear. These methods all rely on the c-fos gene promoter to manipulate the activity of strongly activated neurons and to identify these neurons for subsequent molecular and cellular analysis. The c-fos promoter is rapidly induced within strongly activated neurons (see Box 2 for details) and c-fos mRNA and Fos protein products have been used in numerous papers as markers of neuronal activation in many neuronal phenotypes in many brain areas 34–36. Below we describe the different methods, their application to studying learned behaviours, and the limitations of each method.
Box 2. The c-fos promoter as a marker of neuronal activity during behaviour.
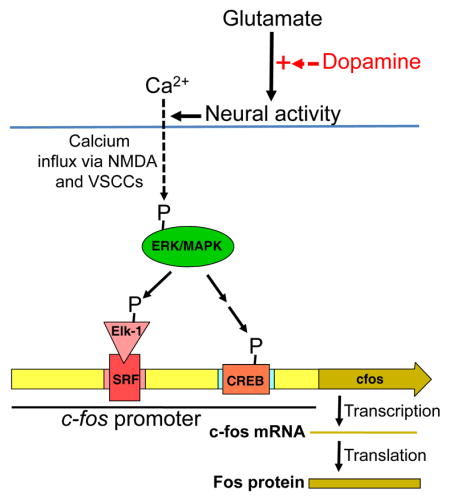
The widespread use of c-fos mRNA and its Fos protein products to identify neuronal activation in brain 34–36 has led to many studies that have examined the detailed molecular and cellular mechanisms of c-fos promoter activation. The relevant literature is immense and is summarized here with only a few selected citations. By necessity, the early studies of c-fos and IEG induction were performed under very artificial conditions using cell or slice culture. These studies were important for identifying candidate signalling mechanisms for IEG induction 34, 134, 135. Many of these mechanisms were also shown in the brain during either development, following chemical or mechanical damage, or following various manipulations that produced non-physiological activation of signal transduction pathways. However, only a few of these mechanisms have a significant role in c-fos promoter activation in the brains of intact behaving rats or mice. In the striatum and hippocampus, c-fos expression is mediated by ERK/MAPK-dependent phosphorylation of Elk-1/SRF and CREB on the c-fos promoter 35, 136, 137, and not by the cAMP pathway 138, 139. Neuronal activation of the ERK/MAPK pathway requires consistent (not sporadic) high levels of calcium influx through NMDA receptors and voltage-sensitive calcium channels (VSCCs) 35, 137, 140, 141. Thus, we hypothesize that c-fos expression in behaving animals reflects a summation or integration of neuronal activity-dependent calcium influx over seconds to minutes, and only strong consistent activity over this timeframe will increase calcium levels enough to induce c-fos. Glutamatergic excitatory input (along with modulation by GABA inhibitory input) plays a major part in inducing strong neuronal activation 35. Drug-induced dopamine release in the striatum is often thought to have a direct pharmacological role in activating neuronal activity and Fos expression, but this is unlikely because dopamine and dopaminergic agonists tend to hyperpolarize striatal neurons in the absence of ongoing glutamatergic input 142. Instead drug-induced dopamine release is thought more likely to enhance the post-synaptic effect of ongoing glutamatergic input on the most strongly activated neurons which increases their neural activity even further 142 to a level sufficient for c-fos promoter activation 35. In contrast drug-induced dopamine attenuates the effect of ongoing glutamatergic input and neural activity of the less activated majority of neurons 142.
Previous attempts to directly examine the association between electrophysiological activity and Fos expression in striatum and hippocampus have shown that the level of Fos expression correlates with the level of synaptic activity and not with the number of action potentials 131, 143. However, results from these studies are difficult to interpret in the context of Fos-related neuronal ensembles activity, because electrophysiological recordings of randomly selected striatal or hippocampal dentate gyrus neurons are almost certainly recordings from the majority of (less activated) neurons and not the neurons that were activated strongly enough to express Fos. In vivo calcium imaging has recently been used to demonstrate a low correlation between spontaneous neuronal activity and Fos expression in single auditory cortex neurons in anaesthetized mice 121. However, these negative data are also difficult to interpret in the context of Fos-related neuronal ensemble activity, because anaesthesia has been shown repeatedly to block the more behaviourally relevant Fos expression induced in awake behaving rodents 144. In the future, it will be important to repeat similar studies with in vivo calcium imaging to appropriately compare neuronal activity with induced c-fos promoter activation in awake behaving rats or mice.
Neuronal ensembles in addiction and relapse
Neuronal ensembles in the nucleus accumbens and prefrontal cortex
Since the 1960s, many studies in humans and laboratory animals have demonstrated that classical and operant conditioning mechanisms play a major role in drug use and relapse 37–40. With repeated drug use, addicted individuals learn to associate drug effects with stimuli or cues in the drug environment (e.g., drug paraphernalia, places of drug taking, and co-users), and over time these cues often promote drug craving and drug seeking 39–41. Drug-related cues are complex combinations of different stimuli that are recognized with a high degree of resolution. Therefore, any neural mechanism capable of encoding these learned associations must have a comparably high degree of resolution. It has been proposed that neuronal ensembles can provide a mechanistic framework for understanding the behavioural and motivational effects of drug-associated cues 15, 42, 43. Indeed, over the past two decades, several studies combining drug self-administration procedures 44 with in vivo electrophysiology have provided correlative evidence for a role of neuronal ensembles in several brain areas (the nucleus accumbens, medial prefrontal cortex (mPFC), ventral pallidum and basolateral amygdala) in cue-induced drug seeking 45–50. There is also limited correlative evidence from studies that compared context-specific locomotor sensitization behaviour with double-labelling of c-fos mRNA and FosB protein (Box 1) in context-specific selection of neuronal ensembles in the nucleus accumbens 15.
The neuronal ensemble hypothesis has had some impact on addiction research, but studies based on this hypothesis still form only a small proportion of neurobiological research on drug addiction. The vast majority of studies on molecular and synaptic plasticity mechanisms of drug reward, relapse and conditioned drug effects assess drug- or cue-induced molecular and cellular alterations in randomly selected neurons or in neurons of a particular cell type, independently of their activation state during behaviour in different animal models of drug addiction 51–57. Therefore, the alterations assessed in these studies were induced largely in the non-activated majority of neurons and not specifically in the neuronal ensembles that were selectively activated during the behaviour. The unique drug-induced or cue-induced molecular and cellular alterations in the minority of activated neurons or neuronal ensembles, which presumably mediate drug-seeking behaviour and conditioned drug effects, were likely missed or masked by drug-induced alterations in the non-activated majority of neurons.
Based on the above considerations, during the last several years we have developed a set of pharmacogenetic, molecular biology and genetic tools to selectively inhibit neuronal ensembles and assess their unique molecular and synaptic alterations. We developed these tools in rats because long-term studies using intravenous drug self-administration procedures 44 and animal models of drug relapse and craving 58–60 are technically very challenging in mice.
The Daun02 inactivation method
The Daun02 method28 was developed to manipulate only those sparsely distributed neurons that are activated by specific stimuli or events without affecting either the surrounding non-activated neurons or neurons that are activated by other stimuli or events. We applied the Daun02 inactivation procedure to selectively inactivate neurons that were previously activated by drug-associated cues or contexts 28 (Figure 2). Below we describe the method, its application to studying drug-related behaviours, as well as its limitations.
Figure 2. The Daun02 inactivation method.

(A) The c-fos-lacZ transgene in the transgenic rats contains a c-fos promoter that regulates transcription of the lacZ coding sequence. Sufficiently strong and persistent neural activity induces fos expression and, subsequently, activates the c-fos promoter. As a result, the expression of lacZ mRNA and its protein product β-galactosidase is increased in these strongly activated neurons but not in the surrounding majority of neurons. The prodrug Daun02 is injected into the brain area of interest and is initially inactive. However, β-galactosidase catalyzes Daun02 to the active product Daunorubicin that induces apoptosis and cell death in only those neurons that were activated strongly enough during behaviour to induce β-galactosidase. (B) The general experimental procedure requires repeated exposures in one context (context A) during the training phase, followed by withdrawal/abstinence or extinction in a different context (context B). On the induction day, specific neuronal ensembles can be reactivated by the training, extinction or exposure to a novel context (context C), along with cues and/or the drug to induce β-galactosidase. Vehicle or Daun02 is injected 90 minutes later (the time of maximal β-galactosidase protein induction after neuronal activation). On the test day, 3 days later, reactivation of these neurons and behavioural effects in the training context (context A) are assessed.
We used c-fos-lacZ transgenic rats, which have a transgene that contains a c-fos promoter to induce transcription of the lacZ coding sequence and translation of the protein product β-galactosidase. This induction occurs only in strongly activated (that is, Fos-positive) neurons, but not in the surrounding non-activated or weakly activated (that is, Fos-negative) neurons 61–63. Once β-galactosidase is induced in neurons that are activated, these neurons can then be inactivated through injection of the inactive prodrug Daun02 28, 64–68. Daun02 is catalyzed by β-galactosidase into daunorubicin, which inactivates the previously activated neurons through two potential mechanisms: apoptotic cell death 65, 66 or blockade of voltage-dependent calcium channels 69. Thus, in our experiments, Daun02 injections disable those neurons that were activated by the cue, context or drug and thus presumably the neuronal ensemble that encodes the association between the cue or context and the drug24, 62, 63. On the test day, typically 3 days after Daun02 injections, we assess whether these injections decrease the ability of the drug-associated cues or contexts, or the ability of the drug itself, to reactivate the same drug-related neuronal ensemble and induce a conditioned response or drug seeking. A critical control condition that is required to show a causal role of neuronal ensembles in the drug-related behaviour is that Daun02 injections after exposure to a non-drug-associated cue or context or to a novel context inactivates neurons distinct from those in the drug-related neuronal ensemble and should, therefore, have no effect on the drug-related behaviour on the test day.
We first used the Daun02 inactivation method 28 to demonstrate a causal involvement for neuronal ensembles in the nucleus accumbens in context-specific sensitization of cocaine-induced locomotion 15, 70, 71. We first demonstrated context-specific activation of accumbens neurons by training a group of rats to associate cocaine (7 daily injections) with one context (A) and another group of rats to associate cocaine with a different context (B). After 7 withdrawal days, we injected rats with cocaine or saline in context A and perfused them 90 min later to assess Fos expression in the nucleus accumbens. Cocaine injections in test context A enhanced (sensitized) cocaine-induced locomotion and accumbens Fos expression in rats that were previously injected with cocaine in the same context A, but not in rats that were previously injected with cocaine in the different context B. Double-labelling immunohistochemistry for Fos and the neuronal marker NeuN showed Fos expression in ~3% of accumbens neurons 72. To assess a causal role for this minority of activated Fos-expressing neurons in context-specific cocaine sensitization, we trained c-fos-lacZ transgenic rats to associate context A with cocaine (7 daily injections). After 7 withdrawal days, we injected separate groups of rats with cocaine in context A or in a novel context B, and then injected Daun02 or vehicle into the accumbens 90 min later. Three days later, on the test day, we found that prior Daun02 inactivation of accumbens neurons attenuated cocaine-induced locomotor sensitization and neuronal activation (assessed by c-fos promoter-induced β-galactosidase expression) when Daun02 was injected following cocaine administration in context A, but not when it was injected following cocaine administration in the novel context B 28. Together, these results indicate that context-specific locomotor sensitization to cocaine is mediated by context-specific selection of accumbens neuronal ensembles that are comprised of a small proportion of sparsely distributed neurons.
In our second application 67 of the Daun02 inactivation method, we demonstrated a causal role for neuronal ensembles in the ventral mPFC in context-induced reinstatement of drug seeking, an animal model of relapse induced by exposure to the drug-associated environment 73. We first trained rats to self-administer (by lever pressing) heroin in context A and extinguished lever pressing in context B. On the test day, 14+ days later, re-exposure to the heroin context (A), but not the extinction context (B), increased heroin seeking and increased Fos expression in ~6% of the ventral mPFC neurons. To assess a causal role for this minority of activated Fos-expressing neurons in context-induced reinstatement, we trained c-fos-lacZ transgenic rats to self-administer heroin in context A and extinguished lever pressing in context B. On induction day, 14+ days later, separate groups of rats were exposed to either the heroin context (A) or the extinction context (B) for 30 min and Daun02 or vehicle was injected into the ventral mPFC 90 min after the beginning of context exposure. On the test day, 3 days later, we found that prior Daun02-induced inactivation of the ventral mPFC decreased context-induced reinstatement and neuronal activation when Daun02 was injected following exposure to the heroin-associated context (A), but not the when it was injected after exposure to the extinction context (B). Of note, the magnitude of inhibition of context-induced reinstatement by Daun02 injections was similar to that observed following ventral mPFC injections of a mixture of GABAA and GABAB agonists (muscimol and baclofen, respectively) to reversibly inactivate this brain area 5–10 min before the reinstatement tests67. Together, these results indicate that a small subset of ventral mPFC neurons form neuronal ensembles that encode the learned association between heroin reward and the context in which the drug is self-administered.
In our most recent application 68 of the Daun02 inactivation method, we demonstrated a causal role for neuronal ensembles in the orbitofrontal cortex (OFC) in incubation of drug craving (as indicated by time-dependent increases in cue-induced drug seeking after withdrawal from the drug) 74–76. We trained rats to self-administer heroin (6-h/d for 10 d; drug infusions were paired with discrete light cue) and assessed cue-induced heroin seeking in extinction tests after 1 or 14 days of withdrawal. Cue-induced heroin seeking increased from 1 day to 14 days (incubation of heroin craving) and was accompanied by increased Fos expression in ~12% of OFC neurons on withdrawal day 14. To assess a causal role for this minority of activated Fos-expressing OFC neurons in heroin craving, we trained c-fos-lacZ transgenic rats to self-administer heroin. On induction day, 11 days later, we re-exposed these rats to the light cue in the heroin-associated context or to a novel context without the light cue for 15 min and injected Daun02 or vehicle into OFC 90 min after the beginning of context exposure. On the test day, 3 days later, we found that prior Daun02 inactivation of OFC neurons decreased cue-induced heroin seeking and OFC neuronal activation when Daun02 was injected following re-exposure to the heroin-associated cues, but not when Daun02 was injected following exposure to the novel context68. Non-selective inactivation of OFC neurons with muscimol and baclofen also decreased cue-induced heroin-seeking on withdrawal day 14 (but not on day 1) 68. These results indicate that heroin-cue-activated OFC neuronal ensembles play a causal role in persistent responding to heroin cues after withdrawal and incubation of heroin craving.
Taken together, the Daun02 inactivation procedure can be used to study the role of neuronal ensembles in the motivational effects of drug cues and contexts. However, the method has some limitations and unresolved issues. The Daun02 method, like all c-fos promoter-based methods, cannot manipulate behaviours that are dependent on a lower level of neuronal ensemble activity than that required to activate the c-fos promoter. Additionally, the method is limited to brain areas in which Fos and β-galactosidase are highly co-expressed (such as the striatum and the mPFC) and cannot be used to assess neuronal ensemble activity in brain areas in which co-expression is moderate or low (for example, the thalamus, unpublished observations). Furthermore, we have yet to assess a time course for Daun02 inactivation beyond three days and do not know the detailed molecular and cellular mechanism involved in Daun02 inactivation. If daunorubicin in activated neurons ablates these neurons through apoptosis, then some collateral effects of this apoptosis might be expected on the surrounding neurons. However, evidence suggests this is unlikely: as shown above, Daun02 inactivation of neuronal ensembles that were activated by non-paired (B) or novel contexts did not decrease lever pressing or neuronal activity in the training context (A) 28, 67, 68; additionally, prior Daun02 inactivation of the cocaine-activated nucleus accumbens ensemble in paired context A had no collateral effect on the ability of intra-accumbens injections of a cocktail of AMPA+picrotoxin to activate all accumbens neurons 28.
FACS sorting of activated Fos-expressing neurons
The studies described above indicate that selectively activated neuronal ensembles in accumbens and cortical areas have a causal role in context-specific sensitization of cocaine-induced locomotion, context-induced reinstatement of heroin seeking, and incubation of heroin craving. Thus, molecular alterations within these neuronal ensembles are likely to have unique and important roles in these drug-related learned behaviours. FACS can be used to analyze and purify Fos-expressing neurons for molecular analysis. In the flow cytometry component of FACS, brain tissue is enzymatically and mechanically dissociated into single cells, which are then fluorescently labelled with antibodies and forced to pass single-file through a narrow flow cell in a flow cytometer. In the cell-sorting component of FACS, cells are sorted as they leave the flow cell according to their light-scattering and immunofluorescent characteristics 77–80. We recently developed a FACS-based method to assess gene expression in activated Fos-expressing neurons 29, 81 (Figure 3). In this method, neurons are identified by labelling with NeuN antibodies, and activated versus non-activated neurons are identified according to their labelling with Fos or β-galactosidase antibodies. Below we describe the method, its application to studying drug-related behaviours, as well as its limitations. We used FACS to purify activated neurons from different brain areas in two drug-induced behavioural models: context-dependent cocaine sensitization and incubation of heroin craving 29, 68.
Figure 3. FACS sorting of activated neurons.

The FACS method is used for assessing unique molecular alterations within activated versus non-activated neurons. (A) In flow cytometry, including FACS, single cells are enzymatically dissociated from brain tissue and fluorescently labeled with different antibodies. Labelled samples are then forced to pass single file through a narrow flow cell. Absorbance of transmitted laser light for each particle is called Forward Scatter (FSC) light, whereas light scattered at a 90-degree angle is called Side Scatter (SSC) light. Each particle (cell or non-cell) is called an ‘event’. (B) Each event is indicated by a black dot in the Scattergram. The cluster of events in the lower part of the Scattergram corresponds to neurons that were subsequently selected (or ‘gated’) by the indicated triangle for further analyses of their fluorescence characteristics. Positively labelled events (e.g., Fos-positive cells) have high fluorescence levels (red circles), whereas negatively labelled events (e.g., Fos-negative cells) have low fluorescence levels (black circles). (C) These events are displayed in a Fluorescence scattergram. Rectangular gates are used to select positive events (e.g., Fos-positive and NeuN-positive cells) and negative events (e.g., Fos-negative and NeuN-positive cells) for collection using FACS. Droplets containing gated events can be programmed to receive an electric charge as they leave the flow cell. Magnetic plates direct the charged droplets and sort them into separate ‘positive’ or ‘negative’ sample tubes for further molecular analysis.
In the first study, we assessed unique alterations of cocaine-induced gene expression in activated versus non-activated striatal neurons29. We used FACS to purify activated (β-galactosidase-expressing) neurons 90 min after injections of cocaine in naïve and cocaine-sensitized c-fos-lacZ transgenic rats rats. We then compared gene expression in these cell populations to gene expression in all neurons from control rats that had received saline injections 29. Microarray and qPCR analyses indicated several unique alterations for gene expression levels of IEGs (markers of activity) and other genes within activated neurons. Expression of the IEGs arc, fosB and nr4a3 was higher in activated neurons from cocaine-injected rats than in non-activated neurons from the same cocaine-injected rats and in all neurons from saline-injected rats. Notably, gene expression was similar in the two control conditions: non-activated neurons from cocaine-injected rats and all neurons from saline-injected rats29.
In the second study, we used the method to assess unique alterations of heroin cue-induced gene expression in activated versus non-activated neurons following FACS purification of activated Fos-expressing neurons from the mPFC and OFC 82. Rats were trained to self-administer heroin as above and then remained in their home cages for 14–30 days. On the test day, we tested half of the rats for cue-induced heroin seeking in an extinction test (a test for incubation of craving after prolonged withdrawal) while the other half remained in their home cage (no-test rats). qPCR analyses indicated several unique alterations in gene expression levels for IEGs and other genes within activated neurons. Cue-induced heroin seeking increased the expression of the IEGs arc, fosB, egr1 and egr2 in activated neurons relative to levels in the non-activated neurons from the same ‘test’ rats or in all neurons from the ‘no test’ rats 82.
Taken together, in both studies IEGs were induced in activated neurons but not in non-activated neurons. This finding, along with our immunohistochemical findings 29, 82, supports the idea of sparse coding, in which only a small proportion of sparsely distributed neurons undergo the molecular and cellular alterations needed to encode conditioned drug effects, whereas the surrounding majority of neurons presumably play a much smaller role. As many of these IEGs are also transcription factors, it is likely that they can induce further alterations of gene expression within activated neurons that may play uniquely important roles in learned behaviours mediated by activated neuronal ensembles.
The FACS-based method has several limitations. We can only identify the relevant neuronal ensembles following their activation by acute drug or cue exposure on the test day. Thus, we cannot assess molecular alterations that were induced in these neurons during self-administration training prior to acute drug or cue exposure on the test day. In addition, it is not possible to manipulate genes selectively in these activated neurons to assess any potential causal roles for these genes in behaviour. We are currently developing methods to overcome these issues. Additionally, until recently, our FACS-based method required pooling the relevant brain areas, such as the striatum and PFC, from 6–10 rats. This makes the method less useful for time- and labour-intensive studies that require intravenous surgery and many weeks of behavioural training (including studies on mechanisms of drug reward and relapse). Furthermore, different sub-regions of striatum and frontal cortex are known to have different roles in the behavioural effects of drugs and non-drug rewards, and the cues and contexts associated with them 55, 83–85. To address this issue, we recently combined our existing FACS method 81 with Arcturus PicoPure RNA Kit and pre-amplification of the target genes to assess gene expression from as few as 5 Fos-positive neurons. These modifications enable us to reliably measure gene expression changes in a limited number of Fos-expressing neurons from a single dorsal striatum of rats injected with saline or methamphetamine 86. We are currently using this improved FACS-method to study unique molecular alterations in activated Fos-expressing accumbens and dorsal striatum neurons using the context-induced reinstatement of drug seeking model.
c-fos-GFP transgenic mice and rats
Alterations in synaptic efficacy, particularly within excitatory synapses, are regarded as the main cellular mechanism underlying learning and memory processes 87, 88, including those involved in drug addiction 51, 89. However, as discussed above, previous studies examined drug-induced or drug–cue-induced global alterations of synaptic efficacy in randomly selected neurons, regardless of their activation state.
c-fos-GFP transgenic mice were developed to assess unique electrophysiological characteristics of activated Fos-expressing cortical neurons during different behavioural states 90–94. These mice can also be used to study unique synaptic alterations in activated Fos-expressing neuronal ensembles during learned behaviours; we describe this use below, including its application to studying drug-related behaviours and its limitations. The transgene of these mice contains a c-fos promoter that induces expression of green fluorescent protein (GFP) to identify activated neurons in brain slice preparations. In our experience, confocal microscopy is necessary to visualize GFP-labelled neurons, because regular epifluorescence microscopy does not provide adequate sensitivity. Once a GFP-labelled neuron is identified, infrared differential interference contrast (DIC) microscopy is used to perform whole-cell patch electrophysiology.
We have used these cfos-GFP transgenic mice to assess unique synaptic alterations within activated neurons in the nucleus accumbens following context-specific cocaine sensitization 95. We previously found that activated Fos-expressing neuronal ensembles in the nucleus accumbens mediate context-specific sensitization of cocaine-induced locomotion 28. The main finding in our study was that cocaine sensitization, but not acute cocaine, produced higher levels of ‘silent synapses’ (synapses that contain functional NMDA receptors but no functional AMPA receptors 96) in activated (GFP-positive) neurons, and not in non-activated or weakly activated (GFP-negative) neurons. Interestingly, the silent synapses induced in activated, GFP-expressing neurons appear different from those previously observed in randomly selected nucleus accumbens neurons following repeated cocaine injections 97, 98. Specifically, NMDA receptors in silent synapses from randomly selected neurons were characterized by high levels of the NR2B subunit 98, whereas this was not the case for silent synapses in our activated, GFP-positive neurons 95. We hypothesize that silent synapses in GFP-positive neurons may be due to AMPA receptor endocytosis resulting from strong activation of these neurons. The data from this study95., together with the finding described above28, suggest that distinct synaptic alterations are induced in the activated nucleus accumbens neurons that mediate context-specific cocaine sensitization.
The c-fos-GFP transgenic mouse is an excellent tool for studying unique synaptic alterations in activated neuronal ensembles following relatively simple behavioural models used in the addiction field, such as locomotor sensitization and conditioned place preference. However, transgenic mice are not ideal subjects for complex studies of drug reward and relapse that are based on intravenous drug self-administration. For this reason, we developed a c-fos-GFP transgenic rat using the genetic construct described earlier 30 (Figure 4). In the initial neurobiological study with these transgenic rats, we adapted the classic reinstatement model of drug relapse 58 to study reinstatement of food seeking as an animal model of relapse during dieting 99. We assessed whether stress-induced reinstatement of palatable food seeking 100, which is dependent on dorsal mPFC activity 101, 102, is associated with unique synaptic alterations in this brain area. We found that reinstatement of food seeking induced by the pharmacological stressor yohimbine 103 was associated with reduced AMPAR/NMDAR current ratios (indicating reduced glutamatergic synaptic efficacy 104) and increased paired-pulse facilitation (indicating decreased synaptic glutamate release 105) in activated GFP-positive but not non-activated or weakly activated GFP-negative neurons 30.
Figure 4. Electrophysiology of activated neurons using the c-fos-GFP rat.
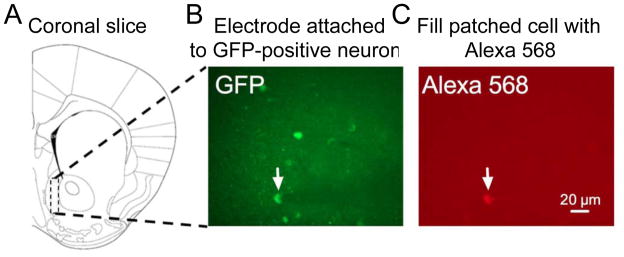
Assessing unique electrophysiological alterations within activated versus non-activated neurons. The cfos-GFP transgene in transgenic rats (or mice) contains a c-fos promoter that regulates transcription of the coding sequence for green fluorescent protein (GFP). Sufficiently strong and persistent neural activity activates the c-fos promoter which induces GFP in these strongly activated neurons but not in the surrounding majority of neurons. (A) Coronal slices are obtained for electrophysiological analysis. (B) GFP expression (induced by drug or cue exposure) can be used to guide the electrode to GFP-positive or GFP-negative neurons and attach it using differential interference contrast (DIC) optics. The arrow indicates a GFP-positive neuron with the shadow of the attached electrode to the right. (C) Alexa 568 in the electrode can diffuse into the attached cell to confirm that recorded cell was GFP-positive.
Taken together, these studies in c-fos-GFP transgenic rats and mice, as well as earlier studies 92, 93 demonstrate that these transgenic rodents are suitable for studying unique synaptic alterations in the minority of activated neuronal ensembles that presumably control learned behaviours. There are, however, several limitations of this approach. One limitation is that, as with the FACS procedure, we cannot assess synaptic alterations that were induced in these neurons during training prior to acute drug or cue exposure on test day. Nor can we manipulate these altered synaptic mechanisms selectively in activated neurons to assess their causal roles in learned behaviours. Another limitation is that combining behavioural studies with synaptic physiology of activated neurons is technically challenging, because of the difficulties associated with identifying a sufficient number of GFP-positive neurons in a slice preparation. This difficulty arises from the fact that only a minority of neurons expresses GFP and this expression is transient, lasting only a few hours. Additionally, to date, we have only used this approach after pharmacological activation (using cocaine or yohimbine) that induces significantly stronger neuronal activation than that induced by exposure to drug or food cues. Thus, we have not yet established that c-fos-GFP rats can be used for studies of conditioned drug effects and cue-induced relapse to drug or food seeking. As is the case with c-fos-lacZ rats, electrophysiological studies with c-fos-GFP rats and mice are limited to behaviours that increase neural activity enough to activate the c-fos promoter and induce a high level of co-expression of Fos and GFP in the brain areas of interest.
Neuronal ensembles in fear conditioning
The neuronal ensembles hypothesis has been the inspiration for many studies on neuronal mechanisms of fear conditioning and extinction 13, 106. As in other neuroscience disciplines, most published work on this topic was derived from correlational studies between behaviour and in vivo electrophysiology 107, 108 or cellular imaging 109–112 (Box 1) methods. Recently, investigators have developed two methods to manipulate putative neuronal ensembles and examine their causal role in conditioned fear 18, 113. We describe these methods below (Figure 5).
Figure 5. Manipulating activated fear-encoding neuronal ensembles in the hippocampus and amygdala.

(A) The c-fos-tTA transgene in transgenic mice contains a c-fos promoter that regulates transcription of the coding sequence for the tet-off transcriptional activator (tTA) protein. Sufficiently strong and persistent neural activity during a particular learned behaviour induces tTA in these strongly activated neurons but not in the surrounding majority of neurons. Doxycycline provided to the mice (commonly through the diet) inactivates tTA transcriptional activity. When doxycycline is removed from the diet, the tTA can bind to the tet operator and activate a second transgene (viral or genomic) that expresses the optogenetic activating protein channel rhodopsin-2 (ChR2) or the pharmacogenetic inactivating DREADD receptor (hM3Dq) in those neurons that were previously activated during the behaviour. Blue light activates and manipulates the ChR2-expressing neurons, whereas clozapine-N-oxide (CNO) activates hM3Dq-expressing neurons that were activated (red) during subsequent behavioural tests. (B) A herpes simplex virus (HSV) transgene contains a constitutively activated HSV IE4/5 promoter that drives the expression of two genes encoding GFP-CREB fusion protein and Cre recombinase, separated by an internal ribosome entry site (IRES) (cds indicates coding DNA sequence). Over-expression of GFP-CREB increases the sensitivity of neurons to synaptic input (red circles). Cre recombinase in the same neurons recognizes loxP DNA sequences in the DTR transgene of transgenic mice to cut out the Stop DNA sequence; this permits constitutive rosa26 promoter-induced expression of diphtheria toxin receptor (DTR) protein. Subsequent injections of Diphtheria toxin ablate DTR-expressing neurons. (C) A herpes simplex virus (HSV) transgene contains two separate genes: one uses a constitutively activated HSV IE4/5 promoter to drive the expression of the gene encoding GFP-CREB fusion protein, and the other gene uses a cytomegalovirus immediate early gene promoter (CMV) to drive expression of the gene (Alstr) that encodes the drosophila Allatostatin receptor. The same neurons (red circles) over-express GFP-CREB, which increases both the sensitivity of neurons (red circles) and the expression of the Allatostatin receptor. Subsequent site-specific injections of the Allatostatin peptide can inactivate these neurons during a behavioural test.
Manipulation of neuronal ensembles in c-fos-tTA transgenic mice
In two recent studies, c-fos-tTA transgenic mice 16, 114 were used in combination with optogenetic or DREADD methods to examine causal roles of neuronal ensembles in fear conditioning 19, 33. As described in Box 1, c-fos induction of the tTA gene is repressed by doxycycline in the drinking water prior to learning. Doxycycline is removed before the first learning session, allowing tTA to be induced in neuronal ensembles that are activated during the learning task. The tTA activator protein can then bind to a tet operator in the promoter of a second transgene and drive the expression of this gene only in neurons that were activated (Fos-positive) during the learning task. Investigators have used optogenetic or DREADD genetic constructs as the second transgenes to selectively reactivate these neurons during tests for the expression of fear learning 19, 33.
Liu et al. 19 used the c-fos-tTA transgenic mice to determine causal involvement of hippocampal neuronal ensembles in Pavlovian fear conditioning. They tested whether reactivation of neuronal ensembles in the dentate gyrus that were activated during learning was sufficient for fear memory recall, operationally defined as increased freezing. The experimental group of consisted of c-fos-tTA mice that were injected with an adeno-associated virus (AAV) expressing ChR2-EYFP and implanted with an optical fibre in the dentate gyrus. Mice were kept on doxycycline during habituation days, so that their basal freezing levels in context A could be determined during light-off and light-on epochs of optical stimulation. The mice showed little freezing during these habituation sessions prior to fear conditioning. Mice were then taken off doxycycline and underwent tone (CS)–shock (UCS) pairing (fear conditioning) in context B to induce ChR2 expression in Fos-positive neurons that were activated during fear conditioning. Mice were given doxycycline again and tested during light-off and light-on epochs in context A. ChR2 activation by optical stimulation induced reactivation of the neurons that had previously been activated during fear conditioning in context B and produced increased freezing behaviour in context A. In an important control condition, after habituation with doxycycline in context A, the authors removed doxycycline and induced ChR2 in neuronal ensembles activated by exposure to a novel context C in the absence of fear conditioning. Doxycycline was then given to the mice before fear conditioning in context B. ChR2-induced activation of context C-related ensembles in these mice did not produce higher levels of freezing in context A, presumably because this ensemble was not associated with fear conditioning19.
The results of this elegant study 19 suggest a causal role for dentate gyrus neuronal ensembles in the formation of stable fear memories. This conclusion would be strengthened if it could be shown that halorhodopsin-dependent inhibition of the neuronal ensembles that were previously activated during fear conditioning in context B (and that presumably encode the fear memory) prevent tone-cue-induced freezing in context B. In other words, although the authors showed that activation of dentate gyrus neuronal ensemble is ‘sufficient’ for reactivating a fear memory, it is unknown whether endogenous activity of this putative ensemble is ‘necessary’ for encoding the fear memory.
In a second study, Garner et al. 33 used the c-fos-tTA transgenic mice with DREADD technology to activate Fos-expressing fear-encoding neurons in the brain. These transgenic mice have two transgenes that are widely expressed in many brain areas. The first transgene is the c-fos-tTA described above. The second transgene contains a tet operator that drives tTA-dependent expression of hM3Dq, an artificial Gq-coupled receptor that binds the drug clozapine-N-oxide (CNO), which is typically injected systemically to activate neurons expressing this receptor 115. One of the experiments of this study33 was similar to that used in the earlier study 19. Specifically, doxycycline was removed for 2 days, after which mice underwent fear conditioning training in context B, so that the hM3Dq protein was induced in neurons that were activated during the fear conditioning. On the test day, the mice were placed in a novel context (A) and CNO was systemically injected to activate hM3Dq and thereby reactivate those neurons that were previously activated during fear conditioning in context B. However, CNO did not induce freezing in context A, suggesting that DREADD-mediated reactivation of neurons paired with fear conditioning was not sufficient to recall the fear memory. The authors also performed several experiments suggesting that co-activation of artificially induced neuronal ensembles can interfere with naturally produced cue-activated neuronal ensembles. It is beyond the scope of this article to describe these other experiments, because they did not directly test a causal role for neuronal ensembles in conditioned fear.
The reasons for the different results between the two studies19, 33 are unknown. Optogenetic activation of neurons may be stronger than DREADD-based activation of neurons. Beyond methodological differences related to the fear conditioning procedures, a very important difference is that one study 19 reactivated neuronal ensembles in only the dentate gyrus, whereas the other 33 reactivated ensembles in multiple brain areas, which may interfere or mask the expression of conditioned fear. Finally, it should be noted that in a different study116 using c-fos-tTA mice, the number of tTA-induced GFP-expressing hippocampal neurons was less than the number of Fos-expressing neurons following context-induced reactivation of fear on test day. This finding suggests that tTA activation in other studies using c-fos-tTA mice may underestimate the number of neurons that are activated during fear conditioning.
Inactivation of CREB-overexpressing neurons
Viral overexpression of CREB combined with diphtheria toxin or activation of allatostatin receptors for subsequent inactivation of CREB-overexpressing neurons has been proposed as a method to study neuronal ensembles in lateral amygdala in fear conditioning 31, 32. Han et al. 4 used herpes simplex virus (HSV) to overexpress both CREB and Cre recombinase transgenes from the same viral construct in a small number of lateral amygdala neurons in transgenic mice carrying a Cre-inducible diphtheria toxin receptor (DTR) transgene. Subsequent injections of diphtheria toxin (which binds to DTR) can then selectively inactivate neurons that express DTR 31. In the first phase of the experiment, test mice that overexpressed CREB and DTR in the same neurons and control mice that overexpressed DTR but not CREB underwent fear conditioning in what was termed weak (one tone–shock pairing) or strong (two tone–shock pairings) training in one context. All mice were tested one day later for expression of fear conditioning (freezing) in a different context. Similar to results in a previous study 117, CREB overexpression in the test mice increased neural responsiveness of the CREB-overexpressing neurons, which leads to their preferential activation during fear learning and enhanced fear expression in the weak but not strong training condition (presumably the strong training condition already produced maximal levels of fear learning) 31. In the subsequent lesioning phase of the experiment, control and test mice were injected systemically with diphtheria toxin to ablate DTR-expressing neurons and subsequently tested for expression of fear conditioning. Diphtheria toxin reduced the expression of fear memory in test mice but not in control mice, in which a similar number of lateral amygdala neurons overexpressing DTR (but not CREB) were inactivated by diphtheria toxin.
In second study, Zhou et al. 32 used a similar strategy. They used HSV to overexpress both CREB and the inhibitory drosophila allatostatin receptor (which is not expressed in rodents) in the same neurons; the peptide allatostatin binds to this receptor to reversibly inactivate neurons. The authors used experimental methods similar to those described above 31 to demonstrate that following auditory fear learning, inactivation of CREB-overexpressing neurons by allatostatin decreased the expression of conditioned fear32.
Although these two studies successfully inactivated CREB-overexpressing neurons and decreased the expression of conditioned fear, the CREB overexpression method is not optimal for studying causal roles for neuronal ensembles in learned behaviours. This is because the ‘neuronal ensembles’ that are inactivated by this method are artificially selected CREB-sensitized neurons rather than neuronal ensembles that were naturally selected by cue or context exposure during fear conditioning training. That is, the neurons overexpressing CREB in these experiments are randomly selected during HSV infection, prior to any learning experience. CREB over-expression in these neurons results in the formation of a hypersensitive cell type with synaptic alterations that make them highly responsive to many stimuli 97, 118, 119. This leads to the formation of artificial ‘neuronal ensembles’ that probably have a very different composition than the putative endogenous neuronal ensembles that would be selected during the same learning experience. For comparison, in the c-fos promoter-based neuronal ensemble inactivation methods described above, only the neurons that are strongly activated by cues, contexts or drugs during learning are selected for subsequent inactivation. A promising future direction would be to combine the diphtheria toxin or allatostatin inactivation methods with c-fos promoter selection of neurons activated during learned behaviours.
Conclusions and future directions
We have discussed recent technical developments that make it possible to determine causal roles of putative activated neuronal ensembles in learned behaviours and to characterize molecular and synaptic physiology of the activated neurons. These methods are unique and largely orthogonal to current mainstream neuroscience research that followed the introduction of optogenetic- and DREADD-based methods to the field 23, 27. This is because the goal of most optogenetic and DREADD studies is to identify causal roles of specific cell types, receptors or cellular signalling molecules in a given brain area or a particular cell-specific projection in learned behaviour, independently of the activation state of the neurons.
The study of the causal involvement of neuronal ensembles in drug addiction and fear is in its infancy, and as discussed above, each method has its own limitations. The only relatively clear-cut demonstration of causal roles of endogenous neuronal ensembles in learned behaviours has come from studies using the Daun02 inactivation procedure, in which inhibition of a small proportion of activated neurons in the nucleus accumbens or cortex inhibited context-specific cocaine locomotor sensitization, context-induced reinstatement of heroin seeking, and incubation of heroin craving 28, 67, 68. By contrast, as discussed above, studies combining optogenetic or DREADD methods with c-fos-tTA mice either only demonstrated the ‘sufficiency’ (but not the necessity) of neuronal ensembles in conditioned fear 19 or did not provide clear evidence for a role of neuronal ensembles in fear conditioning 33. Additionally, although studies using CREB overexpression demonstrated that selective inactivation of a small proportion of activated neurons decreases the expression of fear conditioning 32, 117, the neurons to be activated during fear learning and subsequently inactivated were pre-selected by the HSV viral manipulation. Thus, the neuronal ensembles identified in these studies may not reflect the composition of the endogenous amygdala neuronal ensembles that are normally selected by the learning experience.
There are also important limitations in the methods that have been developed for examining molecular 29, 81 and synaptic physiology 30, 95 alterations that were induced in neuronal ensembles activated during prior learning or on test day. One main limitation is that we cannot assess the basal conditions of the putative neuronal ensembles prior to activation, because we must acutely induce GFP, β-galactosidase or Fos to identify the activated neurons. Thus, we cannot determine whether any observed alterations are due to prior learning during the training phase or due to acute expression of the learned behaviour on the test day. To solve this problem we need to identify neurons that are ‘destined’ to be activated and participate in an ensemble, prior to their activation on test day. The second limitation is that although we can potentially demonstrate causal roles of selectively activated neurons in learned behaviours, there are no tools to manipulate the molecular and synaptic alterations induced selectively in the activated neurons to demonstrate how these molecular or synaptic alterations affect learned behaviours or cell function. A third limitation is that the c-fos promoter-based techniques described above cannot identify groups of neurons that are inhibited or insufficiently activated by cues for c-fos promoter activation to occur. Therefore, an absence of c-fos activation in neurons does not necessarily imply that they were not active and have no role in the behaviour.
It may be possible to overcome the first limitation regarding basal conditions by using the c-fos-tTA mouse system or our recently developed c-fos-tet-Cre recombinase transgenic rat system (unpublished data; FCC, YS, BTH) in which previously activated neurons can be identified with a constitutively expressed molecular marker (e.g., GFP) that persists for many days after the last manipulation when basal conditions are re-established. A similar strategy for identifying previously activated neurons involves tamoxifen-sensitive Cre recombinase induced by either the c-fos or arc promoter 120. Additionally, a recent and promising technology uses transgenic mice with photoactivable GFP that, once activated, can be placed in a long-lasting fluorescent state that permit neurons to be identified at a later time for more detailed analysis using slice electrophysiology 121. One study 122 has recently achieved selective manipulations within activated neurons by using transgenic mice carrying both c-fos-tTA and GFP-GluA1 transgenes 123. The authors used the GFP–GluA1 construct to assess structural changes in dendritic spines in hippocampal neurons that were activated at the time of fear learning. The main finding was that spines on active (Fos-positive) but not those on inactive (Fos-negative) neurons of context-fear-conditioned mice were reduced 24 h after conditioned fear training, and this reduction did not occur in control mice (exposed to the context along or to an unpaired shock). To date, however, none of the published techniques described above have been used to define the interactions between neuronal ensembles in different brain areas that are active at the same time during learning and on test day that may form the circuitry underlying learned behaviour. It will also be important to expand on the present work using other activity-dependent promoters such as for arc, which has been used for two-photon imaging of integrated neural activity in arc-GFP mice 124, as well as for inducing Cre recombinase in activated neurons 120. The arc promoter is similar to the c-fos promoter, but generally has higher levels of basal activity and a lower threshold for activation 125.
Finally, it is perhaps too early to speculate about the clinical implications of a better understanding of the unique molecular and synaptic physiology alterations in behaviourally activated neuronal ensembles. Nevertheless, one potential implication is the shift in direction of medication development — from strategies that target specific receptor, cell type or signalling mechanism that are independent of the neuron’s activity status, to strategies that target specific mechanisms that are observed only in activated neuronal ensembles. For example, drugs like ketamine or memantine that bind preferably to activated NMDA receptors 126–128 can be used to potentially erase (via interference with memory reconsolidation 26, 129) or diminish the motivational impact of memories of drug-associated or fear-associated cues. Thus, giving ketamine or memantine immediately after exposing drug users or PTSD patients to drug- or trauma-associated cues to reactivate neuronal ensembles that encode the drug- or trauma-associated memory may diminish the motivational impact of these cues and decrease relapse.
Acknowledgments
The writing of this article was supported by the National Institute on Drug Abuse, Intramural Research Program. We would like to thank the members of the Hope, Lupica, and Shaham labs who contributed to the development and implementation of the new technologies described in this review.
References
- 1.Hebb D. The organization of behavior, a neuropsychological theory. Wiley Subscription Services, Inc, A Wiley Company; New York: 1949. [Google Scholar]
- 2.Schwindel CD, McNaughton BL. Hippocampal-cortical interactions and the dynamics of memory trace reactivation. Prog Brain Res. 2011;193:163–177. doi: 10.1016/B978-0-444-53839-0.00011-9. [DOI] [PubMed] [Google Scholar]
- 3.Nicolelis MA, Fanselow EE, Ghazanfar AA. Hebb’s dream: the resurgence of cell assemblies. Neuron. 1997;19:219–221. doi: 10.1016/s0896-6273(00)80932-0. [DOI] [PubMed] [Google Scholar]
- 4.Guzowski JF, Knierim JJ, Moser EI. Ensemble dynamics of hippocampal regions CA3 and CA1. Neuron. 2004;44:581–584. doi: 10.1016/j.neuron.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 5.Pennartz CM, Groenewegen HJ, Lopes da Silva FH. The nucleus accumbens as a complex of functionally distinct neuronal ensembles: an integration of behavioural, electrophysiological and anatomical data. Prog Neurobiol. 1994;42:719–761. doi: 10.1016/0301-0082(94)90025-6. [DOI] [PubMed] [Google Scholar]
- 6.Knierim JJ, Zhang K. Attractor dynamics of spatially correlated neural activity in the limbic system. Annu Rev Neurosci. 2012;35:267–285. doi: 10.1146/annurev-neuro-062111-150351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buzsaki G, Moser EI. Memory, navigation and theta rhythm in the hippocampal-entorhinal system. Nat Neurosci. 2013;16:130–138. doi: 10.1038/nn.3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Penner MR, Mizumori SJ. Neural systems analysis of decision making during goal-directed navigation. Progress in neurobiology. 2012;96:96–135. doi: 10.1016/j.pneurobio.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 9.Mountcastle VB. Modality and topographic properties of single neurons of cat’s somatic sensory cortex. J Neurophysiol. 1957;20:408–434. doi: 10.1152/jn.1957.20.4.408. [DOI] [PubMed] [Google Scholar]
- 10.John ER, Schwartz EL. The neurophysiology of information processing and cognition. Annu Rev Psychol. 1978;29:1–29. doi: 10.1146/annurev.ps.29.020178.000245. [DOI] [PubMed] [Google Scholar]
- 11.O’Keefe J. A review of the hippocampal place cells. Prog Neurobiol. 1979;13:419–439. doi: 10.1016/0301-0082(79)90005-4. [DOI] [PubMed] [Google Scholar]
- 12.Carelli RM, Deadwyler SA. Cellular mechanisms underlying reinforcement-related processing in the nucleus accumbens: electrophysiological studies in behaving animals. Pharmacology, biochemistry, and behavior. 1997;57:495–504. doi: 10.1016/s0091-3057(96)00442-x. [DOI] [PubMed] [Google Scholar]
- 13.Maren S. Neurobiology of Pavlovian fear conditioning. Annual review of neuroscience. 2001;24:897–931. doi: 10.1146/annurev.neuro.24.1.897. [DOI] [PubMed] [Google Scholar]
- 14.Guzowski JF, McNaughton BL, Barnes CA, Worley PF. Environment-specific expression of the immediate-early gene Arc in hippocampal neuronal ensembles. Nat Neurosci. 1999;2:1120–1124. doi: 10.1038/16046. [DOI] [PubMed] [Google Scholar]
- 15.Mattson BJ, et al. Context-specific sensitization of cocaine-induced locomotor activity and associated neuronal ensembles in rat nucleus accumbens. The European journal of neuroscience. 2008;27:202–212. doi: 10.1111/j.1460-9568.2007.05984.x. [DOI] [PubMed] [Google Scholar]
- 16.Reijmers LG, Perkins BL, Matsuo N, Mayford M. Localization of a stable neural correlate of associative memory. Science. 2007;317:1230–1233. doi: 10.1126/science.1143839. [DOI] [PubMed] [Google Scholar]
- 17.Guzowski JF, et al. Mapping behaviorally relevant neural circuits with immediate-early gene expression. Current opinion in neurobiology. 2005;15:599–606. doi: 10.1016/j.conb.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 18.Garner A, Mayford M. New approaches to neural circuits in behavior. Learn Mem. 2012;19:385–390. doi: 10.1101/lm.025049.111. [DOI] [PubMed] [Google Scholar]
- 19.Liu X, et al. Optogenetic stimulation of a hippocampal engram activates fear memory recall. Nature. 2012;484:381–385. doi: 10.1038/nature11028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grienberger C, Konnerth A. Imaging calcium in neurons. Neuron. 2012;73:862–885. doi: 10.1016/j.neuron.2012.02.011. [DOI] [PubMed] [Google Scholar]
- 21.Harvey CD, Collman F, Dombeck DA, Tank DW. Intracellular dynamics of hippocampal place cells during virtual navigation. Nature. 2009;461:941–946. doi: 10.1038/nature08499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ziv Y, et al. Long-term dynamics of CA1 hippocampal place codes. Nat Neurosci. 2013;16:264–266. doi: 10.1038/nn.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rogan SC, Roth BL. Remote control of neuronal signaling. Pharmacol Rev. 2011;63:291–315. doi: 10.1124/pr.110.003020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sweatt JD. The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem. 2001;76:1–10. doi: 10.1046/j.1471-4159.2001.00054.x. [DOI] [PubMed] [Google Scholar]
- 25.Alberini CM. Mechanisms of memory stabilization: are consolidation and reconsolidation similar or distinct processes? Trends Neurosci. 2005;28:51–56. doi: 10.1016/j.tins.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 26.Tronson NC, Taylor JR. Molecular mechanisms of memory reconsolidation. Nat Rev Neurosci. 2007;8:262–275. doi: 10.1038/nrn2090. [DOI] [PubMed] [Google Scholar]
- 27.Yizhar O, Fenno LE, Davidson TJ, Mogri M, Deisseroth K. Optogenetics in neural systems. Neuron. 2011;71:9–34. doi: 10.1016/j.neuron.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 28.Koya E, et al. Targeted disruption of cocaine-activated nucleus accumbens neurons prevents context-specific sensitization. Nat Neurosci. 2009;12:1069–1073. doi: 10.1038/nn.2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guez-Barber D, et al. FACS Identifies Unique Cocaine-Induced Gene Regulation in Selectively Activated Adult Striatal Neurons. J Neurosci. 2011;31:4251–4259. doi: 10.1523/JNEUROSCI.6195-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cifani C, et al. Medial prefrontal cortex neuronal activation and synaptic alterations after stress-induced reinstatement of palatable food seeking: a study using c-fos-GFP transgenic female rats. J Neurosci. 2012;32:8480–8490. doi: 10.1523/JNEUROSCI.5895-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Han JH, et al. Selective erasure of a fear memory. Science. 2009;323:1492–1496. doi: 10.1126/science.1164139. [DOI] [PubMed] [Google Scholar]
- 32.Zhou Y, et al. CREB regulates excitability and the allocation of memory to subsets of neurons in the amygdala. Nat Neurosci. 2009;12:1438–1443. doi: 10.1038/nn.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garner AR, et al. Generation of a synthetic memory trace. Science. 2012;335:1513–1516. doi: 10.1126/science.1214985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morgan JI, Curran T. Stimulus-transcription coupling in the nervous system: involvement of the inducible proto-oncogenes fos and jun. Annual review of neuroscience. 1991;14:421–451. doi: 10.1146/annurev.ne.14.030191.002225. [DOI] [PubMed] [Google Scholar]
- 35.Cohen S, Greenberg ME. Communication between the synapse and the nucleus in neuronal development, plasticity, and disease. Annu Rev Cell Dev Biol. 2008;24:183–209. doi: 10.1146/annurev.cellbio.24.110707.175235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Herdegen T, Leah JD. Inducible and constitutive transcription factors in the mammalian nervous system: control of gene expression by Jun, Fos and Krox, and CREB/ATF proteins. Brain Res Brain Res Rev. 1998;28:370–490. doi: 10.1016/s0165-0173(98)00018-6. [DOI] [PubMed] [Google Scholar]
- 37.Goldberg SR. Stimuli associated with drug injections as events that control behavior. Pharmacol Rev. 1976;27:325–340. [PubMed] [Google Scholar]
- 38.Stewart J, de Wit H, Eikelboom R. Role of unconditioned and conditioned drug effects in the self-administration of opiates and stimulants. Psychological review. 1984;91:251–268. [PubMed] [Google Scholar]
- 39.O’Brien CP, Ehrman RN, Ternes JW. Classical conditioning in human opioid dependence. In: Goldberg S, Stolerman I, editors. Behavioral analysis of drug dependence. Academic Press; Orlando: 1986. pp. 329–356. [Google Scholar]
- 40.Wikler A. Dynamics of drug dependence. Implications of a conditioning theory for research and treatment. Arch Gen Psychiatry. 1973;28:611–616. doi: 10.1001/archpsyc.1973.01750350005001. [DOI] [PubMed] [Google Scholar]
- 41.Siegel S. Drug anticipation and drug addiction. The 1998 H. David Archibald Lecture. Addiction. 1999;94:1113–1124. doi: 10.1046/j.1360-0443.1999.94811132.x. [DOI] [PubMed] [Google Scholar]
- 42.Carelli RM. The nucleus accumbens and reward: neurophysiological investigations in behaving animals. Behavioral and cognitive neuroscience reviews. 2002;1:281–296. doi: 10.1177/1534582302238338. [DOI] [PubMed] [Google Scholar]
- 43.Rebec GV, Sun W. Neuronal substrates of relapse to cocaine-seeking behavior: role of prefrontal cortex. Journal of the experimental analysis of behavior. 2005;84:653–666. doi: 10.1901/jeab.2005.105-04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schuster CR, Thompson T. Self administration of and behavioral dependence on drugs. Annu Rev Pharmacol. 1969;9:483–502. doi: 10.1146/annurev.pa.09.040169.002411. [DOI] [PubMed] [Google Scholar]
- 45.Chang JY, Zhang L, Janak PH, Woodward DJ. Neuronal responses in prefrontal cortex and nucleus accumbens during heroin self-administration in freely moving rats. Brain Res. 1997;754:12–20. doi: 10.1016/s0006-8993(97)00012-7. [DOI] [PubMed] [Google Scholar]
- 46.Kiyatkin EA, Rebec GV. Activity of presumed dopamine neurons in the ventral tegmental area during heroin self-administration. Neuroreport. 1997;8:2581–2585. doi: 10.1097/00001756-199707280-00032. [DOI] [PubMed] [Google Scholar]
- 47.Carelli RM, King VC, Hampson RE, Deadwyler SA. Firing patterns of nucleus accumbens neurons during cocaine self-administration in rats. Brain Res. 1993;626:14–22. doi: 10.1016/0006-8993(93)90557-4. [DOI] [PubMed] [Google Scholar]
- 48.Peoples LL, West MO. Phasic firing of single neurons in the rat nucleus accumbens correlated with the timing of intravenous cocaine self-administration. J Neurosci. 1996;16:3459–3473. doi: 10.1523/JNEUROSCI.16-10-03459.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carelli RM, Williams JG, Hollander JA. Basolateral amygdala neurons encode cocaine self-administration and cocaine-associated cues. J Neurosci. 2003;23:8204–8211. doi: 10.1523/JNEUROSCI.23-23-08204.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Root DH, Fabbricatore AT, Ma S, Barker DJ, West MO. Rapid phasic activity of ventral pallidal neurons during cocaine self-administration. Synapse. 2010;64:704–713. doi: 10.1002/syn.20792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bowers MS, Chen BT, Bonci A. AMPA receptor synaptic plasticity induced by psychostimulants: the past, present, and therapeutic future. Neuron. 2010;67:11–24. doi: 10.1016/j.neuron.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Russo SJ, et al. The addicted synapse: mechanisms of synaptic and structural plasticity in nucleus accumbens. Trends Neurosci. 2010;33:267–276. doi: 10.1016/j.tins.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wolf ME, Ferrario CR. AMPA receptor plasticity in the nucleus accumbens after repeated exposure to cocaine. Neuroscience and biobehavioral reviews. 2010;35:185–211. doi: 10.1016/j.neubiorev.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thomas MJ, Kalivas PW, Shaham Y. Neuroplasticity in the mesolimbic dopamine system and cocaine addiction. Br J Pharmacol. 2008;154:327–342. doi: 10.1038/bjp.2008.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kalivas PW, Volkow ND. The neural basis of addiction: a pathology of motivation and choice. Am J Psychiatry. 2005;162:1403–1413. doi: 10.1176/appi.ajp.162.8.1403. [DOI] [PubMed] [Google Scholar]
- 56.Shaham Y, Hope BT. The role of neuroadaptations in relapse to drug seeking. Nat Neurosci. 2005;8:1437–1439. doi: 10.1038/nn1105-1437. [DOI] [PubMed] [Google Scholar]
- 57.Mameli M, Luscher C. Synaptic plasticity and addiction: Learning mechanisms gone awry. Neuropharmacology. 2011 doi: 10.1016/j.neuropharm.2011.01.036. [DOI] [PubMed] [Google Scholar]
- 58.Shaham Y, Shalev U, Lu L, De Wit H, Stewart J. The reinstatement model of drug relapse: history, methodology and major findings. Psychopharmacology. 2003;168:3–20. doi: 10.1007/s00213-002-1224-x. [DOI] [PubMed] [Google Scholar]
- 59.Marchant NJ, Li X, Shaham Y. Recent developments in animal models of drug relapse. Curr Opin Neurobiol. 2013 doi: 10.1016/j.conb.2013.01.003. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pickens CL, et al. Neurobiology of incubation of drug craving. Trends Neurosci. 2011;34:411–420. doi: 10.1016/j.tins.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kasof GM, et al. Spontaneous and evoked glutamate signalling influences Fos-lacZ expression and pyramidal cell death in hippocampal slice cultures from transgenic rats. Brain research Molecular brain research. 1995;34:197–208. doi: 10.1016/0169-328x(95)00158-o. [DOI] [PubMed] [Google Scholar]
- 62.Kasof GM, et al. Kainic acid-induced neuronal death is associated with DNA damage and a unique immediate-early gene response in c-fos-lacZ transgenic rats. J Neurosci. 1995;15:4238–4249. doi: 10.1523/JNEUROSCI.15-06-04238.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kasof GM, Smeyne RJ, Curran T, Morgan JI. Developmental expression of Fos-lacZ in the brains of postnatal transgenic rats. Brain research Developmental brain research. 1996;93:191–197. doi: 10.1016/0165-3806(96)00005-3. [DOI] [PubMed] [Google Scholar]
- 64.Bakina E, Farquhar D. Intensely cytotoxic anthracycline prodrugs: galactosides. Anti-cancer drug design. 1999;14:507–515. [PubMed] [Google Scholar]
- 65.Farquhar D, et al. Suicide gene therapy using E. coli beta-galactosidase. Cancer chemotherapy and pharmacology. 2002;50:65–70. doi: 10.1007/s00280-002-0438-2. [DOI] [PubMed] [Google Scholar]
- 66.Ghosh AK, Khan S, Marini J, Nelson JC, Farquhar D. A daunorubicin β-galactoside prodrug for use in conjunction with gene-directed enzyme prodrug therapy. Tetrahedron Letters. 2000;41:4871–4874. [Google Scholar]
- 67.Bossert JM, et al. Ventral medial prefrontal cortex neuronal ensembles mediate context-induced relapse to heroin. Nat Neurosci. 2011 doi: 10.1038/nn.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fanous S, et al. Role of orbitofrontal cortex neuronal ensembles in the expression of incubation of heroin craving. J Neurosci. 2012;32:11600–11609. doi: 10.1523/JNEUROSCI.1914-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Santone KS, Oakes SG, Taylor SR, Powis G. Anthracycline-induced inhibition of a calcium action potential in differentiated murine neuroblastoma cells. Cancer research. 1986;46:2659–2664. [PubMed] [Google Scholar]
- 70.Badiani A, Robinson TE. Drug-induced neurobehavioral plasticity: the role of environmental context. Behavioural pharmacology. 2004;15:327–339. doi: 10.1097/00008877-200409000-00004. [DOI] [PubMed] [Google Scholar]
- 71.Stewart J, Badiani A. Tolerance and sensitization to the behavioral effects of drugs. Behav Pharmacol. 1993;4:289–312. [PubMed] [Google Scholar]
- 72.Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201–211. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- 73.Crombag H, Bossert JM, Koya E, Shaham Y. Context-induced relapse to drug seeking: a review. Trans R Soc Lond B: Biol Sci. 2008;363:3233–3243. doi: 10.1098/rstb.2008.0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Grimm JW, Hope BT, Wise RA, Shaham Y. Incubation of cocaine craving after withdrawal. Nature. 2001;412:141–142. doi: 10.1038/35084134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shalev U, Morales M, Hope B, Yap J, Shaham Y. Time-dependent changes in extinction behavior and stress-induced reinstatement of drug seeking following withdrawal from heroin in rats. Psychopharmacology. 2001;156:98–107. doi: 10.1007/s002130100748. [DOI] [PubMed] [Google Scholar]
- 76.Neisewander JL, et al. Fos protein expression and cocaine-seeking behavior in rats after exposure to a cocaine self-administration environment. J Neurosci. 2000;20:798–805. doi: 10.1523/JNEUROSCI.20-02-00798.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lobo MK. Molecular profiling of striatonigral and striatopallidal medium spiny neurons past, present, and future. International review of neurobiology. 2009;89:1–35. doi: 10.1016/S0074-7742(09)89001-6. [DOI] [PubMed] [Google Scholar]
- 78.Lobo MK, Karsten SL, Gray M, Geschwind DH, Yang XW. FACS-array profiling of striatal projection neuron subtypes in juvenile and adult mouse brains. Nat Neurosci. 2006;9:443–452. doi: 10.1038/nn1654. [DOI] [PubMed] [Google Scholar]
- 79.Dougherty JD, Geschwind DH. Progress in realizing the promise of microarrays in systems neurobiology. Neuron. 2005;45:183–185. doi: 10.1016/j.neuron.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 80.Karsten SL, Kudo LC, Geschwind DH. Gene expression analysis of neural cells and tissues using DNA microarrays. In: Crawley Jacqueline N, et al., editors. Current protocols in neuroscience. Unit 4. Chapter 4. 2008. p. 28. [DOI] [PubMed] [Google Scholar]
- 81.Guez-Barber D, et al. FACS purification of immunolabeled cell types from adult rat brain. Journal of neuroscience methods. 2012;203:10–18. doi: 10.1016/j.jneumeth.2011.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fanous S, et al. Unique gene alterations are induced in FACS-purified Fos-positive neurons activated during cue-induced relapse to heroin seeking. J Neurochem. 2013;124:100–108. doi: 10.1111/jnc.12074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Everitt BJ, Robbins TW. Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci. 2005;8:1481–1489. doi: 10.1038/nn1579. [DOI] [PubMed] [Google Scholar]
- 84.Kelley AE, Berridge KC. The neuroscience of natural rewards: relevance to addictive drugs. J Neurosci. 2002;22:3306–3311. doi: 10.1523/JNEUROSCI.22-09-03306.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ikemoto S. Dopamine reward circuitry: Two projection systems from the ventral midbrain to the nucleus accumbens-olfactory tubercle complex. Brain Res Rev. 2007;56:27–78. doi: 10.1016/j.brainresrev.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liu QR, et al. Detection of molecular changes in methamphetamine activated neurons of a single rat dorsal striatum using fluorescence-activated cell sorting (FACS) J Neurochem. 2013 doi: 10.1111/jnc.12381. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- 88.Martin SJ, Grimwood PD, Morris RG. Synaptic plasticity and memory: an evaluation of the hypothesis. Annual review of neuroscience. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- 89.Luscher C, Malenka RC. Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron. 2011;69:650–663. doi: 10.1016/j.neuron.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Benedetti BL, Takashima Y, Wen JA, Urban-Ciecko J, Barth AL. Differential Wiring of Layer 2/3 Neurons Drives Sparse and Reliable Firing During Neocortical Development. Cerebral cortex. 2012 doi: 10.1093/cercor/bhs257. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Clem RL, Barth A. Pathway-specific trafficking of native AMPARs by in vivo experience. Neuron. 2006;49:663–670. doi: 10.1016/j.neuron.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 92.Barth AL. Visualizing circuits and systems using transgenic reporters of neural activity. Current opinion in neurobiology. 2007;17:567–571. doi: 10.1016/j.conb.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Barth AL, Gerkin RC, Dean KL. Alteration of neuronal firing properties after in vivo experience in a FosGFP transgenic mouse. J Neurosci. 2004;24:6466–6475. doi: 10.1523/JNEUROSCI.4737-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yassin L, et al. An embedded subnetwork of highly active neurons in the neocortex. Neuron. 2010;68:1043–1050. doi: 10.1016/j.neuron.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Koya E, et al. Silent synapses in selectively activated nucleus accumbens neurons following cocaine sensitization. Nat Neurosci. 2012;15:1556–1562. doi: 10.1038/nn.3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Isaac JT, Crair MC, Nicoll RA, Malenka RC. Silent synapses during development of thalamocortical inputs. Neuron. 1997;18:269–280. doi: 10.1016/s0896-6273(00)80267-6. [DOI] [PubMed] [Google Scholar]
- 97.Brown TE, et al. A silent synapse-based mechanism for cocaine-induced locomotor sensitization. J Neurosci. 2011;31:8163–8174. doi: 10.1523/JNEUROSCI.0016-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Huang YH, et al. In vivo cocaine experience generates silent synapses. Neuron. 2009;63:40–47. doi: 10.1016/j.neuron.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nair SG, Adams-Deutsch T, Epstein DH, Shaham Y. The neuropharmacology of relapse to food seeking: methodology, main findings, and comparison with relapse to drug seeking. Prog Neurobiol. 2009;89:18–45. doi: 10.1016/j.pneurobio.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ghitza UE, Gray SM, Epstein DH, Rice KC, Shaham Y. The anxiogenic drug yohimbine reinstates palatable food seeking in a rat relapse model: a role of CRF(1) receptors. Neuropsychopharmacology. 2006;31:2188–2196. doi: 10.1038/sj.npp.1300964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nair SG, et al. Role of dorsal medial prefrontal cortex dopamine D1-family receptors in relapse to high-fat food seeking induced by the anxiogenic drug yohimbine. Neuropsychopharmacology. 2011;36:497–510. doi: 10.1038/npp.2010.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Calu DJ, et al. Optogenetic inhibition of dorsal medial prefrontal cortex attenuates stress-induced reinstatement of palatable food seeking in female rats. J Neurosci. 2013;33:214–226. doi: 10.1523/JNEUROSCI.2016-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bremner JD, Krystal JH, Southwick SM, Charney DS. Noradrenergic mechanisms in stress and anxiety: II.clinical studies. Synapse. 1996;23:39–51. doi: 10.1002/(SICI)1098-2396(199605)23:1<39::AID-SYN5>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 104.Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- 105.Mennerick S, Zorumski CF. Paired-pulse modulation of fast excitatory synaptic currents in microcultures of rat hippocampal neurons. J Physiol. 1995;488(Pt 1):85–101. doi: 10.1113/jphysiol.1995.sp020948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Quirk GJ, Mueller D. Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology. 2008;33:56–72. doi: 10.1038/sj.npp.1301555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pape HC, Pare D. Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol Rev. 2010;90:419–463. doi: 10.1152/physrev.00037.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Paz R, Pare D. Physiological basis for emotional modulation of memory circuits by the amygdala. Curr Opin Neurobiol. 2013 doi: 10.1016/j.conb.2013.01.008. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Barot SK, Chung A, Kim JJ, Bernstein IL. Functional imaging of stimulus convergence in amygdalar neurons during Pavlovian fear conditioning. PLoS One. 2009;4:e6156. doi: 10.1371/journal.pone.0006156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chung A, Barot SK, Kim JJ, Bernstein IL. Biologically predisposed learning and selective associations in amygdalar neurons. Learn Mem. 2011;18:371–374. doi: 10.1101/lm.2053711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hashikawa K, Matsuki N, Nomura H. Preferential Arc transcription at rest in the active ensemble during associative learning. Neurobiol Learn Mem. 2011;95:498–504. doi: 10.1016/j.nlm.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 112.Purgert RJ, Wheeler DS, McDannald MA, Holland PC. Role of amygdala central nucleus in aversive learning produced by shock or by unexpected omission of food. J Neurosci. 2012;32:2461–2472. doi: 10.1523/JNEUROSCI.5090-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Josselyn SA. Continuing the search for the engram: examining the mechanism of fear memories. J Psychiatry Neurosci. 2010;35:221–228. doi: 10.1503/jpn.100015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Reijmers L, Mayford M. Genetic control of active neural circuits. Frontiers in molecular neuroscience. 2009;2:27. doi: 10.3389/neuro.02.027.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Alexander GM, et al. Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron. 2009;63:27–39. doi: 10.1016/j.neuron.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tayler KK, Tanaka KZ, Reijmers LG, Wiltgen BJ. Reactivation of Neural Ensembles during the Retrieval of Recent and Remote Memory. Current biology : CB. 2013;23:99–106. doi: 10.1016/j.cub.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 117.Han JH, et al. Neuronal competition and selection during memory formation. Science. 2007;316:457–460. doi: 10.1126/science.1139438. [DOI] [PubMed] [Google Scholar]
- 118.Dong Y, et al. CREB modulates excitability of nucleus accumbens neurons. Nat Neurosci. 2006;9:475–477. doi: 10.1038/nn1661. [DOI] [PubMed] [Google Scholar]
- 119.Marie H, Morishita W, Yu X, Calakos N, Malenka RC. Generation of silent synapses by acute in vivo expression of CaMKIV and CREB. Neuron. 2005;45:741–752. doi: 10.1016/j.neuron.2005.01.039. [DOI] [PubMed] [Google Scholar]
- 120.Guenthner CJ, Miyamichi K, Yang HH, Heller HC, Luo L. Permanent Genetic Access to Transiently Active Neurons via TRAP: Targeted Recombination in Active Populations. Neuron. 2013;78:773–784. doi: 10.1016/j.neuron.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Peter M, et al. Transgenic mouse models enabling photolabeling of individual neurons in vivo. PLoS One. 2013;8:e62132. doi: 10.1371/journal.pone.0062132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sanders J, Cowansage K, Baumgartel K, Mayford M. Elimination of dendritic spines with long-term memory is specific to active circuits. J Neurosci. 2012;32:12570–12578. doi: 10.1523/JNEUROSCI.1131-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Matsuo N, Reijmers L, Mayford M. Spine-type-specific recruitment of newly synthesized AMPA receptors with learning. Science. 2008;319:1104–1107. doi: 10.1126/science.1149967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cao VY, et al. In Vivo Two-photon Imaging Of Experience-dependent Molecular Changes In Cortical Neurons. Journal of visualized experiments : JoVE. 2013 doi: 10.3791/50148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kawashima T, et al. Synaptic activity-responsive element in the Arc/Arg3.1 promoter essential for synapse-to-nucleus signaling in activated neurons. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:316–321. doi: 10.1073/pnas.0806518106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Johnson JW, Kotermanski SE. Mechanism of action of memantine. Current opinion in pharmacology. 2006;6:61–67. doi: 10.1016/j.coph.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 127.Sinner B, Graf BM. Ketamine. Handbook of experimental pharmacology. 2008:313–333. doi: 10.1007/978-3-540-74806-9_15. [DOI] [PubMed] [Google Scholar]
- 128.Wood PL. The NMDA receptor complex: a long and winding road to therapeutics. IDrugs : the investigational drugs journal. 2005;8:229–235. [PubMed] [Google Scholar]
- 129.Nader K, Schafe GE, LeDoux JE. The labile nature of consolidation theory. Nat Rev Neurosci. 2000;1:216–219. doi: 10.1038/35044580. [DOI] [PubMed] [Google Scholar]
- 130.Lerea LS, Butler LS, McNamara JO. NMDA and non-NMDA receptor-mediated increase of c-fos mRNA in dentate gyrus neurons involves calcium influx via different routes. J Neurosci. 1992;12:2973–2981. doi: 10.1523/JNEUROSCI.12-08-02973.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sgambato V, Abo V, Rogard M, Besson MJ, Deniau JM. Effect of electrical stimulation of the cerebral cortex on the expression of the Fos protein in the basal ganglia. Neuroscience. 1997;81:93–112. doi: 10.1016/s0306-4522(97)00179-6. [DOI] [PubMed] [Google Scholar]
- 132.Vazdarjanova A, Guzowski JF. Differences in hippocampal neuronal population responses to modifications of an environmental context: evidence for distinct, yet complementary, functions of CA3 and CA1 ensembles. J Neurosci. 2004;24:6489–6496. doi: 10.1523/JNEUROSCI.0350-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hope BT, et al. Induction of a long-lasting AP-1 complex composed of altered Fos-like proteins in brain by chronic cocaine and other chronic treatments. Neuron. 1994;13:1235–1244. doi: 10.1016/0896-6273(94)90061-2. [DOI] [PubMed] [Google Scholar]
- 134.Sheng M, Greenberg ME. The regulation and function of c-fos and other immediate early genes in the nervous system. Neuron. 1990;4:477–485. doi: 10.1016/0896-6273(90)90106-p. [DOI] [PubMed] [Google Scholar]
- 135.Hardingham GE, Chawla S, Johnson CM, Bading H. Distinct functions of nuclear and cytoplasmic calcium in the control of gene expression. Nature. 1997;385:260–265. doi: 10.1038/385260a0. [DOI] [PubMed] [Google Scholar]
- 136.Valjent E, Caboche J, Vanhoutte P. Mitogen-activated protein kinase/extracellular signal-regulated kinase induced gene regulation in brain: a molecular substrate for learning and memory? Molecular neurobiology. 2001;23:83–99. doi: 10.1385/MN:23:2-3:083. [DOI] [PubMed] [Google Scholar]
- 137.Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nature reviews Neuroscience. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- 138.Mattson BJ, et al. Cocaine-induced CREB phosphorylation in nucleus accumbens of cocaine-sensitized rats is enabled by enhanced activation of extracellular signal-related kinase, but not protein kinase A. Journal of neurochemistry. 2005;95:1481–1494. doi: 10.1111/j.1471-4159.2005.03500.x. [DOI] [PubMed] [Google Scholar]
- 139.LaHoste GJ, Yu J, Marshall JF. Striatal Fos expression is indicative of dopamine D1/D2 synergism and receptor supersensitivity. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:7451–7455. doi: 10.1073/pnas.90.16.7451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hardingham GE, Arnold FJ, Bading H. Nuclear calcium signaling controls CREB-mediated gene expression triggered by synaptic activity. Nat Neurosci. 2001;4:261–267. doi: 10.1038/85109. [DOI] [PubMed] [Google Scholar]
- 141.Deisseroth K, Mermelstein PG, Xia H, Tsien RW. Signaling from synapse to nucleus: the logic behind the mechanisms. Current opinion in neurobiology. 2003;13:354–365. doi: 10.1016/s0959-4388(03)00076-x. [DOI] [PubMed] [Google Scholar]
- 142.O’Donnell P. Dopamine gating of forebrain neural ensembles. The European journal of neuroscience. 2003;17:429–435. doi: 10.1046/j.1460-9568.2003.02463.x. [DOI] [PubMed] [Google Scholar]
- 143.Labiner DM, et al. Induction of c-fos mRNA by kindled seizures: complex relationship with neuronal burst firing. J Neurosci. 1993;13:744–751. doi: 10.1523/JNEUROSCI.13-02-00744.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kreuter JD, Mattson BJ, Wang B, You ZB, Hope BT. Cocaine-induced Fos expression in rat striatum is blocked by chloral hydrate or urethane. Neuroscience. 2004;127:233–242. doi: 10.1016/j.neuroscience.2004.04.047. [DOI] [PubMed] [Google Scholar]