

Abstract
The last two decades have witnessed a rise in the “NMDA receptor hypofunction” hypothesis for schizophrenia, a devastating disorder that affects around 1% of the population worldwide. A variety of presynaptic, postsynaptic and regulatory proteins involved in glutamatergic signaling have thus been proposed as potential therapeutic targets. This Review focuses on positive allosteric modulation of metabotropic glutamate 2 receptors (mGlu2Rs) and discusses how recent preclinical epigenetic data may provide a molecular explanation for the discrepant results of clinical studies, further stimulating the field to exploit the promise of mGlu2R as a target for schizophrenia treatment.
Keywords: Schizophrenia, mGlu2R, 5-HT2AR, PAMs, HDAC2, epigenetics
Schizophrenia: limitations with currently available drugs
Schizophrenia is a chronic debilitating mental disorder that affects approximately 1% of the general population. Symptoms vary from patient to patient but are generally categorized into positive (e.g., hallucinations, delusions, and disorganized speech and behavior), negative (e.g., social withdrawal, lack of motivation, flat affect), and cognitive (e.g., impairments in memory, attention, and executive function). These symptoms are typically associated with social and/or occupational dysfunction. Although the natural course is heterogeneous, the illness typically strikes in late adolescence or early adulthood and usually continues throughout life [1, 2]. The prognosis of patients is also variable, but is often poor with high rates of unemployment, homelessness, violence, and suicide [3, 4]. Given the combination of onset in early adulthood and the chronic course, schizophrenia presents a staggering drag on the economy and society. It has been estimated that the cost of schizophrenia in the U.S. in 2002 exceeded $62 billion [5] and the World Health Oganization ranks this disorder among the top 10 causes of disability in developed countries [6].
The classical dopamine (DA) hypothesis (see glossary) has dominated the theories of schizophrenia since the mid-20th century after the observation that first generation, or “typical”, antipsychotic drugs, such as chlorpromazine and haloperidol, are high-affinity antagonists of dopamine D2-receptors. Hyperactivity in the mesolimbic DA pathway was originally proposed to underlie positive symptoms of schizophrenia [7]. While effective against positive symptoms, typical antipsychotic drugs demonstrate limited efficacy against negative symptoms and cognitive impairments which have been shown to contribute to functional impairment and predict poor prognosis [8]. Moreover, these drugs are associated with a number of side effects including hyperprolactinemia, and extrapyramidal symptoms (EPS). The second generation, or “atypical”, antipsychotic drugs were introduced into the clinical practice in the early 1990s in an attempt to improve clinical efficacy and decrease side effects. Second generation drugs (such as clozapine, olanzapine, risperidone and quetiapine), which have less potential to induce EPS or hyperprolactinemia, differ pharmacologically from the typical ones in that they have less affinity for D2-receptors and higher affinity/functional interaction with other monoaminergic receptors, including the serotonin 2A receptor (5-HT2A) [2, 9]. Because of the dual dopamine-serotonin mechanism of action of atypical antipsychotic drugs, a serotonin hypothesis for schizophrenia was also proposed [10]. Although highly effective against a wider range of symptoms than typical agents, atypical antipsychotics still have modest efficacy against negative symptoms. In addition, recent clinical evidence does not support the notion that second generation drugs are superior to first generation in improving neurocognition [11]. Last but not least, atypical agents are associated with side effects, usually metabolic, and in rare cases severe, such as agranulocytosis seen with clozapine [2]. Despite the availability of many typical and atypical antipsychotic drugs, full functional remission is achieved in fewer than 35% of people with schizophrenia [12]. Moreover, most of the responders are “partial responders” with whom negative and cognitive symptoms remain problematic, suggesting that impairment and/or dysregulation in DA and serotonin signaling cannot fully account for the underlying pathology. The limitations of the presently available drugs underscore the need for identification of new antipsychotic compounds aiming new molecular targets. Here we focus on positive allosteric modulation of mGlu2R as a promising therapeutic strategy to treat schizophrenia.
Glutamate and schizophrenia
To account for negative and cognitive symptoms of schizophrenia, the DA hypothesis was expanded to postulate hypoactivity in the mesocortical pathway as well. Despite this expansion, the DA hypothesis still likely oversimplifies the neurocircuitry of schizophrenia and does not explain why the mesocortical DA pathway will be hypoactive while the mesolimbic DA pathway is hyperactive. If anything, this dichotomy suggests that either DA is partially involved in the molecular underpinnings of schizophrenia, or alternatively that multiple defective neurotransmitter systems eventually converge on and disrupt the DA system [7]. In an attempt to account for the shortcomings of the DA hypothesis, the “NMDA receptor hypofunction” hypothesis was proposed by Olney and Farber in 1995 [13] based on the observation that non-competitive NMDA antagonists such as phencyclidine (PCP), and ketamine induce a psychotomimetic state that closely resembles schizophrenia in healthy human individuals [14, 15] and exacerbate preexisting symptoms in schizophrenic patients [16, 17]. Although the pharmacology of cocaine and other amphetamine-like psychostimulants is complex [18, 19], they share the ability to bind dopamine transporters and increase synaptic levels of dopamine. Importantly, unlike amphetamine-like psychostimulants, glutamatergic PCP and ketamine dissociative drugs not only induce positive symptoms but also negative symptoms and cognitive dysfunction that better recapitulate the clinical syndrome of schizophrenia. Subsequently, PCP-like drug models have become widely employed in the search for novel treatments. It was proposed that defective NMDA receptors on cortical gamma aminobutyric acid (GABA) interneurons (Figure 1) render these interneurons less effective in inhibiting glutamate projection neurons that project to the ventral tegmental area (VTA). This disinhibition results in an excessive glutamate tone, which now over-stimulates the DA mesolimbic pathway giving rise to the positive symptoms. An alternative pathway that could be fundamental for cognition might be the functional crosstalk between NMDA and 5-HT2A receptors in pyramidal neurons of prefrontal cortex [20]. Similarly, negative and cognitive symptoms may arise from NMDA receptor hypofunction. Here hyperactive glutamate projection neurons over-activate GABAergic interneurons, located in the VTA, causing them to release excess GABA and overinhibit the mesocortical DA pathway that now becomes unable to adequately supply DA to the prefrontal cortex [7]. Thus, defective glutamate neurocircuitry might actually derive the excess DA in the mesolimbic pathway as well as the deficiency of DA in the mesocortical pathway. Additionally, the resulting glutamate excitotoxicity might drive an ongoing neurodegenerative process responsible for the structural brain changes seen in schizophrenic brains on volumetric MRI [21, 22], although the presence of neurodegeneration in schizophrenia remains controversial [23–27]. Interestingly, group II metabotropic glutamate receptors (mGluRs) are located presynaptically on these hyperactive glutamatergic neurons where they function as autoreceptors to help keep glutamate tone in check [7]. Activation of these receptors can thus attenuate schizophrenia symptoms and might prevent potential neurodegeneration. Recent findings suggest that the mGlu2Rs are also expressed postsynaptically, where they play a key role in the control of the responses induced by atypical antipsychotics [28–31]
Figure 1. Glutamate neurocircuitry implicated in Schizophrenia.
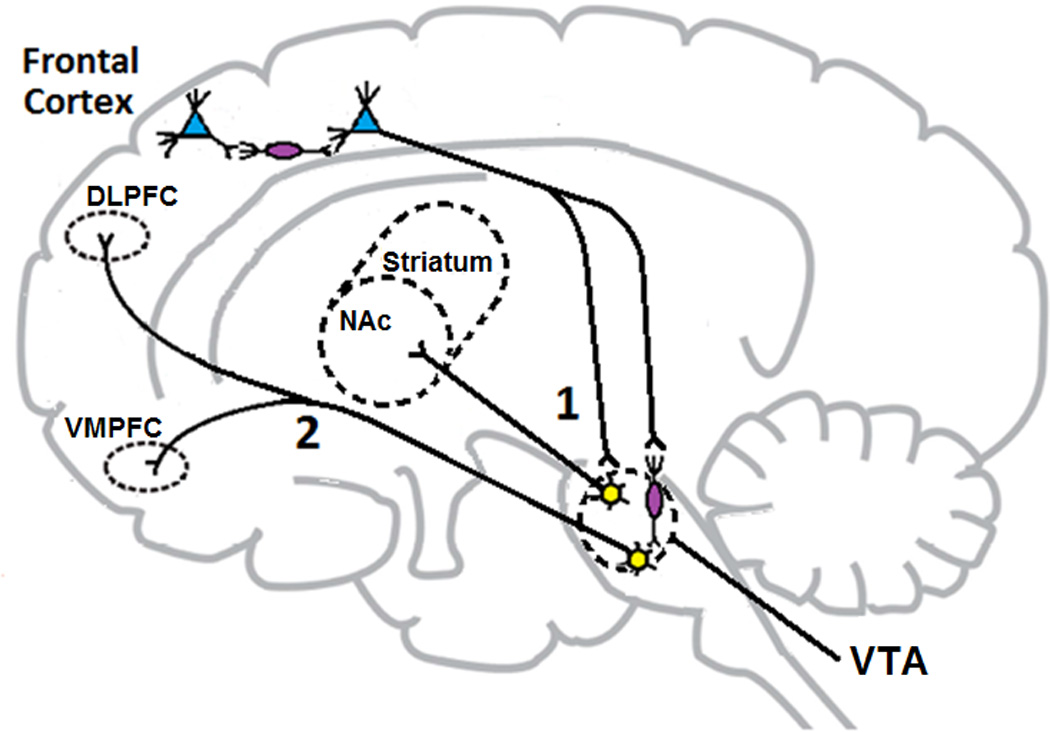
Normally, cortical GABA interneurons (in purple) exert an inhibitory tone on glutamate neurons that project to the VTA. When optimal, this inhibitory tone controls the amount of glutamate released in the VTA and subsequently the activity of the mesolimbic (1) and mesocortical (2) DA pathways. In schizophrenia, a defective NMDA receptor on the cortical GABA interneurons results in disinhibition of cortical brainstem glutamate projections. Excessive glutamate firing leads to overactivation of the mesolimbic DA pathway (1) and excessive release of DA in the nucleus accumbens. This might be responsible for development of positive symptoms in schizophrenic patients. Similarly, negative and cognitive symptoms may arise from NMDA receptor hypofunction. Hyperactive glutamate tone overactivates GABAergic interneurons in the VTA, overinhibiting the mesocortical DA pathway (2) that now becomes unable to adequately supply DA to the prefrontal cortex resulting in “hypofrontality”. Abbreviations: DLPFC, dorsolateral prefrontal cortex; NAc, nucleus accumbens, VMPFC: ventromedial prefrontal cortex; VTA, ventral tegmental area.
Additional evidence for involvement of glutamate in the pathophysiology of schizophrenia comes from postmortem studies, which revealed altered ionotropic glutamate receptor binding and gene expression mainly in the prefrontal cortex and the hippocampus [32–35]. Furthermore, functional neuroimaging studies have shown reduced NMDA binding in the hippocampus of medication-free schizophrenic patients compared to healthy controls [36]. More recently, proton magnetic resonance spectroscopy has revealed increased cortical glutamate levels in individuals with poor response to antipsychotic treatment in first-episode schizophrenia compared with those who were responders, supporting the hypothesis that poor responders to antidopaminergic therapy may have a glutamatergic basis for their psychosis [37]. Lastly, many genes involved in glutamatergic neurotransmission were found to be associated with schizophrenia in a recent genome wide association study (GWAS) involving a consortium of over 200 institutions worldwide in the largest molecular genetic study conducted to date for a neuropsychiatric disorder [38]. Taken together, the above evidence indicates an important role for dysfunctional glutamatergic neurotransmission in the pathophysiology of schizophrenia.
mGluRs and allosteric modulation
mGluRs are class C G protein-coupled receptors (GPCRs) that are characterized by a conserved heptahelical transmembrane domain (TMD), a large N-terminal extracellular domain (ECD), and a C-terminal intracellular domain. The large ECD, characteristic of family C GPCRs, consists of a Venus flytrap domain (VFD), which contains the orthosteric binding site for glutamate, and a cysteine-rich domain (CRD). Another distinguishing feature of class C GPCRs is constitutive homo- or heterodimerization at the cell-surface [39]. It has been demonstrated that class C mGluRs function as homodimers at the plasma membrane in living cells, whereas the class C GABAB receptor needs to form a heterodimeric complex composed of GABAB-R1 and GABAb-R2 to reach the plasma membrane as a functional receptor complex (for review, see [40]). The eight mGluR subtypes identified so far, are classified into three groups based on sequence homology, G protein–coupling, and pharmacology [41]. Group I mGluRs (mGlu1R and mGlu5R) are predominantly coupled to Gq/11 and activate the phospholipase C enzyme. Group II mGluRs (mGlu2R and mGlu3R) and group III (mGlu4R, mGlu6R, mGlu7R and mGlu8R) are coupled to Gi/o proteins and typically inhibit adenylyl cyclase activity [42]. Like the majority GPCRs, mGluRs have been classically targeted through their “orthosteric” site (i.e., the binding site recognized by the endogenous ligand). However, since all mGluR orthosteric ligands bind to the VFD, which is highly conserved among all eight mGluRs, subtype selectivity has been difficult to achieve especially for mGluR members from the same group. Moreover, CNS penetration for many of those orthosteric compounds has been limited by their poor physicochemical properties owing to their glutamate-like structures [43]. The difficulty to identify suitable glutamate analogs with adequate subtype selectivity and pharmacokinetic properties triggered the search for an alternative means to target mGluRs. A promising approach has been the use of allosteric modulators; ligands that bind to sites non-overlapping and spatially distinct from, but conformationally linked to, the orthosteric site (Figure 2) [44]. Mutagenesis studies have revealed that the majority of allosteric modulators identified for mGluRs bind to the TMD, which is relatively less conserved among mGluR subtypes than the VFD [45]. This has been recently confirmed by crystal structures for TMDs of both mGlu1R and mGlu5R bound by allosteric modulators [46, 47].
Figure 2. Allosteric vs Orthosteric.
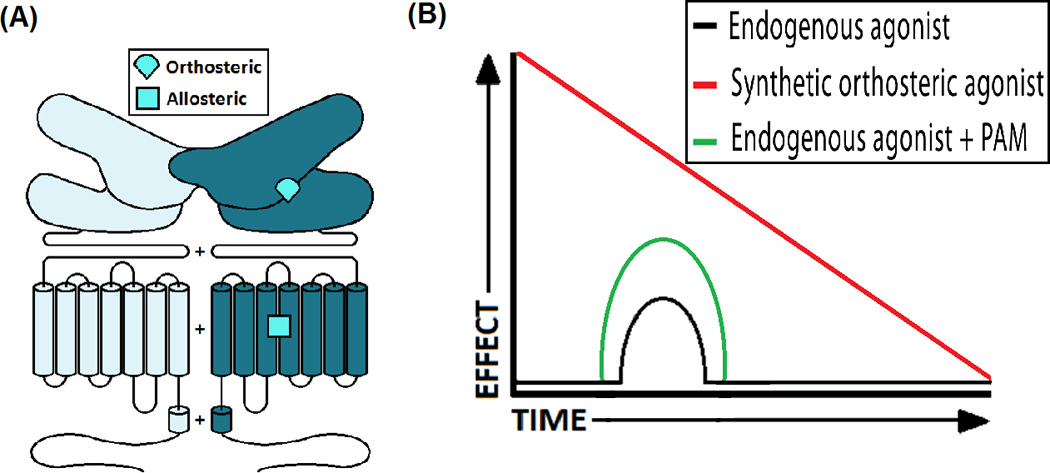
(A) A schematic of the dimeric structure of a class C GPCR. mGluRs, as well as other receptors in this class, are homodimers that use the VFD exclusively to bind orthosteric ligands. The TMD may have one or more potential pockets for allosteric ligands. (B) A synthetic orthosteric agonist often produces a bigger effect than the endogenous agonist however its effects may decline by time due to receptor desensitization and/or downregulation. On the other hand, PAM enhances the action of the endogenous agonist on its receptor in a more physiologic temporal pattern and thus is less likely to cause receptor desensitization and/or downregulation.
Allosteric modulators can modify the action of the orthosteric ligand by modulating its affinity and/or efficacy (Figure 3), either in a positive (in case of positive allosteric modulators or PAMs) or a negative direction (in case of negative allosteric modulators or NAMs). This phenomenon is referred to as “cooperativity” [48]. PAMs potentiate the response to an agonist and cause a leftward (and often an upward) shift in the concentration-response curve for the orthosteric agonist. Depending on the degree of stimulus-response coupling in an assay, a potentiator can behave as a pure PAM; an allosteric ligand that elicits no detectable response in the absence of the orthosteric agonist, or an ago-PAM that can directly elicit a response like an agonist [49]. NAMs, noncompetitively antagonize agonists and cause a rightward (and often downward) shift in the agonist concentration-response curves (Figure 3). Neutral allosteric ligands (previously referred to as silent allosteric modulators or SAMs) occupy the allosteric site without affecting the agonist responses on their own, however they can block the allosteric effects of both PAMs and NAMs [44].
Figure 3. Modes of action of allosteric modulators.
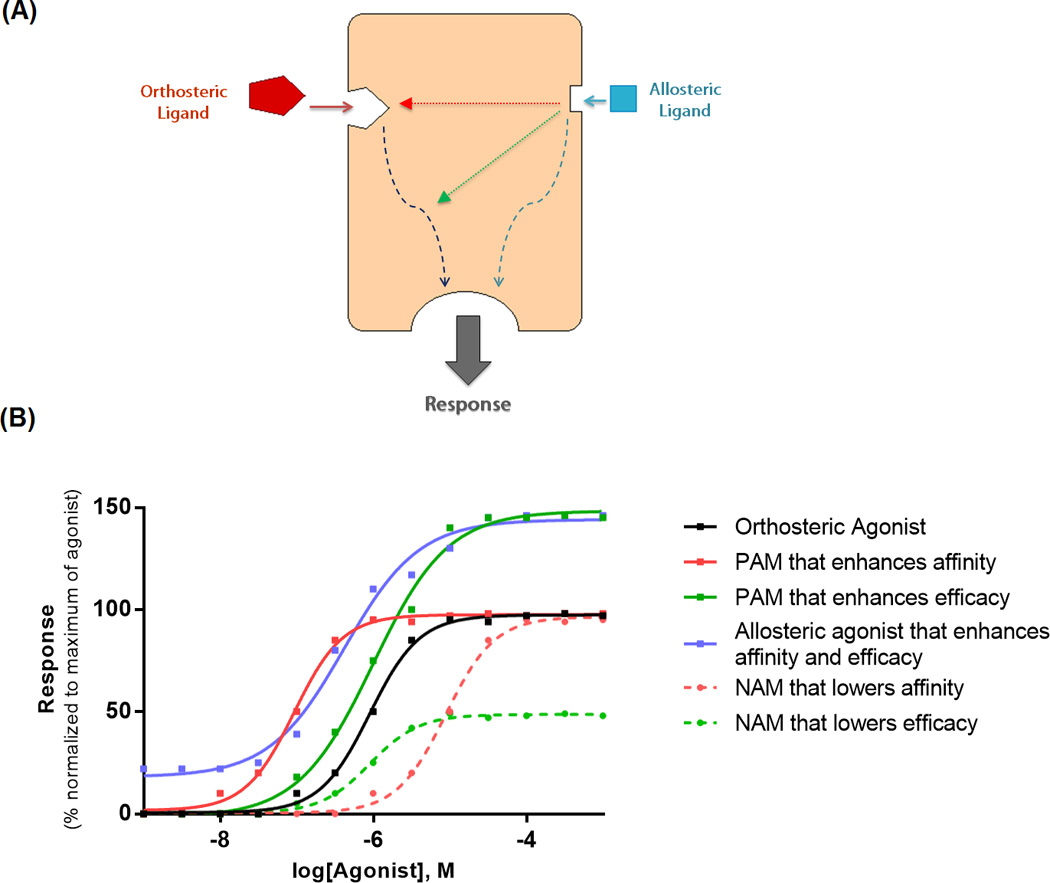
(A) The allosteric ligand binds to a site topographically distinct from the orthosteric binding site and modulates affinity (red) and/or efficacy (green) of the orthosteric ligand. Some allosteric ligands are capable of directly eliciting a response on their own (blue). Figure adapted from Conn et al. [35]. (B) Simple schematic representation of effects mediated by different allosteric ligands on the functional response of an orthosteric agonist. The first (solid red) is a PAM that purely enhances orthosteric agonist affinity as evident by an increase in potency (lower EC50) and leftward shift of concentration-response curve compared to the orthosteric agonist alone (solid black). The second (solid green) is a PAM that purely enhances orthosteric agonist efficacy as evident by an increase in Emax and upward shift in its concentration-response curve. The third (blue) demonstrates allosteric agonism that modulates both the affinity and efficacy. The fourth (dashed red) is a NAM that lowers the orthosteric agonist affinity and thus shifts its concentration-response curve to the right. The fifth (dashed green) is a NAM that lowers the orthosteric agonist efficacy and shifts its concentration-response curve downward. Figure adapted from Wootten et al. [101].
Advantages of allosteric modulators
Pure allosteric modulators have the advantage over synthetic orthosteric agonists in their ability to preserve the temporal and spatial patterns of receptor signaling since their effects are dependent on the presence of the endogenous ligand. This “state-dependent” mechanism of action explains, for example, why the ability of mGlu2R PAMs to inhibit striatal excitatory postsynaptic potentials (EPSPs) shows dependence on frequency of presynaptic stimulation of corticostriatal afferents. Unlike agonists, mGlu2R PAMs inhibit EPSPs only at high frequency stimulation indicating that excessive synaptic glutamate release is required for mGlu2R PAMS to exert their modulatory effect [50]. Because PAMs will not continuously activate the receptor, receptor desensitization and/or downregulation is less likely to occur, as opposed to synthetic orthosteric agonists. In addition, since no effect is expected at saturating concentrations above that determined by cooperativity, an allosteric drug will have a greater potential to fine-tune physiological responses (Figure 2) with less risk of toxicity [51]. This saturability phenomenon is referred to as the “ceiling level” of the allosteric effect. Lastly, mGluR allosteric modulators generally possess better physicochemical properties and blood brain barrier (BBB) permeability compared to orthosteric drugs, an important feature considering the therapeutic potential of mGluRs in neuropsychiatric disorders [52]
Challenges with allosteric modulators
Despite the above-mentioned advantages, a number of characteristics unique to GPCR allosteric modulators may represent a challenge in the translation of data from such agents. For example, the magnitude and direction of the allosteric effect mediated by the same modulator acting on the same receptor can vary depending on the orthosteric ligand that is used to probe receptor function, a phenomenon referred to as “probe dependence” [51]. Although examples have not been discovered yet for mGluRs [52], the use of glutamate as the orthosteric probe in in vitro assays might be more favorable since results are more likely to be of physiological relevance. Accordingly, any results obtained from assays using non-native agonists as probes should be interpreted cautiously since an allosteric drug may behave differently in vivo in the presence of the endogenous agonist [53]. Moreover, potential species differences in the responses to allosteric drugs may exist since the allosteric sites, presumably having undergone less evolutionary pressure to accommodate an endogenous ligand compared to orthosteric sites, are more likely to show sequence divergence between species [48]. Another challenge is that a number of mGluR allosteric modulator classes are prone to “molecular switches”, by which subtle structural changes within the scaffold can dramatically change the pharmacology of compounds within a class, switching them for example from NAMs to PAMs, PAMs to NAMs, or even change their receptor-subtype selectivity [54–56]. This raises questions over metabolism and pharmacology of metabolites as well [57]. All the above-mentioned complexities, together with the potential implications of mGluR heterodimer formation [58], are issues that need to be addressed before advancing any mGluR allosteric modulator into the clinic.
Potential for biased signaling
Although GPCR activation was first described by a classical two-state model where the receptor exists in an equilibrium between an active and an inactive state, recent evidence supports an alternative multi-state model where GPCRs can adopt multiple conformational states with each state possibly activating a discrete subset of cellular behaviors of a broader spectrum than just G protein coupling or second messenger activation [48, 59]. The ability of ligands to stabilize some unique conformational states activating certain cellular pathways and not others has been termed “biased signaling”, functional selectivity”, and “stimulus trafficking” [48, 60]. Biased allosteric modulation has been demonstrated for a number of mGluR allosteric ligands [61–63]. For instance, the gadolinium ion (Gd3+), an allosteric modulator of mGlu1αR, potentiates Gs-linked cAMP production yet inhibits Gq/11-linked Ca2+ mobilization when administered with glutamate [64]. Biased signaling can also involve non-G protein-mediated pathways such as the recruitment of β-arrestin. For instance, TRV130, a G protein-biased agonist of the µ-opioid receptor with minimal β -arrestin recruitment, was shown in mice to produce analgesic effects comparable to morphine with less respiratory depression and gastrointestinal dysfunction [65]. This strongly suggests that biased allosteric agonism and modulation may help selectively target signaling pathways critical for therapeutic efficacy while simultaneously excluding others associated with adverse effects (Figure 4).
Figure 4. Schematic for biased allosteric modulation.
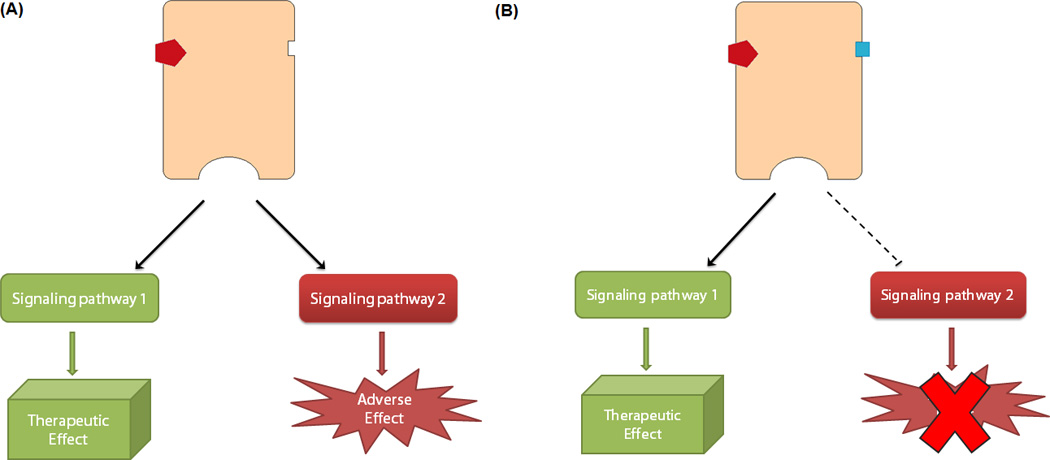
(A) An orthosteric agonist turns the receptor “on” activating signaling pathway 1, associated with therapeutic benefits, and simultaneously pathway 2, associated with adverse effects. (B) Co-binding of an allosteric modulator can bias the signaling of the agonist-bound receptor, selectively engaging pathway 1 while inhibiting pathway 2.
mGlu2R and schizophrenia
Group II mGluRs are widely expressed in the brain with generally similar distribution patterns in human and rodent brains [66]. mGlu2R is particularly expressed in regions known to be implicated in schizophrenia such as the prefrontal cortex, hippocampus, striatum, thalamus, and amygdala [67]. Compared to mGlu2R, mGlu3R shows a considerably more diffuse distribution pattern in the brain with both showing an overlapping cortical distribution with the 5-HT2A receptor [54]. At the cellular level, although mGlu2R is also found postsynaptically [30, 68], both receptors are located presynaptically, where they function as autoreceptors inhibiting glutamate release and modulating synaptic transmission [69]. In contrast to mGlu2R, mGlu3R is additionally expressed by astrocytes [70] where it might also be involved in a feedback mechanism to modulate neuronal excitability.
While the mGlu3R gene (GRM3) has been suggested by several independent groups [71–73] as well as by the recent GWAS data [38] to be implicated in schizophrenia, the majority of findings from postmortem human brains have not shown significant changes in mGlu3R expression in schizophrenia [74–77]. However, when testing either mGlu2 receptor density or mGlu2 mRNA expression in postmortem human brain samples, several reports have suggested both down-regulation [74, 78, 79] and up-regulation [75, 80] of mGlu2R (GRM2) expression in either antipsychotic-free or antipsychotic-treated schizophrenia patients as revealed by absence or presence of antipsychotics in postmortem toxicological analysis (see also [66, 77, 81, 82] for studies suggesting absence of changes in mGlu2R expression in postmortem human brains of schizophrenic subjects). A potential explanation for these discrepancies is that the expression and function of mGlu2R in schizophrenic subjects might be related to both the profound effects of age on mGlu2R density in cortical regions [74, 77] and of antipsychotic drug treatment on the promoter activity of the GRM2 gene (for further discussion, see below).
Group II mGluR agonists have been shown to reverse the effects of PCP on locomotion [83, 84], working memory, stereotypy, and cortical glutamate efflux [83] and to suppress the head-twitch response induced by 2,5-Dimethoxy-4-iodoamphetamine (DOI), a hallucinogenic 5-HT2A agonist, in rats [68]. Due to the lack of subtype-selective orthosteric ligands, it has been difficult to determine whether the antipsychotic-like effects of mGlu2/3R agonists are mediated via mGlu2R, mGlu3R, or both. Both experiments with knockout (KO) mice and experiments using selective mGlu2R PAMs suggested that these effects are predominantly mGlu2R–mediated. Indeed, the inhibitory effects of mGlu2/3R agonists on PCP- and amphetamine-evoked hyperactivity are absent in mGlu2R–KO but not in mGlu3R–KO mice [85, 86].
Recent evidence, however, suggests that mGlu3R may be a potential therapeutic target for schizophrenia-associated cognitive dysfunction since the mGlu3R NAMs VU0469942 and VU0477950 reverse the positive effects of the mGlu2/3R orthosteric agonist LY379268 on synaptic plasticity and learning abilities in mice [87].
Crosstalk between mGlu2 and 5-HT2A receptors
Not only does mGlu2R seem to be the target for many glutamate antipsychotics, but it has also been found to crosstalk with 5-HT2A receptor, a key target for atypical antipsychotics [30, 74, 88–93]. Interestingly, mGlu2R has been shown to be necessary for pharmacological and behavioral effects induced by hallucinogenic 5-HT2A agonists since these effects were abolished in mGlu2R–KO mice [94]. On the other hand, the locomotor activity induced by the mGlu2/3R antagonist LY341495 is attenuated in 5-HT2A-KO mice [74]. Moreover, chronic administration of the hallucinogenic 5-HT2A agonist 2,5-Dimethoxy-4-bromoamphetamine (DOB) in mice attenuates the behavioral effects of the mGlu2/3R agonist LY379268 [95] whereas chronic treatment by LY341495 decreases 5-HT2A binding and the hallucinogenic effects of LSD [96].
Indeed, mGlu2R and 5-HT2A were found to co-localize in mouse and human cortical pyramidal neurons and form a heterocomplex with unique signaling properties [30, 74]. Fribourg and colleagues [30] elucidated the functional coupling between mGlu2R which signals via a Gi heterotrimeric G protein, and 5-HT2AR which signals via Gq protein. Compared to the homomeric responses of each receptor to its endogenous neurotransmitter, heteromerization of mGlu2R with 5-HT2AR potentiates glutamate-induced mGlu2R–coupled Gi signaling and attenuates the 5-HT-induced 5-HT2AR-coupled Gq signaling. Moreover, drugs that stabilize the active or inactive conformation in one receptor cause the opposite conformation of the partner receptor. This inverse conformational coupling of the two receptors as parts of a heteromer unifies the mechanism of action of antipsychotic drugs targeting these receptors (Figure 5). Using the PCP-like drug MK801 as a model of psychosis, it was demonstrated that the mGlu2R–dependent antipsychotic-like behavioral effects of LY379268 were absent in 5-HT2AR-KO mice, while the 5-HT2AR-dependent antipsychotic-like behavioral effects of clozapine were absent in mGlu2R–KO mice. In schizophrenia, where the relative expression of these two receptors is dysregulated, a combination of an inverse agonist of 5-HT2AR and a strong agonist of mGlu2R may prove beneficial, as suggested by Fribourg and colleagues [30]. Collectively, these data suggest that a common target for psychedelics, atypical and glutamate antipsychotics is the 5-HT2A–mGlu2R complex, for which designing a bivalent ligand might be a rational therapeutic strategy.
Figure 5. Gi-Gq Balance Model of the Mechanism of Action of Antipsychotic and Psychedelic Drugs through the mGluR2/2AR Complex.
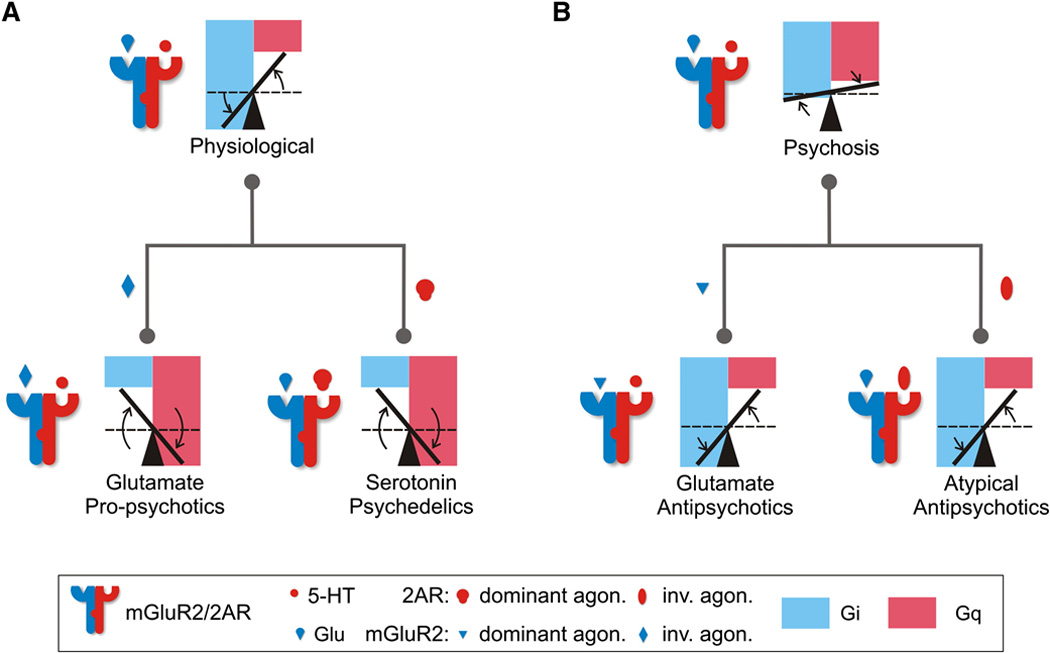
(A) Heteromerization of mGlu2R and 5-HT2AR establishes an optimal Gi-Gq signaling balance in response to the endogenous neurotransmitters, glutamate and serotonin (an increase in Gi and a decrease in Gq when compared to the homomeric receptor signaling; dashed lines). Psychedelics (mGlu2R inverse agonists and 5-HT2AR agonists) invert the signaling balance through the complex tipping the balance from being in favor of Gi signaling (normal complex) to becoming in favor of Gq signaling (propsychotic). (B) Disruption of the optimal balance in psychotic states can be compensated for by antipsychotics (mGlu2R agonists and 5-HT2AR inverse agonists) that tip the balance in favor of Gi signaling thus restoring the physiological signaling. Figure reprinted from Fribourg et al. [21].
Clinical effects of mGlu2/3R agonists
In 2007, a phase II proof-of-concept clinical trial compared pomaglumetad methionil (LY2140023 monohydrate; a prodrug of the mGlu2/3R agonist LY404039) to the atypical antipsychotic olanzapine or placebo. The study showed a positive effect for pomaglumetad against positive and negative symptoms of schizophrenia compared to placebo. Although the efficacy was not as significant as for olanzapine, pomaglumetad was safe and well-tolerated and, importantly, was neither associated with extrapyramidal symptoms nor with metabolic abnormalities [97]. Interestingly, a pharmacogenetic analysis identified 23 single nucleotide polymorphisms (SNPs) significantly associated with pomaglumetad response, sixteen of which were located in the 5-HT2A receptor (HTR2A) gene [98]. However, subsequent clinical trials with pomaglumetad showed either inconclusive results [99], or results that are no different from placebo [100]. Importantly, results form a recent post-hoc analysis suggest that the therapeutic effects of pomaglumetad depend on previous exposure to either typical or atypical antipsychotic drugs [101]. These findings and their basic mechanism based on previous preclinical findings [102] are discussed in Box 1
Box 1. Can epigenetics explain discrepant clinical data?
Interestingly, recent preclinical and postmortem human brain findings may provide an epigenetic explanation of the apparently discordant results with both mGlu2/3R agonists and mGlu2R PAMs. It was demonstrated that chronic atypical antipsychotic therapy induces a highly selective down-regulation of mGlu2R expression in mouse frontal cortex (Figure I). This effect was mediated via a signaling mechanism that involved 5-HT2A receptor-dependent modulation of the promoter activity of the Histone deacetylase 2 (Hdac2) gene. Thus, the presence of serotonin resulted in inhibition of HDAC2 promoter activity, an effect that was reversed in vitro and in mouse frontal cortex by clozapine. Importantly, chronic treatment with atypical antipsychotics induced a 5-HT2A receptor-dependent up-regulation and increased binding of HDAC2 to the mGlu2R promoter. These epigenetic changes occurred in association with repressive histone modifications at the promoter region of the mGlu2R gene in mouse and human frontal cortex [102]. Because chronic antipsychotic drug treatment induced a 5-HT2A receptor-dependent decrease in mGlu2R transcription and its electrophysiological properties, together these findings suggest that previous medication with atypical antipsychotic drugs may prevent the therapeutic responses to mGlu2R in schizophrenia patients. This is supported by results from a recent post-hoc analysis disclosed by Eli Lilly investigators where the subgroup of patients who were earlier in disease course (<3 years of illness) responded to pomaglumetad, as opposed to patients late in disease (>10 years of illness). More interestingly, in patients with previous exposure to predominantly D2-blocking drugs, the efficacy of pomaglumetad was comparable to risperidone, whereas it did not differ from placebo in patients previously treated predominantly with 5-HT2A antagonists [101]. These findings also suggest that inhibition of HDAC2 may reverse the repressive epigenetic changes induced by chronic atypical antipsychotic drugs and hence improve the antipsychotic properties of mGlu2/3R agonists and mGlu2R PAMs.
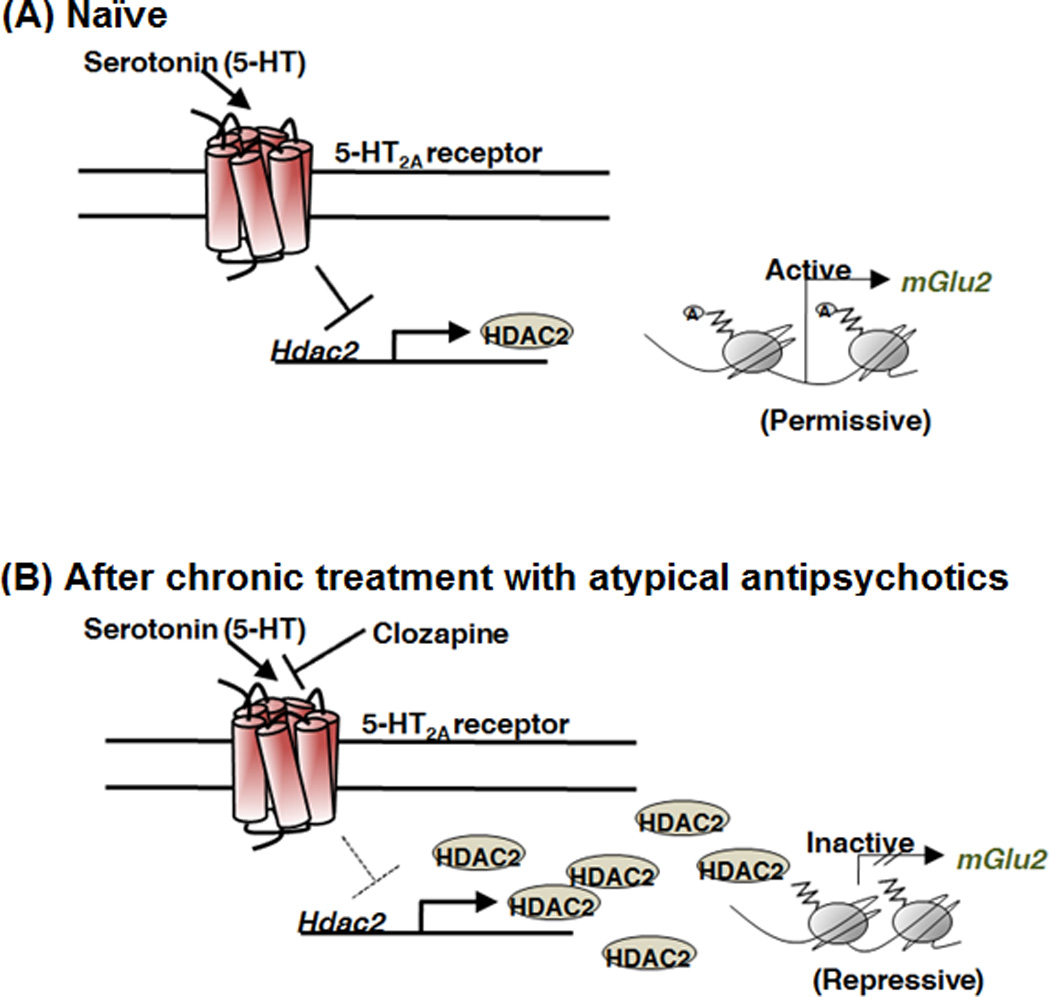
Figure I (in box 1). Chronic atypical antipsychotic therapy downregulates mGlu2R. Schematic model of the effect of chronic atypical antipsychotic treatment to the epigenetic status of the mGlu2R (Grm2) gene. (A) Activation of the 5-HT2A receptor by the endogenous neurotransmitter serotonin represses the promoter activity of the HDAC2 gene in mouse and in human frontal cortex. (B) Atypical antipsychotic drugs, such as clozapine and risperidone, reverse the 5-HT2A receptor-dependent repression of HDAC2, an effect that is associated with increased HDAC2 promoter activity and repressive histone modifications at the mGlu2R promoter. This epigenetic effect of chronic atypical antipsychotic treatment may limit the therapeutic effects of mGlu2/3R agonists such as pomaglumetad.
Promise of mGluR positive allosteric modulators
LY487379, reported by researchers at Eli Lilly in 2003, was the first identified mGlu2R–selective PAM [103]. LY487379 showed comparable efficacy to mGlu2/3R agonists in preclinical psychosis models [104, 105]. However, the poor bioavailability, modest potency, and short duration of action limited the further in vivo characterization of compounds within the LY487379 class. Soon after, the biphenyl-indanone class was reported [106, 107]. Biphenyl-indanone A (BINA) became the prototype mGlu2R PAM for in vivo studies after it was shown to be a potent mGlu2R–selective PAM with robust long-lasting in vivo activity [108]. BINA blocks the PCP-induced hyperlocomotor activity [108, 109] as well as the DOB-induced head twitch response [89]. Pharmacologic magnetic resonance imaging (phMRI) revealed that BINA blocks PCP-induced blood oxygenation level-dependent (BOLD) activation in the rat brain. This allowed defining BINA’s mechanism of action at the brain circuitry level where its effect was apparent in the prefrontal cortex, caudaute–putamen, nucleus accumbens, and mediodorsal thalamus, structures linked to schizophrenia [109]. After the introduction of LY487379 and BINA, a variety of structurally distinct classes of mGlu2R PAMs were discovered, many of which proved to have in vivo activity as well [110]. Early mutagenesis studies identified three amino acid residues in transmembrane (TM) segments IV and V of mGlu2R to be critical for the activity of LY487379 and several other mGlu2R PAMs [111, 112]. A more recent extensive study identified additional residues in TM III, V, and VI to play an important role in the activity of mGlu2R PAMs [113]. Interestingly, 3 residues in TM IV of mGlu2R have been found to be essential for the heteromerization of 5-HT2A-mGlu2R [68] indicating that TMD region of mGlu2R is implicated in different types of allosteric interactions. However, the implication of mGlu2R involvement in heteromeric receptor complexes on PAM pharmacology remains unknown. Although glutamate-induced signaling through mGlu2R can be potentiated by a PAM, mGlu2R in its heteromeric state may not necessarily respond to a PAM in the same manner. Moreover, it is not known yet whether mGlu2R PAMs can crosstalk and alter the signaling of 5-HT2A through the 5-HT2A–mGlu2 receptor heteromer.
So far, two mGlu2R PAMs have made it into clinical trials. The first compound is ADX71149, from Addex and Janssen, which showed the first successful clinical proof-of-concept in a Phase IIa study. Data reported in late 2012, demonstrated safety and tolerability and identified patients with residual negative symptoms as the population most likely to benefit from adjunctive treatment with ADX71149 [114]. The second compound, AZD8529, from Astrazeneca, failed to separate from placebo in a phase IIa study, unlike the active control risperidone [115]. However, it is worth mentioning that this study tested a single dose of AZD8529 as a monotherapy, thus further investigation is warranted.
One more ongoing effort by Janssen is to identify potential positron emission tomography (PET) tracers for imaging mGlu2R [116]. Preliminary evaluation of [11C]JNJ-42491293 in rat brain demonstrated specific and reversible binding to an mGlu2R allosteric site [117]. Subsequent evaluation in 20 healthy male subjects confirmed radioactivity uptake consistent with reported distribution of the mGlu2R in human brain [118]. These encouraging results would likely allow quantifying mGlu2R in human brain as well as for assessment of occupancy by drug candidates targeting this allosteric site.
mGlu5R is another receptor within the mGluR superfamily for which positive allosteric modulation is a potential treatment strategy for schizophrenia. The efficacy of mGlu5R PAMs in animal models of schizophrenia and cognitive dysfunction were thought to be mediated by mGlu5R–induced potentiation of NMDA receptors [119, 120]. However, a recent study has challenged this hypothesis by showing that VU0409551, a novel mGlu5R PAM with robust antipsychotic and cognition-enhancing effects in animal models, exhibits biased allosteric modulation by selectively potentiating mGlu5R coupling to Gq-mediated signaling but not mGlu5R modulation of NMDA receptors [61]. This raises an interesting question regarding the signaling mechanisms underlying the antipsychotic effects of mGlu2R PAMs and whether they could exert some of their beneficial effects through NMDA-independent signaling as well. It would also be interesting to compare mGlu2R vs. mGlu5R PAMs, particularly which classes of drugs are superior for treatment of cognitive dysfunction associated with schizophrenia.
Recent findings suggest that ligands that interact with putative receptor heteromers, such as µ-opioid-mGlu5R and µ-δ opioid receptor heteromer, modulate unique phenotypes in rodent models [121, 122]. Since the heteromer between mGlu2R and 5-HT2AR has been suggested to function as a primary molecular target responsible for the therapeutic effects of both atypical and glutamate antipsychotic drugs (Figure 5), further medicinal chemistry work with bivalent compounds that specifically target the 5-HT2AR-mGlu2R heteromer inducing an antipsychotic-like pattern of G protein signaling in frontal cortex pyramidal neurons may provide a route for the development of new and more effective agents for the treatment of schizophrenia.
Concluding remarks
This review has summarized recent findings suggesting that mGlu2R activation may provide a novel therapeutic strategy for treatment of schizophrenia and help avoid the adverse effects associated with currently available antipsychotic drugs. As pharmacologic tools, PAMs might be superior to orthosteric agonists since they have a unique ability to modulate glutamate release in a “state-dependent” manner that helps fine-tune physiological responses. An important challenge in the field is related to the potential adverse signaling effects induced by mGluR PAMs in the absence of orthosteric agonists [123]. This is particularly important considering the recent findings that mGluRs with VFTs deleted, when reconstituted into nanodiscs, are able to couple to and activate heterotrimeric G proteins in the presence of PAMs [124]. Whether the expression of alternatively spliced variants that generate truncated isoforms of mGluRs [125] might lead to unfavorable PAM-dependent signaling events in the absence of endogenous agonists, needs to be explored. Despite the advances in our knowledge of medicinal chemistry and basic mechanism of action of mGluRs, many questions are yet to be addressed with both biophysical approaches and whole animal experimental systems (Box 2). Only time and additional research will provide us with answers.
Box 2. Outstanding questions.
How do mGlu2R PAMs modulate mGlu2R in its heteromeric states?
Do mGlu2R PAMs have the ability to crosstalk to 5-HT2A receptors?
Can an mGlu2R PAM be a part of a bivalent ligand targeting the 5-HT2A–mGlu2 receptor complex implicated in schizophrenia? And if yes, can a bivalent PET tracer be used diagnostically for quantifying such receptor complex?
Would an HDAC2 inhibitor improve the therapeutic efficacy of mGlu2R antipsychotics in patients with a history of chronic atypical antipsychotic therapy?
What is the right patient population that is likely to benefit from mGlu2R allosteric modulation?
Highlights.
mGlu2 activation is a potential therapeutic strategy for schizophrenia.
Unlike orthosteric agonists, PAMs have a unique ability to fine-tune physiological responses.
Chronic atypical antipsychotic therapy may reduce the efficacy of mGlu2 antipsychotics.
Glossary Box
- Allosteric site
a binding site on a receptor macromolecule that is non-overlapping and spatially distinct from, but conformationally linked to, the orthosteric binding site
- Dopamine (DA) hypothesis
the most classical schizophrenia hypothesis that attributes psychotic symptoms to hyperactive DA signaling in the brain
- Epigenetics
literally means "above" or "on top of" genetics. It refers to external modifications to DNA that turn genes "on" or "of" without changes in DNA sequence
- Excitatory postsynaptic potential (EPSP)
a temporary depolarization of postsynaptic neuron membrane potential that makes it more likely for this neuron to trigger an action potential
- Glutamate
the main excitatory neurotransmitter in the brain
- G protein-coupled receptor (GPCR)
an integral plasma membrane protein that senses molecules outside the cell and transduce those extracellular signals to intracellular relay proteins, the heterotrimeric GTP-binding proteins (G proteins)
- Heteromer
structural assembly composed of two or more different components
- Histone
Evolutionary conserved proteins found in eukaryotic cell nuclei that package DNA into structural units called nucleosomes. Covalent modifications at the N-terminal tail of histones correlate with open or closed states of chromatin, depending on the type of modification, and thus lead to changes in gene expression
- Histone deacetylase (HDAC)
a class of enzymes that remove acetyl groups from histones allowing the histones to wrap the DNA more tightly and repress gene transcription
- Metabotropic glutamate receptors (mGluRs)
GPCRs that bind glutamate and function to modulate, rather than mediate, synaptic transmission. This is to differentiate them from “ionotropic” glutamate receptors like NMDA, which are ion channels mediating excitatory neurotransmission
- N-methyl-D-aspartate (NMDA) receptor
is a glutamate receptor and ion channel protein found in nerve cells. It is involved in synaptic transmission and plays an important role in learning and memory
- NMDA hypofunction hypothesis
a glutamate-based hypothesis which postulates that reduced activation of NMDA glutamate receptor underlies development of different schizophrenia symptoms
- Orthosteric site
the binding site on a receptor macromolecule that is recognized by the endogenous agonist for that receptor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.American Psychiatric Association. Diagnostic and statistical manual of mental disorders, (DSM-5®) American Psychiatric Pub; 2013. [Google Scholar]
- 2.Freedman R. Schizophrenia. N. Engl. J. Med. 2003;349:1738–1749. doi: 10.1056/NEJMra035458. [DOI] [PubMed] [Google Scholar]
- 3.Pompili M, et al. Suicide risk in schizophrenia: learning from the past to change the future. Ann. Gen. Psychiatry. 2007;6:10. doi: 10.1186/1744-859X-6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tamminga C, Holcomb H. Phenotype of schizophrenia: a review and formulation. Mol. Psychiatry. 2005;10:27–39. doi: 10.1038/sj.mp.4001563. [DOI] [PubMed] [Google Scholar]
- 5.Wu EQ, et al. The economic burden of schizophrenia in the United States in 2002. J. Clin. Psychiatry. 2005 doi: 10.4088/jcp.v66n0906. [DOI] [PubMed] [Google Scholar]
- 6.Mathers C, et al. The global burden of disease: 2004 update. World Health Organization. 2008 [Google Scholar]
- 7.Schwartz TL, et al. Glutamate neurocircuitry: theoretical underpinnings in schizophrenia. Frontiers in Pharmacology. 2012:3. doi: 10.3389/fphar.2012.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Milev P, et al. Predictive values of neurocognition and negative symptoms on functional outcome in schizophrenia: a longitudinal first-episode study with 7-year follow-up. Am. J. Psychiatry. 2005;162:495–506. doi: 10.1176/appi.ajp.162.3.495. [DOI] [PubMed] [Google Scholar]
- 9.Meltzer HY. Update on typical and atypical antipsychotic drugs. Annu. Rev. Med. 2013;64:393–406. doi: 10.1146/annurev-med-050911-161504. [DOI] [PubMed] [Google Scholar]
- 10.Eggers AE. A serotonin hypothesis of schizophrenia. Med. Hypotheses. 2013;80:791–794. doi: 10.1016/j.mehy.2013.03.013. [DOI] [PubMed] [Google Scholar]
- 11.Keefe RS, et al. Neurocognitive effects of antipsychotic medications in patients with chronic schizophrenia in the CATIE Trial. Arch. Gen. Psychiatry. 2007;64:633–647. doi: 10.1001/archpsyc.64.6.633. [DOI] [PubMed] [Google Scholar]
- 12.Haro JM, et al. Cross-national clinical and functional remission rates: Worldwide Schizophrenia Outpatient Health Outcomes (W-SOHO) study. Br. J. Psychiatry. 2011;199:194–201. doi: 10.1192/bjp.bp.110.082065. [DOI] [PubMed] [Google Scholar]
- 13.Olney JW, Farber NB. Glutamate receptor dysfunction and schizophrenia. Arch. Gen. Psychiatry. 1995;52:998–1007. doi: 10.1001/archpsyc.1995.03950240016004. [DOI] [PubMed] [Google Scholar]
- 14.Krystal JH, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans: psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen. Psychiatry. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- 15.Malhotra AK, et al. NMDA receptor function and human cognition: the effects of ketamine in healthy volunteers. Neuropsychopharmacology. 1996;14:301–307. doi: 10.1016/0893-133X(95)00137-3. [DOI] [PubMed] [Google Scholar]
- 16.Lahti AC, et al. Ketamine activates psychosis and alters limbic blood flow in schizophrenia. Neuroreport. 1995;6:869–872. doi: 10.1097/00001756-199504190-00011. [DOI] [PubMed] [Google Scholar]
- 17.Malhotra AK, et al. Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacology. 1997;17:141–150. doi: 10.1016/S0893-133X(97)00036-5. [DOI] [PubMed] [Google Scholar]
- 18.Steeds H, et al. Drug models of schizophrenia. Therapeutic Advances in Psychopharmacology. 2014 doi: 10.1177/2045125314557797. 2045125314557797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kalivas PW. Cocaine and amphetamine-like psychostimulants: neurocircuitry and glutamate neuroplasticity. Dialogues Clin. Neurosci. 2007;9:389–397. doi: 10.31887/DCNS.2007.9.4/pkalivas. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yuen EY, et al. Activation of 5-HT2A/C receptors counteracts 5-HT1A regulation of n-methyl-D-aspartate receptor channels in pyramidal neurons of prefrontal cortex. J. Biol. Chem. 2008;283:17194–17204. doi: 10.1074/jbc.M801713200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis KL, et al. Ventricular enlargement in poor-outcome schizophrenia. Biol. Psychiatry. 1998;43:783–793. doi: 10.1016/s0006-3223(97)00553-2. [DOI] [PubMed] [Google Scholar]
- 22.Pantelis C, et al. Neuroanatomical abnormalities before and after onset of psychosis: a cross-sectional and longitudinal MRI comparison. The Lancet. 2003;361:281–288. doi: 10.1016/S0140-6736(03)12323-9. [DOI] [PubMed] [Google Scholar]
- 23.Rapoport JL, et al. Childhood-onset schizophrenia: progressive ventricular change during adolescence. Arch. Gen. Psychiatry. 1997;54:897–903. doi: 10.1001/archpsyc.1997.01830220013002. [DOI] [PubMed] [Google Scholar]
- 24.Jacobsen LK, et al. Progressive reduction of temporal lobe structures in childhood-onset schizophrenia. Am. J. Psychiatry. 1998;155:678–685. doi: 10.1176/ajp.155.5.678. [DOI] [PubMed] [Google Scholar]
- 25.Mathalon DH, et al. Progressive brain volume changes and the clinical course of schizophrenia in men: a longitudinal magnetic resonance imaging study. Arch. Gen. Psychiatry. 2001;58:148–157. doi: 10.1001/archpsyc.58.2.148. [DOI] [PubMed] [Google Scholar]
- 26.Gur RE, et al. A follow-up magnetic resonance imaging study of schizophrenia: relationship of neuroanatomical changes to clinical and neurobehavioral measures. Arch. Gen. Psychiatry. 1998;55:145–152. doi: 10.1001/archpsyc.55.2.145. [DOI] [PubMed] [Google Scholar]
- 27.Weinberger DR, McClure RK. Neurotoxicity, neuroplasticity, and magnetic resonance imaging morphometry: what is happening in the schizophrenic brain? Arch. Gen. Psychiatry. 2002;59:553–558. doi: 10.1001/archpsyc.59.6.553. [DOI] [PubMed] [Google Scholar]
- 28.Ohishi H, et al. Distribution of a metabotropic glutamate receptor, mGluR2, in the central nervous system of the rat and mouse: an immunohistochemical study with a monoclonal antibody. Neurosci. Res. 1998;30:65–82. doi: 10.1016/s0168-0102(97)00120-x. [DOI] [PubMed] [Google Scholar]
- 29.Neki A, et al. Pre-and postsynaptic localization of a metabotropic glutamate receptor, mGluR2, in the rat brain: an immunohistochemical study with a monoclonal antibody. Neurosci. Lett. 1996;202:197–200. doi: 10.1016/0304-3940(95)12248-6. [DOI] [PubMed] [Google Scholar]
- 30.Fribourg M, et al. Decoding the signaling of a GPCR heteromeric complex reveals a unifying mechanism of action of antipsychotic drugs. Cell. 2011;147:1011–1023. doi: 10.1016/j.cell.2011.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moreno JL, et al. Identification of three residues essential for 5-hydroxytryptamine 2A–metabotropic glutamate 2 (5-HT2A.mGlu2) receptor heteromerization and its psychoactive behavioral function. J. Biol. Chem. 2012;287:44301–44319. doi: 10.1074/jbc.M112.413161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akbarian S, et al. Selective alterations in gene expression for NMDA receptor subunits in prefrontal cortex of schizophrenics. J. Neurosci. 1996;16:19–30. doi: 10.1523/JNEUROSCI.16-01-00019.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao X, et al. Ionotropic glutamate receptors and expression of N-methyl-D-aspartate receptor subunits in subregions of human hippocampus: effects of schizophrenia. Am. J. Psychiatry. 2000;157:1141–1149. doi: 10.1176/appi.ajp.157.7.1141. [DOI] [PubMed] [Google Scholar]
- 34.Kerwin R, et al. Quantitative autoradiographic analysis of glutamate binding sites in the hippocampal formation in normal and schizophrenic brain post mortem. Neuroscience. 1990;39:25–32. doi: 10.1016/0306-4522(90)90219-t. [DOI] [PubMed] [Google Scholar]
- 35.Nishikawa T, et al. Increased [3 H] kainic acid binding in the prefrontal cortex in schizophrenia. Neurosci. Lett. 1983;40:245–250. doi: 10.1016/0304-3940(83)90046-0. [DOI] [PubMed] [Google Scholar]
- 36.Pilowsky L, et al. First in vivo evidence of an NMDA receptor deficit in medication-free schizophrenic patients. Mol. Psychiatry. 2006;11:118–119. doi: 10.1038/sj.mp.4001751. [DOI] [PubMed] [Google Scholar]
- 37.Egerton A, et al. Anterior cingulate glutamate levels related to clinical status following treatment in first-episode schizophrenia. Neuropsychopharmacology. 2012;37:2515–2521. doi: 10.1038/npp.2012.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kniazeff J, et al. Dimers and beyond: The functional puzzles of class C GPCRs. Pharmacol. Ther. 2011;130:9–25. doi: 10.1016/j.pharmthera.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 40.González-Maeso J. Family a GPCR heteromers in animal models. Frontiers in Pharmacology. 2014:5. doi: 10.3389/fphar.2014.00226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakanishi S. Molecular diversity of glutamate receptors and implications for brain function. Science. 1992;258:597–603. doi: 10.1126/science.1329206. [DOI] [PubMed] [Google Scholar]
- 42.Moreno JL, et al. Group II metabotropic glutamate receptors and schizophrenia. Cellular and Molecular Life Sciences. 2009;66:3777–3785. doi: 10.1007/s00018-009-0130-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gasparini F, Spooren W. Allosteric modulators for mGlu receptors. Curr. Neuropharmacol. 2007;5:187–194. doi: 10.2174/157015907781695900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Christopoulos A, et al. International union of basic and clinical pharmacology. XC. multisite pharmacology: recommendations for the nomenclature of receptor allosterism and allosteric ligands. Pharmacol. Rev. 2014;66:918–947. doi: 10.1124/pr.114.008862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gregory KJ, et al. Pharmacology of metabotropic glutamate receptor allosteric modulators: structural basis and therapeutic potential for CNS disorders. Oligomerization and Allosteric Modulation in G-Protein Coupled Receptors. 2012;115:61. doi: 10.1016/B978-0-12-394587-7.00002-6. [DOI] [PubMed] [Google Scholar]
- 46.Wu H, et al. Structure of a Class C GPCR Metabotropic Glutamate Receptor 1 Bound to an Allosteric Modulator. Science. 2014;344:58–64. doi: 10.1126/science.1249489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dore AS, et al. Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature. 2014;511:557–562. doi: 10.1038/nature13396. [DOI] [PubMed] [Google Scholar]
- 48.Conn PJ, et al. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nature Reviews Drug Discovery. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Langmead CJ. Ligand properties and behaviours in an allosteric age. Trends Pharmacol. Sci. 2012;33:621. doi: 10.1016/j.tips.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 50.Johnson M, et al. Allosteric modulators of metabotropic glutamate receptors: lessons learnt from mGlu1, mGlu2 and mGlu5 potentiators and antagonists. Biochem. Soc. Trans. 2004;32:881–887. doi: 10.1042/BST0320881. [DOI] [PubMed] [Google Scholar]
- 51.Christopoulos A. Advances in G protein-coupled receptor allostery: from function to structure. Mol. Pharmacol. 2014;86:463–478. doi: 10.1124/mol.114.094342. [DOI] [PubMed] [Google Scholar]
- 52.Yin S, Niswender CM. Progress toward advanced understanding of metabotropic glutamate receptors: structure, signaling and therapeutic indications. Cell. Signal. 2014;26:2284–2297. doi: 10.1016/j.cellsig.2014.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nickols HH, Conn PJ. Development of allosteric modulators of GPCRs for treatment of CNS disorders. Neurobiol. Dis. 2014;61:55–71. doi: 10.1016/j.nbd.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lamb JP, et al. Discovery of molecular switches within the ADX-47273 mGlu 5 PAM scaffold that modulate modes of pharmacology to afford potent mGlu 5 NAMs, PAMs and partial antagonists. Bioorg. Med. Chem. Lett. 2011;21:2711–2714. doi: 10.1016/j.bmcl.2010.11.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sheffler DJ, et al. Development of a novel, CNS-penetrant, metabotropic glutamate receptor 3 (mGlu 3) NAM probe (ML289) derived from a closely related mGlu 5 PAM. Bioorg. Med. Chem. Lett. 2012;22:3921–3925. doi: 10.1016/j.bmcl.2012.04.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou Y, et al. Discovery of N-aryl piperazines as selective mGluR5 potentiators with improved in vivo utility. ACS Medicinal Chemistry Letters. 2010;1:433–438. doi: 10.1021/ml100181a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wood MR, et al. “Molecular switches” on mGluR allosteric ligands that modulate modes of pharmacology. Biochemistry (N. Y. ) 2011;50:2403–2410. doi: 10.1021/bi200129s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Doumazane E, et al. A new approach to analyze cell surface protein complexes reveals specific heterodimeric metabotropic glutamate receptors. FASEB J. 2011;25:66–77. doi: 10.1096/fj.10-163147. [DOI] [PubMed] [Google Scholar]
- 59.Kenakin T. New concepts in drug discovery: collateral efficacy and permissive antagonism. Nature Reviews Drug Discovery. 2005;4:919–927. doi: 10.1038/nrd1875. [DOI] [PubMed] [Google Scholar]
- 60.Urban JD, et al. Functional selectivity and classical concepts of quantitative pharmacology. J. Pharmacol. Exp. Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- 61.Rook JM, et al. Biased mGlu 5-Positive Allosteric Modulators Provide In Vivo Efficacy without Potentiating mGlu 5 Modulation of NMDAR Currents. Neuron. 2015 doi: 10.1016/j.neuron.2015.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sheffler DJ, Conn PJ. Allosteric potentiators of metabotropic glutamate receptor subtype 1a differentially modulate independent signaling pathways in baby hamster kidney cells. Neuropharmacology. 2008;55:419–427. doi: 10.1016/j.neuropharm.2008.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang Y, et al. Allosteric potentiators of metabotropic glutamate receptor subtype 5 have differential effects on different signaling pathways in cortical astrocytes. J. Pharmacol. Exp. Ther. 2005;315:1212–1219. doi: 10.1124/jpet.105.090308. [DOI] [PubMed] [Google Scholar]
- 64.Tateyama M, Kubo Y. Dual signaling is differentially activated by different active states of the metabotropic glutamate receptor 1alpha. Proc. Natl. Acad. Sci. U. S. A. 2006;103:1124–1128. doi: 10.1073/pnas.0505925103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.DeWire SM, et al. A G protein-biased ligand at the mu-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J. Pharmacol. Exp. Ther. 2013;344:708–717. doi: 10.1124/jpet.112.201616. [DOI] [PubMed] [Google Scholar]
- 66.Ghose S, et al. Differential expression of metabotropic glutamate receptors 2 and 3 in schizophrenia: a mechanism for antipsychotic drug action? Am. J. Psychiatry. 2009;166:812–820. doi: 10.1176/appi.ajp.2009.08091445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marek GJ. Metabotropic glutamate 2/3 (mGlu 2/3) receptors, schizophrenia and cognition. Eur. J. Pharmacol. 2010;639:81–90. doi: 10.1016/j.ejphar.2010.02.058. [DOI] [PubMed] [Google Scholar]
- 68.Moreno JL, et al. Identification of three residues essential for 5-hydroxytryptamine 2A–metabotropic glutamate 2 (5-HT2A.mGlu2) receptor heteromerization and its psychoactive behavioral function. J. Biol. Chem. 2012;287:44301–44319. doi: 10.1074/jbc.M112.413161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schoepp DD. Unveiling the functions of presynaptic metabotropic glutamate receptors in the central nervous system. J. Pharmacol. Exp. Ther. 2001;299:12–20. [PubMed] [Google Scholar]
- 70.Aronica E, et al. Expression and functional role of mGluR3 and mGluR5 in human astrocytes and glioma cells: opposite regulation of glutamate transporter proteins. Eur. J. Neurosci. 2003;17:2106–2118. doi: 10.1046/j.1460-9568.2003.02657.x. [DOI] [PubMed] [Google Scholar]
- 71.Egan MF, et al. Variation in GRM3 affects cognition, prefrontal glutamate, and risk for schizophrenia. Proc. Natl. Acad. Sci. U. S. A. 2004;101:12604–12609. doi: 10.1073/pnas.0405077101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mössner R, et al. Further evidence for a functional role of the glutamate receptor gene GRM3 in schizophrenia. European Neuropsychopharmacology. 2008;18:768–772. doi: 10.1016/j.euroneuro.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 73.Chen Q, et al. A case-control study of the relationship between the metabotropic glutamate receptor 3 gene and schizophrenia in the Chinese population. Schizophr. Res. 2005;73:21–26. doi: 10.1016/j.schres.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 74.González-Maeso J, et al. Identification of a serotonin/glutamate receptor complex implicated in psychosis. Nature. 2008;452:93–97. doi: 10.1038/nature06612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ghose S, et al. Metabotropic glutamate receptor 2 and 3 gene expression in the human prefrontal cortex and mesencephalon in schizophrenia. Int. J. Neurosci. 2008;118:1609–1627. doi: 10.1080/00207450802330702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ohnuma T, et al. Expression of the human excitatory amino acid transporter 2 and metabotropic glutamate receptors 3 and 5 in the prefrontal cortex from normal individuals and patients with schizophrenia. Mol. Brain Res. 1998;56:207–217. doi: 10.1016/s0169-328x(98)00063-1. [DOI] [PubMed] [Google Scholar]
- 77.Richardson-Burns SM, et al. Metabotropic glutamate receptor mRNA expression in the schizophrenic thalamus. Biol. Psychiatry. 2000;47:22–28. doi: 10.1016/s0006-3223(99)00207-3. [DOI] [PubMed] [Google Scholar]
- 78.Kordi-Tamandani DM, et al. Evaluation of hypermethylation and expression pattern of GMR2, GMR5, GMR8, and GRIA3 in patients with schizophrenia. Gene. 2013;515:163–166. doi: 10.1016/j.gene.2012.10.075. [DOI] [PubMed] [Google Scholar]
- 79.Bullock WM, et al. Altered expression of genes involved in GABAergic transmission and neuromodulation of granule cell activity in the cerebellum of schizophrenia patients. 2008 doi: 10.1176/appi.ajp.2008.07121845. [DOI] [PubMed] [Google Scholar]
- 80.Gupta DS, et al. Metabotropic glutamate receptor protein expression in the prefrontal cortex and striatum in schizophrenia. Synapse. 2005;57:123–131. doi: 10.1002/syn.20164. [DOI] [PubMed] [Google Scholar]
- 81.Crook JM, et al. Comparative analysis of group II metabotropic glutamate receptor immunoreactivity in Brodmann's area 46 of the dorsolateral prefrontal cortex from patients with schizophrenia and normal subjects. Mol. Psychiatry. 2002;7:157–164. doi: 10.1038/sj.mp.4000966. [DOI] [PubMed] [Google Scholar]
- 82.Frank E, et al. Density of metabotropic glutamate receptors 2 and 3 (mGluR2/3) in the dorsolateral prefrontal cortex does not differ with schizophrenia diagnosis but decreases with age. Schizophr. Res. 2011;128:56–60. doi: 10.1016/j.schres.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 83.Moghaddam B, Adams BW. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science. 1998;281:1349–1352. doi: 10.1126/science.281.5381.1349. [DOI] [PubMed] [Google Scholar]
- 84.Cartmell J, et al. The metabotropic glutamate 2/3 receptor agonists LY354740 and LY379268 selectively attenuate phencyclidine versus d-amphetamine motor behaviors in rats. J. Pharmacol. Exp. Ther. 1999;291:161–170. [PubMed] [Google Scholar]
- 85.Fell MJ, et al. Evidence for the role of metabotropic glutamate (mGlu)2 not mGlu3 receptors in the preclinical antipsychotic pharmacology of the mGlu2/3 receptor agonist (−)-(1R,4S,5S,6S)-4-amino-2-sulfonylbicyclo[3.1.0]hexane-4,6-dicarboxylic acid (LY404039) J. Pharmacol. Exp. Ther. 2008;326:209–217. doi: 10.1124/jpet.108.136861. [DOI] [PubMed] [Google Scholar]
- 86.Woolley M, et al. The mGlu2 but not the mGlu3 receptor mediates the actions of the mGluR2/3 agonist, LY379268, in mouse models predictive of antipsychotic activity. Psychopharmacology (Berl. ) 2008;196:431–440. doi: 10.1007/s00213-007-0974-x. [DOI] [PubMed] [Google Scholar]
- 87.Walker AG, et al. Metabotropic glutamate receptor 3 activation is required for long-term depression in medial prefrontal cortex and fear extinction. Proc. Natl. Acad. Sci. U. S. A. 2015;112:1196–1201. doi: 10.1073/pnas.1416196112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gewirtz JC, Marek GJ. Behavioral evidence for interactions between a hallucinogenic drug and group II metabotropic glutamate receptors. Neuropsychopharmacology. 2000;23:569–576. doi: 10.1016/S0893-133X(00)00136-6. [DOI] [PubMed] [Google Scholar]
- 89.Benneyworth MA, et al. A selective positive allosteric modulator of metabotropic glutamate receptor subtype 2 blocks a hallucinogenic drug model of psychosis. Mol. Pharmacol. 2007;72:477–484. doi: 10.1124/mol.107.035170. [DOI] [PubMed] [Google Scholar]
- 90.Bespalov A, et al. Habituation deficits induced by metabotropic glutamate receptors 2/3 receptor blockade in mice: reversal by antipsychotic drugs. J. Pharmacol. Exp. Ther. 2007;320:944–950. doi: 10.1124/jpet.106.110684. [DOI] [PubMed] [Google Scholar]
- 91.Gewirtz JC, et al. Modulation of DOI-induced increases in cortical BDNF expression by group II mGlu receptors. Pharmacology Biochemistry and Behavior. 2002;73:317–326. doi: 10.1016/s0091-3057(02)00844-4. [DOI] [PubMed] [Google Scholar]
- 92.Winter J, et al. Serotonergic/glutamatergic interactions: the effects of mGlu2/3 receptor ligands in rats trained with LSD and PCP as discriminative stimuli. Psychopharmacology (Berl. ) 2004;172:233–240. doi: 10.1007/s00213-003-1636-2. [DOI] [PubMed] [Google Scholar]
- 93.Zhai Y, et al. Group II metabotropic glutamate receptor modulation of DOI-induced c-fos mRNA and excitatory responses in the cerebral cortex. Neuropsychopharmacology. 2003;28:45–52. doi: 10.1038/sj.npp.1300013. [DOI] [PubMed] [Google Scholar]
- 94.Moreno JL, et al. Metabotropic glutamate mGlu2 receptor is necessary for the pharmacological and behavioral effects induced by hallucinogenic 5-HT2A receptor agonists. Neurosci. Lett. 2011;493:76–79. doi: 10.1016/j.neulet.2011.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Benneyworth MA, et al. Chronic phenethylamine hallucinogen treatment alters behavioral sensitivity to a metabotropic glutamate 2/3 receptor agonist. Neuropsychopharmacology. 2008;33:2206–2216. doi: 10.1038/sj.npp.1301600. [DOI] [PubMed] [Google Scholar]
- 96.Moreno JL, et al. Chronic treatment with LY341495 decreases 5-HT 2A receptor binding and hallucinogenic effects of LSD in mice. Neurosci. Lett. 2013;536:69–73. doi: 10.1016/j.neulet.2012.12.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Patil ST, et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat. Med. 2007;13:1102–1107. doi: 10.1038/nm1632. [DOI] [PubMed] [Google Scholar]
- 98.Liu W, et al. Pharmacogenetic analysis of the mGlu2/3 agonist LY2140023 monohydrate in the treatment of schizophrenia. The Pharmacogenomics Journal. 2012;12:246–254. doi: 10.1038/tpj.2010.90. [DOI] [PubMed] [Google Scholar]
- 99.Kinon BJ, et al. A multicenter, inpatient, phase 2, double-blind, placebo-controlled dose-ranging study of LY2140023 monohydrate in patients with DSM-IV schizophrenia. J. Clin. Psychopharmacol. 2011;31:349–355. doi: 10.1097/JCP.0b013e318218dcd5. [DOI] [PubMed] [Google Scholar]
- 100.Hopkins CR. Is There a Path Forward for mGlu2 Positive Allosteric Modulators for the Treatment of Schizophrenia? ACS Chemical Neuroscience. 2013;4:211–213. doi: 10.1021/cn400023y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kinon BJ, et al. Exploratory analysis for a targeted patient population responsive to the metabotropic glutamate 2/3 receptor agonist pomaglumetad methionil in schizophrenia. Biol. Psychiatry. 2015 doi: 10.1016/j.biopsych.2015.03.016. [DOI] [PubMed] [Google Scholar]
- 102.Kurita M, et al. HDAC2 regulates atypical antipsychotic responses through the modulation of mGlu2 promoter activity. Nat. Neurosci. 2012;15:1245–1254. doi: 10.1038/nn.3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Johnson MP, et al. Discovery of allosteric potentiators for the metabotropic glutamate 2 receptor: synthesis and subtype selectivity of N-(4-(2-methoxyphenoxy) phenyl)-N-(2, 2, 2-trifluoroethylsulfonyl) pyrid-3-ylmethylamine. J. Med. Chem. 2003;46:3189–3192. doi: 10.1021/jm034015u. [DOI] [PubMed] [Google Scholar]
- 104.Galici R, et al. A selective allosteric potentiator of metabotropic glutamate (mGlu) 2 receptors has effects similar to an orthosteric mGlu2/3 receptor agonist in mouse models predictive of antipsychotic activity. J. Pharmacol. Exp. Ther. 2005;315:1181–1187. doi: 10.1124/jpet.105.091074. [DOI] [PubMed] [Google Scholar]
- 105.Johnson MP, et al. Metabotropic glutamate 2 receptor potentiators: receptor modulation, frequency-dependent synaptic activity, and efficacy in preclinical anxiety and psychosis model (s) Psychopharmacology (Berl. ) 2005;179:271–283. doi: 10.1007/s00213-004-2099-9. [DOI] [PubMed] [Google Scholar]
- 106.Bonnefous C, et al. Biphenyl-indanones: allosteric potentiators of the metabotropic glutamate subtype 2 receptor. Bioorg. Med. Chem. Lett. 2005;15:4354–4358. doi: 10.1016/j.bmcl.2005.06.062. [DOI] [PubMed] [Google Scholar]
- 107.Pinkerton AB, et al. Allosteric potentiators of the metabotropic glutamate receptor 2 (mGlu2). Part 3: Identification and biological activity of indanone containing mGlu2 receptor potentiators. Bioorg. Med. Chem. Lett. 2005;15:1565–1571. doi: 10.1016/j.bmcl.2005.01.077. [DOI] [PubMed] [Google Scholar]
- 108.Galici R, et al. Biphenyl-indanone A, a positive allosteric modulator of the metabotropic glutamate receptor subtype 2, has antipsychotic- and anxiolytic-like effects in mice. J. Pharmacol. Exp. Ther. 2006;318:173–185. doi: 10.1124/jpet.106.102046. [DOI] [PubMed] [Google Scholar]
- 109.Hackler E, et al. Selective potentiation of the metabotropic glutamate receptor subtype 2 blocks phencyclidine-induced hyperlocomotion and brain activation. Neuroscience. 2010;168:209–218. doi: 10.1016/j.neuroscience.2010.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cid J, et al. Metabotropic Glutamate Receptor 2 Activators. In: Celanire S, Poli S, editors. Small Molecule Therapeutics for Schizophrenia. Cham: Springer International Publishing; 2015. pp. 101–142. [Google Scholar]
- 111.Schaffhauser H, et al. Pharmacological characterization and identification of amino acids involved in the positive modulation of metabotropic glutamate receptor subtype 2. Mol. Pharmacol. 2003;64:798–810. doi: 10.1124/mol.64.4.798. [DOI] [PubMed] [Google Scholar]
- 112.Rowe BA, et al. Transposition of three amino acids transforms the human metabotropic glutamate receptor (mGluR)-3-positive allosteric modulation site to mGluR2, and additional characterization of the mGluR2-positive allosteric modulation site. J. Pharmacol. Exp. Ther. 2008;326:240–251. doi: 10.1124/jpet.108.138271. [DOI] [PubMed] [Google Scholar]
- 113.Farinha A, et al. Molecular determinants of positive allosteric modulation of the human metabotropic glutamate receptor 2. Br. J. Pharmacol. 2015 doi: 10.1111/bph.13065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Addex Therapeutics. Addex Reports Top-line Data from a Successful Phase 2a Clinical Study with ADX71149 in Schizophrenia Patients, 2015. 2012 [Google Scholar]
- 115.Litman RE, et al. Poster# S239 AZD8529, A POSITIVE ALLOSTERIC MODULATOR AT THE MGLUR2 RECEPTOR, DOES NOT IMPROVE SYMPTOMS IN SCHIZOPHRENIA: A PROOF OF PRINCIPLE STUDY. Schizophr. Res. 2014;153:S176. doi: 10.1016/j.schres.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 116.Andres J, et al. Synthesis, Evaluation, and Radio labeling of New Potent Positive Allosteric Modulators of the Metabotropic Glutamate Receptor 2 as Potential Tracers for Positron Emission Tomography Imaging. J. Med. Chem. 2012;55:8685–8699. doi: 10.1021/jm300912k. [DOI] [PubMed] [Google Scholar]
- 117.Celen S, et al. Preliminary biological evaluation of [11C]JNJ42491293 as a radioligand for PET imaging of mGluR2 in brain. J NUCL MED MEETING ABSTRACTS. 2012;53:286. [Google Scholar]
- 118.Van Laere K, et al. Biodistribution, dosimetry and kinetic modeling of [11C]JNJ-42491293, a PET tracer for the mGluR2 receptor in the human brain. J NUCL MED MEETING ABSTRACTS. 2012;53:355. [Google Scholar]
- 119.Ayala JE, et al. mGluR5 positive allosteric modulators facilitate both hippocampal LTP and LTD and enhance spatial learning. Neuropsychopharmacology. 2009;34:2057–2071. doi: 10.1038/npp.2009.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Homayoun H, et al. Functional Interaction Between NMDA and mGluS Receptors: Effects on Working Memory, Instrumental Learning, Motor Behaviors, and Dopamine Release. Neuropsychopharmacology. 2004 doi: 10.1038/sj.npp.1300417. [DOI] [PubMed] [Google Scholar]
- 121.Akgun E, et al. Ligands that interact with putative MOR-mGluR5 heteromer in mice with inflammatory pain produce potent antinociception. Proc. Natl. Acad. Sci. U. S. A. 2013;110:11595–11599. doi: 10.1073/pnas.1305461110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gomes I, et al. Identification of a mu-delta opioid receptor heteromer-biased agonist with antinociceptive activity. Proc. Natl. Acad. Sci. U. S. A. 2013;110:12072–12077. doi: 10.1073/pnas.1222044110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rook JM, et al. Unique signaling profiles of positive allosteric modulators of metabotropic glutamate receptor subtype 5 determine differences in in vivo activity. Biol. Psychiatry. 2013;73:501–509. doi: 10.1016/j.biopsych.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.El Moustaine D, et al. Distinct roles of metabotropic glutamate receptor dimerization in agonist activation and G-protein coupling. Proc. Natl. Acad. Sci. U. S. A. 2012;109:16342–16347. doi: 10.1073/pnas.1205838109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sartorius LJ, et al. Alternative splicing of human metabotropic glutamate receptor 3. J. Neurochem. 2006;96:1139–1148. doi: 10.1111/j.1471-4159.2005.03609.x. [DOI] [PubMed] [Google Scholar]