

Abstract
Post-translational acetylation of lysines is most extensively studied in histones, but this modification is also found in many other proteins and is implicated in a wide range of biological processes in both the cell nucleus and the cytoplasm. Like phosphorylation, acetylation patterns and levels are often altered in cancer, therefore small molecule inhibition of enzymes that regulate acetylation and deacetylation offers much potential for inhibiting cancer cell growth, as does disruption of interactions between acetylated residues and ‘reader’ proteins. For more than a decade now, histone deacetylase (HDAC) inhibitors have been investigated for their ability to increase acetylation and promote expression of tumor suppressor genes. However, emerging evidence suggests that acetylation can also promote cancer, in part by enhancing the functions of oncogenic transcription factors. In this review we focus on how acetylation of both histone and non-histone proteins may drive cancer, and we will discuss the implications of such changes on how patients are assigned to therapeutic agents. Finally, we will explore what the future holds in the design of small molecule inhibitors for modulation of levels or functions of acetylation states.
Introduction
From transcriptional regulation to metabolic functions, protein acetylation is involved in several processes that keep a cell working properly. Acetylation is a dynamic process that involves the removal of a hydrogen atom on the episilon NH3+ side chain of lysines followed by the transfer of an acetyl group from acetyl-CoA (AcCoA). This exchange neutralizes the positive charge on the lysine and also changes the structure of the R-group on this amino acid, leading to various effects on the protein modified. Lysine acetylation chemically blocks other modifications, such as methylation or ubiquitination, for example, which can in turn increased protein stability, alter subcellular localization, or change the spectrum of interacting proteins. As such, acetylation provides a rich regulatory ‘switch’.
Acetylation levels are regulated by a balance in the activities of acetyltransferases and deacetylases. Although originally termed histone acetyltransferases (HATs), due to their actions towards abundant histone substrates, lysine acetyltransferases (KATs) are located both in the nucleus and in the cytoplasm, and they have many non-histone substrates as well. Deacetylases similarly have multiple substrates, but they are still primarily referred to as HDACs rather than KDACs. Several excellent reviews on HDAC families and their functions are available 1–3, so we will focus mostly on acetylation and KATs in this review.
Histone Acetylation and Chromatin Regulation
In the nucleus, DNA is packaged into chromatin. The basic unit of chromatin is the nucleosome, which consists of 146 bp of DNA and histones, the proteins that provide the scaffold that DNA is wrapped around. Histones contain a globular domain that promotes histone-histone interactions within the nucleosome and also provides a binding surface for DNA. In addition, they contain tail domains that protrude out of the nucleosome, where they influence histone-histone interactions, interactions between histones and DNA, and between histones and other proteins. Although both the globular domains and the tail domains can be modified, the histone tails are particularly rich in modifications, including methylation, acetylation, phosphorylation, ubiquitination, and sumoylation. The many sites and types of modification provide a wealth of variable combinations, which in turn provides huge regulatory potential for remodeling chromatin states to either facilitate or inhibit gene transcription, DNA replication, repair, or recombination.
Acetylation has long been associated with chromatin opening and active gene transcription. Both individual nucleosomes and higher order chromatin folding can block access of RNA polymerase and other factors to gene promoters. Acetylation affects chromatin folding as the addition of the acetyl group neutralizes the positive charge of the lysine, weakening bonds between histones and the negatively charged DNA backbone, as well as the bonds between neighboring nucleosomes, allowing for more relaxed chromatin structures (Figure 1A). In addition, acetylation at specific lysine residues on particular histones can promote binding of regulatory factors involved in specific steps of the transcription process. For example, Histone H3 lysine 9 acetylation (H3K9ac), catalyzed largely by Gcn5/ PCAF, 4 is enriched at gene promoters, whereas H3K27ac, catalyzed largely by CBP/p300, is enriched at enhancer sequences. 5 These modifications promote binding of other factors through interactions with KAc reader domains, which are often located in other chromatin modifying proteins, including acetyltransferases, methyltransferases, and ATP-dependent chromatin remodelers such as Swi/Snf. 6–8
Figure 1. Mechanisms of action of acetylation.
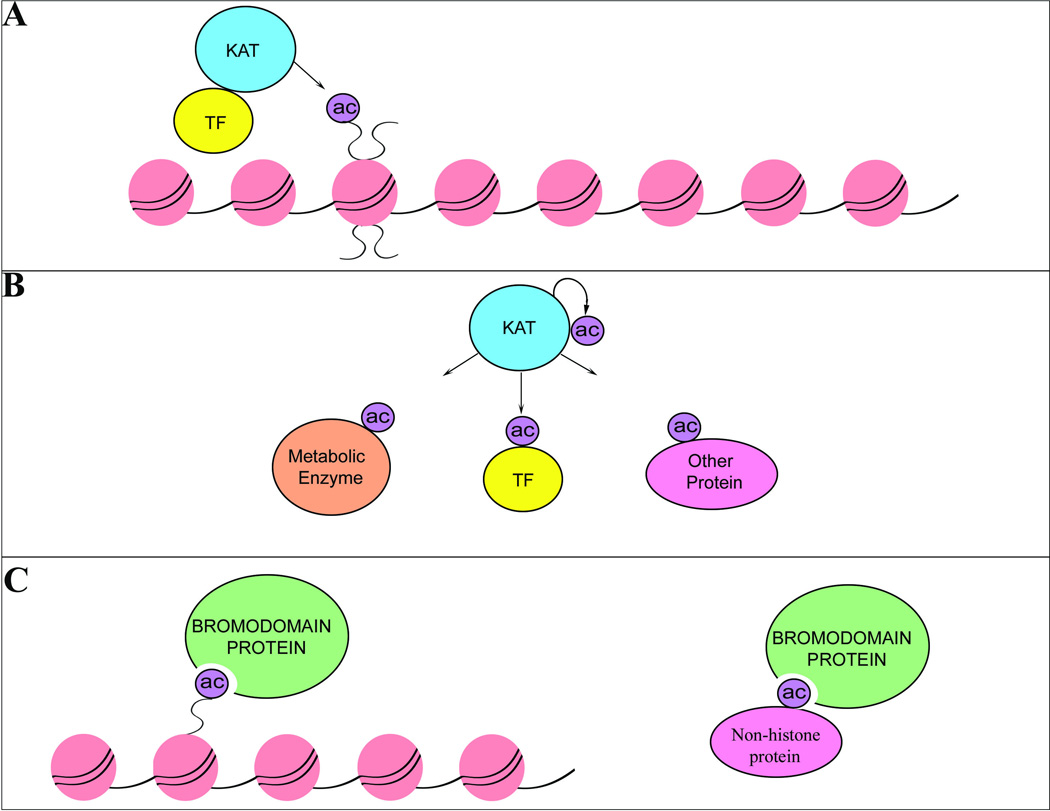
A. KATs target both tails and globular domains of all 4 histone proteins. B. KATs acetylate non-histone proteins including transcription factors (TF) as well as metabolic enzymes and other nuclear and cytoplasmic proteins. C. Bromodomain-containing proteins bind to acetyl-lysines on histone tails and on non-histone proteins.
Readers of Acetyl-lysines: Bromodomains and YEATS domains
Bromodomains were the first, and until recently, the only, acetyl-lysine binding domains described. 9,10 These domains are highly conserved across evolution and many specifically bind acetylated lysines, while only poorly binding non-acetylated lysines, thus ‘reading’ the acetylation status of histones or other proteins. 10 As such, bromodomains provide bridges for histone-protein and protein-protein interactions (Figure 1C). The bromodomain family is split into many branches, each with different structural characteristics that provide specificity for different acetylation states or proteins. 11 Although these families have wide variations in sequence, bromodomains contain a conserved binding site that is lined with a loop region linking 4 α-helices, which allows for binding to acetylated lysines. The sequence differences, variation in length of the loop region, as well as the post-translational modifications on the residues adjacent to the acetylated lysine provide specificity for substrate binding. 11,12 Intriguingly, many KATs themselves possess bromodomains, or are associated into stable complexes with bromodomain containing proteins, leading to the possibility of feed forward loops wherein they reinforce their own interactions with chromatin or with other proteins to facilitate even more acetylation. Such KATs include GCN5, PCAF, and CBP/p300.
Bromodomains have also been implicated in a wide range of diseases including cancer. 13,14,15,16 For example, Santillan et al found that the HAT and bromodomain of CBP were both important for maintenance of the cancer phenotype in leukemias caused by translocations of the MLL gene. 17 In this case, the bromodomain promoted increased proliferation of the leukemic cells. In another example, the Brd4 and Brd3 genes, members of the BET bromodomain family, are fused with the gene encoding nuclear protein in testis (NUT) in NUT midline carcinomas (NMC). 18 The resulting fusion protein, BRD-NUT, binds to acetylated histones through the bromodomain, leading to inappropriate gene expression 19, including expression of the oncoprotein c-Myc. 20 The translocation of IgH and Myc common in Burkitts lymphoma and multiple myeloma causes Myc expression to be amplified. This expression is maintained by binding of Brd4 to Igh enhancers that drives the expression of the Igh-Myc fusion protein in these cancers. 21,22 As we will see later, small molecule inhibitors of BET domains are showing clinical promise in a variety of Myc-driven cancers.
Interestingly, Lange et al found that the double PHD finger domain of DPF3, a member of the BAF chromatin remodeling complex, recognizes acetylated lysines on histones H3 and H4 as well as methylated lysines. 23,24 To date this is the only PHD domain family member identified that recognizes acetylated lysines.
New studies have identified the YEATS domain containing proteins as another possible family of acetyl-lysine readers. 25 In particular, the YEATS domain of the AF9 protein binds to H3K9ac, and these interactions promote recruitment of AF9 interacting proteins to genomic loci enriched with this modification. AF9 is part of a super elongation complex (SEC), and it also interacts strongly with the Dot1L histone methyltransferase. Thus, H3K9ac, which is largely mediated by Gcn5/PCAF, promotes both transcriptional elongation and methylation of H3 at K79 by Dot1L. Although only a few YEATS domain proteins have been analyzed for KAc binding so far, their conserved structure predicts that many if not all will bind this modification. Like bromodomains, though, different YEATS domains may display differential binding to specific sites of KAc. For example, the YEATS domain in the ENL protein binds to H3K27ac with greater affinity than H3K9ac, in contrast to the preferred binding of AF9 to H3K9ac. Interestingly, ENL is part of an alternative SEC and H3K27ac is enriched at enhancer regions. Taken together, these findings raise the possibility that differential sites of H3 acetylation direct different transcription related complexes to active enhancers, promoters, or gene bodies. The importance of such differential recruitment is highlighted by the fact that MLL-AF9 fusions lead to misdirection of DOT1L activity and inappropriate gene expression patterns associated with leukemogenesis. 26
Although they both bind acetyl-lysine, the structures of YEATS domains and bromodomains are quite different. Small molecule inhibitors of YEATS domains, then, could provide a new avenue for development of cancer therapies in the future.
KATs and Cancer
KATs are divided into families based on their structure and sequence similarity. The best characterized include the GNAT family, which includes GCN5 and PCAF, the p300/CBP family, and the MYST family, which includes Tip60 and Moz/Morf (Figure 2). In addition to the histone acetyltransferase domain, KATs possess several domains that facilitate interactions with other proteins, including reader domains for acetylation and other modifications. Together these domains allow for specificity and diversity in KAT substrates. Substrates of these enzymes as well as their roles in biological functions are continuing to be defined. However, several, such as CBP/p300, have already been implicated in cancer development and progression. 27–30
Figure 2. Selected KAT families.
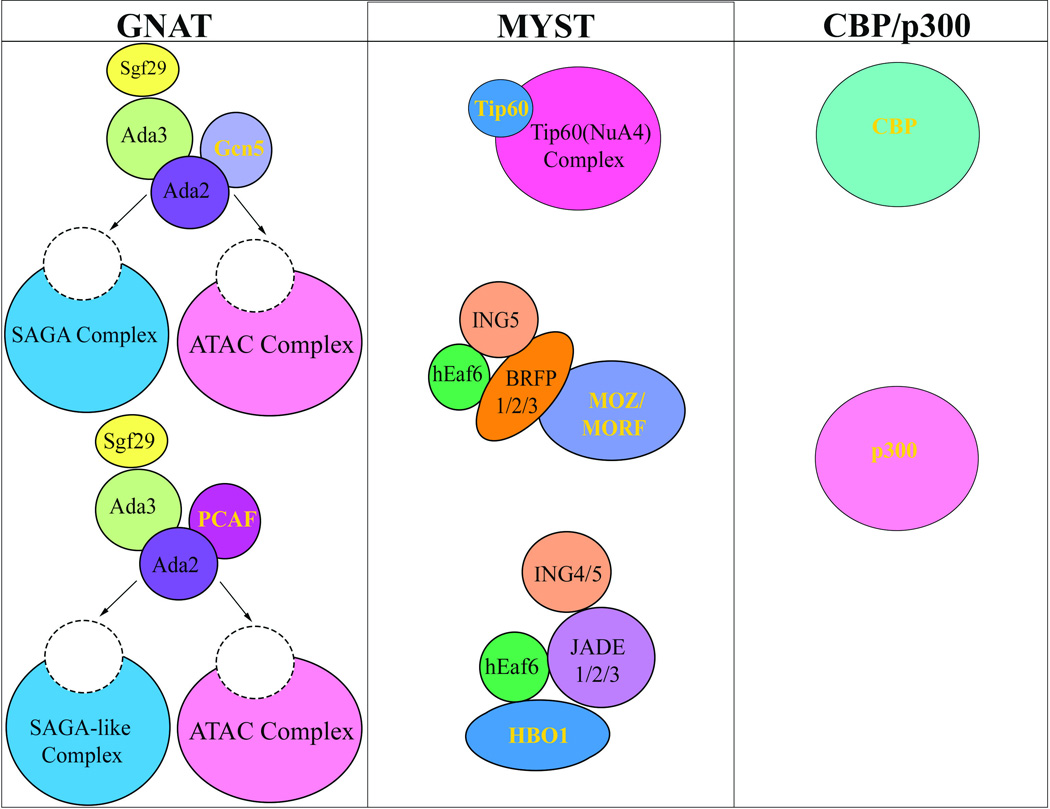
Lysine acetyltransferases are classified into different families dependent on their structure. The major families are GNAT, MYST, and CBP/p300. Not all subunits in complexes are represented.
All KATs examined to date have important functions in cellular differentiation and embryo development. 31 Since many cancers represent failures in differentiation, it is not surprising that several KATs have also been associated with oncogenesis.
GCN5
GCN5 (aka GCN5L2 and KAT2A; from here on referred to as GCN5) is the evolutionarily conserved KAT catalytic subunit of the SAGA and ATAC chromatin modifying complexes. 8 GCN5 was first identified in yeast as a transcriptional adaptor protein, and the Tetrahymena protein was later identified as the first HAT linked to transcriptional regulation. 8 In isolation, Gcn5 preferentially acetylates H3K9 and H3K14, both marks of active transcription. 4 As part of SAGA, it acetylates additional sites in H3 and also H2B. Like most HATs, GCN5 does not bind to DNA but is recruited to chromatin by sequence-specific DNA binding transcription factors. For example, GCN5 and another component of SAGA, TRRAP, have been linked to the Myc oncoprotein in mammalian cells. 32, 33 Myc is the most overexpressed gene in cancer, and it is involved in transcription of genes implicated in a wide range of cellular processes including growth, invasion, metastasis, and apoptosis. 34 The SAGA complex is recruited to chromatin by Myc, where GCN5 acts as a coactivator 35, creating an open chromatin conformation that allows for transcription of Myc target genes. 36, 37, 38 Myc is also a target of GCN5 acetyltransferase activity at K323 39, and acetylation of this site increases the stability of the Myc protein. Myc often acts in concert with another transcription factor important in the regulation of cell growth, E2F1. Interestingly GCN5 also interacts with E2F1 and is important for its functions in gene regulation. For example, in small cell lung cancer, E2F1 recruits GCN5 to acetylate H3K9, facilitating transcription of the E2F1, cyclin E, and cyclin D1 target genes 40, all of which promote cellular proliferation and tumor growth.
GCN5 may also contribute to oncogenesis through modification of non-histone proteins. For example, in many cases of acute lymphoblastic leukemia (ALL), a translocation event fuses E2A with the pre-B-cell leukemia transcription factor 1 (PBX1). The resulting oncoprotein, E2A-PBX1, aberrantly activates HOX contributing to the failure of differentiation of the leukemic cells. 41 Gcn5 acetylates and stabilizes this oncoprotein in ALL cells, further increasing target gene expression. 42 Gcn5 also interacts with tumor promoting proteins such as Pygopus homolog 2 (Pygo2), which is a component of the Wnt/beta-catenin pathway. Pygo2 directly interacts with GCN5 in the SAGA complex, bridging an interaction with beta-catenin. 43 Pygo2 enhances the growth of breast cancer stem-like cells and its HAT interacting domain is necessary for this function.
Gcn5 is also part of the ATAC KAT complex, which is distinct from SAGA. 44,45 Although ATAC has not yet been implicated in cancer, it contains a YEATS domain protein, YEATS2, which likely serves as a KAc reader. 25 ChIP-Seq data indicates that ATAC binds to both enhancer elements and promoters, whereas SAGA preferentially binds to promoters. 45 It will be interesting to determine if this differential distribution reflects the differential binding of bromodomains in Gcn5 and other SAGA components and the YEATS domain in YEATS2.
PCAF
The acetyltransferase p300/CBP associated factor, PCAF, was first described by its competition with the oncoprotein E1A for binding to p300/CBP. 46 PCAF is a paralog of Gcn5 that resides in a SAGA-like complex 46 and an alternative ATAC complex 44,47 mutually exclusive of Gcn5. Both Gcn5 and PCAF also directly bind to p300/CBP. 48 Gcn5 and PCAF share some redundant functions, exhibited by the necessity to knockout both Gcn5 and PCAF in order to completely abolish H3K9 acetylation in MEFs 5; however they also possess separate and distinct functions. 49 The phenotype of Gcn5 and PCAF knockout mice differ, as PCAF null mice show no obvious abnormal phenotypes, but Gcn5 null embryos die early in gestation. 50,51 PCAF was one of the first KATs shown to acetylate non-histone proteins, namely p53. 52, 53 PCAF acetylation of p53 increases DNA binding of p53 to its target genes. The effects on p53 as well as the ability to displace E1A indicate PCAF might function as a tumor suppressor.
More recent data also indicates that PCAF may have an oncogenic function as well, through acetylation of additional non-histone substrates. PCAF influences metabolic processes by acetylating three lysines in ATP-citrate lyase (ACLY), which cleaves citrate in rapidly dividing cells to produce AcCoA, a building block for de novo lipid synthesis and an obligate cofactor for lysine acetylation. Cancer cells preferentially utilize this alternative method of lipid biosynthesis to build membranes and promote tumor growth. ACLY is overexpressed in lung cancer. 54 Lin et al showed that PCAF-mediated acetylation of ACLY stabilizes the protein by blocking lysine ubiquitination, leading to increased ACLY protein levels, increased cell proliferation, and increased lipid biosynthesis. 54
PCAF is also involved in the Hedgehog (Hh) signaling pathway, which has been implicated in several cancers including medulloblastoma and glioblastoma. The Helin group found that PCAF is recruited by Gli, a downstream signaling molecule in the Hh pathway, to the promoters of Hh target genes. PCAF acts as a coactivator of the expression of hedgehog target genes by acetylating H3K9. 55 Loss of PCAF also reduces the tumor promoting activity of neural stem cells in vivo. 55 PCAF may therefore be a therapeutic target for medulloblastoma and glioblastoma patients. PCAF was also shown to be necessary for Gli dependent transcriptional activation in the TGFbeta pathway. 56 Additionally, PCAF is up regulated in prostate cancer cells leading to AR transcriptional activity and cell growth. 57 PCAF has also been shown to acetylate and stabilize beta-catenin in colon cancer cells. 58 Taken together PCAF makes an attractive potential target for new cancer therapies.
CBP/p300
CBP was first identified as the coactivator of the transcription factor CREB 59, while p300 was initially identified to interact with the adenovirus transforming protein E1A. 60 These two proteins were later found to be highly related, sharing 75% sequence similarity. 61 Deletion of CBP/p300 specifically reduces acetylation on H3K18ac and H3K27ac in MEFs. 5 CBP/p300 also acetylate transcription factors, such as p53 62 and AR 63, thereby inhibiting their polyubiquitination and increasing their stability. CBP/p300 are large proteins with multiple functional domains, and they are involved in many different transcription programs. In fact, CBP/p300 have been reported to interact with more than 400 different cellular proteins to date 64, including factors important to cancer development and progression such as HIF-1, beta-catenin, c-Myc, c-Myb, CREB, E1, E6, p53, AR, and ER. Interactions between these transcription factors and CBP/p300 are crucial for efficient transcription of their downstream target genes, in keeping with the functions of CBP/p300 as transcriptional co-activators.
Like Gcn5 and PCAF, CBP and p300 have both shared and distinct functions. For example, CBP and p300 control the outcomes of Wnt/beta-catenin signaling in hematopoietic cells. CBP is essential for HSC self-renewal, while p300 is required for hematopoietic differentiation. 65 In addition, CBP activity is required for maximal HIF-2 signaling 66, while p300 positively regulates the transactivation of HIF-1. 67 Furthermore, p300, but not CBP, is the dominant co-regulator for androgen-regulated gene expression since 47% of androgen-regulated genes are p300-dependent, while only 0.3% are CBP-dependent. 68 Consistent with this finding, knockdown of p300, but not CBP, leads to an increase of caspase-dependent apoptosis in androgen-dependent and castration-resistant prostate cancer cells. 69
Aberrations in both CBP and p300 show strong implications in cancer initiation and progression. Inactivation of one allele of CBP causes Rubinstein-Taybi syndrome (RTS), and RTS patients have an increased susceptibility for tumor development. 70 In addition, mice heterozygous for CBP invariably develop myelodysplastic/myeloproliferative neoplasm 71. Genetic alterations and somatic mutations/deletions of CBP/p300 are correlated with transitional cell carcinoma of the bladder 72, follicular lymphoma (41%), diffuse large B-cell lymphoma (39%) 73, relapsed ALL 74, and microsatellite instability(MSI)-positive colon cancer cells. 75
Although many studies indicate that CBP/p300 function as tumor suppressors, other studies indicate that CBP/p300 promote the tumor progression. Chromosomal translocations involving MOZ or MLL fused with CBP or p300 are associated with AML through gain-of-function. 76 In addition, interaction of c-Myb with p300 is required for the induction of AML by AML oncogenes 77,78. High CBP/p300 expression is associated with poor prognosis in small cell lung cancer. 79 High expression of p300 was also positively associated with higher grade, distant metastasis and later clinical stages of nasopharyngeal carcinoma. 80 Furthermore, the subcellular localization of p300 is also associated with cancer progression. Decreased expression of nuclear p300 and gain of cytoplasmic p300 expression are associated with disease progression and worse prognosis of melanoma patients. 81 Moreover, CBP/p300 is implicated in resistance in certain cancer types. P300 expression is reduced in doxorubicin-resistant bladder cancer cells and knockdown of p300 leads bladder cancer cells resistant to doxorubicin. 82 However, CBP/p300 are required in the IL-4 mediated castration resistance of prostate cancer cells. Downregulation of CBP/p300 abolished IL-4 mediated AR activation. 83 Overall, the changes in genetic alterations, expression level, and subcellular localization of CBP/p300 are associated with progression, prognosis, and resistance of many types of cancers.
Onco-viruses, such as HTLV-1, SV40, and HPV, induce cancer, mediated, at least in part, by manipulating CBP/p300 function. HTLV-1 is crucial for developing adult T-cell leukemia. 84 HTLV-1 Tax protein interacts with CBP/p300 to activate gene expression regulated by ATF/CREB and NF-kB pathways, but it inhibits p53-mediated gene expression. 85,86 In addition, transforming protein SV40 large T antigen (SVT) interacts with CBP/p300 87, which is required by SVT-mediated transformation 88,89, and increases CBP/p300 protein levels through increasing the loading of CBP/p300-mRNA onto polysomes. 90 Furthermore, the high-risk HPV E6 protein interacts with CBP/p300 directly and blocks p300-mediated p53 acetylation and p53-dependent gene transcription. 91 Moreover, adenovirus E1A protein also interacts with CBP/p300 92 and alters gene expression by reducing CBP/p300 recruitment on the promoters of differentiation-specific genes but increasing the CBP/p300 loading on the promoters of genes involved in cell proliferation. 93
CBP/p300 can also promote tumorigenesis independently of its effects on gene transcription. p300 acetylates S-phase kinase-associated protein 2 (Skp2), which is a driving factor in tumorigenesis, leading to increased Skp2 stability and cytoplasmic retention. Cytoplasmic Skp2 enhances cellular migration through ubiquitination and destruction of E-cadherin. 94
Tip60
Tip60 is the acetyltransferase component of a multiprotein Tip60 complex that includes up to 16 subunits. The Tip60 complex also contains an ATPase, p400/TRAPP, and helicases, Tip49a and Tip49b. 95,96 The Tip60 complex preferentially acetylates H2AK5, H3K14, and H4K5/8/12/16. 97 Tip60 plays important roles not only in transcription but also in double-strand DNA break repair and apoptosis. DNA damage induces accumulation of Tip60 98, which acetylates and activates ATM kinase activity at sites of DNA damage. 99–101 In addition, Tip60 also acetylates nucleosomal phosphor-H2Av, the Drosophila homolog of human H2A.X, which results in its exchange with unmodified H2Av at DNA DSB loci. 102 Furthermore, Tip60 complex induces transcription of DNA repair and apoptosis genes by interacting with E2F1 and p53, respectively. 103,104,105 Tip60 also acetylates the Abelson murine leukemia viral oncogene homolog (Abl) and activates transcription-independent apoptotic activity of Abl 106 As to the transcription related function, Tip60 is identified as co-activator of AR, ER, PR, c-Myc, and more. 107,108,109,39,110 However, Tip60 is also reported to repress STAT3, c-Myb, and beta-catenin dependent transcription in part through recruiting HDACs. 111, 112,113
The majority of studies investigating the relationship of Tip60 and cancer show that Tip60 functions as a tumor suppressor. Tip60 has a haplo-insufficient tumor suppressor activity in both mouse and human. 114 Tip60 heterozygosity impairs Myc-induced DNA damage repair in B cells and accelerates the onset of Myc-induced B-cell lymphomas when breeding the Tip60 heterozygous mice with Eu-myc transgenic mice, which develop spontaneous lymphoma and leukemia. 115 In human cancer samples, Tip60 mRNA or protein level was down regulated in colon, lung, mammary, head-and-neck carcinomas, metastatic prostate cancer, metastatic melanoma, lymphomas, and AML. 116,117,114,118,112,119,113 In addition, reduced Tip60 expression was associated with poor prognosis in primary and metastatic melanoma patients. 119 However, Tip60 was over expressed in cisplatin-resistant human lung cancer cells and knockdown of Tip60 expression rendered cells sensitive to cisplatin treatment. 120 Moreover, in hormore refractory or androgen independent prostate cancer, Tip60 predominantly accumulates in the nucleus rather than displays a more diffuse distribution pattern observed in benign prostate hyperplasia or primary prostate cancer. 116 Consistent with this finding, it is also reported that Tip60 is up regulated in castration-resistant LNCaP derivative CxR cells, which results in an increase of acetylated androgen receptor localizing in the nucleus and activation of androgen-dependent gene transcription even in the absence of androgen. 121
Other KATs
Many other KATs have been also linked to cancer. HBO1, a member of the MYST family is overexpressed in several breast cancer cell lines. HBO1 increases mammosphere formation when phosphorylated by cyclin E/CDK2 122, suggesting that HBO1 overexpression may lead to a more cancer stem cell like phenotype. The most common translocation in AML fuses the KAT Moz, another MYST family member, with CBP. 123 This oncoprotein causes disruption of the association between the differentiation factor AML1 and MOZ, thereby inhibiting AML1 induced gene transcription. 124 Lack of AML1 target gene transcription leads to a more undifferentiated state characteristic of cancer cells.
Acetylation of Non-histone Substrates
Over 6800 lysine acetylation sites have been identified in mammals 125 with 3600 sites on 1750 proteins identified in humans. 126 The acetylation of non-histone proteins initiates diverse outcomes (Figure 1B). For example, transcription factors such as E2F1 127, c-Myc 39, and beta-catenin 58 are stabilized by acetylation, while acetylation of PTEN decreases its stability. 128 Acetylation may also affect the subcellular localization of a protein, as in the case of SRY, where acetylation triggers translocation to the nucleus. 129 The DNA binding activity 53 of p53, as well as its stability 130, increases upon acetylation. Acetylation of one of the residues of the chaperone protein Heat-shock protein 90 (Hsp90) prevents association with both client proteins and its co-chaperones. 131,132 This modification not only affects the function of Hsp90 but also reduces the activity of its client proteins such as tyrosine kinase Chk1. 123
Acetylation is being recognized as increasingly important in metabolism. In addition to mitochondria being the source of AcCoA, several mitochondrial proteins are subject to acetylation. Acetylation of the glycolytic enzyme Phosphoglycerate mutase-1 (PGAM1) stimulates its activity 133 whereas acetylation of the mitochondrial matrix protein AcCoA synthetase 2 (AceSC2) inactivates the enzyme. 134 Zhao et al found that almost every enzyme involved in the TCA cycle, glycolysis, gluconeogenesis, fatty acid metabolism, the urea cycle, and glycogen metabolism is acetylated in the liver. 135 Obviously acetylation has a profound impact on metabolism. As cancer cells have an altered metabolism that gives them a proliferative advantage, perhaps manipulation of these acetylation events might provide another avenue for therapy development.
The proteins mentioned here constitute only a subset of the many proteins that are regulated in some way by acetylation. In thinking about the functions of KATs in both normal cells and cancer, it is important to keep in mind that effects on the non-histone substrates may be as important, or even more important, as effects on histones. Clearly, additional work is needed to identify the full range of acetylated proteins in the cell and the consequences of changes in acetylation states in cancer and other diseases.
Therapies targeting KATs
Given the wide-reaching importance of acetylation to cellular function, inhibitors of both KATs and HDACs have potential as anti-cancer therapies. HDAC inhibitors have been used in clinical trials for some time now, with varied results. 2, 3, 136 These drugs show some effect in leukemias and other blood borne cancers, especially in combination with other epigenetic inhibitors, such as decitabine. 137 However, they have largely failed in solid tumors. One intriguing possibility is that acetylation might augment the functions of Myc and other oncogenic proteins that drive tumor progression and growth. Use of an HDAC inhibitor in such cases might actually be detrimental rather than helpful. Genetic studies in mice indicate that deletion of HDACs enhance the severity of Myc driven lymphomas or solid tumors in mice 138, consistent with this idea. Clearly we need to know more about how acetylation contributes to cancer initiation and progression, so that more rationale therapies can be designed.
Regulation of KAT Activity
Normal means of KAT regulation provide ideas for development of KAT inhibitors. Interestingly, KATs are regulated by multiple mechanisms. Many KATs are not fully active unless associated with their partner proteins in KAT complexes. 139,140 Association into the complex can affect not only the level of activity but also substrate specificity. 47 Gcn5, for example, has very limited activity and substrate range alone, but when part of the full SAGA complex, it efficiently acetylates multiple sites in histones H3 and H2B. 139 KATs themselves are also subject to posttranslational modifications that affect their activity, stability, and subcellular localization. Autoacetylation of p300 and MOF enhances their enzymatic activity. 141,142 The nuclear localization signal in PCAF can be autoacetylated or acetylated by p300 in the cytoplasm, increasing PCAF activity and promoting its localization to the nucleus. 143 Some KATs, including GCN5 and PCAF, are also stabilized upon binding of their coenzyme, AcCoA. 144
The many layers of regulation ensure that KAT activity is spatially and temporally restricted in the cell and in the organism, and they also provide specific vulnerabilities for development of small molecule inhibitors. For example, small molecules that interfere with AcCoA or substrate binding would directly impair KAT acetyltransferase activity. Such inhibition would affect acetylation of histones and associated transcription of oncogenes or downstream oncogene targets (Figure 3A). It would also affect acetylation of non-histone substrates, such as p53 and AR, thereby affecting the stability and activity of these transcription factors (Figure 3A). In practice, however, it can be difficult to develop inhibitors that specifically affect HATs and not other enzymes that utilize AcCoA.
Figure 3. The mechanisms of small molecule inhibitors targeting lysine acetylation.
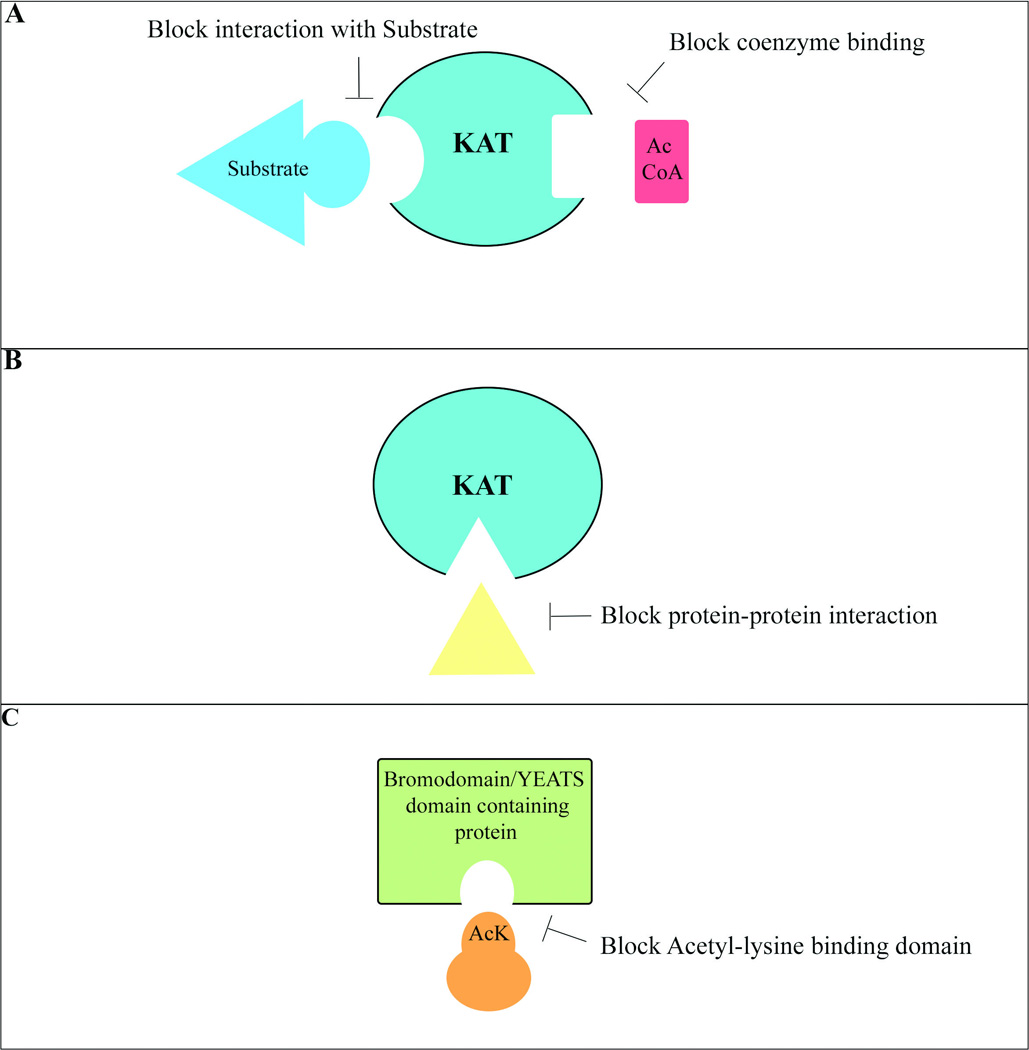
A. Small molecule KAT inhibitors can impair KAT acetyltransferase activity by interfering with AcCoA or substrate binding. The substrates include histone and non-histone proteins, such as p53 and AR. B. Small molecule KAT inhibitors can block interactions between KATs and other proteins, such as beta-catenin and HIF, which would affect transcription of downstream genes. C. Bromodomain inhibitors act as acetyl-lysine mimics that occupy the acetylated lysine binding site in bromodomain-containing proteins.
Alternatively, small molecule inhibitors might be designed to block interactions between KATs and other proteins, such as beta-catenin and HIF, which would affect transcription of downstream genes. Although beta-catenin is not classified as a transcription factor, it directly interacts with the transcription factor TCF/LEF to regulate downstream target genes (Figure 3B). Similarly, KAT inhibitors might be designed to decrease KAT or transcription factor occupancy at target genes by interfering with interactions with histones or the transcriptional machinery.
We will summarize the current status of small molecule inhibitor development for the main three KATs families that have so far shown some inhibitory effect on cancer cell growth in vitro or on xenograft growth in vivo.
GCN5-PCAF Inhibitors
So far, several small molecule inhibitors of GCN5 and PCAF have been developed and characterized in vitro. In most cases, these molecules are thought to act as substrate or AcCoA mimics. The first identified GCN5 inhibitor, α-methylene-γ-butyrolactone 3 (MB-3), which decreases GCN5 acetyltransferase activity potently, was reported to decrease acetylation and stabilization of the E2A-PBX1 oncoprotein in leukemia cells. 42 However, the effect of MB-3 on leukemia cell growth and proliferation is not clear. A series of thiazole derivatives, such as cyclopentylidene-[4-(4’-chlorophenyl)thiazol-2-yl)hydrazone (CPTH2) 145, CPTH6 146, and BF1 147, also decrease GCN5 activity in vitro. This inhibition is reversed upon increased concentration of H3, but not AcCoA 145, suggesting that thiazole derivatives inhibit acetylation by competing with substrate binding to GCN5. CPTH6 has been shown to impair viability and cell cycle progression, reflected by accumulation in G0/G1 phase, of leukemia cells. 146 This year, a novel thiazole-based HAT inhibitor BF1, was reported to inhibit the HAT activity of recombinant GCN5 and p300. BF1 treatment reduced overall levels of H3 acetylation in HeLa, neuroblastoma, and glioblastoma cells. However, BF1 administration had only modest effects on the viability of neuroblastoma cells, and had even less effect on the viability of HeLa or glioblastoma cells. 147
The first selective PCAF inhibitor to be identified and synthesized was the bisubstrate compound, H3-CoA-20 (IC50, 0.3uM). 148 However, this bisubstrate inhibitor lacks cell-permeability, which limits its development as a therapeutic drug. Anacardic acid, a chemical compound from the cashew nut shell, is a potent inhibitor of PCAF and p300 (IC50, 5 and 8.5 µM, respectively). 149 Anacardic acid potentiated apoptosis induced by cytokine and chemotherapeutic agents, which correlated with the down regulation of various gene products that mediate proliferation, survival, invasion and angiogenesis regulated by NF-kB in a series of human cancer cell lines 150 (Table 1). One anacardic acid derivative, 6d, showed a twofold higher inhibitory effect than anacardic acid on PCAF HAT activity and histone acetylation in liver hepatocellular carcinoma HepG2 cells. 151 Garcinol, a potential anti-cancer agent 152 from Garcinia indica fruit rind, is a potent inhibitor of p300 and PCAF (IC50, 5uM and 7uM respectively). 153 Garcinol inhibits KATs by binding both the AcCoA binding site and the histone binding site, 154 and it suppresses proliferation or induces apoptosis of various cancer cells153, including HeLa cells, breast cancer cells 155–157, lung cancer cells 158, pancreatic cancer cells 159, hepatocellular carcinoma(HCC) cells 160, and head and neck squamous carcinoma(HNSCC) cells 161 in vitro. Moreover, Garcinol has been reported to inhibit the growth of human HCC and HNSCC xenograft tumors in athymic nu/nu mice. 160,161 In addition, isothiazolones-based inhibitors for PCAF and p300, CCT077791 and CCT077792, inhibit the growth of a panel of human tumor cell lines. 162
Table 1.
KATs inhibitors.
| Targeted KAT |
Molecule | Mechanism | Category | Affected cells or tumors |
|---|---|---|---|---|
| GCN5 | MB-3 | NA | Synthetic compound | leukemia cells 42 |
| GCN5/p300 | CPTH2, CPTH6, BF1 | competes with substrate | thiazole derivatives | leukemia cells, neuroblastoma cells 145–147 |
| PCAF/p300 | anacardic acid | NA | Natural compound, phenolic lipid | myeloid KBM-5 cells, T-cell lymphoma Jurkat cells, lung adenocarcinoma H1299 cells, embryonic kidney A293 cells, prostate cancer Du145 cells, squamous cell carcinoma SCC4 cells 150 |
| Garcinol | inhibits AcCoA and histone binding | Natural compound, polyisoprenylated benzophenone | HeLa cells, breast cancer cells, lung cancer cells, pancreatic cancer cells, HCC cells, and HNSCC cells, HCC and HNSCC xenografts 153,155–157,158,159,160,161 | |
| CCT077791 and CCT077792 | NA | isothiazolones derivative | colon tumor HCT116 and HT29 cells 162 | |
| PCAF | H3-CoA-20 | inhibits AcCoA and substrate binding | bisubstrate inhibitor | NA |
| 6d | NA | anacardic acid derivative | HCC HepG2 cells 151 | |
| CBP/p300 | curcumin | promotes CBP/p300 degradation, inhibits KAT activity | Natural compound, diarylheptanoid | HeLa cells 165 |
| prostate cancer cells, prostate tumor xenograft 167 | ||||
| chetomin | blocks interaction of HIF-1 and CBP/p300 | Natural compound, antibiotic metabolite | glioma xenograft 170 | |
| C646 | inhibits AcCoA and substrate binding | Synthetic compound based on benzoic acid | AML1-ETO(+) AML cells, melanoma cells, TC1 or AE17 xenograft tumors 172,173,174 | |
| KCN1 | blocks interaction of HIF-1 and CBP/p300 | Arylsufonamide | subcutaneous malignant glioma tumor xenograft 177 | |
| HBS | blocks interaction of HIF-1 and CBP/p300 | protein domain mimetic | renal cell carcinoma xenograft 179 | |
| CBP | ICG-001 | blocks CBP/beta-catenin interaction | Synthetic compound | Colon cancer cells and xenograft, pre-B ALL cells, primary ALL xenograft 168,169 |
| p300 | Lys-CoA | inhibits AcCoA and substrate binding | bisubstrate inhibitor | NA |
| Plumbagin(RTK1) | inhibits AcCoA and histone binding | Natural compound | NA | |
| NK13650 | NA | microbial metabolite | prostate cancer cells 176 | |
| L002 | interacts with p300 catalytic domain | NA | leukemia and lymphoma cell, MDA-MB-468 xenograft 178 | |
| Windorphen | blocks CBP/beta-catenin interaction | Acrylaldehyde derivative | colon adenocarcinoma SW480 cells, prostate cancer DU145 and PC3 cells 180 | |
| CH1iB | mimics C-terminal domain of HIF-1, blocks interaction of HIF-1 and CBP/p300 | Synthetic compound | HPV(+) HNSCC cells 181 | |
| Tip60 | anacardic acid | NA | phenolic lipid | HeLa cells, squamous cell carcinoma SQ20B and SCC35 cells 182,183 |
| 6-alkylsalicylates | anacardic acid analog | |||
| pentamidine | NA | bisbenzamidine derivative | HeLa cells 184 | |
| NU9056 | NA | isothiazolones derivative | prostate cancer cells 185 | |
| TH1834 | inhibits AcCoA binding | Synthetic compound | breast cancer cells 186 |
NA: Non Applicable; HCC: hepatocellular carcinoma; HNSCC: head and neck squamous carcinoma.
Unfortunately, the fact that these compounds are usually only effective in vitro when administered in micromolar concentrations, along with ambiguities regarding their specificity, greatly limits their utility as possible anticancer agents. Development of additional types of inhibitors, perhaps directed at bromodomains in Gcn5 and PCAF, might offer more promise in the future.
CBP/p300 Inhibitors
Several studies have focused on potential therapies targeting CBP/p300, more so than any other type of KAT. Many kinds of CBP/p300 inhibitors have been identified or synthesized, and their effects have been tested on tumor cell growth in vitro or in vivo. The first identified selective p300 inhibitor was the bisubstrate inhibitor, Lys-CoA (IC50, 0.5uM). 148 However, Lys-CoA lacks cell-permeability, which can be somewhat improved by conjugation to a cell permeabilizing peptide. 163 Another well-studied CBP/p300 inhibitor is curcumin, which inhibits the KAT activity of purified p300 (IC50, 25uM) in vitro. 164 Curcumin inhibits p300-mediated acetylation of p53 and induces apoptosis in HeLa cells. 165 It can also affect cell proliferation of prostate cancer cells through modulation of aberrantly activated Wnt signaling. 166 In addition, curcumin suppresses CBP/p300 occupancy at AR target genes and inhibits xenograft tumor growth of androgen sensitive cells and CRPC cells. Co-treatment with curcumin and androgen deprivation reduces tumor growth and delays the onset of castration-resistant tumors. 167
ICG-001 is a specific CBP/beta-catenin antagonist that blocks CBP/beta-catenin interactions (IC50, 3uM) but not p300/beta-catenin interactions. 168 Treatment with ICG-001 leads to the differentiation of pre-B ALL cells and eradication of drug-resistant primary leukemia when used in combination with conventional therapy in vitro (VDL (Vincristine, Dexamethasone and L-Aparaginase) or Nilotinib for Philadelphia chromosome negative or positive ALL cells, respectively). ICG-001 also significantly improves the survival of NOD/SCID mice engrafted with primary ALL. 169
Chetomin, a natural small molecule that disrupts the interaction of HIF-1 with CBP/p300, abolishes the differentiation inhibitory effect of HIF-1alpha. Combined administration of chetomin with forskolin, a differentiation inducer, significantly suppresses malignant glioma growth in vitro and in vivo in a xenograft model. 170 Another CBP/p300 inhibitor, C646, (IC50, 1.6uM) 171 induces cell cycle arrest and apoptosis selectively in AML1-ETO-positive AML cells. 172 C646 also promotes sensitivity to DNA damaging agents, leading to enhanced apoptosis of melanoma cells after combination treatment with cisplatin. 173 Furthermore, C646 treatment in Foxp3+ T regulatory cells increased T cell receptor-induced apoptosis of T regulatory cells, limited tumor growth in immunocompetent but not in immunodeificient mice. 174
Besides these well-established inhibitors for CBP/p300 HAT activity or CBP/p300 interaction with transcription factors, researchers continue to screen or synthesize novel compounds to inhibit CBP/p300 function. Plumbagin (RTK1), a natural compound isolated from Plumbago rosea root extract, inhibits specifically p300-mediated acetylation of p53. 175 NK13650A and NK13650B (IC50, 11 and 22 nM, respectively), a microbial metabolite, inhibits p300 HAT activity and inhibited hormone-dependent and-independent growth of prostate cancer cells. 176 Arylsulfonamide KCN1 interferes with the interaction between HIF-1alpha and CBP/p300 (IC50, ~590nM), and potently inhibits the growth of subcutaneous malignant glioma tumor xenografts. 177 L002 (IC50, 1.98uM), interacting with the active site of the p300 catalytic domain, potently suppressed tumor growth of MDA-MB-468 xenografts as well as proliferation of leukemia and lymphoma cell lines. 178 HBS, a protein domain mimic, targets the interaction of the HIF-1alpha with p300/CBP and suppresses tumor growth in a renal cell carcinoma murine xenograft model. 179 Windorphen, a selective small molecule targeting beta-catenin and inhibiting p300, robustly and selectively kills cancer cells that harbor Wnt activating mutations. 180 CH1iB, a novel small-molecule inhibitor of p300 targeting HPV16 E6-p300 interaction, reactivates p53 and enhances the anticancer effect of cis-platinum on HPV-positive head and neck squamous cell carcinoma cells. 181
Tip60 Inhibitors
Interestingly, Tip60 is down regulated in primary cancer cells but becomes up regulated in drug resistant cancer cells. 119,120 This discrepancy may be explained by the regulatory processes used by cells to decide between life and death. Since Tip60 is crucial for the DNA repair process, its down regulation causes cells to accumulate genomic instabilities induced by oncofactors, and can eventually lead to uncontrolled cellular growth. However, after treatment of cells with therapeutic DNA damage inducing agents, drug-resistant cells exhibit higher levels of DNA repair that requires Tip60. Thus, inhibition of Tip60 activity might provide a potential strategy to treat DNA damaging drug-resistant cancers. Other agents also affect Tip60 functions in DNA repair. Anacardic acid and its analogs, 6-alkylsalicylates, inhibit Tip60 182 and sensitize HeLa cells to ionizing radiation. 183 In addition, pentamidine, a bisbenzamidine derivative that inhibits Tip60 acetyltransferase activity, also increases the radio-sensitivity of HeLa cells. 184 Moreover, two newly identified Tip60 inhibitors, NU9056 (IC50, 2uM) and TH1834, are reported to inhibit cell proliferation and induce apoptosis in prostate cancer cells and breast cancer cells, respectively. 185,186
Limitations of KAT Inhibitors
Almost all of the studies on KAT inhibitors are currently still in a preclinical phase. Only curcumin has been moved forward into clinical trials as a potential anti-cancer therapy. According to data currently available in the ClinicalTrials.gov database, about 35 registered clinical trials are ongoing in cancer patients, including colorectal cancer, pancreatic cancer, lung cancer, and leukemia patients, with curcumin alone or combined with other chemotherapy agents. Better agents, with greater specificity and better pharmokinetic properties are sorely needed.
An alternative to inhibiting KATs directly is to inhibit readers of KAc. Bromodomain inhibitors are showing much promise in early trials as effective therapies for a variety of tumor types. Bromodomain inhibitors act as acetyl-lysine mimics that occupy the acetylated lysine binding site in bromodomain-containing proteins (Figure 3C), such as BRD4 and BRD2. 187 They affect the gene expression profile of leukemia or solid tumor cells, highlighted by downregulation of Myc expression, which results in suppression of tumor cell growth and proliferation. 188,189 A few bromodomain inhibitors, such as I-BET762, OTX015, CP-0610, and TEN-010, are in clinical phase I trials to test their efficacy on various hematological malignancies and solid tumors. 190 Combinational treatment using both a BRD4 inhibitor and an HDAC inhibitor has been reported to synergistically induce apoptosis of leukemia cells and improve the survival of mice engrafted with leukemia cells. 191 Such combination therapies may also prove greatly beneficial. Since most KATs contain bromodomains, combinational treatment of inhibitors targeting these domains together with BET bromodomain inhibitors might also have synergic effects on inhibiting tumor growth, by limiting both the expression and the function of oncoproteins such as c-Myc. Combination therapies are attractive as they may allow use of the two agents in low dose, thereby increasing effectiveness while also reducing side effects or toxicities associated with use of single inhibitors in high dose.
Conclusion and Perspectives
Although aberrations of KATs have been reported widely in human cancers, our understanding of the functions of KATs in tumor development and progression is still limited. While some studies show that KATs act as tumor suppressors, other studies indicate that KATs promote tumor progression by promoting oncogene expression or function, providing a strong, rationale for the development of KAT inhibitors as anti-cancer agents. The paradox that both KAT and HDAC inhibitors may be beneficial as anti-cancer agents illustrates the necessity of defining the full range of functions of these enzymes in specific cancer settings. Alterations in the balance of these enzyme activities might lead to hallmark changes in histone modification status that could provide cancer biomarkers for predicting prognosis and for determining best treatment options for cancer patients. 192 A greater understanding of the functions of KATs and HDACs in both normal cells and in cancer will also allow better design of combination therapies using inhibitors for these enzymes together with other epigenetic agents, such as bromodomain inhibitors.
Acknowledgements
The authors thank Evangelia Koutelou for the critical reading of this manuscript and Andria Schibler for advice and contributions to artwork. This work was partially supported by NIH R01067718 to SYRD.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. The Biochemical journal. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nature reviews. Genetics. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim HJ, Bae SC. Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. American journal of translational research. 2011;3:166–179. [PMC free article] [PubMed] [Google Scholar]
- 4.Kuo MH, et al. Transcription-linked acetylation by Gcn5p of histones H3 and H4 at specific lysines. Nature. 1996;383:269–272. doi: 10.1038/383269a0. [DOI] [PubMed] [Google Scholar]
- 5.Jin Q, et al. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. The EMBO journal. 2011;30:249–262. doi: 10.1038/emboj.2010.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakamura T, et al. huASH1 protein, a putative transcription factor encoded by a human homologue of the Drosophila ash1 gene, localizes to both nuclei and cell-cell tight junctions. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:7284–7289. doi: 10.1073/pnas.97.13.7284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaeser MD, Aslanian A, Dong MQ, Yates JR, 3rd, Emerson BM. BRD7, a novel PBAF-specific SWI/SNF subunit, is required for target gene activation and repression in embryonic stem cells. The Journal of biological chemistry. 2008;283:32254–32263. doi: 10.1074/jbc.M806061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brownell JE, et al. Tetrahymena histone acetyltransferase A: a homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell. 1996;84:843–851. doi: 10.1016/s0092-8674(00)81063-6. [DOI] [PubMed] [Google Scholar]
- 9.Haynes SR, et al. The bromodomain: a conserved sequence found in human, Drosophila and yeast proteins. Nucleic acids research. 1992;20:2603. doi: 10.1093/nar/20.10.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dhalluin C, et al. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399:491–496. doi: 10.1038/20974. [DOI] [PubMed] [Google Scholar]
- 11.Zeng L, Zhou MM. Bromodomain: an acetyl-lysine binding domain. FEBS letters. 2002;513:124–128. doi: 10.1016/s0014-5793(01)03309-9. [DOI] [PubMed] [Google Scholar]
- 12.Filippakopoulos P, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Asangani IA, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014;510:278–282. doi: 10.1038/nature13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patel AJ, et al. BET bromodomain inhibition triggers apoptosis of NF1-associated malignant peripheral nerve sheath tumors through Bim induction. Cell reports. 2014;6:81–92. doi: 10.1016/j.celrep.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bandopadhayay P, et al. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20:912–925. doi: 10.1158/1078-0432.CCR-13-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zou Z, et al. Brd4 maintains constitutively active NF-kappaB in cancer cells by binding to acetylated RelA. Oncogene. 2014;33:2395–2404. doi: 10.1038/onc.2013.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Santillan DA, et al. Bromodomain and histone acetyltransferase domain specificities control mixed lineage leukemia phenotype. Cancer research. 2006;66:10032–10039. doi: 10.1158/0008-5472.CAN-06-2597. [DOI] [PubMed] [Google Scholar]
- 18.French CA, et al. BRD4-NUT fusion oncogene: a novel mechanism in aggressive carcinoma. Cancer research. 2003;63:304–307. [PubMed] [Google Scholar]
- 19.French CA, et al. BRD-NUT oncoproteins: a family of closely related nuclear proteins that block epithelial differentiation and maintain the growth of carcinoma cells. Oncogene. 2008;27:2237–2242. doi: 10.1038/sj.onc.1210852. [DOI] [PubMed] [Google Scholar]
- 20.Grayson AR, et al. MYC, a downstream target of BRD-NUT, is necessary and sufficient for the blockade of differentiation in NUT midline carcinoma. Oncogene. 2014;33:1736–1742. doi: 10.1038/onc.2013.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Delmore JE, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mertz JA, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:16669–16674. doi: 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lange M, et al. Regulation of muscle development by DPF3, a novel histone acetylation and methylation reader of the BAF chromatin remodeling complex. Genes & development. 2008;22:2370–2384. doi: 10.1101/gad.471408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeng L, et al. Mechanism and regulation of acetylated histone binding by the tandem PHD finger of DPF3b. Nature. 2010;466:258–262. doi: 10.1038/nature09139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y, Wen H, Xi Y, Tanaka K, Wang H, Peng D, Ren Y, Jin Q, Dent SYR, Li W, Li H, Shi X. AF9 YEATS Domain Links Histone Acetylation to Dot1L H3K9 Methylation. Cell. 2014 doi: 10.1016/j.cell.2014.09.049. (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bernt KM, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer cell. 2011;20:66–78. doi: 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhoumik A, Ronai Z. ATF2: a transcription factor that elicits oncogenic or tumor suppressor activities. Cell cycle. 2008;7:2341–2345. doi: 10.4161/cc.6388. [DOI] [PubMed] [Google Scholar]
- 28.Endo M, Su L, Nielsen TO. Activating transcription factor 2 in mesenchymal tumors. Human pathology. 2014;45:276–284. doi: 10.1016/j.humpath.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 29.Lv G, et al. MicroRNA-451 regulates activating transcription factor 2 expression and inhibits liver cancer cell migration. Oncology reports. 2014;32:1021–1028. doi: 10.3892/or.2014.3296. [DOI] [PubMed] [Google Scholar]
- 30.Shah M, et al. A role for ATF2 in regulating MITF and melanoma development. PLoS genetics. 2010;6:e1001258. doi: 10.1371/journal.pgen.1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Butler JS, Koutelou E, Schibler AC, Dent SY. Histone-modifying enzymes: regulators of developmental decisions and drivers of human disease. Epigenomics. 2012;4:163–177. doi: 10.2217/epi.12.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McMahon SB, Van Buskirk HA, Dugan KA, Copeland TD, Cole MD. The novel ATM-related protein TRRAP is an essential cofactor for the c-Myc and E2F oncoproteins. Cell. 1998;94:363–374. doi: 10.1016/s0092-8674(00)81479-8. [DOI] [PubMed] [Google Scholar]
- 33.McMahon SB, Wood MA, Cole MD. The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-Myc. Molecular and cellular biology. 2000;20:556–562. doi: 10.1128/mcb.20.2.556-562.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Molecular and cellular biology. 1999;19:1–11. doi: 10.1128/mcb.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flinn EM, et al. Recruitment of Gcn5-containing complexes during c-Myc-dependent gene activation. Structure and function aspects. The Journal of biological chemistry. 2002;277:23399–23406. doi: 10.1074/jbc.M201704200. [DOI] [PubMed] [Google Scholar]
- 36.Liu X, Tesfai J, Evrard YA, Dent SY, Martinez E. c-Myc transformation domain recruits the human STAGA complex and requires TRRAP and GCN5 acetylase activity for transcription activation. The Journal of biological chemistry. 2003;278:20405–20412. doi: 10.1074/jbc.M211795200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kenneth NS, et al. TRRAP and GCN5 are used by c-Myc to activate RNA polymerase III transcription. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:14917–14922. doi: 10.1073/pnas.0702909104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang N, et al. MYC interacts with the human STAGA coactivator complex via multivalent contacts with the GCN5 and TRRAP subunits. Biochimica et biophysica acta. 2014;1839:395–405. doi: 10.1016/j.bbagrm.2014.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Patel JH, et al. The c-MYC oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Molecular and cellular biology. 2004;24:10826–10834. doi: 10.1128/MCB.24.24.10826-10834.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen L, et al. Lysine acetyltransferase GCN5 potentiates the growth of non-small cell lung cancer via promotion of E2F1, cyclin D1, and cyclin E1 expression. The Journal of biological chemistry. 2013;288:14510–14521. doi: 10.1074/jbc.M113.458737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu Q, Kamps MP. Heterodimerization of Hox proteins with Pbx1 and oncoprotein E2a-Pbx1 generates unique DNA-binding specifities at nucleotides predicted to contact the N-terminal arm of the Hox homeodomain--demonstration of Hox-dependent targeting of E2a-Pbx1 in vivo. Oncogene. 1997;14:75–83. doi: 10.1038/sj.onc.1200799. [DOI] [PubMed] [Google Scholar]
- 42.Holmlund T, Lindberg MJ, Grander D, Wallberg AE. GCN5 acetylates and regulates the stability of the oncoprotein E2A-PBX1 in acute lymphoblastic leukemia. Leukemia. 2013;27:578–585. doi: 10.1038/leu.2012.265. [DOI] [PubMed] [Google Scholar]
- 43.Chen J, et al. Pygo2 associates with MLL2 histone methyltransferase and GCN5 histone acetyltransferase complexes to augment Wnt target gene expression and breast cancer stem-like cell expansion. Molecular and cellular biology. 2010;30:5621–5635. doi: 10.1128/MCB.00465-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang YL, Faiola F, Xu M, Pan S, Martinez E. Human ATAC Is a GCN5/PCAF-containing acetylase complex with a novel NC2-like histone fold module that interacts with the TATA-binding protein. The Journal of biological chemistry. 2008;283:33808–33815. doi: 10.1074/jbc.M806936200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krebs AR, Karmodiya K, Lindahl-Allen M, Struhl K, Tora L. SAGA and ATAC histone acetyl transferase complexes regulate distinct sets of genes and ATAC defines a class of p300-independent enhancers. Molecular cell. 2011;44:410–423. doi: 10.1016/j.molcel.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang XJ, Ogryzko VV, Nishikawa J, Howard BH, Nakatani Y. A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature. 1996;382:319–324. doi: 10.1038/382319a0. [DOI] [PubMed] [Google Scholar]
- 47.Nagy Z, et al. The metazoan ATAC and SAGA coactivator HAT complexes regulate different sets of inducible target genes. Cellular and molecular life sciences : CMLS. 2010;67:611–628. doi: 10.1007/s00018-009-0199-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu W, Edmondson DG, Roth SY. Mammalian GCN5 and P/CAF acetyltransferases have homologous amino-terminal domains important for recognition of nucleosomal substrates. Molecular and cellular biology. 1998;18:5659–5669. doi: 10.1128/mcb.18.10.5659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nagy Z, Tora L. Distinct GCN5/PCAF-containing complexes function as co-activators and are involved in transcription factor and global histone acetylation. Oncogene. 2007;26:5341–5357. doi: 10.1038/sj.onc.1210604. [DOI] [PubMed] [Google Scholar]
- 50.Yamauchi T, et al. Distinct but overlapping roles of histone acetylase PCAF and of the closely related PCAF-B/GCN5 in mouse embryogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:11303–11306. doi: 10.1073/pnas.97.21.11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu W, et al. Loss of Gcn5l2 leads to increased apoptosis and mesodermal defects during mouse development. Nature genetics. 2000;26:229–232. doi: 10.1038/79973. [DOI] [PubMed] [Google Scholar]
- 52.Sakaguchi K, et al. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes & development. 1998;12:2831–2841. doi: 10.1101/gad.12.18.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu L, et al. p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Molecular and cellular biology. 1999;19:1202–1209. doi: 10.1128/mcb.19.2.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin R, et al. Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth. Molecular cell. 2013;51:506–518. doi: 10.1016/j.molcel.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Malatesta M, et al. Histone acetyltransferase PCAF is required for Hedgehog-Gli-dependent transcription and cancer cell proliferation. Cancer research. 2013;73:6323–6333. doi: 10.1158/0008-5472.CAN-12-4660. [DOI] [PubMed] [Google Scholar]
- 56.Nye MD, et al. The Transcription Factor GLI1 Interacts with SMAD Proteins to Modulate Transforming Growth Factor beta-Induced Gene Expression in a p300/CREB-binding Protein-associated Factor (PCAF)-dependent Manner. The Journal of biological chemistry. 2014;289:15495–15506. doi: 10.1074/jbc.M113.545194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gong AY, et al. miR-17-5p targets the p300/CBP-associated factor and modulates androgen receptor transcriptional activity in cultured prostate cancer cells. BMC cancer. 2012;12:492. doi: 10.1186/1471-2407-12-492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ge X, Jin Q, Zhang F, Yan T, Zhai Q. PCAF acetylates {beta}-catenin and improves its stability. Molecular biology of the cell. 2009;20:419–427. doi: 10.1091/mbc.E08-08-0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chrivia JC, et al. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- 60.Stein RW, Corrigan M, Yaciuk P, Whelan J, Moran E. Analysis of E1A-mediated growth regulation functions: binding of the 300-kilodalton cellular product correlates with E1A enhancer repression function and DNA synthesis-inducing activity. Journal of virology. 1990;64:4421–4427. doi: 10.1128/jvi.64.9.4421-4427.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Arany Z, Sellers WR, Livingston DM, Eckner R. E1A-associated p300 and CREB-associated CBP belong to a conserved family of coactivators. Cell. 1994;77:799–800. doi: 10.1016/0092-8674(94)90127-9. [DOI] [PubMed] [Google Scholar]
- 62.Shi D, et al. CBP and p300 are cytoplasmic E4 polyubiquitin ligases for p53. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:16275–16280. doi: 10.1073/pnas.0904305106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhong J, et al. p300 acetyltransferase regulates androgen receptor degradation and PTEN-deficient prostate tumorigenesis. Cancer research. 2014;74:1870–1880. doi: 10.1158/0008-5472.CAN-13-2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bedford DC, Kasper LH, Fukuyama T, Brindle PK. Target gene context influences the transcriptional requirement for the KAT3 family of CBP and p300 histone acetyltransferases. Epigenetics : official journal of the DNA Methylation Society. 2010;5:9–15. doi: 10.4161/epi.5.1.10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rebel VI, et al. Distinct roles for CREB-binding protein and p300 in hematopoietic stem cell self-renewal. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:14789–14794. doi: 10.1073/pnas.232568499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen R, et al. The acetylase/deacetylase couple CREB-binding protein/Sirtuin 1 controls hypoxia-inducible factor 2 signaling. The Journal of biological chemistry. 2012;287:30800–30811. doi: 10.1074/jbc.M111.244780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gu J, Milligan J, Huang LE. Molecular mechanism of hypoxia-inducible factor 1alpha -p300 interaction. A leucine-rich interface regulated by a single cysteine. The Journal of biological chemistry. 2001;276:3550–3554. doi: 10.1074/jbc.M009522200. [DOI] [PubMed] [Google Scholar]
- 68.Ianculescu I, Wu DY, Siegmund KD, Stallcup MR. Selective roles for cAMP response element-binding protein binding protein and p300 protein as coregulators for androgen-regulated gene expression in advanced prostate cancer cells. The Journal of biological chemistry. 2012;287:4000–4013. doi: 10.1074/jbc.M111.300194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Santer FR, et al. Inhibition of the acetyltransferases p300 and CBP reveals a targetable function for p300 in the survival and invasion pathways of prostate cancer cell lines. Molecular cancer therapeutics. 2011;10:1644–1655. doi: 10.1158/1535-7163.MCT-11-0182. [DOI] [PubMed] [Google Scholar]
- 70.Miller RW, Rubinstein JH. Tumors in Rubinstein-Taybi syndrome. American journal of medical genetics. 1995;56:112–115. doi: 10.1002/ajmg.1320560125. [DOI] [PubMed] [Google Scholar]
- 71.Zimmer SN, et al. Mice heterozygous for CREB binding protein are hypersensitive to gamma-radiation and invariably develop myelodysplastic/myeloproliferative neoplasm. Experimental hematology. 2012;40:295–306. e295. doi: 10.1016/j.exphem.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gui Y, et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nature genetics. 2011;43:875–878. doi: 10.1038/ng.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pasqualucci L, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471:189–195. doi: 10.1038/nature09730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mullighan CG, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 2011;471:235–239. doi: 10.1038/nature09727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ionov Y, Matsui S, Cowell JK. A role for p300/CREB binding protein genes in promoting cancer progression in colon cancer cell lines with microsatellite instability. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:1273–1278. doi: 10.1073/pnas.0307276101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang F, Marshall CB, Ikura M. Transcriptional/epigenetic regulator CBP/p300 in tumorigenesis: structural and functional versatility in target recognition. Cellular and molecular life sciences : CMLS. 2013;70:3989–4008. doi: 10.1007/s00018-012-1254-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pattabiraman DR, Sun J, Dowhan DH, Ishii S, Gonda TJ. Mutations in multiple domains of c-Myb disrupt interaction with CBP/p300 and abrogate myeloid transforming ability. Molecular cancer research : MCR. 2009;7:1477–1486. doi: 10.1158/1541-7786.MCR-09-0070. [DOI] [PubMed] [Google Scholar]
- 78.Pattabiraman DR, et al. Interaction of c-Myb with p300 is required for the induction of acute myeloid leukemia (AML) by human AML oncogenes. Blood. 2014;123:2682–2690. doi: 10.1182/blood-2012-02-413187. [DOI] [PubMed] [Google Scholar]
- 79.Gao Y, et al. Expression of p300 and CBP is associated with poor prognosis in small cell lung cancer. International journal of clinical and experimental pathology. 2014;7:760–767. [PMC free article] [PubMed] [Google Scholar]
- 80.Liao ZW, et al. High expression of p300 is linked to aggressive features and poor prognosis of nasopharyngeal carcinoma. Journal of translational medicine. 2012;10:110. doi: 10.1186/1479-5876-10-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rotte A, et al. Decreased expression of nuclear p300 is associated with disease progression and worse prognosis of melanoma patients. PloS one. 2013;8:75405. doi: 10.1371/journal.pone.0075405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Takeuchi A, et al. p300 mediates cellular resistance to doxorubicin in bladder cancer. Molecular medicine reports. 2012;5:173–176. doi: 10.3892/mmr.2011.593. [DOI] [PubMed] [Google Scholar]
- 83.Lee SO, et al. Interleukin-4 activates androgen receptor through CBP/p300. The Prostate. 2009;69:126–132. doi: 10.1002/pros.20865. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 84.Yoshida M, Miyoshi I, Hinuma Y. Isolation and characterization of retrovirus from cell lines of human adult T-cell leukemia and its implication in the disease. Proceedings of the National Academy of Sciences of the United States of America. 1982;79:2031–2035. doi: 10.1073/pnas.79.6.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bex F, Yin MJ, Burny A, Gaynor RB. Differential transcriptional activation by human T-cell leukemia virus type 1 Tax mutants is mediated by distinct interactions with CREB binding protein and p300. Molecular and cellular biology. 1998;18:2392–2405. doi: 10.1128/mcb.18.4.2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Suzuki T, Uchida-Toita M, Yoshida M. Tax protein of HTLV-1 inhibits CBP/p300-mediated transcription by interfering with recruitment of CBP/p300 onto DNA element of E-box or p53 binding site. Oncogene. 1999;18:4137–4143. doi: 10.1038/sj.onc.1202766. [DOI] [PubMed] [Google Scholar]
- 87.Eckner R, et al. Association of p300 and CBP with simian virus 40 large T antigen. Molecular and cellular biology. 1996;16:3454–3464. doi: 10.1128/mcb.16.7.3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Valls E, de la Cruz X, Martinez-Balbas MA. The SV40 T antigen modulates CBP histone acetyltransferase activity. Nucleic acids research. 2003;31:3114–3122. doi: 10.1093/nar/gkg418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ahuja D, Saenz-Robles MT, Pipas JM. SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene. 2005;24:7729–7745. doi: 10.1038/sj.onc.1209046. [DOI] [PubMed] [Google Scholar]
- 90.Saenz Robles MT, et al. Two independent regions of simian virus 40 T antigen increase CBP/p300 levels, alter patterns of cellular histone acetylation, and immortalize primary cells. Journal of virology. 2013;87:13499–13509. doi: 10.1128/JVI.02658-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Patel D, Huang SM, Baglia LA, McCance DJ. The E6 protein of human papillomavirus type 16 binds to and inhibits co-activation by CBP and p300. The EMBO journal. 1999;18:5061–5072. doi: 10.1093/emboj/18.18.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Arany Z, Newsome D, Oldread E, Livingston DM, Eckner R. A family of transcriptional adaptor proteins targeted by the E1A oncoprotein. Nature. 1995;374:81–84. doi: 10.1038/374081a0. [DOI] [PubMed] [Google Scholar]
- 93.Ferrari R, et al. Epigenetic reprogramming by adenovirus e1a. Science. 2008;321:1086–1088. doi: 10.1126/science.1155546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Inuzuka H, et al. Acetylation-dependent regulation of Skp2 function. Cell. 2012;150:179–193. doi: 10.1016/j.cell.2012.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ikura T, et al. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell. 2000;102:463–473. doi: 10.1016/s0092-8674(00)00051-9. [DOI] [PubMed] [Google Scholar]
- 96.Fuchs M, et al. The p400 complex is an essential E1A transformation target. Cell. 2001;106:297–307. doi: 10.1016/s0092-8674(01)00450-0. [DOI] [PubMed] [Google Scholar]
- 97.Kimura A, Horikoshi M. Tip60 acetylates six lysines of a specific class in core histones in vitro. Genes to cells : devoted to molecular & cellular mechanisms. 1998;3:789–800. doi: 10.1046/j.1365-2443.1998.00229.x. [DOI] [PubMed] [Google Scholar]
- 98.Legube G, et al. Tip60 is targeted to proteasome-mediated degradation by Mdm2 and accumulates after UV irradiation. The EMBO journal. 2002;21:1704–1712. doi: 10.1093/emboj/21.7.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sun Y, Jiang X, Chen S, Fernandes N, Price BD. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:13182–13187. doi: 10.1073/pnas.0504211102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sun Y, Jiang X, Price BD. Tip60: connecting chromatin to DNA damage signaling. Cell cycle. 2010;9:930–936. doi: 10.4161/cc.9.5.10931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sun Y, et al. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nature cell biology. 2009;11:1376–1382. doi: 10.1038/ncb1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kusch T, et al. Acetylation by Tip60 is required for selective histone variant exchange at DNA lesions. Science. 2004;306:2084–2087. doi: 10.1126/science.1103455. [DOI] [PubMed] [Google Scholar]
- 103.Van Den Broeck A, Nissou D, Brambilla E, Eymin B, Gazzeri S. Activation of a Tip60/E2F1/ERCC1 network in human lung adenocarcinoma cells exposed to cisplatin. Carcinogenesis. 2012;33:320–325. doi: 10.1093/carcin/bgr292. [DOI] [PubMed] [Google Scholar]
- 104.Sykes SM, et al. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Molecular cell. 2006;24:841–851. doi: 10.1016/j.molcel.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Molecular cell. 2006;24:827–839. doi: 10.1016/j.molcel.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 106.Jiang Z, et al. Tip60-mediated acetylation activates transcription independent apoptotic activity of Abl. Molecular cancer. 2011;10:88. doi: 10.1186/1476-4598-10-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Brady ME, et al. Tip60 is a nuclear hormone receptor coactivator. The Journal of biological chemistry. 1999;274:17599–17604. doi: 10.1074/jbc.274.25.17599. [DOI] [PubMed] [Google Scholar]
- 108.Gaughan L, Brady ME, Cook S, Neal DE, Robson CN. Tip60 is a co-activator specific for class I nuclear hormone receptors. The Journal of biological chemistry. 2001;276:46841–46848. doi: 10.1074/jbc.M103710200. [DOI] [PubMed] [Google Scholar]
- 109.Gaughan L, Logan IR, Cook S, Neal DE, Robson CN. Tip60 and histone deacetylase 1 regulate androgen receptor activity through changes to the acetylation status of the receptor. The Journal of biological chemistry. 2002;277:25904–25913. doi: 10.1074/jbc.M203423200. [DOI] [PubMed] [Google Scholar]
- 110.Jeong KW, et al. Recognition of enhancer element-specific histone methylation by TIP60 in transcriptional activation. Nature structural & molecular biology. 2011;18:1358–1365. doi: 10.1038/nsmb.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Xiao H, Chung J, Kao HY, Yang YC. Tip60 is a co-repressor for STAT3. The Journal of biological chemistry. 2003;278:11197–11204. doi: 10.1074/jbc.M210816200. [DOI] [PubMed] [Google Scholar]
- 112.Zhao H, Jin S, Gewirtz AM. The histone acetyltransferase TIP60 interacts with c-Myb and inactivates its transcriptional activity in human leukemia. The Journal of biological chemistry. 2012;287:925–934. doi: 10.1074/jbc.M111.279950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chevillard-Briet M, et al. Interplay between chromatin-modifying enzymes controls colon cancer progression through Wnt signaling. Human molecular genetics. 2014;23:2120–2131. doi: 10.1093/hmg/ddt604. [DOI] [PubMed] [Google Scholar]
- 114.Gorrini C, et al. Tip60 is a haplo-insufficient tumour suppressor required for an oncogene-induced DNA damage response. Nature. 2007;448:1063–1067. doi: 10.1038/nature06055. [DOI] [PubMed] [Google Scholar]
- 115.Harris AW, et al. The E mu-myc transgenic mouse. A model for high-incidence spontaneous lymphoma and leukemia of early B cells. The Journal of experimental medicine. 1988;167:353–371. doi: 10.1084/jem.167.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Halkidou K, et al. Expression of Tip60, an androgen receptor coactivator, and its role in prostate cancer development. Oncogene. 2003;22:2466–2477. doi: 10.1038/sj.onc.1206342. [DOI] [PubMed] [Google Scholar]
- 117.ME LL, et al. New p53 related genes in human tumors: significant downregulation in colon and lung carcinomas. Oncology reports. 2006;16:603–608. doi: 10.3892/or.16.3.603. [DOI] [PubMed] [Google Scholar]
- 118.Mattera L, et al. The p400/Tip60 ratio is critical for colorectal cancer cell proliferation through DNA damage response pathways. Oncogene. 2009;28:1506–1517. doi: 10.1038/onc.2008.499. [DOI] [PubMed] [Google Scholar]
- 119.Chen G, Cheng Y, Tang Y, Martinka M, Li G. Role of Tip60 in human melanoma cell migration, metastasis, and patient survival. The Journal of investigative dermatology. 2012;132:2632–2641. doi: 10.1038/jid.2012.193. [DOI] [PubMed] [Google Scholar]
- 120.Miyamoto N, et al. Tip60 is regulated by circadian transcription factor clock and is involved in cisplatin resistance. The Journal of biological chemistry. 2008;283:18218–18226. doi: 10.1074/jbc.M802332200. [DOI] [PubMed] [Google Scholar]
- 121.Shiota M, et al. Tip60 promotes prostate cancer cell proliferation by translocation of androgen receptor into the nucleus. The Prostate. 2010;70:540–554. doi: 10.1002/pros.21088. [DOI] [PubMed] [Google Scholar]
- 122.Duong MT, et al. Hbo1 is a cyclin E/CDK2 substrate that enriches breast cancer stem-like cells. Cancer research. 2013;73:5556–5568. doi: 10.1158/0008-5472.CAN-13-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Borrow J, et al. The translocation t(8;16)(p11;p13) of acute myeloid leukaemia fuses a putative acetyltransferase to the CREB-binding protein. Nature genetics. 1996;14:33–41. doi: 10.1038/ng0996-33. [DOI] [PubMed] [Google Scholar]
- 124.Kitabayashi I, Aikawa Y, Nguyen LA, Yokoyama A, Ohki M. Activation of AML1-mediated transcription by MOZ and inhibition by the MOZ-CBP fusion protein. The EMBO journal. 2001;20:7184–7196. doi: 10.1093/emboj/20.24.7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rauh D, et al. An acetylome peptide microarray reveals specificities and deacetylation substrates for all human sirtuin isoforms. Nature communications. 2013;4:2327. doi: 10.1038/ncomms3327. [DOI] [PubMed] [Google Scholar]
- 126.Choudhary C, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 127.Martinez-Balbas MA, Bauer UM, Nielsen SJ, Brehm A, Kouzarides T. Regulation of E2F1 activity by acetylation. The EMBO journal. 2000;19:662–671. doi: 10.1093/emboj/19.4.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Okumura K, et al. PCAF modulates PTEN activity. The Journal of biological chemistry. 2006;281:26562–26568. doi: 10.1074/jbc.M605391200. [DOI] [PubMed] [Google Scholar]
- 129.Thevenet L, et al. Regulation of human SRY subcellular distribution by its acetylation/deacetylation. The EMBO journal. 2004;23:3336–3345. doi: 10.1038/sj.emboj.7600352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Li M, Luo J, Brooks CL, Gu W. Acetylation of p53 inhibits its ubiquitination by Mdm2. The Journal of biological chemistry. 2002;277:50607–50611. doi: 10.1074/jbc.C200578200. [DOI] [PubMed] [Google Scholar]
- 131.Yu X, et al. Modulation of p53, ErbB1, ErbB2, and Raf-1 expression in lung cancer cells by depsipeptide FR901228. Journal of the National Cancer Institute. 2002;94:504–513. doi: 10.1093/jnci/94.7.504. [DOI] [PubMed] [Google Scholar]
- 132.Scroggins BT, et al. An acetylation site in the middle domain of Hsp90 regulates chaperone function. Molecular cell. 2007;25:151–159. doi: 10.1016/j.molcel.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hallows WC, Yu W, Denu JM. Regulation of glycolytic enzyme phosphoglycerate mutase-1 by Sirt1 protein-mediated deacetylation. The Journal of biological chemistry. 2012;287:3850–3858. doi: 10.1074/jbc.M111.317404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:10224–10229. doi: 10.1073/pnas.0603968103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhao S, et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Khan O, La Thangue NB. HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunology and cell biology. 2012;90:85–94. doi: 10.1038/icb.2011.100. [DOI] [PubMed] [Google Scholar]
- 137.Kalac M, et al. HDAC inhibitors and decitabine are highly synergistic and associated with unique gene-expression and epigenetic profiles in models of DLBCL. Blood. 2011;118:5506–5516. doi: 10.1182/blood-2011-02-336891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Santoro F, et al. A dual role for Hdac1: oncosuppressor in tumorigenesis, oncogene in tumor maintenance. Blood. 2013;121:3459–3468. doi: 10.1182/blood-2012-10-461988. [DOI] [PubMed] [Google Scholar]
- 139.Grant PA, et al. Yeast Gcn5 functions in two multisubunit complexes to acetylate nucleosomal histones: characterization of an Ada complex and the SAGA (Spt/Ada) complex. Genes & development. 1997;11:1640–1650. doi: 10.1101/gad.11.13.1640. [DOI] [PubMed] [Google Scholar]
- 140.Balasubramanian R, Pray-Grant MG, Selleck W, Grant PA, Tan S. Role of the Ada2 and Ada3 transcriptional coactivators in histone acetylation. The Journal of biological chemistry. 2002;277:7989–7995. doi: 10.1074/jbc.M110849200. [DOI] [PubMed] [Google Scholar]
- 141.Thompson PR, et al. Regulation of the p300 HAT domain via a novel activation loop. Nature structural & molecular biology. 2004;11:308–315. doi: 10.1038/nsmb740. [DOI] [PubMed] [Google Scholar]
- 142.Sun B, et al. Regulation of the histone acetyltransferase activity of hMOF via autoacetylation of Lys274. Cell research. 2011;21:1262–1266. doi: 10.1038/cr.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Santos-Rosa H, Valls E, Kouzarides T, Martinez-Balbas M. Mechanisms of P/CAF auto-acetylation. Nucleic acids research. 2003;31:4285–4292. doi: 10.1093/nar/gkg655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Herrera JE, Bergel M, Yang XJ, Nakatani Y, Bustin M. The histone acetyltransferase activity of human GCN5 and PCAF is stabilized by coenzymes. The Journal of biological chemistry. 1997;272:27253–27258. doi: 10.1074/jbc.272.43.27253. [DOI] [PubMed] [Google Scholar]
- 145.Chimenti F, et al. A novel histone acetyltransferase inhibitor modulating Gcn5 network: cyclopentylidene-[4-(4'-chlorophenyl)thiazol-2-yl)hydrazone. Journal of medicinal chemistry. 2009;52:530–536. doi: 10.1021/jm800885d. [DOI] [PubMed] [Google Scholar]
- 146.Trisciuoglio D, et al. CPTH6, a thiazole derivative, induces histone hypoacetylation and apoptosis in human leukemia cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:475–486. doi: 10.1158/1078-0432.CCR-11-0579. [DOI] [PubMed] [Google Scholar]
- 147.Secci D, et al. Synthesis of a novel series of thiazole-based histone acetyltransferase inhibitors. Bioorganic & medicinal chemistry. 2014;22:1680–1689. doi: 10.1016/j.bmc.2014.01.022. [DOI] [PubMed] [Google Scholar]
- 148.Lau OD, et al. HATs off: selective synthetic inhibitors of the histone acetyltransferases p300 and PCAF. Molecular cell. 2000;5:589–595. doi: 10.1016/s1097-2765(00)80452-9. [DOI] [PubMed] [Google Scholar]
- 149.Balasubramanyam K, Swaminathan V, Ranganathan A, Kundu TK. Small molecule modulators of histone acetyltransferase p300. The Journal of biological chemistry. 2003;278:19134–19140. doi: 10.1074/jbc.M301580200. [DOI] [PubMed] [Google Scholar]
- 150.Sung B, et al. Anacardic acid (6-nonadecyl salicylic acid), an inhibitor of histone acetyltransferase, suppresses expression of nuclear factor-kappaB-regulated gene products involved in cell survival, proliferation, invasion, and inflammation through inhibition of the inhibitory subunit of nuclear factor-kappaBalpha kinase, leading to potentiation of apoptosis. Blood. 2008;111:4880–4891. doi: 10.1182/blood-2007-10-117994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ghizzoni M, Boltjes A, Graaf C, Haisma HJ, Dekker FJ. Improved inhibition of the histone acetyltransferase PCAF by an anacardic acid derivative. Bioorganic & medicinal chemistry. 2010;18:5826–5834. doi: 10.1016/j.bmc.2010.06.089. [DOI] [PubMed] [Google Scholar]
- 152.Saadat N, Gupta SV. Potential role of garcinol as an anticancer agent. Journal of oncology. 2012;2012:647206. doi: 10.1155/2012/647206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Balasubramanyam K, et al. Polyisoprenylated benzophenone, garcinol, a natural histone acetyltransferase inhibitor, represses chromatin transcription and alters global gene expression. The Journal of biological chemistry. 2004;279:33716–33726. doi: 10.1074/jbc.M402839200. [DOI] [PubMed] [Google Scholar]
- 154.Arif M, et al. Mechanism of p300 specific histone acetyltransferase inhibition by small molecules. Journal of medicinal chemistry. 2009;52:267–277. doi: 10.1021/jm800657z. [DOI] [PubMed] [Google Scholar]
- 155.Ye X, et al. Garcinol, an acetyltransferase inhibitor, suppresses proliferation of breast cancer cell line MCF-7 promoted by 17beta-estradiol. Asian Pacific journal of cancer prevention : APJCP. 2014;15:5001–5007. doi: 10.7314/apjcp.2014.15.12.5001. [DOI] [PubMed] [Google Scholar]
- 156.Ahmad A, et al. Apoptosis-inducing effect of garcinol is mediated by NF-kappaB signaling in breast cancer cells. Journal of cellular biochemistry. 2010;109:1134–1141. doi: 10.1002/jcb.22492. [DOI] [PubMed] [Google Scholar]
- 157.Ahmad A, et al. Garcinol regulates EMT and Wnt signaling pathways in vitro and in vivo, leading to anticancer activity against breast cancer cells. Molecular cancer therapeutics. 2012;11:2193–2201. doi: 10.1158/1535-7163.MCT-12-0232-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yu SY, et al. Induction of p21(Waf1/Cip1) by garcinol via downregulation of p38-MAPK signaling in p53-independent H1299 lung cancer. Journal of agricultural and food chemistry. 2014;62:2085–2095. doi: 10.1021/jf4037722. [DOI] [PubMed] [Google Scholar]
- 159.Ahmad A, et al. Garcinol-induced apoptosis in prostate and pancreatic cancer cells is mediated by NF- kappaB signaling. Frontiers in bioscience. 2011;3:1483–1492. doi: 10.2741/e349. [DOI] [PubMed] [Google Scholar]
- 160.Sethi G, et al. Inhibition of STAT3 dimerization and acetylation by garcinol suppresses the growth of human hepatocellular carcinoma in vitro and in vivo. Molecular cancer. 2014;13:66. doi: 10.1186/1476-4598-13-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Li F, et al. Garcinol, a polyisoprenylated benzophenone modulates multiple proinflammatory signaling cascades leading to the suppression of growth and survival of head and neck carcinoma. Cancer prevention research. 2013;6:843–854. doi: 10.1158/1940-6207.CAPR-13-0070. [DOI] [PubMed] [Google Scholar]
- 162.Stimson L, et al. Isothiazolones as inhibitors of PCAF and p300 histone acetyltransferase activity. Molecular cancer therapeutics. 2005;4:1521–1532. doi: 10.1158/1535-7163.MCT-05-0135. [DOI] [PubMed] [Google Scholar]
- 163.Zheng Y, et al. Synthesis and evaluation of a potent and selective cell-permeable p300 histone acetyltransferase inhibitor. Journal of the American Chemical Society. 2005;127:17182–17183. doi: 10.1021/ja0558544. [DOI] [PubMed] [Google Scholar]
- 164.Marcu MG, et al. Curcumin is an inhibitor of p300 histone acetylatransferase. Medicinal chemistry. 2006;2:169–174. doi: 10.2174/157340606776056133. [DOI] [PubMed] [Google Scholar]
- 165.Balasubramanyam K, et al. Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. The Journal of biological chemistry. 2004;279:51163–51171. doi: 10.1074/jbc.M409024200. [DOI] [PubMed] [Google Scholar]
- 166.Shah S, Prasad S, Knudsen KE. Targeting pioneering factor and hormone receptor cooperative pathways to suppress tumor progression. Cancer research. 2012;72:1248–1259. doi: 10.1158/0008-5472.CAN-11-0943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Teiten MH, et al. Anti-proliferative potential of curcumin in androgen-dependent prostate cancer cells occurs through modulation of the Wingless signaling pathway. International journal of oncology. 2011;38:603–611. doi: 10.3892/ijo.2011.905. [DOI] [PubMed] [Google Scholar]
- 168.Emami KH, et al. A small molecule inhibitor of beta-catenin/CREB-binding protein transcription [corrected] Proceedings of the National Academy of Sciences of the United States of America. 2004;101:12682–12687. doi: 10.1073/pnas.0404875101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Gang EJ, et al. Small-molecule inhibition of CBP/catenin interactions eliminates drug-resistant clones in acute lymphoblastic leukemia. Oncogene. 2014;33:2169–2178. doi: 10.1038/onc.2013.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lu H, et al. Hypoxia-inducible factor-1alpha blocks differentiation of malignant gliomas. The FEBS journal. 2009;276:7291–7304. doi: 10.1111/j.1742-4658.2009.07441.x. [DOI] [PubMed] [Google Scholar]
- 171.Bowers EM, et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chemistry & biology. 2010;17:471–482. doi: 10.1016/j.chembiol.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Gao XN, et al. A histone acetyltransferase p300 inhibitor C646 induces cell cycle arrest and apoptosis selectively in AML1-ETO-positive AML cells. PloS one. 2013;8:e55481. doi: 10.1371/journal.pone.0055481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Yan G, et al. Selective inhibition of p300 HAT blocks cell cycle progression, induces cellular senescence, and inhibits the DNA damage response in melanoma cells. The Journal of investigative dermatology. 2013;133:2444–2452. doi: 10.1038/jid.2013.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Liu Y, et al. Inhibition of p300 impairs Foxp3(+) T regulatory cell function and promotes antitumor immunity. Nature medicine. 2013;19:1173–1177. doi: 10.1038/nm.3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ravindra KC, et al. Inhibition of lysine acetyltransferase KAT3B/p300 activity by a naturally occurring hydroxynaphthoquinone, plumbagin. The Journal of biological chemistry. 2009;284:24453–24464. doi: 10.1074/jbc.M109.023861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Tohyama S, et al. Discovery and characterization of NK13650s, naturally occurring p300-selective histone acetyltransferase inhibitors. The Journal of organic chemistry. 2012;77:9044–9052. doi: 10.1021/jo301534b. [DOI] [PubMed] [Google Scholar]
- 177.Yin S, et al. Arylsulfonamide KCN1 inhibits in vivo glioma growth and interferes with HIF signaling by disrupting HIF-1alpha interaction with cofactors p300/CBP. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:6623–6633. doi: 10.1158/1078-0432.CCR-12-0861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Yang H, et al. Small-molecule inhibitors of acetyltransferase p300 identified by high-throughput screening are potent anticancer agents. Molecular cancer therapeutics. 2013;12:610–620. doi: 10.1158/1535-7163.MCT-12-0930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kushal S, et al. Protein domain mimetics as in vivo modulators of hypoxia-inducible factor signaling. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:15602–15607. doi: 10.1073/pnas.1312473110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hao J, et al. Selective small molecule targeting beta-catenin function discovered by in vivo chemical genetic screen. Cell reports. 2013;4:898–904. doi: 10.1016/j.celrep.2013.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Xie X, et al. Targeting HPV16 E6-p300 interaction reactivates p53 and inhibits the tumorigenicity of HPV-positive head and neck squamous cell carcinoma. Oncogene. 2014;33:1037–1046. doi: 10.1038/onc.2013.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Ghizzoni M, et al. 6-alkylsalicylates are selective Tip60 inhibitors and target the acetyl-CoA binding site. European journal of medicinal chemistry. 2012;47:337–344. doi: 10.1016/j.ejmech.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Sun Y, Jiang X, Chen S, Price BD. Inhibition of histone acetyltransferase activity by anacardic acid sensitizes tumor cells to ionizing radiation. FEBS letters. 2006;580:4353–4356. doi: 10.1016/j.febslet.2006.06.092. [DOI] [PubMed] [Google Scholar]
- 184.Kobayashi J, Kato A, Ota Y, Ohba R, Komatsu K. Bisbenzamidine derivative, pentamidine represses DNA damage response through inhibition of histone H2A acetylation. Molecular cancer. 2010;9:34. doi: 10.1186/1476-4598-9-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Coffey K, et al. Characterisation of a Tip60 specific inhibitor, NU9056, in prostate cancer. PloS one. 2012;7:e45539. doi: 10.1371/journal.pone.0045539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Gao C, et al. Rational design and validation of a Tip60 histone acetyltransferase inhibitor. Scientific reports. 2014;4:5372. doi: 10.1038/srep05372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Prinjha RK, Witherington J, Lee K. Place your BETs: the therapeutic potential of bromodomains. Trends in pharmacological sciences. 2012;33:146–153. doi: 10.1016/j.tips.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 188.Loven J, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Gao L, et al. Androgen receptor promotes ligand-independent prostate cancer progression through c-Myc upregulation. PloS one. 2013;8:e63563. doi: 10.1371/journal.pone.0063563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nature reviews. Drug discovery. 2014;13:337–356. doi: 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- 191.Fiskus W, et al. Highly active combination of BRD4 antagonist and histone deacetylase inhibitor against human acute myelogenous leukemia cells. Molecular cancer therapeutics. 2014;13:1142–1154. doi: 10.1158/1535-7163.MCT-13-0770. [DOI] [PubMed] [Google Scholar]
- 192.Kurdistani SK. Histone modifications as markers of cancer prognosis: a cellular view. British journal of cancer. 2007;97:1–5. doi: 10.1038/sj.bjc.6603844. [DOI] [PMC free article] [PubMed] [Google Scholar]