

Abstract
Mitochondrial calcium is thought to play an important role in the regulation of cardiac bioenergetics and function. The entry of calcium into the mitochondrial matrix requires that the divalent cation pass through the inner mitochondrial membrane via a specialized pore known as the mitochondrial calcium uniporter (MCU). Here, we use mice deficient for MCU expression to rigorously assess the role of mitochondrial calcium in cardiac function. Mitochondria isolated from MCU-/- mice have reduced matrix calcium levels, impaired calcium uptake and a defect in calcium-stimulated respiration. Nonetheless, we find that the absence of MCU expression does not affect basal cardiac function at either 12 or 20 months of age. Moreover, the physiological response of MCU-/- mice to isoproterenol challenge or transverse aortic constriction appears similar to control mice. Thus, while mitochondria derived from MCU-/- mice have markedly impaired mitochondrial calcium handling, the hearts of these animals surprisingly appear to function relatively normally under basal conditions and during stress.
Introduction
To achieve its ongoing energy demand, the heart relies almost exclusively on ATP production from the mitochondria. Cardiac myocytes are particularly well constructed for this energetic task with approximately ten to twenty thousand mitochondria per myocyte and with the mitochondria comprising roughly a third of the cardiac myocyte cellular volume [1]. In certain situations, for instance with exercise or after adrenergic stimulation, the heart sees a dramatic increase in its energetic demand. Experimental evidence has demonstrated that during such episodes, the ratio of ADP/ATP remains relatively constant [2, 3]. This suggests that during these periods of suddenly increased demand, the heart has the capacity to rapidly augment ATP production.
One mechanism for how ATP production could be rapidly augmented came from observations in the 1970s that various mitochondrial matrix enzymes were regulated by calcium. In particular, the key metabolic enzymes pyruvate dehydrogenase, oxoglutarate dehydrogenase and isocitrate dehydrogenase all demonstrated calcium-induced activation [4]. These observations led to the hypothesis that entry of mitochondrial calcium across the inner membrane results in a rise in matrix calcium, which activates matrix dehydrogenases, leading to an increase in ATP production necessary to meet the increase in demand. Recent studies have also demonstrated that calcium also activates specific components of the electron transport chain including Complex V [5].
For many years, the molecular details of how calcium enters the mitochondria remained elusive. Pharmacological, biochemical and electrophysiological evidence all pointed to an inner mitochondrial membrane protein that acted as a calcium selective pore, relying on the large mitochondrial membrane potential as a driving force for calcium entry. Nonetheless, the molecular identity of the mitochondrial calcium uniporter (MCU) was unknown until several years ago, when two groups independently identified a 40 kD protein as the long sought uniporter [6, 7]. Both groups were able to demonstrate that knockdown of MCU attenuated mitochondrial calcium uptake.
In order to characterize the physiological role of mitochondrial calcium, we recently described a MCU mouse knockout model [8]. Remarkably, in a mixed genetic background, MCU-/- mice were viable and except for a smaller size, displayed remarkably few outwardly discernable phenotypes. We did observe that when challenged with an increase in workload, the skeletal muscle of MCU-/- mice appeared impaired and were unable to perform as well as their wild type littermates. Given the significant experimental data concerning the role of mitochondrial calcium in the heart [9, 10], we sought to further characterize the cardiac function of our MCU-/- mice under basal and stress conditions. Here, we report that the absence of MCU expression surprisingly does not alter basal cardiac function, nor does it seemingly markedly impair the ability of the heart to respond to pharmacological or physiological stress.
Methods
Detailed methods are available in the online supplement.
Results and Discussion
MCU protein expression could readily be detected in mitochondria isolated from the hearts of wild type mice, although this expression, as expected, was seemingly absent in mitochondria obtained from the hearts of MCU-/- mice (Figure 1A). The absence of MCU also resulted in the corresponding reduction of EMRE (essential MCU regulator), a known component of the uniporter complex [11]. Previous in vitro observations have also noted that reducing MCU expression results in the destabilization of EMRE [11]. In contrast, the level of other mitochondrial proteins, such as cytochrome oxidase subunit 4 (COX4), were unchanged by MCU deletion. Consistent with a lack of MCU expression, compared to WT mitochondria, mitochondria from MCU-/- animals had a defect in their ability to take up exogenous calcium (Figure 1B). While in some experiments, we saw evidence consistent with very modest calcium uptake at very low concentrations of calcium addition (Figure 1B), this was not seen reproducibly and this uptake was often not inhibited by Ru360 addition (Supplementary Figure 1A). We believe this signal most likely represents non-specific calcium binding to cellular membranes rather than true mitochondrial calcium uptake. Consistent with a lack of calcium uptake in MCU-/- mitochondria, while ADP stimulated respiration was similar in WT and MCU-/- cardiac mitochondria (Supplemental Figure 1B), no calcium-dependent respiration was observed in MCU-/- mitochondria (Figure 1C). We next sought to analyze basal calcium levels in mitochondria isolated from WT or MCU-/- mice. We noted that matrix calcium levels were significantly reduced in MCU-/- mitochondria (Figure 1D), consistent with data we had previously obtained using skeletal muscle mitochondria [8]. Basal levels of ATP were, however, similar in WT and MCU-/- hearts (Supplementary Figure 1C). Thus, mitochondria isolated from MCU-/- mice lack MCU expression, have markedly impaired calcium uptake and lower resting calcium levels, yet have seemingly preserved basal mitochondrial function and energetics.
Figure 1.
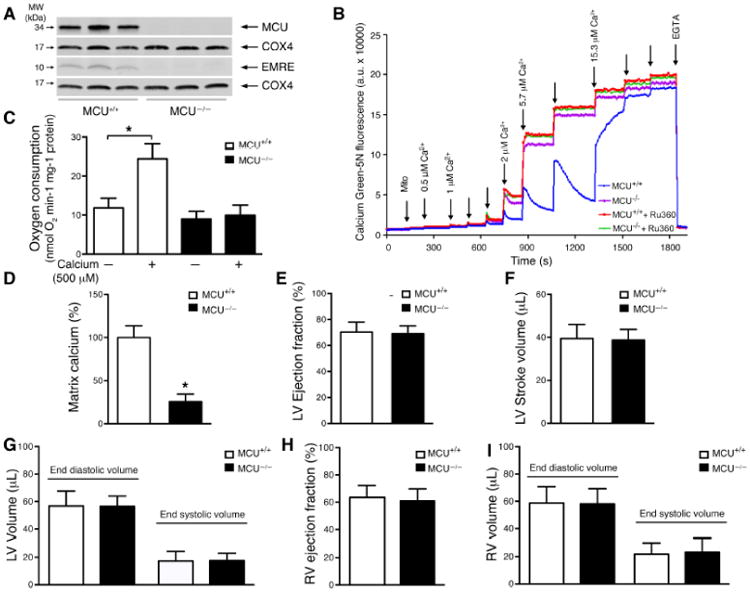
Lack of the mitochondrial calcium uniporter does not alter basal heart function despite the loss of rapid mitochondrial calcium uptake and lower mitochondrial matrix calcium. A) Western blot analysis of MCU and EMRE protein in heart mitochondria (n= 3 mice for each genotype). COX4 was used as a loading control. B) Mitochondrial calcium uptake of isolated cardiac mitochondria. Shown is one representative trace from at least three similar experiments with WT and MCU-/- mitochondria. Ru360 (3 μM) is a pharmacological inhibitor of MCU pore activity. EGTA (20 mM) was added at the end of the experiment. C) Mitochondrial oxygen consumption measured basally with glutamate/malate as substrates (-) and after addition of 500 μM calcium (*p<0.0001 by one-way ANOVA; n=5 WT and n=3 MCU-/- mice). D) Levels of matrix calcium measured in WT and MCU-/- heart mitochondria (*p<0.01 by t-test; n=3 WT and n=3 MCU-/- mice). (E-I) MRI analysis of 20-month old WT and MCU-/- mice assessing LV E) ejection fraction F) stroke volume and G) end diastolic and systolic volumes. Right ventricular H) ejection fraction as well as I) end diastolic and systolic volumes were also measured (for MRI analysis n=10 WT and n=9 MCU-/- mice). The absence of a (*) indicates a non-significant statistical relationship.
We next sought to understand the physiological effects of MCU deletion in the heart. Using echocardiography, we analyzed the hearts of 12-month old female MCU-/- mice or their WT littermates. Basal ejection fraction (WT 59.2±1.6% and MCU-/- 61.0±2.2%, n=11 WT and n=10 MCU-/-) and fractional shortening (WT 31.3±1.1% and MCU-/- 32.5±1.5%) were indistinguishable between these two genotypes. Analysis of the ventricular wall dimensions revealed that MCU-/- mice and WT animals also had similar wall thickness (diastole WT 0.87±0.008 mm and MCU-/- 0.87±0.009 mm, systole WT 1.22±0.01 mm and MCU-/- 1.20±0.02 mm).
While our echocardiographic analysis of one-year old mice failed to detect a significant difference in the basal cardiac function of MCU-/- mice, we reasoned that the effects of MCU deletion might be time dependent. We therefore sought to analyze the function of 20 months old female mice. Again, even in these older mice, using more sensitive MRI determinations, we noted no differences in baseline left ventricular (LV) ejection fraction (Figure 1E). Similarly, stroke volumes were indistinguishable between the two genotypes (Figure 1F). MRI-based determination of end diastolic and end systolic volumes also revealed no measurable differences in LV chamber dimensions (Figure 1G). Furthermore, assessment of right ventricular (RV) function and volumes revealed that MCU deletion also had no significant effect on right-sided ejection fraction or chamber size (Figure 1H&I). Finally, we re-analyzed the results by normalizing stroke volume, ventricular volumes and wall thickness to body weight to correct for the slightly smaller size of the MCU-/- mice. Nonetheless, after this modification there were still no significant differences between WT and MCU-/- (Supplementary Figure 2A-E).
Taken together these results suggest that mice lacking MCU expression have remarkably preserved basal cardiac function over at least the first 20 months of life. We therefore next sought to assess whether the absence of MCU expression might modulate heart function during increased workload. We have previously shown that MCU-/- cardiomyocytes do not uptake calcium into the mitochondria after β-adrenergic stimulation by isoproterenol, unlike their WT counterparts [8]. Therefore, we sought to analyze the response of 12 month old mice to isoproterenol using miniature pressure volume Millar catheters. When genotypes were compared, we observed no differences in the left ventricular cardiac output of WT and MCU-/- 12-month old female mice at baseline and following isoproterenol (Figure 2A). Although WT mice exhibited a significant increase in cardiac output following isoproterenol and the MCU-/- mice showed a more modest increase that did not reach significance, no significant difference could be detected between WT and MCU-/- after adrenergic stimulation. Similar relationships were seen with both heart rate and (Figure 2B) and dP/dTmax (Figure 2C). Our previously published results further suggested a diminished capacity in MCU-/- mice to withstand increased stress in skeletal muscle [8]. We therefore subjected animals to the more chronic stress paradigm of surgical transverse aortic constriction (TAC). When TAC surgeries were performed on WT and MCU-/- male mice, we noted similar overall survival with 10 of 16 WT and 11 of 16 MCU-/- mice surviving until 8 weeks post-surgery. Echocardiographic analysis of these surviving mice showed a similar time-dependent decline in ejection fraction (Figure 2D) and fractional shortening (Figure 2E). Assessment of chamber size showed similar increases in LV anterior wall thickness (Figure 2F&G). Finally, determination of heart weight to body weight ratios, as well as histological staining for collagen deposition confirmed similar levels of hypertrophy and fibrosis in WT and MCU-/- mice following TAC (Supplementary Figure 3).
Figure 2.
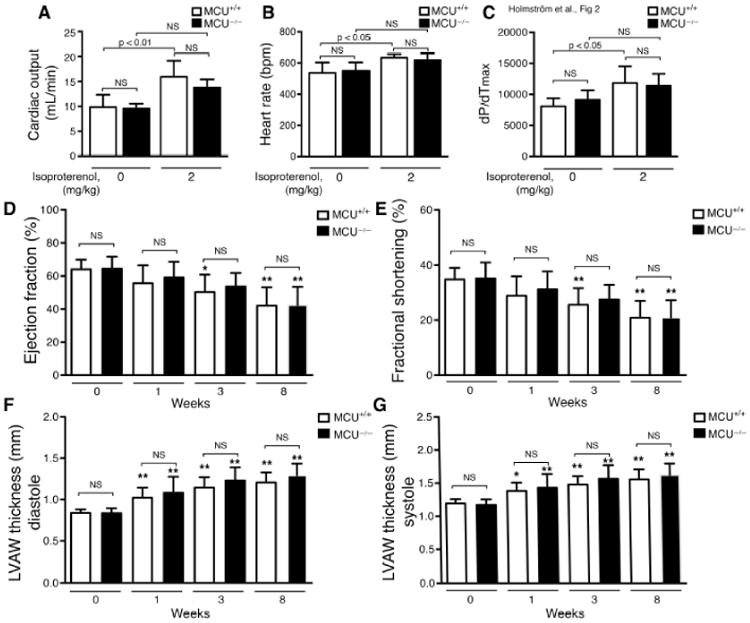
Lack of MCU expression does not markedly alter heart function upon stress. (A-C) LV function was assessed by miniature pressure volume catheters in WT and MCU-/- mice following pharmacological stimulation. Parameters assessed include A) cardiac output, B) heart rate and C) dP/dTmax (n=6 WT and n=5 MCU-/- mice). (D-G) Transverse aortic constriction was performed on three to five-month old WT and MCU-/- mice. Echocardiography was performed before surgery and 1, 3, 8 weeks after the procedure. Parameters assessed include D) ejection fraction, E) fractional shortening and F) left ventricle anterior wall (LVAW) thickness at diastole and G) systole (for TAC functional analysis, n= 10 WT and n=11 MCU-/- mice). No statistically significant differences were observed between WT and MCU-/- mice at any time point. *p<0.05 and **p<0.01 when compared to the initial value (week zero) within the same genotype.
In summary, we have analyzed the cardiac phenotype of mice lacking MCU expression under basal conditions and under various stress conditions. Unexpectedly, we saw no marked cardiac defects in mice without MCU expression. This lack of an overt cardiac phenotype is somewhat surprising given the significant literature suggesting an important role for mitochondrial calcium in regulating cardiac function [9, 10]. Moreover, in our initial description of the MCU-/- mice, we did observe a clear defect in skeletal muscle power generation [8]. The differences between heart and skeletal muscle phenotypes might relate to an earlier observations where patch clamp recordings of the inner mitochondrial membrane revealed that MCU-dependent currents were roughly 30-fold lower in heart tissue compared to skeletal muscle [12]. Since we show that MCU protein is abundantly expressed in heart, these differences in activity presumably relate to tissue-specific differences in the regulation of putative negative regulators of MCU including MCUb and MICU1, or the relative role for other components such as EMRE [13]. A recent fascinating report in which a dominant negative form of MCU was expressed in the heart noted that these animals had a markedly impaired chronotropic response to isoproterenol [14]. In our case, comparison within genotypes revealed that while our WT mice had a significant increase in heart rate after isoproterenol (average heart rate at baseline 537 bpm, with isoproterenol 634 bpm; n=6, p< 0.014), the response in MCU-/- mice did not reach significance (average heart rate at baseline 548 bpm, with isoproterenol 617 bpm; n=5, p<0.15). Similarly, while WT mice had a significant isoproterenol-stimulated increase in cardiac output and dP/dTmax, this again was not evident in the hearts of MCU-/- mice. This lack of statistical significance may be a result of our study being underpowered, or it may reflect slight impairment in the physiological response of MCU-/- mice to isoproterenol. Interestingly, our WT mice had a much more modest chronotropic response to isoproterenol than the control mice used in the aforementioned study [14]. Such differences in the response between control groups presumably reflect variations in experimental conditions, including differences in the genetic background of the mice used. In addition to such experimental differences, it should be noted that we (Figure 1A) and others have found that in the absence of MCU, other components of the uniporter complex also have reduced stability and hence expression [11, 15]. These destabilizing effects would presumably not be seen when expressing a non-functional dominant negative MCU, which lacks pore function, yet still retains the ability to act as a structural scaffold for EMRE and other components. Such structural differences in the uniporter complex may also contribute to differences observed in the chronotropic response between the two models. Finally, as we note here, cardiac mitochondria appear to have reduced but not entirely absent levels of mitochondrial calcium (Figure 1D). As we noted before, our mouse model was created using a gene trap strategy [8], and hence the possibility remains that low levels of MCU are still expressed, although this would have to be below the level of Western blot detection. Such low level expression is a possible explanation for the potential modest calcium uptake seen on occasion with very low concentrations of calcium addition (Figure 1B), although as discussed, we favor alternative explanations. Perhaps more likely, it would appear that additional mechanisms of presumably slow calcium uptake are possible. Elucidation of these additional pathways will undoubtedly be important in further understanding the role of mitochondrial calcium in cardiac function.
Supplementary Material
Highlights.
-MCU expression is required for rapid calcium uptake in cardiac mitochondria.
-Hearts lacking MCU have normal basal cardiac function
-Deletion of MCU does not alter the response to transverse aortic constriction
Acknowledgments
We thank Hong San for assistance with the TAC surgery, Zuxi Yu for help with pathology and Ilsa I Rovira for help for the manuscript. This work was supported by a grant from the Leducq Foundation and with NIH Intramural Funds.
Footnotes
Disclosures: None declared
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Barth E, Stammler G, Speiser B, Schaper J. Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J Mol Cell Cardiol. 1992;24:669–81. doi: 10.1016/0022-2828(92)93381-s. [DOI] [PubMed] [Google Scholar]
- 2.Neely JR, Denton RM, England PJ, Randle PJ. The effects of increased heart work on the tricarboxylate cycle and its interactions with glycolysis in the perfused rat heart. Biochem J. 1972;128:147–59. doi: 10.1042/bj1280147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Katz LA, Swain JA, Portman MA, Balaban RS. Relation between phosphate metabolites and oxygen consumption of heart in vivo. Am J Physiol. 1989;256:H265–74. doi: 10.1152/ajpheart.1989.256.1.H265. [DOI] [PubMed] [Google Scholar]
- 4.McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev. 1990;70:391–425. doi: 10.1152/physrev.1990.70.2.391. [DOI] [PubMed] [Google Scholar]
- 5.Glancy B, Balaban RS. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry. 2012;51:2959–73. doi: 10.1021/bi2018909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–5. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–40. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15:1464–72. doi: 10.1038/ncb2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Griffiths EJ, Balaska D, Cheng WH. The ups and downs of mitochondrial calcium signalling in the heart. Biochim Biophys Acta. 2010;1797:856–64. doi: 10.1016/j.bbabio.2010.02.022. [DOI] [PubMed] [Google Scholar]
- 10.Williams GS, Boyman L, Lederer WJ. Mitochondrial calcium and the regulation of metabolism in the heart. J Mol Cell Cardiol. 2015;78:35–45. doi: 10.1016/j.yjmcc.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sancak Y, Markhard AL, Kitami T, Kovacs-Bogdan E, Kamer KJ, Udeshi ND, et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science. 2013;342:1379–82. doi: 10.1126/science.1242993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fieni F, Lee SB, Jan YN, Kirichok Y. Activity of the mitochondrial calcium uniporter varies greatly between tissues. Nat Commun. 2012;3:1317. doi: 10.1038/ncomms2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Foskett JK, Philipson B. The mitochondrial Ca(2+) uniporter complex. J Mol Cell Cardiol. 2015;78:3–8. doi: 10.1016/j.yjmcc.2014.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu Y, Rasmussen TP, Koval OM, Joiner ML, Hall DD, Chen B, et al. The mitochondrial uniporter controls fight or flight heart rate increases. Nat Commun. 2015;6:6081. doi: 10.1038/ncomms7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kamer KJ, Mootha VK. MICU1 and MICU2 play nonredundant roles in the regulation of the mitochondrial calcium uniporter. EMBO Rep. 2014;15:299–307. doi: 10.1002/embr.201337946. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.