

Abstract
The obligate intracellular bacterium Chlamydia trachomatis is the most common cause of bacterial sexually transmitted disease in the United States and the leading cause of preventable blindness worldwide. Transfer of cultured Chlamydia-specific CD8+ T cells or vaccination with recombinant virus expressing a MHCI-restricted Chlamydia antigen confers protection, yet surprisingly a protective CD8+ T cell response is not stimulated following natural infection. In this study we demonstrate that the presence of excess IL12 and IFNγ contributes to poor memory CD8+ T cell development during C. trachomatis infection of mice. IL12 is required for CD8+ T cell expansion but drives effector CD8+ T cells into a short-lived fate whereas IFNγ signaling impairs the development of effector memory cells. We show that transient blockade of IL12 and IFNγ during priming promotes the development of memory precursor effector CD8+ T cells and increases the number of memory T cells that participate in the recall protection against subsequent infection. Overall, this study identifies key factors shaping memory development of Chlamydia-specific CD8+ T cells that will inform future vaccine development against this and other pathogens.
Introduction
There remains a pressing need for vaccines that induce robust CD8+ T cell-mediated immunity, specifically to combat pathogens that replicate within cells and evade the protective mechanisms mediated by antibodies. A typical CD8+ T cell response is characterized by a rapid expansion of rare Ag-specific T cells that contribute to the elimination of a specific pathogen. A vast majority of these cells then contract to maintain homeostasis of the immune system (1). The cells that survive contraction remain stable over time and mediate immunological memory (2). Stimulation of robust memory is critical for any successful vaccine. When effector CD8+ T cells expand and differentiate during a primary response, they do not equally acquire memory cell properties (3, 4). The signals that promote memory cells development are not thoroughly understood and usually occur early in the immune response (4, 5). Here we investigate the factors that drive memory CD8+ T cell fate following infection with the obligate intracellular bacterial pathogen, Chlamydia trachomatis.
C. trachomatis infects over 100 million people worldwide annually (WHO, 2008), and is both the most prevalent bacterial genital tract infection and the leading cause of preventable blindness. Chronic C. trachomatis genital tract infections lead to pelvic inflammatory disease (PID), which can cause fallopian tube scarring, infertility, and ectopic pregnancy (6, 7). Although human infection with C. trachomatis stimulates multiple elements of the immune system, these responses often fail to clear the infection or prevent subsequent reinfection (8). As with other pathogens that cause chronic infectious diseases, this lack of immune protection suggests a failure in adaptive immunity–specifically the memory responses that should provide long-lasting protection against reinfection. Therefore, an effective Chlamydia vaccine must induce a memory response better than that stimulated during natural infection.
Although antibody and CD4+ T cells clearly are required for full immunity to C. trachomatis (9, 10), CD8+ T cells should also be a major component of adaptive immunity against this pathogen. C. trachomatis infects epithelial cells in the genital tract, a cell type that expresses MHCI but not usually MHCII. Because C. trachomatis translocates a subset of its proteins into the host cell cytosol it allows for MHCI processing of these proteins and subjects the cell to recognition by CD8+ T cells (11, 12). CD8+ T cells have been shown to protect against infection when cultured ex vivo and transferred into naïve animals, and immunization with recombinant vaccinia viruses expressing CD8+ T cell antigens from C. trachomatis also confers protection in mice (12). Yet during natural infection of mice, the CD8+ T cell response does not play a significant protective role (13, 14). Previous studies from our laboratory have shown that CD8+ T cells respond well to primary C. trachomatis infection, but the memory cells that result from initial infection are impaired in their ability to respond to subsequent encounters with the pathogen (15, 16).
To better understand the failure of CD8+ T cell memory development following C. trachomatis infection, we compared the Ag-specific CD8+ T cells induced by C. trachomatis (poor recall) with those of the same antigen specificity induced by recombinant vaccinia virus expressing a C. trachomatis antigen, CrpA (robust recall) (16). We found that the proinflammtory cytokines IL12 and IFNγ drive effector CD8+ T cells stimulated by C. trachomatis into a short-lived fate (TSLEC) and impair the development of effecter memory cells. Transient blockade of these cytokines during priming increases the frequency of memory precursor CD8+ T cells (TMPEC) and memory CD8+ T cell numbers. Overall, this study identified factors that are critical for CD8+ T cell memory development following C. trachomatis infection, which should aid in vaccine development against this and other pathogens responsible for chronic infections.
Materials and Methods
Mice
C57BL/6J, B6.PL-Thy1a/CyJ (CD90.1 congenic), B6.129S7-Ifngr1tm1Agt/J (IFNγR−/−), and B6.129S1-Il12rb1tm1Jm/J (IL12Rβ−/−) were purchased from The Jackson Laboratory (Bar Harbor, ME). Tbet−/− mice (C57BL/6 background) were kindly provided by L. Glimcher (Harvard School of Public Health) (17). Tbet+/− mice were generated by crossing C57BL/6J and Tbet−/− mice. PDL1−/− mice (C57BL/6 background) have been described before and were generously provided by A. Sharpe (Harvard Medical School) (18). To generate Chlamydia-specific CD8+ TCR transgenic mice specific for CrpA63-71, we cloned the rearranged genomic TCRα and TCRβ sequences from Chlamydia-specific CD8+ T cell clone NR23.4 into expression vectors (19). The cloned TCR constructs were then linearized and injected into C57BL/6 fertilized oocytes. TCR tg founders were identified by PCR. Although NR23.4 transgenes were integrated into the genome of these founders, possible competition from endogenous TCR rearrangements inhibited efficient expression of the NR23.4 TCR. In order to restrict TCR expression, we crossed these mice onto a RAG1−/− background (NR23.4 mice). The rearranged TCR from NR23.4 uses the Vα4JTA13 and Vβ8.2Jβ2.5 receptor chains. NR23.4 IL12Rβ−/− and NR23.4 IFNγR−/− were generated by crossing NR23.4 mice with IL12Rβ−/− and IFNγR−/− mice, respectively. Mice were maintained within the Harvard Medical School Center for Animal Resources and Comparative Medicine. All experiments in this report were approved by Harvard’s Institutional Animal Care and Use Committee.
Growth, isolation, and detection of bacteria and virus
C. trachomatis serovar L2 (434/Bu; ATCC) was propagated within McCoy cell monolayers grown in Eagle’s MEM (Invitrogen) supplemented with 10% FCS, 1.5 g/L sodium bicarbonate, 0.1 mM nonessential amino acids, and 1 mM sodium pyruvate. Infected monolayers were disassociated from flasks using 0.05 % trypsin/EDTA and sonicated to disrupt the inclusion. Elementary bodies (EBs) were purified by density gradient centrifugation as previously described (20). Aliquots were stored at −80 °C in sucrose-phosphate-glutamate buffer (SPG) and thawed immediately before use. Construction of the recombinant vaccinia virus expressing the Chlamydia CrpA protein (VacCrpA) has been described previously (12). Virus preparations were treated with an equal volume of 0.25 mg/ml trypsin for 30 min at 37° C and diluted in PBS before infecting mice.
Preparation of IL2-anti-IL2 complexes
IL2-anti-IL2 complexes were prepared as previously described (23–25). 1.5 μg carrier-free mouse recombinant IL2 (eBioscience) and 50 μg anti-IL2 monoclonal antibody (S4B6, BioXCell) were mixed in 10 μl HBSS at room temperature for 15 minutes before adding 190 μl HBSS for each injection. Control groups were treated with IgG2a isotype control antibodies (2A3, BioXCell).
Infection of mice and preparation of tissue
For systemic infection, mice were infected i.v. with 107 inclusion-forming units (IFU) of C. trachomatis in 200 μl SPG, 2×103 PFU of VacCrpA in 200 μl PBS, or 103 CFU of LmCrpA in 200 μl of PBS, unless otherwise noted. To infect in the genital tract, mice were treated s.c. with 2.5 mg medroxyprogesterone acetate (Pfizer) and then infected one week later transcervically with 5×106 IFU of C. trachomatis or 5×105 PFU of VacCrpA as described previously (26). At specific times post-infection, the iliac lymph nodes, spleen, and uterine horns were excised. Uteri were dissected free of the mesometrium and then finely minced with scalpels. Minced tissues were enzymatically dissociated in HBSS/Ca2+/Mg2+ containing 1 mg/ml type XI collagenase and 50 Kunitz/ml DNase for 30 minutes at 37° C, washed in Ca2+/Mg2+-free PBS containing 5 mM EDTA, and then ground between frosted microscope slides prior to filtration through a 70-cm mesh (27). Single cell suspensions of secondary lymphoid organs (SLO) were prepared by grinding the tissue between frosted microscope slides. Red blood cells in the splenocytes were lysed using ammonium chloride.
Flow cytometry
Cells were immediately stained for surface and activation markers or stimulated for 4–5 hours with 10μM CrpA63-71 peptide in the presence of brefeldin A (Biolegend) for intracellular cytokine staining. The Db/ASFVNPIYL (CrpA63-71) MHC tetramer was generated at the National Institutes of Health Tetramer Facility. Antibodies were purchased from Biolegend except for CD16/CD32 (2.4G2; Bio X-Cell), anti-CD62L-PE-Texas Red (Invitrogen), anti-CD95 (BD Biosciences), anti-CD127 (ebiosciences), anti-CD4 Qdot605 (Invitrogen), anti-IL18Rα (R&D systems), and anti-IFNγ APC-Cy7 (BD Biosciences). Cells were pre-incubated with CD16/CD32 (2.4G2) before staining with tetramer and fluorochrome (APC, APC-Cy7, FITC, PE, PerCP, PerCP-Cy5.5, PE-Cy7, Pacific Blue, PE-Texas Red) conjugated antibodies against mouse B220 (RA3-6B2), CD4 (RM4-5 or GK1.5), CD8 (53-6.7), CD90.1 (OX-7), CD90.2 (53-2.1), CD27 (Lg.3A10), PDL1 (10F.9G2), CD3 (17A2), CD11b (M1/70), killer cell lectin-like receptor G1 (KLRG1) (2F1), CD25 (PC61), CD95 (Jo2), IL18Rα (112614), CD122 (TM-β1), CD326 (G8.8), CD11c (N418), MHC I-Ab (AF6-120.1), or CD127 (A7R34). LIVE/DEAD fixable aqua dead cell stain kit (Invitrogen) was used along with other antibodies to exclude dead cells from analyses. For intracellular staining, cells were permeabilized with the Cytofix/Cytoperm Plus Kit according to the manufacturer’s instructions (BD Biosciences) and stained with anti-IFNγ (XMG 1.2). The absolute cell number in each sample was determined using AccuCheck Counting Beads (Invitrogen). Data were collected on a LSRII (BD Bioscience) and analyzed using FlowJo (Tree Star Industries, Ashland, OR). CrpA-specific CD8+ T cells were gated as LIVE/DEAD−CD4−B220−CD11b−MHC I-Ab−CD3+CD8+CrpA-tetramer+.
Detection of BrdU uptake
To determine proliferation rate of T cells, mice were injected i.p. with 1 mg of BrdU daily on days 30–35 post-inoculation (p.i.). On day 35, splenocytes were isolated, surface stained, fixed, permeabilized, and stained with anti-BrdU mAb as recommended by the manufacturer of the BrdU flow kit (BD Biosciences).
Cytokine detection and depletion
Serum was extracted from peripheral blood and cytokine levels in the serum were determined as recommended by the luminex kit (Millipore) or by ELISA as previously described (12). To deplete cytokines, mice were injected i.p. with 200 μg anti-IFNγ (XMG1.2) together with 200 μg anti-IL12 (C17.8), or isotype control (200 μg HRPN and 200 μg 2A3) in 200 μl PBS on day 4 p.i. Serum was extracted from these mice on day 7 p.i. and cytokine levels in the serum were determined by ELISA to check the efficiency of depletion protocol. IFNγ level is below the limit of detection in depletion antibody treated mice. IL12 level is significantly lower in mice treated with depletion antibodies (12.99±0.02% of the levels in control mice) compared to control antibody treated mice (p < 0.01). All isotype and neutralizing antibodies were purchased from Bio-X-Cell.
Transfer of T cells
For transfer of transgenic cells, C. trachomatis-specific CD8+ T cells were isolated from the SLOs of donor NR23.4 mice. Recipient mice were injected with 104–105 cells i.v. into tail veins one day before infection. For transfer of immune T cells, SLOs were isolated on day 28 p.i., and homogenized into single cell suspensions. CD8+ T cells were isolated using Dynal Mouse CD8 Negative Isolation Kit according to manufacturer’s instructions (Invitrogen). Isolated cells were then labeled with 10 μM CFSE (Invitrogen) as previously described (28). Unless otherwise stated, 5×106 CD8+ T cells were injected i.v. into naïve mice 4 hours prior to transcervical infection.
Quantitative PCR
The levels of C. trachomatis or VacCrpA in the spleens or the uteri of infected mice were quantified using a previously described quantitative PCR assay (qPCR) (29, 30). Briefly, total nucleic acid from infected spleen or uterus homogenates was prepared using the QIAamp DNA mini kit (Qiagen). Chlamydia 16S DNA, vaccinia ribonucleotide reductase (Vvl4L), and mouse GAPDH DNA content of individual samples were then quantified by qPCR on an ABI 7000 sequence detection system (Applied Biosystems) using primer pairs and dual-labeled probes (IDT or Applied Biosystems). Standard curves were generated from known amounts of Chlamydia, vaccinia, or mouse DNA, and these curves were used to calculate the amount (in pg) of Chlamydia DNA or vaccinia DNA per unit weight (in μg) of mouse DNA in the samples.
Statistical analysis
A two-tailed Mann-Whitney U test was applied to determine statistical significance for bacterial burdens among groups. All other data were evaluated for statistical significance with an unpaired two-tailed t test. Differences were considered statistically significant if the P value was <0.05. *: p < 0.05; **: p < 0.01. Results were shown as mean ± standard error.
Results
CD8+ T cells induced by C. trachomatis contract more than those induced by VacCrpA
VacCrpA is known to induce a more robust CrpA-specific recall response than C. trachomatis (16). To determine why C. trachomatis induces an impaired CD8+ T cell population that fails to efficiently participate in recall, we compared CrpA-specific CD8+ T cells induced by C. trachomatis to those induced by VacCrpA. To rule out the impact of pathogen level on memory CD8+ T cell development, the VacCrpA challenge dose was carefully titrated so that similar numbers of CrpA-specific CD8+ T cells were induced at the peak of expansion following either C. trachomatis or VacCrpA infection (Fig. 1A). Two thousand PFU of VacCrpA and 107 IFU of C. trachomatis yielded no significant differences in the number of CrpA-specific CD8+ T cells at the peak of the primary response (day 7). Therefore these doses were used to challenge animals throughout this study. Significantly more CrpA-specific CD8+ T cells were recovered from VacCrpA infected mice than C. trachomatis infected mice at later times when stable memory had formed (Fig. 1B), indicating that CrpA-specific CD8+ T cells induced by C. trachomatis contracted more than the cells induced by VacCrpA. A similar proportion of memory CrpA-specific CD8+ T cells from C. trachomatis- or VacCrpA-infected mice secreted IFNγ following ex vivo restimulation (Fig. 1C), suggesting that the memory cells that did survive the contraction following C. trachomatis infection were similarly functional compared to cells stimulated by VacCrpA infection. Although these memory cells proliferated as efficiently as VacCrpA-induced memory cells as measured by BrdU uptake (Fig. 1D), they expressed higher level of CD95 (FasR) (Fig. 1E), suggesting that these cells are more prone to apoptosis. Moreover, compared to CrpA-specific memory CD8+ T cells from VacCrpA infected mice, CrpA-specific cells from C. trachomatis infected mice expressed lower levels of CD27, CD122 (IL2/IL15 receptor β), and IL18Rα, all of which are critical for the recall capacity of memory CD8+ T cells (Fig. 1F–H) (31–34). Overall, these data suggest that although CrpA-specific CD8+ T cells are expanded by C. trachomatis or VacCrpA infection to a similar extent, C. trachomatis-stimulated cells contract more and the memory cells that do survive the contraction are of lower quality.
Figure 1.

C. trachomatis induces impaired memory CD8+ T cells. (A) CrpA-specific CD8+ T cell numbers in the spleen of systemically infected mice on day 7 p.i are shown. (B) CrpA-specific CD8+ T cell numbers in the spleen of mice systemically infected with C. trachomatis or 2×103 PFU of VacCrpA on indicated days p.i. are shown. (C) IFNγ+ %, (D) BrdU+ %, (E) CD95 MFI, (F) CD27+ %, (G) CD122 MFI, and (H) IL18Rα MFI of CrpA-specific CD8+ T cells in the spleen on days 26–30 p.i. are shown. Data are representative of at least two experiments, each with 5–7 mice per group.
IL2-α-IL2 complexes are not sufficient to rescue the recall capacity of CD8+ T cells stimulated by C. trachomatis
The kinetics of CrpA-specific CD8+ T cells stimulated by C. trachomatis infection were indicative of “helpless” CD8+ T cells that are not durable and wane over time (35–39). One of the mechanisms by which helper T cells mediate help is to instruct DCs to produce cytokines that induce up-regulation of IL2Rα (CD25) on Ag-specific CD8+ T cells, rendering them more responsive to IL2 (40, 41). We therefore explored whether CD25 expression is differentially stimulated following infection with C. trachomatis vs. VacCrpA. Since CD25 expression peaks early, when the number of endogenous CrpA-specific CD8+ T cells is too low to be reliably detected, we transferred CrpA-specific transgenic cells before infection and examined the expression of CD25 on these cells. On day 3 p.i., significantly fewer CrpA-specific transgenic cells expressed CD25 as they divided in C. trachomatis-infected mice compared to VacCrpA-infected mice. A similar trend was observed on day 4 p.i. By day 6 p.i., the C. trachomatis stimulated cells had caught up, suggesting that the induction of CD25 on CrpA-specific CD8+ T cells was delayed in mice infected with C. trachomatis compared to those infected with VacCrpA (Fig. 2A).
Figure 2.

IL2-anti-IL2 immune complex treatment is not sufficient to rescue the blunted recall response. (A) CFSEdimCD25+ % of NR23.4 cells in the spleen are shown. (B) Mice were infected i.v. with C. trachomatis, treated with IL2-anti-IL2 immune complex (IL2 IC) or isotype control antibodies on days 3 and 5 p.i. (early) or on days 24 and 26 p.i. (late), and re-challenged with VacCrpA on day 28 p.i. CrpA-specific CD8+ T cell numbers 5 days after secondary challenge are shown. Representative data from two experiments are shown, each with 5–7 mice per group.
Previous reports have shown that IL2 signaling not only augments the accumulation of CD8+ T cells, but also programs the ability of memory cells to expand upon secondary challenge (40–43). To test whether IL2 signaling differences were responsible for the poor recall capacity of Chlamydia-stimulated memory cells, we treated mice with IL2-anti-IL2 (IL2/S4B6) complexes that have been shown to increase the recall capacity of CD8+ T cells (44). Mice were infected with C. trachomatis, treated with IL2-anti-IL2 complexes or isotype control antibodies on days 3 and 5 (early) or on days 24 and 26 (late), and then rechallenged with VacCrpA on day 28. The numbers of CrpA-specific CD8+ T cells in these mice were determined 5 days later. Numbers of CrpA-specific CD8+ T cells were similar among all rechallenged groups and were significantly lower than the primary control group (Fig. 2B), suggesting that stimulating IL2 signaling early or late during memory development was not sufficient to rescue the recall capacity of CrpA-specific CD8+ T cells. Overall, these data suggest that the delayed up-regulation of CD25 is not the primary reason why CrpA-specific CD8+ T cells induced by C. trachomatis fail to efficiently participate in the recall response.
C. trachomatis-stimulated effector CD8+ T cells are enriched for the short-lived phenotype
Early in priming the differential expression of KLRG1 and CD127 (IL7Rα) has been shown to mark two effector T cell populations with distinct memory potential. The CD127lowKLRG1high short-lived effector cells (TSLEC) do not gain memory T cell potential. Rather, it is primarily the descendents of CD127highKLRG1low memory precursor effector cell (TMPEC) that participate in secondary responses upon reinfection (45). We hypothesized that C. trachomatis infection may favor the development of TSLEC since the kinetics of the Chlamydia-specific CD8+ T cell response resembles the kinetics of short-lived effector cells. To test whether C. trachomatis and VacCrpA differentially stimulate TSLEC vs. TMPEC among the CrpA-specific CD8+ T cells, we compared CD127 and KLRG1 expression on CrpA tetramer+ CD8+ T cells following infection with C. trachomatis vs. VacCrpA. At the peak of expansion, more CrpA-specific CD8+ T cells induced by C. trachomatis were TSLEC than those induced by VacCrpA (Fig. 3A, 3B). In contrast, VacCrpA infection favored the formation of TMPEC (Fig. 3A, 3B). We quantified the total number of CrpA-specific TSLEC and TMPEC cells and found that this trend also held true over the time course of infection (Fig. 3C, 3D). Together, these data suggest that C. trachomatis infection favors the formation of TSLEC CD8+ T cells in contrast to VacCrpA infection.
Figure 3.

CrpA-specific CD8+ T cells induced by C. trachomatis infection show TSLEC characteristics. Mice were infected i.v. with C. trachomatis or VacCrpA. (A) The percentage of TSLEC (CD127−KLRG1+) and TMPEC (CD127+KLRG1−) among CrpA-specific CD8+ T cells on day 7, (B) representative flow cytometry analysis of KLRG1 and CD127 expression on CrpA-specific CD8+ T cells on day 7, (C) the absolute number of CrpA-specific TSLEC and (D) TMPEC overtime, are shown. Data are representative of at least three experiments, each with 5–7 mice per group.
Transient reduction of IFNγ and IL12 levels increases the proportion of memory precursor cells
One of the mechanisms that drive effector cells into a short-lived fate during viral infection is overwhelming inflammation (45). To assess whether C. trachomatis and VacCrpA infected mice experience differential levels of inflammation, we measured levels of several cytokines in serum of these mice, including IFNγ, IL12, IL6, IL10, IL7, and IL2. Among the cytokines tested, the levels of two pro-inflammatory cytokines, IFNγ and IL12, were higher in serum from Chlamydia-infected mice compared to serum from VacCrpA-infected mice between day 2 and day 5 p.i. (Fig. 4A, 4B). To test whether the increased levels of these cytokines in C. trachomatis infected mice is responsible for the dominance of the TSLEC phenotype in the pathogen-specific CD8+ T cells, we treated infected mice with a single dose of neutralizing antibodies against IFNγ and IL12 or isotype control antibodies on day 4 p.i. This transient treatment did not alter C. trachomatis burden (data not shown) or the absolute number of CrpA-specific CD8+ T cells at the peak of expansion (Fig. 4C). However, this treatment did reduce the percentage and number of TSLEC and increased the percentage and number of TMPEC among CrpA-specific CD8+ T cells at the peak of expansion (Fig. 4D, 4E). A previous report from our laboratory has shown that the development of Chlamydia-specific TEM (CD127+CD62low) is inhibited during C. trachomatis infection (16). Cytokine-neutralizing Ab treatment increased the percentage and number of TEM among CrpA-specific CD8+ T cells without sacrificing the development of TCM (Fig. 4F, 4G), suggesting that these two cytokines also inhibit the development of TEM. More importantly, when CrpA-specific CD8+ T cell number was quantified a month after inoculation, more CrpA-specific CD8+ T cells were recovered from neutralizing-Ab-treated mice (Fig. 4H). Similar percentage of CrpA-specific CD8+ T cells secreted the effecter cytokine, IFNγ, in mice treated with depletion and control antibodies (Fig. 4I). Overall, these data suggest that transient ablation of pro-inflammatory cytokines early during priming increases the number of T cells that survive contraction without affecting the functionality of memory T cells, consistent with an overall increase of memory potential at the peak of expansion.
Figure 4.

Transient reduction of pro-inflammatory cytokines early during priming improves the development of memory CrpA-specific CD8+ T cells. (A, B) Serum IFNγ (A) and IL12p70 (B) levels in mice systemically infected with C. trachomatis or VacCrpA are shown. (C–I) Mice were infected i.v. with C. trachomatis and treated i.p. with isotype control or IFNγ and IL12 neutralizing antibodies (αIFNγ+αIL12) on day 4 p.i. (C) CrpA-specific CD8+ T cell numbers, (D) representative flow cytometry analysis of KLRG1 and CD127 expression on CrpA-specific CD8+ T cells, (E) total numbers of CrpA-specific TSLEC and TMPEC, (F) representative flow cytometry analysis of CD62L and CD127 expression on CrpA-specific CD8+ T cells, (G) total numbers of CrpA-specific TEM and TCM in the spleen on day 7 p.i are shown. (H) CrpA-specific CD8+ T cell numbers in the spleen and (I) IFNγ + % among CrpA-specific CD8+ T cells on day 28 p.i. are shown. Data are representative of at least two experiments, each with 5–7 mice per group.
Genetically reducing Tbet expression increases the memory potential of Chlamydia-induced CD8+ T cells
IFNγ and IL12 are known to regulate the expression of Tbet, a transcription factor critical for regulating CD8+ T cell memory development (45, 46). To determine whether IFNγ and IL12 regulate memory development through Tbet following Chlamydia infection, we assessed the phenotype of CrpA-specific CD8+ T cells in Tbet+/− mice. We chose Tbet+/− mice instead of Tbet−/− mice to avoid the impact of the complete loss of Tbet on Th1 CD4+ T cell development with the resulting increase in C. trachomatis burden (data not shown). Moreover, the dose dependency of Tbet on CD8+ T cell memory development has been previously described (45). Consistent with the cytokine-neutralizing data, a genetic reduction in the expression of Tbet increased the number of CrpA-specific CD8+ T cells on day 21 p.i., when a stable memory pool had formed (Fig. 5A), without significantly altering C. trachomatis burden (data not shown) or the number of CrpA-specific CD8+ T cells at the peak of expansion (Fig. 5A). Consistent with the cytokine neutralization results, reducing Tbet expression also altered the TMPEC vs. TSLEC ratio in favor of TMPEC at the peak of CD8+ T cell expansion (Fig. 5B) and when a stable memory pool had formed (Fig. 5C). Moreover, reducing Tbet expression resulted in an overall increase of CD127+ memory T cells within which the formation of CD62Llow TEM population was favored (Fig. 5D). Taken together with the IFNγ and IL12 ablation experiments (Fig. 4), these results suggest that pro-inflammatory cytokines, IFNγ and IL12, modulate Tbet to alter pathogen-specific CD8+ T cell development following C. trachomatis infection.
Figure 5.

Tbet+/− mice have more TMPEC and fewer TSLEC. C57BL/6 (B6) or Tbet+/− mice were infected i.v. with C. trachomatis. (A) CrpA-specific CD8+ T cell numbers on days 7 and 21 p.i., (B) numbers of CrpA-specific TSLEC and TMPEC on day 7, (C) TSLEC % vs. TMPEC %, and (D) TEM % vs. TCM % among CrpA-specific CD8+ T cells on day 21 p.i. are shown. Data are representative of two experiments, each with 5–6 mice per group.
IL12 is critical for TMPEC vs. TSLEC development while IFNγ affects TEM vs. TCM formation among C. trachomatis-specific CD8+ T cells
We next explored whether cell intrinsic IFNγ or IL12 signaling in pathogen-specific CD8+ cells is responsible for altering TSLEC vs. TMPEC or TEM vs. TCM development following C. trachomatis infection. We crossed Chlamydia-specific CD8+ TCR transgenic mice (NR23.4) onto the IL12Rβ−/− or IFNγR−/− backgrounds to create CrpA-specific CD8+ T cells that do not respond to IL12 or IFNγ. The transgenic cells lacking IL12Rβ did not expand as efficiently as wild-type cells (Fig. 6A), suggesting that IL12 signaling is required for efficient expansion of pathogen-specific CD8+ T cells following C. trachomatis infection. Nevertheless, the transgenic cells that do not respond to IL12 did shift toward a TMPEC phenotype (Fig. 6B), consistent with the cytokine depletion experiments described above. IL12 signaling did not seem to affect TEM vs. TCM development since the percentages of TEM and TCM were similar between wild-type and IL12Rβ−/− cells (Fig. 6C). In contrast, similar numbers of wild-type and IFNγR−/− transgenic cells were recovered at the peak of expansion (Fig. 6D), suggesting that IFNγ signaling is not required for pathogen-specific CD8+ T cell expansion following C. trachomatis infection. The transgenic cells that do not respond to IFNγ did not show an obvious shift toward TMPEC (Fig. 6E) but did show an increase of TEM numbers (Fig. 6F). Overall, these data suggest that IL12 signaling is important for TMPEC vs. TSLEC differentiation while IFNγ is involved in TEM vs. TCM development.
Figure 6.
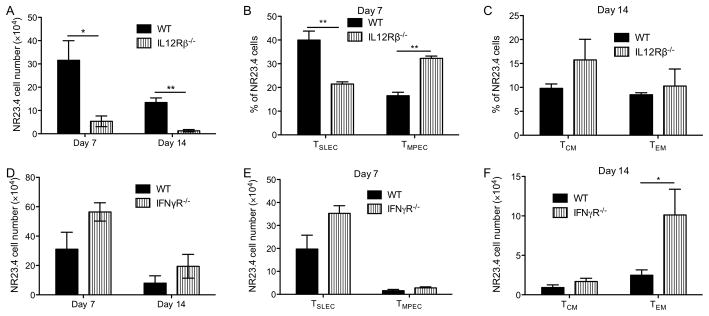
Cell autonomous IL12 signaling is required for expansion and TSLEC vs. TMPEC formation while IFNγ signaling affects TEM vs. TCM formation in Chlamydia-specific CD8+ T cells. CD90.1 C57BL/6 mice received CD90.2 wild-type (WT), IL12Rβ−/− (A–C), or IFNγR−/− (D–F) NR23.4 one day before i.v. infection with C. trachomatis. (A, D) NR23.4 cell numbers in the spleen on day 7and 14 p.i., (B) TSLEC % and TMPEC % among NR23.4 cells on day 7 p.i., (C) TEM % and TCM % among NR23.4 cells on day 14 p.i., (E) TSLEC and TMPEC NR23.4 cell numbers on day 7 p.i., and (F) TEM and TCM NR23.4 cell numbers on day 21 p.i. are shown. Data are representative of two experiments, each with 5–6 mice per group.
Transient ablation of pro-inflammatory cytokines during mucosal infection also favors the formation of memory precursor CD8+ T cells
To test whether reducing pro-inflammatory cytokine signaling can improve Chlamydia-specific CD8+ T cell memory development following mucosal infection, we conducted cytokine depletion experiments in mice infected with C. trachomatis in the genital tract. Transient reduction of IFNγ and IL12 did not significantly alter Chlamydia burden in the uterus of infected mice (Fig. 7A). This treatment did shift CD8+ T cells in the spleens towards a TMPEC phenotype (Fig. 7B, C). A similar trend was observed in the uterine tissues and draining lymph nodes although the differences did not reach statistical significance (Fig. 7B, C). Transient pro-inflammatory cytokine ablation also increased the number of TEM cells in the spleens and uterine tissues of mucosally infected mice without affecting the numbers of TCM cells (Fig. 7D, E). Overall, in mice infected in the genital tract, transient reduction of IFNγ and IL12 levels shifted the CD8+ T cells towards a TMPEC and a TEM phenotype, consistent with what was observed in systemically infected mice.
Figure 7.
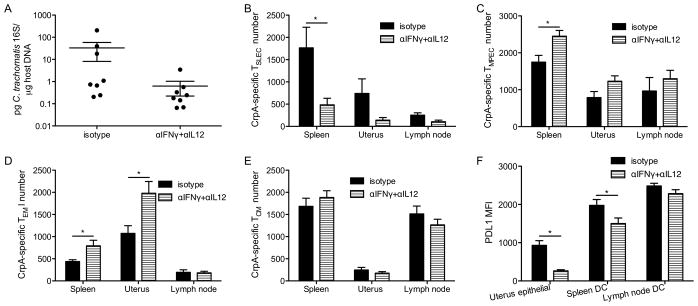
Transient reduction of pro-inflammatory cytokine signaling during transcervical infection improves memory development of CD8+ T cells. Mice were transcervically infected with C. trachomatis, treated with isotype control or IFNγ and IL12 neutralizing antibodies on day 4 p.i. (A) Chlamydia burden in the uterus on day 7 p.i., (B) CrpA-specific TSLEC, (C) TMPEC numbers on day 7 p.i., and (D) CrpA-specific TEM, (E) TCM numbers on day 14 p.i. are shown. (F) PDL1 MFI of uterine epithelial cells (CD326+), and dendritic cells (CD3−CD11c+) in the spleen and lymph node on day 7 p.i. is shown. Data are representative of at least two experiments, each with 6–8 mice per group.
A recent report from our lab has shown that the PDL1-PD1 pathway also contributes to the suppression of CD8+ T cell memory development during Chlamydia infection of the genital tract (15). To test whether reducing pro-inflammatory cytokines modulates memory development by regulating PDL1 expression, we transcervically infected mice with C. trachomatis, treated the mice with IFNγ and IL12 neutralizing antibodies, and then determined PDL1 expression on various cell populations. We found that neutralization of the pro-inflammatory cytokines reduced PDL1 expression on uterine epithelial cells (Fig. 7F). The numbers of uterine dendritic cells were too few to reliably examine the differences in PDL1 expression among groups; however, we did observe a reduction of PDL1 level on splenic dendritic cells in mice treated with the neutralizing antibody compared to the control mice. A similar trend was observed in dendritic cells from the draining lymph node, although the difference did not reach statistical significance (Fig. 7F). Together, these results suggest that the improvement in memory CD8+ T cells development may be driven through a reduction of PDL1 expression.
Reducing inflammation during priming increases the protective capacity of Chlamydia-specific CD8+ T cells
To test whether reducing IFNγ and IL12 levels during priming increases the recall and protective capacity of Chlamydia-specific CD8+ T cells, we treated systemically infected mice with IFNγ and IL12 neutralizing antibodies or isotype control antibodies, waited a month for memory T cells to develop in these mice, then isolated and transferred similar numbers of purified CD8+ T cells from these two groups of mice into naïve mice. The recipient mice were then challenged transcervically with C. trachomatis. Five days later, more CrpA-specific CD8+ donor T cells were recovered from uteri of mice that had been given T cells from donor mice that experienced lower levels of pro-inflammatory cytokines during priming (Fig. 8A). The donor cells that experienced lower levels of pro-inflammatory cytokines proliferated more in SLOs and uterine tissues (Fig. 8B). These cells also showed downregulation of CD62L in SLOs (Fig. 8C), enabling them to migrate to infected tissues. Moreover, CD8+ T cells from neutralizing antibody treated donors conferred significantly more protection against transcervical Chlamydia infection than CD8+ T cells from isotype-control antibody treated donor mice (Fig. 8D). Since the number but not functionality of memory CrpA-specific CD8+ T cells increases in depleted mice (Fig. 4H, 4I), we believe the increased protective capacity conferred by the transferred CD8+ T cells from depleted mice is due to the increased percentage and therefore number of Chlamydia-specific CD8+ T cells among transferred cells but not alteration in per cell functionality. Finally, to determine whether transient reduction of pro-inflammatory cytokine signaling also increases the protection conferred by Chlamydia-specific CD8+ T cells primed in the genital tract, we transcervically inoculated mice with C. trachomatis, treated the mice with IFNγ and IL12 neutralizing antibodies or isotype control antibodies on day 4 p.i., allowed the mice rest for a month, and then re-challenged these mice or naïve mice transcervically with VacCrpA. Five days later, the vaccinia burden was determined in the uterine tissue of these mice. In this heterologous challenge experiment, the protective effect of primary C. trachomatis infection against secondary VacCrpA infection should be mainly mediated by the cross-reactive CrpA-specific CD8+ T cells. Consistent with the memory CD8+ T cell transfer experiment (Fig. 8D), transient reduction of IFNγ and IL12 levels during primary mucosal infection renders the CrpA-specific CD8+ T cells more protective against secondary infection (Fig. 8E). Overall, these results suggest that transient dampening of IFNγ and IL12 levels during priming not only shifts the phenotype of Chlamydia-specific CD8+ T cells to favor memory formation but also increases protection conferred by these cells against a secondary challenge.
Figure 8.

Reducing pro-inflammatory cytokine signaling during priming increased the protective capacity of CD8+ T cells following transcervical Chlamydia infection. (A–D) CD90.1 mice were i.v. infected with C. trachomatis and treated with isotype control or antibodies to deplete cytokines on day 4 p.i. On day 28 p.i., CD8+ T cells were purified from pooled secondary lymphoid organs, CFSE-labeled, and transferred into naïve CD90.2 mice. The recipient mice and control mice that did not receive any T cells (no transfer) were transcervically infected with C. trachomatis. (A) The number of donor cells, (B) CFSEdim % among donor cells, (C) CD62L MFI of donor cells, and (D) Chlamydia burden in the uterus on day 6 p.i. are shown. (E) Mice were transcervically infected with C. trachomatis, and treated with isotype control or antibodies to deplete cytokines on day 4 p.i. On day 28 p.i., these mice and naïve mice (primary) were challenged transcervically with VacCrpA. Viral burden in the uterus on day 6 p.i is shown. Data are representative of at least two experiments, each with 6–7 mice per group.
Discussion
C. trachomatis specific CD8+ T cells can confer protection in mice following immunization with recombinant vaccinia viruses expressing CD8+ T cell antigens or when transferred into naïve mice from ex vivo culture (12). In a primate trachoma model, the protective immunity elicited by a live-attenuated trachoma vaccine also has been shown to be mediated by CD8+ T cells (47). Yet memory CD8+ T cells capable of participating in secondary protection are not stimulated during natural C. trachomatis infection in mice. With the goal of understanding why C. trachomatis infection does not stimulate protective CD8+ T cells, we compared CD8+ T cells generated by C. trachomatis infection to those generated by VacCrpA. We demonstrated that the pro-inflammatory cytokines IL12 and IFNγ drive C. trachomatis-specific CD8+ T cells into a short-lived fate and hinder TEM development. A transient blockade of these cytokines during priming not only shifts CD8+ effector T cells towards a memory precursor phenotype but also increases memory T cell numbers after stable memory has formed.
Helpless CD8+ T cells typically fail to be efficiently maintained, and those that are maintained tend to have elevated KLRG1 expression, and reduced CD127 and CD27 expression (38, 39). We observed all of these characteristics in Chlamydia-specific memory CD8+ T cells, suggesting that a lack of CD4+ T cell help might contribute to the “faulty” memory development of C. trachomatis stimulated CD8+ T cells. However, our data suggest that the “faulty” CD8+ T cell memory development following C. trachomatis infection might not result from a lack of direct CD4+ T cell help. A number of mechanisms have been described to mediate help for CD8+ T cells. For example, CD4+ T cells can license APCs to become more potent in activating CD8+ T cells. However, no significant differences in stimulatory or inhibitory co-receptor expression on CD8+ T cells or their ligand expression on APCs were noted following C. trachomatis vs. VacCrpA infection (data not shown). CD4+ T cells can also directly interact with CD8+ T cells through interactions of membrane-bound molecules, such as CD40-CD40L (48), or through soluble factors, such as IL2 (25) and/or IL15 (49). Although we did not observe differences in CD40 expression on CD8+ T cells (data not shown), we did observe delayed IL2Rα expression on C. trachomatis-stimulated CD8+ T cells. Nevertheless, boosting IL2 mediated signaling by IL2-anti-IL2 immune complex treatment did not rescue the CD8+ T cell response.
Naïve T cell activation, effector differentiation, and subsequent memory T cell development are regulated by TCR signals, costimulation, and inflammation, which are usually referred to as signals 1, 2, and 3. To understand the mechanisms underpinning “faulty” memory CD8+ T cell development during C. trachomatis infection, we compared these three signals experienced by CrpA-specific CD8+ T cells when stimulated by C. trachomatis vs. VacCrpA. Although both pathogens express CrpA, it is not straightforward to control for the level of antigen presentation given their different replication niches. We chose to challenge with doses of C. trachomatis and VacCrpA that expand CrpA-specific CD8+ T cells to a similar extent, and compared the strength of signal 2 and 3 under these conditions. We found no significant differences in costimulatory or inhibitory molecule (CD28, 4-1BB, OX40, and PD1) expression on CrpA-specific CD8+ T cells or their ligand expression on APCs following infection with C. trachomatis vs. VacCrpA (data not shown), suggesting that signal 2 potency was similar.
Accumulating evidence suggests that although signal 3 provided by pro-inflammatory cytokines, mainly IL12, and Type 1 and 2 IFNs, promote antigen-specific CD8+ T cell expansion (50–53), they can also induce terminal differentiation and thus shorten the lifespan of these cells (45, 54). This effect appears to be pathogen specific. For instance, IL12 promotes terminal maturation at the expense of memory precursor subpopulation differentiation following Listeria infection and during Toxoplasma vaccination (54–57). In contrast, no significant differences in TSLEC/TMPEC formation between wild-type and IL12Rβ−/− T cells were observed in the context of lymphocytic choriomeningitis virus (LCMV), vesicular stomatitis virus or vaccinia virus infection (55). We observed a significant switch from TSLEC to TMPEC phenotype in IL12−/− mice (data not shown) and in transgenic cells lacking IL12Rβ following C. trachomatis infection. IFNγ production during the first 24 hours of infection has been shown to regulate the program of CD8+ T cell contraction during Listeria infection through down-regulation of IL7R (58–60). IFNγR is also required in a CD8+ T cell autonomous manner for memory CD8+ T cell formation during LCMV infection (61). We did not observe significant differences in TSLEC vs. TMPEC formation between wild-type and IFNγR-deficient Chlamydia-specific CD8+ T cells. Overall, we found that IL12 but not IFNγ is critical for TSLEC vs. TMPEC fate determination of C. trachomatis stimulated CD8+ T cells.
The terminal differentiation of effector CD8+ T cells during infection is inextricably linked to antigen dose, duration of antigenic stimulation, and inflammatory stimuli. In the case of C. trachomatis, clearance largely depends on IFNγ secreted by T cells (10, 26). Because IL12, IFNγ, and Tbet play a protective role during C. trachomatis infection, a comparison of T cell responses in mice that lack these molecules is complicated by differences in Chlamydia burden and therefore antigen load. Therefore, in this study we 1) transiently treated animals with antibodies to neutralize cytokines, 2) determined the developmental phenotypes of transgenic cells lacking receptors for these cytokines, and 3) challenged Tbet heterozygous animals to avoid significantly changing antigen load while still manipulating the level of inflammation. Although we cannot rule out the impact of subtle changes in antigen load/duration on effector CD8+ T cell differentiation, we did not observe a significant change in bacterial burden in all three experimental manipulations described above. Yet, we observed a shift of the Chlamydia-specific CD8+ T cells towards a memory precursor phenotype. Overall our study indicates that excess induction of IL12, not excess antigen load, during priming might drive terminal differentiation of effector Chlamydia-specific CD8+ T cells.
Memory T cell populations are heterogeneous and two of the best characterized subsets are TEM and TCM (62). TEM cells are thought to provide immediate effector function at the portal of pathogen entry but exhibit reduced proliferative capacity (63). TCM cells migrate through SLOs and are efficient in homeostatic renewal and secondary proliferative responses (64). The results of experiments comparing the protective capacities of TCM and TEM have been mixed and might depend on the route of infection, pathogen dose, or tropism (64–67). We found that reducing IFNγ signaling promotes TEM formation without sacrificing TCM formation. This increase of TEM cell numbers is associated with increased protection conferred by memory CD8+ T cells against either C. trachomatis or a heterologous vaccinia virus genital tract challenge. Future experiments comparing the per cell protective capacity of TEM vs. TCM CD8+ T cells stimulated by C. trachomatis will further clarify the role of each memory population in protection against this pathogen.
Developing effective vaccines is critical for preventing infection and/or immunopathology induced by C. trachomatis. It is important to note that preferentially inducing the TMPEC CD8+ T cells might be as critical as inducing a large number of CD8+ T cells. Our data show that pro-inflammation cytokine signaling has a negative impact on memory CD8+ T cell development following C. trachomatis infection. Thus, future vaccine design for C. trachomatis will benefit from a careful choice of antigens/adjuvants and their doses such that there is a balance in the cytokine milieu that favors effector cell expansion without driving CD8+ T cells into terminal differentiation.
Acknowledgments
Source of support: This work was supported by the National Institutes of Health grants AI39558 (M.N.S.) and AI062827 (M.N.S). This work was also supported by a cooperative agreement from the US National Institutes of Health (National Institute of Allergy and Infectious Diseases; U19 AI113187) establishing the Epidemiology and Prevention Interdisciplinary Center for Sexually Transmitted Diseases.
We thank Dr. Sarah Fankhauser, Dr. Caterina Nogueira, and other members of the Starnbach lab for helpful discussions and their assistance with experiments. We thank Dr. Rebeccah Lijek for critical comments on this manuscript. We thank the NIH tetramer core facility for providing the tetramers.
Abbreviations used in this article
- KLRG1
killer cell lectin-like receptor G1
- TSLEC
short-lived effector T cells
- TMPEC
memory precursor effector cells
- IFU
inclusion-forming units
- SLO
secondary lymphoid organ
- p.i
post-infection
- TEM
effecter memory T cells
- TCM
central memory T cells
- LCMV
lymphocytic choriomeningitis virus
References
- 1.Zhang N, Bevan MJ. CD8(+) T cells: foot soldiers of the immune system. Immunity. 35:161–168. doi: 10.1016/j.immuni.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sallusto F, Lanzavecchia A, Araki K, Ahmed R. From vaccines to memory and back. Immunity. 33:451–463. doi: 10.1016/j.immuni.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Joshi NS, Kaech SM. Effector CD8 T cell development: a balancing act between memory cell potential and terminal differentiation. J Immunol. 2008;180:1309–1315. doi: 10.4049/jimmunol.180.3.1309. [DOI] [PubMed] [Google Scholar]
- 4.Kaech SM, Wherry EJ. Heterogeneity and cell-fate decisions in effector and memory CD8+ T cell differentiation during viral infection. Immunity. 2007;27:393–405. doi: 10.1016/j.immuni.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang JT, V, Palanivel R, Kinjyo I, Schambach F, Intlekofer AM, Banerjee A, Longworth SA, Vinup KE, Mrass P, Oliaro J, Killeen N, Orange JS, Russell SM, Weninger W, Reiner SL. Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science. 2007;315:1687–1691. doi: 10.1126/science.1139393. [DOI] [PubMed] [Google Scholar]
- 6.Cohen CR, Brunham RC. Pathogenesis of Chlamydia induced pelvic inflammatory disease. Sex Transm Infect. 1999;75:21–24. doi: 10.1136/sti.75.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scholes D, Stergachis A, Heidrich FE, Andrilla H, Holmes KK, Stamm WE. Prevention of pelvic inflammatory disease by screening for cervical chlamydial infection. N Engl J Med. 1996;334:1362–1366. doi: 10.1056/NEJM199605233342103. [DOI] [PubMed] [Google Scholar]
- 8.Batteiger BE, Xu F, Johnson RE, Rekart ML. Protective immunity to Chlamydia trachomatis genital infection: evidence from human studies. J Infect Dis. 201(Suppl 2):S178–189. doi: 10.1086/652400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roan NR, Starnbach MN. Immune-mediated control of Chlamydia infection. Cell Microbiol. 2008;10:9–19. doi: 10.1111/j.1462-5822.2007.01069.x. [DOI] [PubMed] [Google Scholar]
- 10.Su H, Feilzer K, Caldwell HD, Morrison RP. Chlamydia trachomatis genital tract infection of antibody-deficient gene knockout mice. Infect Immun. 1997;65:1993–1999. doi: 10.1128/iai.65.6.1993-1999.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grotenbreg GM, Roan NR, Guillen E, Meijers R, Wang JH, Bell GW, Starnbach MN, Ploegh HL. Discovery of CD8+ T cell epitopes in Chlamydia trachomatis infection through use of caged class I MHC tetramers. Proc Natl Acad Sci U S A. 2008;105:3831–3836. doi: 10.1073/pnas.0711504105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Starnbach MN, Loomis WP, Ovendale P, Regan D, Hess B, Alderson MR, Fling SP. An inclusion membrane protein from Chlamydia trachomatis enters the MHC class I pathway and stimulates a CD8+ T cell response. J Immunol. 2003;171:4742–4749. doi: 10.4049/jimmunol.171.9.4742. [DOI] [PubMed] [Google Scholar]
- 13.Morrison RP, Feilzer K, Tumas DB. Gene knockout mice establish a primary protective role for major histocompatibility complex class II-restricted responses in Chlamydia trachomatis genital tract infection. Infection & Immunity. 1995;63:4661–4668. doi: 10.1128/iai.63.12.4661-4668.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morrison SG, Su H, Caldwell HD, Morrison RP. Immunity to murine Chlamydia trachomatis genital tract reinfection involves B cells and CD4(+) T cells but not CD8(+) T cells. Infect Immun. 2000;68:6979–6987. doi: 10.1128/iai.68.12.6979-6987.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fankhauser SC, Starnbach MN. PD-L1 limits the mucosal CD8+ T cell response to Chlamydia trachomatis. J Immunol. 192:1079–1090. doi: 10.4049/jimmunol.1301657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loomis WP, Starnbach MN. Chlamydia trachomatis infection alters the development of memory CD8+ T cells. J Immunol. 2006;177:4021–4027. doi: 10.4049/jimmunol.177.6.4021. [DOI] [PubMed] [Google Scholar]
- 17.Peng SL, Szabo SJ, Glimcher LH. T-bet regulates IgG class switching and pathogenic autoantibody production. Proc Natl Acad Sci U S A. 2002;99:5545–5550. doi: 10.1073/pnas.082114899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, Klemm M, Kuchroo VK, Freeman GJ, Sharpe AH. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci U S A. 2004;101:10691–10696. doi: 10.1073/pnas.0307252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kouskoff V, Signorelli K, Benoist C, Mathis D. Cassette vectors directing expression of T cell receptor genes in transgenic mice. J Immunol Methods. 1995;180:273–280. doi: 10.1016/0022-1759(95)00002-r. [DOI] [PubMed] [Google Scholar]
- 20.Howard L, Orenstein NS, King NW. Purification on renografin density gradients of Chlamydia trachomatis grown in the yolk sac of eggs. Appl Microbiol. 1974;27:102–106. doi: 10.1128/am.27.1.102-106.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lauer P, Chow MY, Loessner MJ, Portnoy DA, Calendar R. Construction, characterization, and use of two Listeria monocytogenes site-specific phage integration vectors. J Bacteriol. 2002;184:4177–4186. doi: 10.1128/JB.184.15.4177-4186.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Angelakopoulos H, Loock K, Sisul DM, Jensen ER, Miller JF, Hohmann EL. Safety and shedding of an attenuated strain of Listeria monocytogenes with a deletion of actA/plcB in adult volunteers: a dose escalation study of oral inoculation. Infect Immun. 2002;70:3592–3601. doi: 10.1128/IAI.70.7.3592-3601.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Castro I, Dee MJ, Malek TR. Transient Enhanced IL-2R Signaling Early during Priming Rapidly Amplifies Development of Functional CD8+ T Effector-Memory Cells. J Immunol. 189:4321–4330. doi: 10.4049/jimmunol.1202067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kamimura D, Bevan MJ. Naive CD8+ T cells differentiate into protective memory-like cells after IL-2 anti IL-2 complex treatment in vivo. J Exp Med. 2007;204:1803–1812. doi: 10.1084/jem.20070543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Williams MA, Tyznik AJ, Bevan MJ. Interleukin-2 signals during priming are required for secondary expansion of CD8+ memory T cells. Nature. 2006;441:890–893. doi: 10.1038/nature04790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gondek DC, Olive AJ, Stary G, Starnbach MN. CD4+ T cells are necessary and sufficient to confer protection against Chlamydia trachomatis infection in the murine upper genital tract. J Immunol. 189:2441–2449. doi: 10.4049/jimmunol.1103032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Collins MK, Tay CS, Erlebacher A. Dendritic cell entrapment within the pregnant uterus inhibits immune surveillance of the maternal/fetal interface in mice. J Clin Invest. 2009;119:2062–2073. doi: 10.1172/JCI38714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gondek DC, Roan NR, Starnbach MN. T cell responses in the absence of IFN-gamma exacerbate uterine infection with Chlamydia trachomatis. J Immunol. 2009;183:1313–1319. doi: 10.4049/jimmunol.0900295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bernstein-Hanley I, Balsara ZR, Ulmer W, Coers J, Starnbach MN, Dietrich WF. Genetic analysis of susceptibility to Chlamydia trachomatis in mouse. Genes Immun. 2006;7:122–129. doi: 10.1038/sj.gene.6364285. [DOI] [PubMed] [Google Scholar]
- 30.Freyschmidt EJ, Mathias CB, MacArthur DH, Laouar A, Narasimhaswamy M, Weih F, Oettgen HC. Skin inflammation in RelB (−/−) mice leads to defective immunity and impaired clearance of vaccinia virus. J Allergy Clin Immunol. 2007;119:671–679. doi: 10.1016/j.jaci.2006.12.645. [DOI] [PubMed] [Google Scholar]
- 31.Harty JT, V, Badovinac P. Shaping and reshaping CD8+ T-cell memory. Nat Rev Immunol. 2008;8:107–119. doi: 10.1038/nri2251. [DOI] [PubMed] [Google Scholar]
- 32.Hikono H, Kohlmeier JE, Takamura S, Wittmer ST, Roberts AD, Woodland DL. Activation phenotype, rather than central- or effector-memory phenotype, predicts the recall efficacy of memory CD8+ T cells. J Exp Med. 2007;204:1625–1636. doi: 10.1084/jem.20070322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ingram JT, Yi JS, Zajac AJ. Exhausted CD8 T cells downregulate the IL-18 receptor and become unresponsive to inflammatory cytokines and bacterial co-infections. PLoS Pathog. 7:e1002273. doi: 10.1371/journal.ppat.1002273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Surh CD, Boyman O, Purton JF, Sprent J. Homeostasis of memory T cells. Immunol Rev. 2006;211:154–163. doi: 10.1111/j.0105-2896.2006.00401.x. [DOI] [PubMed] [Google Scholar]
- 35.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 36.Janssen EM, Droin NM, Lemmens EE, Pinkoski MJ, Bensinger SJ, Ehst BD, Griffith TS, Green DR, Schoenberger SP. CD4+ T-cell help controls CD8+ T-cell memory via TRAIL-mediated activation-induced cell death. Nature. 2005;434:88–93. doi: 10.1038/nature03337. [DOI] [PubMed] [Google Scholar]
- 37.Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2003;421:852–856. doi: 10.1038/nature01441. [DOI] [PubMed] [Google Scholar]
- 38.Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 2003;300:337–339. doi: 10.1126/science.1082305. [DOI] [PubMed] [Google Scholar]
- 39.Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 2003;300:339–342. doi: 10.1126/science.1083317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Obar JJ, Molloy MJ, Jellison ER, Stoklasek TA, Zhang W, Usherwood EJ, Lefrancois L. CD4+ T cell regulation of CD25 expression controls development of short-lived effector CD8+ T cells in primary and secondary responses. Proc Natl Acad Sci U S A. 107:193–198. doi: 10.1073/pnas.0909945107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wiesel M, Joller N, Ehlert AK, Crouse J, Sporri R, Bachmann MF, Oxenius A. Th cells act via two synergistic pathways to promote antiviral CD8+ T cell responses. J Immunol. 185:5188–5197. doi: 10.4049/jimmunol.1001990. [DOI] [PubMed] [Google Scholar]
- 42.Kalia V, Sarkar S, Subramaniam S, Haining WN, Smith KA, Ahmed R. Prolonged interleukin-2Ralpha expression on virus-specific CD8+ T cells favors terminal-effector differentiation in vivo. Immunity. 32:91–103. doi: 10.1016/j.immuni.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 43.Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, Rao A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity. 32:79–90. doi: 10.1016/j.immuni.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hamilton SE, Schenkel JM, Akue AD, Jameson SC. IL-2 complex treatment can protect naive mice from bacterial and viral infection. J Immunol. 185:6584–6590. doi: 10.4049/jimmunol.1001215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27:281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Intlekofer AM, Takemoto N, Kao C, Banerjee A, Schambach F, Northrop JK, Shen H, Wherry EJ, Reiner SL. Requirement for T-bet in the aberrant differentiation of unhelped memory CD8+ T cells. J Exp Med. 2007;204:2015–2021. doi: 10.1084/jem.20070841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Olivares-Zavaleta N, Whitmire WM, Kari L, Sturdevant GL, Caldwell HD. CD8+ T Cells Define an Unexpected Role in Live-Attenuated Vaccine Protective Immunity against Chlamydia trachomatis Infection in Macaques. J Immunol. 192:4648–4654. doi: 10.4049/jimmunol.1400120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bourgeois C, Rocha B, Tanchot C. A role for CD40 expression on CD8+ T cells in the generation of CD8+ T cell memory. Science. 2002;297:2060–2063. doi: 10.1126/science.1072615. [DOI] [PubMed] [Google Scholar]
- 49.Oh S, Perera LP, Terabe M, Ni L, Waldmann TA, Berzofsky JA. IL-15 as a mediator of CD4+ help for CD8+ T cell longevity and avoidance of TRAIL-mediated apoptosis. Proc Natl Acad Sci U S A. 2008;105:5201–5206. doi: 10.1073/pnas.0801003105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Agarwal P, Raghavan A, Nandiwada SL, Curtsinger JM, Bohjanen PR, Mueller DL, Mescher MF. Gene regulation and chromatin remodeling by IL-12 and type I IFN in programming for CD8 T cell effector function and memory. J Immunol. 2009;183:1695–1704. doi: 10.4049/jimmunol.0900592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aichele P, Unsoeld H, Koschella M, Schweier O, Kalinke U, Vucikuja S. CD8 T cells specific for lymphocytic choriomeningitis virus require type I IFN receptor for clonal expansion. J Immunol. 2006;176:4525–4529. doi: 10.4049/jimmunol.176.8.4525. [DOI] [PubMed] [Google Scholar]
- 52.Curtsinger JM, Johnson CM, Mescher MF. CD8 T cell clonal expansion and development of effector function require prolonged exposure to antigen, costimulation, and signal 3 cytokine. J Immunol. 2003;171:5165–5171. doi: 10.4049/jimmunol.171.10.5165. [DOI] [PubMed] [Google Scholar]
- 53.Curtsinger JM, Lins DC, Mescher MF. Signal 3 determines tolerance versus full activation of naive CD8 T cells: dissociating proliferation and development of effector function. J Exp Med. 2003;197:1141–1151. doi: 10.1084/jem.20021910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pearce EL, Shen H. Generation of CD8 T cell memory is regulated by IL-12. J Immunol. 2007;179:2074–2081. doi: 10.4049/jimmunol.179.4.2074. [DOI] [PubMed] [Google Scholar]
- 55.Keppler SJ, Theil K, Vucikuja S, Aichele P. Effector T-cell differentiation during viral and bacterial infections: Role of direct IL-12 signals for cell fate decision of CD8(+) T cells. Eur J Immunol. 2009;39:1774–1783. doi: 10.1002/eji.200839093. [DOI] [PubMed] [Google Scholar]
- 56.Takemoto N, Intlekofer AM, Northrup JT, Wherry EJ, Reiner SL. Cutting Edge: IL-12 inversely regulates T-bet and eomesodermin expression during pathogen-induced CD8+ T cell differentiation. J Immunol. 2006;177:7515–7519. doi: 10.4049/jimmunol.177.11.7515. [DOI] [PubMed] [Google Scholar]
- 57.Wilson DC, Matthews S, Yap GS. IL-12 signaling drives CD8+ T cell IFN-gamma production and differentiation of KLRG1+ effector subpopulations during Toxoplasma gondii Infection. J Immunol. 2008;180:5935–5945. doi: 10.4049/jimmunol.180.9.5935. [DOI] [PubMed] [Google Scholar]
- 58.Badovinac VP, Harty JT. Adaptive immunity and enhanced CD8+ T cell response to Listeria monocytogenes in the absence of perforin and IFN-gamma. J Immunol. 2000;164:6444–6452. doi: 10.4049/jimmunol.164.12.6444. [DOI] [PubMed] [Google Scholar]
- 59.Badovinac VP, Porter BB, Harty JT. CD8+ T cell contraction is controlled by early inflammation. Nat Immunol. 2004;5:809–817. doi: 10.1038/ni1098. [DOI] [PubMed] [Google Scholar]
- 60.Badovinac VP, Tvinnereim AR, Harty JT. Regulation of antigen-specific CD8+ T cell homeostasis by perforin and interferon-gamma. Science. 2000;290:1354–1358. doi: 10.1126/science.290.5495.1354. [DOI] [PubMed] [Google Scholar]
- 61.Whitmire JK, Eam B, Benning N, Whitton JL. Direct interferon-gamma signaling dramatically enhances CD4+ and CD8+ T cell memory. J Immunol. 2007;179:1190–1197. doi: 10.4049/jimmunol.179.2.1190. [DOI] [PubMed] [Google Scholar]
- 62.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 63.Masopust D, Vezys V, Marzo AL, Lefrancois L. Preferential localization of effector memory cells in nonlymphoid tissue. Science. 2001;291:2413–2417. doi: 10.1126/science.1058867. [DOI] [PubMed] [Google Scholar]
- 64.Wherry EJ, Teichgraber V, Becker TC, Masopust D, Kaech SM, Antia R, von Andrian UH, Ahmed R. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol. 2003;4:225–234. doi: 10.1038/ni889. [DOI] [PubMed] [Google Scholar]
- 65.Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, Palmer DC, Antony PA, Hwang ST, Rosenberg SA, Waldmann TA, Restifo NP. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci U S A. 2005;102:9571–9576. doi: 10.1073/pnas.0503726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nolz JC, Harty JT. Protective capacity of memory CD8+ T cells is dictated by antigen exposure history and nature of the infection. Immunity. 34:781–793. doi: 10.1016/j.immuni.2011.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Olson JA, McDonald-Hyman C, Jameson SC, Hamilton SE. Effector-like CD8(+) T cells in the memory population mediate potent protective immunity. Immunity. 38:1250–1260. doi: 10.1016/j.immuni.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]