

Abstract
Background
Exposure to traffic pollution particulate matter, predominantly diesel exhaust particles (DEP), increases risk for asthma and asthma exacerbation, however the underlying mechanisms remain poorly understood.
Objective
To examine the impact of DEP exposure on the generation and persistence of allergen-specific memory T-cells in asthma and translate these findings by determining the impact of early DEP exposure on the prevalence of allergic asthma in children.
Methods
The impact of DEP on HDM-specific memory responses was determined using an asthma model. Data from children enrolled in the Cincinnati Childhood Allergy and Air Pollution Study (CCAAPS) birth cohort were analyzed to determine the impact of the DEP exposure on asthma outcomes.
Results
DEP co-exposure with HDM resulted in persistent Th2/Th17 CD127+ effector/memory cells in the lungs, spleen and lymph nodes of adult and neonatal mice. After 7 weeks of rest, a single exposure to HDM resulted in airway hyperresponsiveness and increased levels of Th2 cytokines in only mice that had been previously exposed to both HDM and DEP versus HDM alone. Based on these data, we examined whether DEP exposure was similarly associated increased asthma prevalence in children in the presence or absence of allergen exposure/sensitization in the CCAAPS birth cohort. Early life exposure to high DEP was associated with significantly increased asthma prevalence among allergic children, but not among non-allergic children.
Conclusion
These findings suggest that DEP exposure results in accumulation of allergen specific Th2/Th17 cells in the lungs, potentiating secondary allergen recall responses and promoting the development of allergic asthma.
Keywords: allergic asthma, traffic pollution, house dust mite, diesel exhaust particle, memory, recall, children
INTRODUCTION
A recent comprehensive and systematic review of worldwide traffic emissions and health science by a Special Panel convened by the Health Effects Institute found sufficient evidence that exposure to traffic-related air pollution (TRAP) causes asthma exacerbation in children1. A key component of traffic related particulate matter (PM) is diesel exhaust particles (DEP), which is the main contributor to TRAP-related asthma exacerbations in children2, 3. These primary ultra-fine DEP particles (diameter < 1.0μm) can reach small airways including the alveolar/gas exchange regions of the lung exacerbating respiratory disease symptoms3. This exposure is highly significant because in large cities in North America, up to 45% of the population resides in zones that are most impacted by TRAP1. Further, over 30% of schools are located in high TRAP exposure areas4. Even short-term exposure to high diesel traffic was able to reduce airway function in asthmatics5. Similarly, we recently reported that, in allergic children with asthma, higher DEP exposure is associated with increased asthma severity6.
Allergic asthma is generally regarded as a Th2 disease characterized by elevated levels of eosinophils and Th2 cytokines (IL4, IL5, IL13) but severe asthma is often characterized by mixed Th2/Th17 responses7. In mice, DEP alone had no impact on AHR, but co-exposure with HDM exacerbated allergic airway responses including allergen-specific IgE, eosinophilia and airway hyperresponsiveness (AHR)8. Transfer of antigen-specific, IL17A and IL13 double producing CD4+ effector T-cells into BALB/c mice triggered more severe inflammation upon allergen challenge compared to transfer of conventional Th2 or Th17 cells, highlighting the potential role of this novel cell subset in allergic asthma severity9. Repeated DEP exposure promoted accumulation of Th17 and Th2/Th17 co-producer cells in the lungs of exposed mice6. Neutralization of IL-17A alleviated DEP-mediated exacerbation of HDM-induced AHR supporting a role for IL17A in severe asthma6.
Herein, we examined the impact of DEP exposure on allergen specific memory and recall responses. Generation and maintenance of memory T cells in the lungs is still poorly understood and has been studied predominantly in the context of viral infections10. Memory T-cells are long-lived antigen-specific T-cells that arise during expansion of effector T-cells and survive the contraction phase of the effector response. Based primarily on studies focusing on CD8+ memory T-cells, three sub-populations have been described: central memory, effector memory and tissue resident memory T-cells11. Memory T-cells express high surface levels of CD44 and most express CD127, the receptor for IL-7, which plays a central role in CD4+ T cell homeostatic proliferation12. Central memory T-cells circulate in secondary lymphoid organs (spleen, lymph nodes) and express CD62L and CCR7. Effector/memory T-cells down-regulate CD62L and CCR7 in order to leave lymphoid tissues and then express tissue-specific integrins and chemokine receptors11, 13. These infiltrating effector-memory T-cells differ from tissue-resident memory T-cells that are generated locally and express CD69, an early activation marker11, 14, 15
In the present study, we assess the impact DEP-mediated asthma exacerbations on the generation and persistence of memory T-cells. Since the nature and size of the effector response influences the nature and size of the memory T-cell pool16–18, we hypothesized that the increased accumulation of effector/memory Th2 cells in the lungs of mice co-exposed to HDM plus DEP will result in the persistence of more HDM-specific memory Th2 cells in the lungs upon resolution of the effector Th2 response, potentiating future recall responses. We further hypothesized that if DEP exposure potentiates recall responses to allergen, then early life DEP exposure may promote the development of allergic asthma in children.
Materials and Methods
For a complete description of the materials and methods used in the murine experiments, see this article’s Online Repository at www.jacionline.org.
CCAAPS Cohort
CCAAPS is an ongoing prospective birth cohort study that has been described previously19–22. For this analysis, 578 children from CCAAPS who had at least one SPT result between ages 1–4 and an asthma diagnosis at age 7 were included. DEP exposure levels were estimated from the birth record address; high DEP was defined as the top quartile as described previously21. Early life aeroallergen sensitization and early HDM sensitization were defined as a positive SPT for any of the 15 aeroallergens or a positive SPT to HDM, respectively, at one exam between the ages of 1 and 4. Asthma diagnosis at age 7 was based on reported symptoms and spirometric testing23, based on American Thoracic Society criteria23. This study was approved by the Institutional Review Board committees of Cincinnati Children’s Hospital Medical Center and the University of Cincinnati.
Statistical analysis
For the human data, statistical analyses were performed in SAS. The differences in the proportion of children with asthma with respect to DEP exposure (low versus high) and early persistent atopy (no versus yes) were evaluated with Chi-squared tests. A p-value <0.05 was considered significant. We also evaluated a logistic model adjusted for race, sex and mother’s education level. For the murine studies, statistical analyses were done using PRISM software (GraphPad Software Inc., La Jolla, CA). Statistical significance was assessed using one-way ANOVA followed by a Bonferroni post-test on the relevant groups.
RESULTS
DEP associated neutrophilia persists after HDM-induced Th2 responses return to baseline
First we determined how co-exposure to DEP affects the resolution of HDM-induced lung inflammation, by assessing eosinophil and Th2 cytokine BALF levels 8 and 30 days after the last exposure (Fig. 1A). As we have previously shown6, 24, DEP co-exposure exacerbates HDM-induced lung inflammation (Fig. 1B). One week later, HDM-mediated induction of BALF eosinophilia had largely subsided and DEP related neutrophilia represented the major inflammatory cell type in the BALF (Fig. 1C, 1D). BALF levels of IL-5 and eotaxin-1 (CCL11) were also significantly ablated whereas CXCL1 and CXCL5 levels remained elevated 8 days after the last exposure (See Fig. E1, B–C in the online repository). At 30 days post-exposure, neutrophils BALF levels were still significantly elevated in DEP exposed mice (Fig. 1C). In contrast, eosinophils and Th2 cytokines were no longer present in the BALF of HDM+DEP exposed mice (Fig. 1D–F). Exposure to DEP alone did not induce AHR, but HDM and DEP co-exposure significantly exacerbated AHR 24h after the last exposure (Fig. 1G). By 30 days post-exposure AHR had returned to baseline (Fig. 1G). Consistent with decreasing BALF Th2 cytokine levels and AHR, goblet cell numbers (assessed by CLCA3 expression) and mucus production were also diminished (Fig. 1H and Fig. E1, D in the online repository). Taken together, these results suggest that allergic Th2 responses have resolved within one month after the last exposure even in mice co-exposed to HDM and DEP.
Figure 1. DEP exacerbation of HDM-induced airway Th2 responses returns to baseline within a month.

(A) Protocol: Balb/c mice were exposed intra-tracheally (i.t) to 50μl of saline (S), DEP (D; 150μg) and/or HDM (H; 10μg) three times a week times over a 3-week period. Mice were sacrificed 1, 8 and 30 days after the last exposure and (B) total BALF cell levels as well as (C) BALF neutrophil and (D) eosinophil counts were assessed (3 separate experiments with a total of n=11–16 mice/group (day 1), n=7–12 mice/group (day 8), n=5–8 mice/groups (day 30)). BALF levels of (E) IL4 and (F) IL13 were assessed by multiplex and IL13/IL13Rα2 ELISA respectively. (G) Airway resistance to methacholine challenges was assessed by FlexiVent (***p < 0.001, **p< 0.01, *p<0.05; 1-way ANOVA with Bonferroni’s multiple comparison test; n.s.= not significant). (H) Representative photomicrographs of CLCA3-stained lung sections (scale: 100μm).
DEP and HDM co-exposure promotes a persistent increase in lung effector/memory Th2-cells
Co-exposure to HDM and DEP significantly increased lung cell numbers compared to mice exposed to either DEP or HDM alone (Fig. 2A). This accumulation of inflammatory cells in the lungs lasted for over a week but lung cell numbers returned to baseline within a month (Fig. 2A). Effector T-cells, defined as CD4+CD44+CD62L- T-cells, were significantly increased in mice co-exposed to HDM and DEP compared to HDM alone (Fig. 2B). One month after the last HDM+DEP exposure, an increased proportion of CD44+ effector/memory CD4+ T-cells was still observed in the lungs. A similar expression pattern was observed for CD69 (Fig. 2C), in accordance with its expression by CD44+ effector cells and tissue memory cells14.
Figure 2. Persistent increase in lung effector/memory T-cells following HDM+DEP exposures.
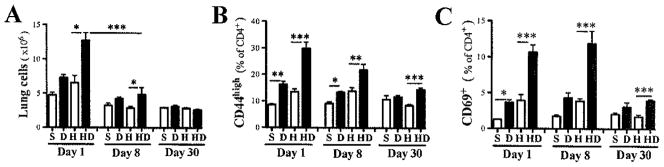
Characterization of lung T-cells 1, 8 and 30 days following the last exposure to saline (S), DEP(D), HDM(H) or HDM+DEP(DH) by FACS. (A) Total lung cells. (B) CD4+CD44+CD62L- T-cells and (C) CD69 expression by effector/memory T-cells (2 separate experiments with 2–8 mice/group; ***p<0.001, **p<0.01, *p<0.05; 1-way ANOVA with Bonferroni’s multiple comparison test).
To establish the nature of these lung effector/memory T-cells, we assessed by flow cytometry their ability to make IFNγ, IL13 and/or IL17A following ex-vivo stimulation with PMA and ionomycin in the presence of Brefeldin A. One month after the last exposure, the proportion of IL13+ Th2 lung cells was significantly greater in HDM and DEP co-exposed mice versus mice exposed only to HDM (Fig. 3A). The proportion of IL17A+ Th17 cells was increased in mice exposed to DEP, either alone or in combination with HDM (Fig. 3A). Interestingly, co-exposure to HDM and DEP resulted in higher levels of IL13 and IL17A double positive T cells compared to mice exposed to HDM alone (Fig. 3B). IFNγ+ Th1 cell levels remained unchanged (Fig. 3A). The CD4+CD44+ T-cells expressed higher levels of IL7R (CD127) than naïve CD4+CD44- T-cells pointing to a memory phenotype notably among CD4+ T-cells from mice pre-exposed to HDM plus DEP (Fig. 3C). Together these findings suggest the presence of higher numbers of HDM-specific memory Th2 cells in the lung of HDM and DEP co-exposed mice versus mice exposed to HDM alone. To support this, we assessed HDM recall responses in vitro. Lung cells from HDM and DEP co-exposed mice secreted significantly more IL4 and IL17A compared to mice exposed to HDM alone (Fig. 3D). Similar findings were made in mice that were sacrificed 45 days after the last allergen exposure (See Fig. E2 in the online repository).
Figure 3. Increased presence of memory Th2 and Th17 cells in the lungs one month after coexposures to HDM and DEP.
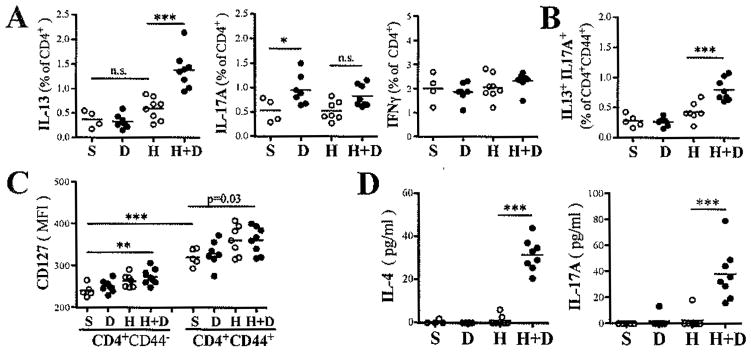
(A) Th1, Th2 and Th17 lungs cells were identified by FACS as CD4+ T-cells expressing respectively IFNγ, IL13, and IL17A, respectively, following ex vivo stimulation with PMA/ionomycin for 4h. (B) Percentage of IL13+IL17+ double producers among CD4+CD44+ T-cells and (C) IL7 receptor mean fluorescence intensity (CD127 MFI) on naïve and CD4+CD44+ T-cells. (C) IL4 and IL17A were detected by ELISA in supernatant of lung cells stimulated with HDM (30μg/ml) for 5 days (n=4–8 mice/group; ***p<0.001, *p<0.05, 1-way ANOVA with Bonferroni’s multiple comparison test; p value calculated by T-test; n.s. = not significant).
Increased HDM recall responses in lymphoid organs is not dependent on the continued presence of DEP
In contrast to HDM, which is rapidly degraded, DEP or at least its carbon core persists within phagocytic cells for weeks25. We assessed how long DEP persisted in lungs following a single exposure of DEP and found numerous DEP positive cells in the lungs 3 months later. Further, over half of all BALF macrophages still contained DEP, albeit less on a per cell basis (See Fig. E3 in the online repository). Similar observations were made in mice that were sacrificed 45 days after the last allergen exposure (See Fig. E2 in the online repository).
While lymph node cells containing DEP were observed 45 days after the last DEP exposure, the size and cellular count of these lymph nodes were comparable to lymph nodes from saline exposed mice (See Fig. E4, A–B in the online repository). Upon re-stimulation with HDM, only mice that were exposed to both HDM and DEP secreted large amounts of cytokines, most notably IL4 and IL17A (Fig. E4, C in the online repository). Similarly, upon HDM re-stimulation, splenocytes from mice exposed to HDM+DEP secreted significantly more IL4 and IL17A compared to mice exposed only to HDM (Fig. E4, D in the online repository).
Since lungs, lymph nodes and to a lesser extend spleens from DEP (±HDM)-exposed mice contain DEP loaded phagocytic cells, we couldn’t exclude that the presence of these DEP positive cells within our cultures were responsible for the observed increase in IL4 and IL17A cytokine production rather than the presence of higher amounts of Th2/Th17 effector/memory cells. To address this, we used a highly enriched population of CD4+ splenocytes from mice exposed 45 days prior to either saline, DEP, HDM or HDM+DEP, and restimulated them in vitro with plate bound anti-CD3 (Fig. E4, E in the online repository). After 3 days of culture, significantly more IL4 and IL17A was secreted by CD4+ T-cells originating from HDM and DEP co-exposed mice compared to HDM exposed mice (Fig. E4, E in the online repository), supporting the presence of more Th2 and Th17 cells in the spleens of HDM and DEP co-exposed mice.
DEP and HDM co-exposure promotes increased HDM recall responses in vivo
Based on our findings demonstrating that 6–7 weeks after the last allergen exposure, mice previously co-exposed to HDM and DEP have significantly more HDM-specific T-cells present both locally and in lymphoid tissues, we exposed these mice to a single dose of HDM and assessed allergic responses 40h later (Fig. 4A). Mice previously exposed to saline, DEP, or HDM alone did not mount an allergic response to this HDM challenge. In contrast, mice previously co-exposed to HDM and DEP developed increased AHR (Fig. 4B).
Figure 4. Primary exposure to HDM and DEP potentiates secondary HDM-specific responses in vivo.

(A) Protocol: A single i.t. challenge with either saline or 10μg of HDM was performed 7 weeks after the last exposure to either saline, DEP, HDM or HDM+DEP. (B) AHR was assessed 40h after HDM recall challenge (2-way ANOVA). (C) HDM-specific IgG1 and IgE levels 7 weeks after primary exposure. (D) Total BALF cells, (E) total BALF neutrophil and (F) eosinophil counts (n=4–10 mice/group; ***p < 0.001, **p< 0.01, *p<0.05; 1-way ANOVA with Bonferroni’s multiple comparison test; n.s. = not significant).
We have previously demonstrated that DEP not only exacerbates allergic T-cells responses but also B-cell responses as evidenced by increased HDM-specific IgG1 and IgE plasma levels in mice co-exposed to HDM and DEP compared to mice exposed only to DEP6. Similar observations were made 7 weeks after 9 exposures to either HDM or HDM and DEP (Fig. 4C), suggesting continued production of HDM-specific IgE and IgG1 antibodies.
In addition to the chronic neutrophilic inflammation observed in the lungs of mice exposed to DEP, secondary HDM challenge induced additional cellular recruitment into the airways (Fig. 4D, 4E). In mice previously co-exposed to HDM and DEP, this secondary HDM challenge also induced a significant recruitment of eosinophils into the BALF (Fig. 4F). Accordingly, these HDM and DEP pre-exposed mice had elevated eotaxin-1 BALF levels following HDM recall compared to mice, which had only been exposed to HDM (Fig. 5A).
Figure 5. Increased presence of HDM-specific memory cells 7 weeks after coexposures to HDM+ DEP results in increased Th2/Th17 responses upon secondary HDM challenge.
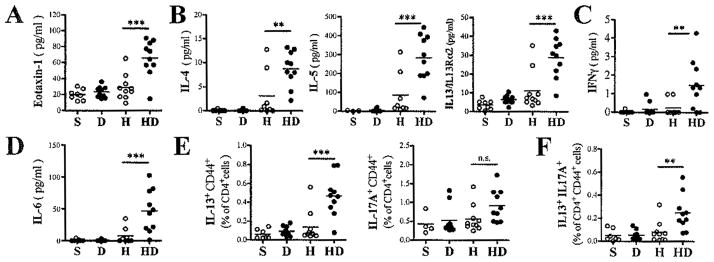
BALF levels of (A) Eotaxin-1, (B) Th2 cytokines (IL4, IL5 and IL13), (C) Th1 cytokine IFNγ and (D) Th17 related cytokines IL6 (n=8–10 mice/group). (E) Th2 and Th17 lungs cells were identified by FACS as CD4+ CD44+ T-cells expressing respectively IL13, and IL17A following ex vivo stimulation with PMA/ionomycin for 4h. (F) Percentage of IL13+IL17+ double producing CD4+CD44+ T-cells. (n=4–6 mice/group; ***p < 0.001, **p< 0.01, *p<0.05; 1way ANOVA with Bonferroni’s multiple comparison test).
BALF Th2 cytokine (IL4, IL5, IL13) levels after secondary HDM challenge were elevated in mice with primary exposure to HDM and DEP compared to only HDM (Fig. 5B). Similarly, BALF levels of IFNγ were also increased in mice previously exposed to HDM and DEP (Fig. 5C). IL17A BALF levels were mostly below detection levels, but the pro-Th17 cytokine IL6 was also elevated in mice previously exposed to both HDM and DEP (data not shown and Fig. 5D). IFNγ positive cells were not predominantly CD4+ T-cells whereas IL13 and IL17A positive cells were mostly CD4+CD44+ effector T-cells (data not shown). Th2 and Th17 cells numbers were increased in the lungs of mice previously exposed to both HDM and DEP (Fig. 5E). Accordingly, effector/memory T-cells co-expressing IL13 and IL17A were more numerous in the lungs of mice previously exposed to both HDM and DEP compared to mice previously exposed to only HDM (Fig. 5F). Taken together, these results demonstrate that the increased accumulation of HDM-specific memory cells in the lungs of mice, which had been previously exposed to both HDM and DEP versus only HDM, promotes increased HDM recall responses in vivo.
Primary neonatal co-exposure to HDM and DEP promotes increased HDM recall responses
Since exposure to DEP increases severity of allergic asthma in young mice and prenatal exposure increases asthma susceptibility24, 26, we next examined the impact of DEP co-exposure on recall responses in neonatal mice. Three-day old neonatal mice were subjected to 9 daily intranasal exposures to either saline, DEP, HDM, or HDM and DEP; then following a 4-week rest period, the mice were challenged twice with HDM (See Fig. E5, A in the online repository).
Airway resistance was significantly increased in mice previously exposed to both HDM and DEP compared to HDM alone (Fig. E5, B in the online repository). Total BALF cells counts were similarly increased following HDM recall exposures in mice previously exposed to HDM or HDM and DEP including similar eosinophil BALF levels between the two groups despite increased eotaxin-1 BALF levels in the HDM and DEP pre-exposed mice (data not shown and Fig. E5, C in the online repository). In accordance with elevated AHR, mice previously exposed to HDM and DEP had higher BALF levels of Th2 cytokines (IL4, IL5 and IL13) (Fig. E5, D in the online repository). IL6 and IL17A BALF levels were also significantly increased in mice pre-exposed to HDM and DEP compared to mice with primary exposure to HDM alone (Fig. E5, E in the online repository). HDM sensitization in neonatal mice appeared weaker than in adult mice and did not demonstrate increased HDM-specific IgG1 levels in neonates co-exposed to HDM and DEP (Fig. E5, F in the online repository). Overall, the results in neonatal mice paralleled our observations in adult mice supporting that exposure to DEP promotes asthma development in the context of allergen exposure and allergic sensitization.
Exposure to high DEP in infants contributed to sensitization earlier in life
We then hypothesized that if DEP exposure potentiates recall responses to allergen, early life DEP exposure may promote the development of allergic asthma in children. To test this hypothesis, we used the CCAAPS birth cohort, an existing birth cohort of 762 children, 578 with known exposure levels to traffic related pollution, notably elemental carbon attributable to traffic (ECAT), sensitization to aeroallergens and development of asthma.
First, we examined whether ECAT exposure impacted allergic sensitization. Overall, birth ECAT level was not associated with aeroallergen sensitization during the first 4 years of life (70% vs 68%, p=0.66). However, when each year was analyzed independently, children exposed to high ECAT at birth were more likely to be aeroallergen sensitized at age 2 (47% vs 35%, p=0.011) and maintained this high level of sensitization at ages 3 (47%) and 4 (48%) while the proportion of children sensitized to an aeroallergen and exposed to low ECAT increased gradually from age 1 to 4 (18%, 35%, 42%, 51%, respectively, Fig. 6A). This suggests that while high ECAT exposure at birth does not contribute to overall higher rates of sensitization, it does contribute to sensitization earlier in life.
Figure 6. Exposure to high birth ECAT contributed to significantly higher asthma prevalence at age 7.
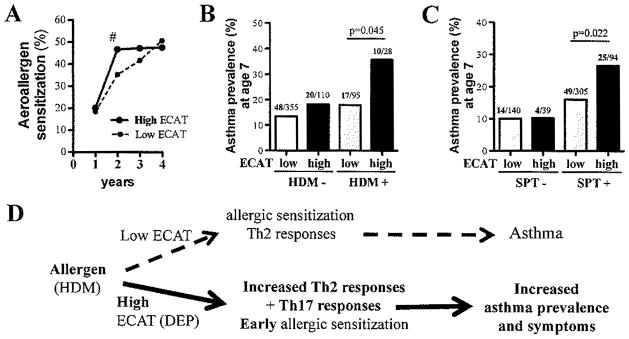
(A) Children exposed to high ECAT levels are more likely to be sensitized to an aeroallergen at age 2 compared to low ECAT exposed children (35% vs 47%, # p=0.011). Children exposed to high ECAT (top quartile) that were (B) sensitized to HDM or (C) SPT+ to any of the 15 aeroallergens tested in the first 4 years of life were significantly more likely to develop asthma by age 7 (p=0.045, p=0.022, respectively). All p-values were calculated from chi-squared tests using SAS; numbers of asthmatics / total cases in each group are presented above asthma prevalence bars. (D) Proposed model.
Exposure to high DEP in infants contributed to significantly higher asthma prevalence at age 7
We next examined children co-exposed to aeroallergen and ECAT versus children exposed to allergen alone to determine whether co-exposure to high levels of ECAT during the first year of life (similar to DEP plus HDM co-exposure in our mouse model) would result in increased prevalence of asthma at age 7. First, we specifically examined HDM sensitization (proxy for HDM exposure) and ECAT co-exposure (Fig. 6B). Among children sensitized to HDM by age 4, co-exposure to high ECAT at birth (defined as the top 25%) doubled the risk of asthma development by age 7 compared to those exposed to low ECAT (defined as the lower 75%) (36% versus 18%, p=0.045). HDM sensitization did not significantly increase the risk of asthma in children exposed to low levels of DEP (14% vs 18%, p=0.277). To assess if our findings with HDM sensitization could be extended to other allergens, we included sensitization to any aeroallergen at ages 1 through 4. We found a significant increase in asthma prevalence at age 7 (16% vs 29%, p=0.02) in children co-exposed to high levels of ECAT at birth compared to children co-exposed to low ECAT (Figure 6C).
In our logistic model adjusted for race, sex and mother’s education level, we observed a significant increasing prevalence of asthma moving from low ECAT, SPT- to high ECAT, SPT+ (OR 1.4, 95%CI 1.1–1.8, p=0.009). When we restricted this adjusted analysis to just those subjects that were SPT+, we still observed a trend of increased asthma prevalence with high ECAT exposure (OR 1.5, 95%CI 0.8–2.7, p=0.18) even though the sample size was reduced by 31% in this restricted analysis. While early atopy and high birth ECAT exposure are independent risk factors for asthma development at age 7 (10 vs 19%, p=0.01 and 14% vs 22%, p=0.04, respectively), co-exposure of aeroallergen sensitization at ages 1–4 and high ECAT at birth results in almost 3-fold higher risk of asthma compared to those that are SPT- (10% vs 27%, p=0.038; Figure 6C). The results were similar when average ECAT levels for the first 7 years of life were used and when HDM was excluded from the definition of early atopy (data not shown). However, there was no difference in asthma prevalence when we evaluated current ECAT exposure level, indicating that high birth ECAT is driving this association. Taken together, these findings support that the impact of ECAT exposure is not specific to HDM co-exposure, but rather to any environmental allergen and suggests that exposure to high levels of ECAT at birth increases susceptibility for allergic asthma.
DISCUSSION
In this study, we demonstrate that DEP exposure promotes increased numbers and persistence of allergen-specific memory T-cells in murine lungs. These memory T-cells, which are poised to produce increased quantities of Th2 cytokines (IL4, IL13) and IL17A the prototypical Th17 cytokine, generate a strong and rapid response upon secondary exposure to allergen in adult and neonatal mice. In allergen exposed and sensitized children in the CCAAPS birth cohort, co-exposure to high ECAT in the first year of life was associated with earlier allergen sensitization and increased prevalence of asthma. Collectively, these data support that exposure to DEP/ECAT results in early sensitization and accumulation of allergen specific Th2/Th17 memory/effector cells in the lungs, thereby potentiating secondary allergen recall responses and the development of allergic asthma (Fig. 6D).
These effects of DEP/ECAT at different points in the allergic inflammatory cascade act to amplify the adverse health effects of DEP/ECAT exposure. Indeed, we have previously demonstrated that increased exposure to traffic pollution in the CCAAPS birth cohort increases wheezing at age 321. We now report that early life exposure to high DEP/ECAT levels is associated with increased prevalence of asthma at age 7 in the context of aeroallergen sensitivity. While allergic sensitization is a known risk factor for asthma, it alone does not necessarily lead to allergic disease27. After adjusting for sex, race and mother’s education we observed a 50% increasing trend in asthma risk in SPT+ children exposed to high ECAT compared to low ECAT, further supporting our results. Taken together with the limited impact of DEP exposure on humoral immunity in children and neonatal mice, we propose that co-exposure to common aeroallergen (HDM) and traffic related ultrafine particulate matter (DEP) early in life potentiate primary and secondary effector/memory Th2 and Th17 responses that promote the development of childhood asthma (Fig. 6D). We did not observe an increase in allergic sensitization by age 4 among high ECAT-exposed children. It is likely that other exposures negate the impact of DEP on sensitization as time ensues. However, earlier sensitization indicates that the high DEP/ECAT-exposed children are likely to have memory effector cells parked in their lungs earlier, specifically during the critical 1st year of life during which exposures may be most harmful. Indeed, these children had the highest prevalence of asthma suggesting that the timing of the exposure and sensitization are critical to the overall outcome of asthma. In this study, the most relevant DEP/ECAT exposure was in the first year of life; there was no difference in asthma prevalence when current ECAT exposure level was used.
Repeated co-exposure to both HDM and DEP induced a significantly stronger effector Th2 response, characterized by increased BALF levels of Th2 cytokines and eosinophils, than exposure to HDM alone, resulting in increased AHR (Fig. 1). When these mice are no longer exposed to HDM, this HDM-specific response is no longer needed and most effector T-cells die. Indeed, 8 days after the last exposure, Th2 cytokines are only detectable in HDM and DEP co-exposed mice reflecting the time needed to return to baseline for Th2 effector responses of different amplitude (Fig. 1). It is well established that following the contraction phase, memory CD8+ T-cell populations are maintained whereas memory CD4+ T-cells slowly decline over time10, 28. The evidence linking a stronger effector T-cell response to the formation of a larger memory T-cell pool has been mostly limited to CD8+ T-cells17, 29. However, the nature and size of the CD4+ primary response also influences the nature and size of the memory CD4+ T-cell pool18, 30. Since HDM and DEP co-exposure results in a greater accumulation of CD4+ effector T-cells in the lungs compared to exposure to HDM alone, and assuming a similar rate of contraction of the effector pool, it is not surprising that the contraction phase would result in higher levels of HDM-specific memory T-cells in the lungs. Indeed, a month after the last exposure significantly more Th2 cells were present in the lungs resulting in stronger HDM recall responses (Fig. 3). Similar results were observed 45 days after the last exposure, thus the observations are not likely to be due to a delayed contraction phase (See Fig. E2 in the online repository).
We extended our findings to neonatal mice since early life exposures to DEP have been postulated to be most relevant. Similar to our observations in adult mice, AHR and airway inflammation (BALF cell counts, Th2 cytokines) were significantly increased in mice co-exposed to HDM and DEP compared to HDM alone. In a previous report, neonatal exposure to nebulized OVA resulted in tolerance (absence of AHR) whereas co-exposure to the air pollutant residual oil fly ash (ROFA) a component of ambient particulate matter derived from oil-burning power plants, resulted in elevated AHR both during primary challenge and following secondary exposure 10 days later35. In contrast, when neonatal mice were repeatedly pre-exposed to combustion-derived particulate matter (CDPM) before being co-exposed to CPDM and HDM, or CPDM and OVA in OT-II mice, tolerance was observed in these young C57Bl/6 mice36. However, when the CPDM and OVA co-exposed OT-II neonates were allowed to reach adulthood, a secondary OVA recall response generated AHR only in mice previously co-exposed to CPDM and OVA as neonates36. This was associated with the accumulation of CD4+ T-cells expressing IL4 or IL17A in the lungs, similar to our findings36.
We have previously demonstrated that IL17A contributes to asthma exacerbation and that co-exposure to HDM and DEP increases IL17A and IL13 double producing T-cells6. These double producing cells have been shown to be more pathogenic than classic Th2 cells and exacerbate chronic allergic asthma9. A recent study revealed that asthma patients have a higher frequency of dual Th2/Th17 cells in their BALF compared to healthy controls and that these cells are more resistant to steroids in vitro37. Additionally, asthmatic with a predominant Th2/Th17 phenotype had more severe airway hyperreactivity than Th2 asthmatics37. In the present study, we demonstrate that HDM and DEP co-exposure results in persistent accumulation of memory Th2 cells in the lungs, as well as increased IL17A+IL13+CD44+CD4+ T-cells. The increased presence of these double producing CD4+ T-cells 6 weeks after the last HDM and DEP co-exposure taken together with the data demonstrating increased HDM-induced IL17A and IL4 secretion in lung cells from previously mice exposed to both HDM and DEP, suggest that these double producing memory cells may play a significant role in the increased AHR observed following secondary HDM challenge (Fig. 4).
In summary, our data demonstrate that DEP and HDM co-exposure result in increased accumulation of HDM-specific memory Th2 and Th17 cells in the lungs, potentiating secondary recall responses and promoting asthma development. This may contribute to the increased prevalence of allergic asthma observed in children with early exposure to high levels of traffic related DEP. This raises the possibility for asthma prevention by minimizing DEP exposure in young children and or by counteracting the impact of DEP exposure with early intervention (perhaps by targeting IL-17A) in high risk sensitized and DEP-exposed children.
Supplementary Material
Key Messages.
DEP persists in murine lungs months after exposure and is associated with elevated BALF IL17A levels and chronic pulmonary neutrophilia.
DEP exposure exacerbates HDM-induced allergic airway responses in neonatal and adult mice, resulting in increased effector/memory T-cell accumulation in the lungs and potentiating HDM-recall responses in vitro and in vivo.
Exposure to high DEP in early life and co-exposure to aeroallergen sensitization by age 4 promotes the development of allergic asthma at age 7.
Exposure to DEP at birth is associated with earlier sensitization in young children.
Acknowledgments
Supported by NHLBI R01HL097135 (TDLC, GKKH), NIEHS T32 ES010957 (EBB), and R01ES011170 (GKL).
The DEP was kindly provided by Ian Gilmour (EPA, Research Triangle Park, NC 27711). We thank Seth Reighard, Reda Baig, Paige Bolcas, and Stacey Bass for technical assistance and Cynthia Chappell for editorial assistance.
Abbreviations
- AHR
airway hyperresponsiveness
- BALF
bronchoalveolar lavage fluid
- CCAAPS
Cincinnati Childhood Allergy and Air Pollution Study
- DEP
diesel exhaust particle
- ECAT
estimates of elemental carbon attributable to traffic, proxy for DEP exposure
- HDM
house dust mite extract
- PM
particulate matter
- SPT
Skin prick test
- TRAP
traffic-related air pollution
Footnotes
Conflict of interest: The authors have declared that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.HEI Panel on the Health Effects of Traffic-Related Air Pollution. Traffic-Related Air Pollution: A Critical Review of the Literature on Emissions, Exposure, and Health Effects. Boston, MA: Health Effects Institute; 2010. HEI Special Report 17. [Google Scholar]
- 2.Spira-Cohen A, Chen LC, Kendall M, Lall R, Thurston GD. Personal exposures to traffic-related air pollution and acute respiratory health among Bronx schoolchildren with asthma. Environ Health Perspect. 2011;119:559–65. doi: 10.1289/ehp.1002653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guarnieri M, Balmes JR. Outdoor air pollution and asthma. Lancet. 2014;383:1581–92. doi: 10.1016/S0140-6736(14)60617-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Appatova AS, Ryan PH, LeMasters GK, Grinshpun SA. Proximal exposure of public schools and students to major roadways: a nationwide US survey. Journal of Environmental Planning and Management. 2008;51:631–46. [Google Scholar]
- 5.McCreanor J, Cullinan P, Nieuwenhuijsen MJ, Stewart-Evans J, Malliarou E, Jarup L, et al. Respiratory effects of exposure to diesel traffic in persons with asthma. N Engl J Med. 2007;357:2348–58. doi: 10.1056/NEJMoa071535. [DOI] [PubMed] [Google Scholar]
- 6.Brandt EB, Kovacic MB, Lee GB, Gibson AM, Acciani TH, Le Cras TD, et al. Diesel exhaust particle induction of IL-17A contributes to severe asthma. J Allergy Clin Immunol. 2013;132:1194–204. e2. doi: 10.1016/j.jaci.2013.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lloyd CM, Hessel EM. Functions of T cells in asthma: more than just T(H)2 cells. Nat Rev Immunol. 2010;10:838–48. doi: 10.1038/nri2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maes T, Provoost S, Lanckacker EA, Cataldo DD, Vanoirbeek JA, Nemery B, et al. Mouse models to unravel the role of inhaled pollutants on allergic sensitization and airway inflammation. Respir Res. 2010;11:7. doi: 10.1186/1465-9921-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang YH, Voo KS, Liu B, Chen CY, Uygungil B, Spoede W, et al. A novel subset of CD4(+) T(H)2 memory/effector cells that produce inflammatory IL-17 cytokine and promote the exacerbation of chronic allergic asthma. J Exp Med. 2010;207:2479–91. doi: 10.1084/jem.20101376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kurtulus S, Tripathi P, Hildeman DA. Protecting and rescuing the effectors: roles of differentiation and survival in the control of memory T cell development. Front Immunol. 2012;3:404. doi: 10.3389/fimmu.2012.00404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mueller SN, Gebhardt T, Carbone FR, Heath WR. Memory T cell subsets, migration patterns, and tissue residence. Annu Rev Immunol. 2013;31:137–61. doi: 10.1146/annurev-immunol-032712-095954. [DOI] [PubMed] [Google Scholar]
- 12.Seddon B, Tomlinson P, Zamoyska R. Interleukin 7 and T cell receptor signals regulate homeostasis of CD4 memory cells. Nat Immunol. 2003;4:680–6. doi: 10.1038/ni946. [DOI] [PubMed] [Google Scholar]
- 13.Farber DL, Yudanin NA, Restifo NP. Human memory T cells: generation, compartmentalization and homeostasis. Nat Rev Immunol. 2014;14:24–35. doi: 10.1038/nri3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Turner DL, Bickham KL, Thome JJ, Kim CY, D’Ovidio F, Wherry EJ, et al. Lung niches for the generation and maintenance of tissue-resident memory T cells. Mucosal Immunol. 2013 doi: 10.1038/mi.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shin H, Iwasaki A. Tissue-resident memory T cells. Immunol Rev. 2013;255:165–81. doi: 10.1111/imr.12087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jenkins MK, Moon JJ. The role of naive T cell precursor frequency and recruitment in dictating immune response magnitude. J Immunol. 2012;188:4135–40. doi: 10.4049/jimmunol.1102661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Redeker A, Welten SP, Arens R. Viral inoculum dose impacts memory T-cell inflation. Eur J Immunol. 2014;44:1046–57. doi: 10.1002/eji.201343946. [DOI] [PubMed] [Google Scholar]
- 18.Iwamura C, Shinoda K, Endo Y, Watanabe Y, Tumes DJ, Motohashi S, et al. Regulation of memory CD4 T-cell pool size and function by natural killer T cells in vivo. Proc Natl Acad Sci U S A. 2012;109:16992–7. doi: 10.1073/pnas.1203494109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.LeMasters GK, Wilson K, Levin L, Biagini J, Ryan P, Lockey JE, et al. High prevalence of aeroallergen sensitization among infants of atopic parents. J Pediatr. 2006;149:505–11. doi: 10.1016/j.jpeds.2006.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ryan PH, Lemasters GK, Biswas P, Levin L, Hu S, Lindsey M, et al. A comparison of proximity and land use regression traffic exposure models and wheezing in infants. Environ Health Perspect. 2007;115:278–84. doi: 10.1289/ehp.9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ryan PH, Bernstein DI, Lockey J, Reponen T, Levin L, Grinshpun S, et al. Exposure to traffic-related particles and endotoxin during infancy is associated with wheezing at age 3 years. Am J Respir Crit Care Med. 2009;180:1068–75. doi: 10.1164/rccm.200808-1307OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reponen T, Vesper S, Levin L, Johansson E, Ryan P, Burkle J, et al. High environmental relative moldiness index during infancy as a predictor of asthma at 7 years of age. Ann Allergy Asthma Immunol. 2011;107:120–6. doi: 10.1016/j.anai.2011.04.018. [DOI] [PubMed] [Google Scholar]
- 23.Reponen T, Lockey J, Bernstein DI, Vesper SJ, Levin L, Khurana Hershey GK, et al. Infant origins of childhood asthma associated with specific molds. J Allergy Clin Immunol. 2012;130:639–44. e5. doi: 10.1016/j.jaci.2012.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Acciani TH, Brandt EB, Khurana Hershey GK, Le Cras TD. Diesel exhaust particle exposure increases severity of allergic asthma in young mice. Clin Exp Allergy. 2013;43:1406–18. doi: 10.1111/cea.12200. [DOI] [PubMed] [Google Scholar]
- 25.Park EJ, Roh J, Kang MS, Kim SN, Kim Y, Choi S. Biological responses to diesel exhaust particles (DEPs) depend on the physicochemical properties of the DEPs. PLoS One. 2011;6:e26749. doi: 10.1371/journal.pone.0026749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Manners S, Alam R, Schwartz DA, Gorska MM. A mouse model links asthma susceptibility to prenatal exposure to diesel exhaust. J Allergy Clin Immunol. 2013 doi: 10.1016/j.jaci.2013.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hamilton RG. Allergic sensitization is a key risk factor for but not synonymous with allergic disease. J Allergy Clin Immunol. 2014;134:360–1. doi: 10.1016/j.jaci.2014.02.022. [DOI] [PubMed] [Google Scholar]
- 28.Homann D, Teyton L, Oldstone MB. Differential regulation of antiviral T-cell immunity results in stable CD8+ but declining CD4+ T-cell memory. Nat Med. 2001;7:913–9. doi: 10.1038/90950. [DOI] [PubMed] [Google Scholar]
- 29.Hou S, Hyland L, Ryan KW, Portner A, Doherty PC. Virus-specific CD8+ T-cell memory determined by clonal burst size. Nature. 1994;369:652–4. doi: 10.1038/369652a0. [DOI] [PubMed] [Google Scholar]
- 30.Moon JJ, Chu HH, Pepper M, McSorley SJ, Jameson SC, Kedl RM, et al. Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity. 2007;27:203–13. doi: 10.1016/j.immuni.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mollo SB, Zajac AJ, Harrington LE. Temporal requirements for B cells in the establishment of CD4 T cell memory. J Immunol. 2013;191:6052–9. doi: 10.4049/jimmunol.1302033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Teijaro JR, Turner D, Pham Q, Wherry EJ, Lefrancois L, Farber DL. Cutting edge: Tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J Immunol. 2011;187:5510–4. doi: 10.4049/jimmunol.1102243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shiow LR, Rosen DB, Brdickova N, Xu Y, An J, Lanier LL, et al. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature. 2006;440:540–4. doi: 10.1038/nature04606. [DOI] [PubMed] [Google Scholar]
- 34.Shinoda K, Tokoyoda K, Hanazawa A, Hayashizaki K, Zehentmeier S, Hosokawa H, et al. Type II membrane protein CD69 regulates the formation of resting T-helper memory. Proc Natl Acad Sci U S A. 2012;109:7409–14. doi: 10.1073/pnas.1118539109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hamada K, Goldsmith CA, Goldman A, Kobzik L. Resistance of very young mice to inhaled allergen sensitization is overcome by coexposure to an air-pollutant aerosol. Am J Respir Crit Care Med. 2000;161:1285–93. doi: 10.1164/ajrccm.161.4.9906137. [DOI] [PubMed] [Google Scholar]
- 36.Saravia J, You D, Thevenot P, Lee GI, Shrestha B, Lomnicki S, et al. Early-life exposure to combustion-derived particulate matter causes pulmonary immunosuppression. Mucosal Immunol. 2014;7:694–704. doi: 10.1038/mi.2013.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Irvin C, Zafar I, Good J, Rollins D, Christianson C, Gorska MM, et al. Increased frequency of dual-positive T2/T17 cells in bronchoalveolar lavage fluid characterizes a population of patients with severe asthma. J Allergy Clin Immunol. 2014 doi: 10.1016/j.jaci.2014.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.