

Abstract
Cellular inhibitor of apoptosis proteins 1 and 2 (c-IAP1/2) play central roles in signal transduction mediated by numerous receptors that participate in inflammatory and immune responses. In certain pathways, such as activation of NF-κB, their degradation is a major regulatory event and is physiologically induced by activation of receptors. Additionally, a number of synthetic compounds have been developed that also target the c-IAPs and induce their degradation. However, the extent of a synthetic IAP antagonist's ability to mirror the transcriptional program by a physiological signal remains unclear. Here we take a systems approach to compare the transcriptional programs triggered by activation of CD30, a well-characterized receptor previously shown to induce the degradation of the c-IAPs, to SM-164, a synthetic IAP antagonist that specifically triggers c-IAP degradation. Employing a technique that allows the specific analysis of newly transcribed RNA, we have generated comparative transcriptome profiles for CD30 activation and SM-164 treatment. Analysis of these profiles revealed that the genes regulated by each stimulus were not completely shared, indicating novel functions of IAP antagonists and consequences of c-IAP1/2 degradation. The data identified a role for c-IAP1/2 in the regulation of the ribosome and protein synthesis, which was subsequently confirmed by biological assays. These findings expand our knowledge of the roles of c-IAP1/2 in signaling and provide insight into the mechanism of synthetic IAP antagonists, furthering our understanding of their therapeutic potential.
Keywords: CD30, Smac mimetic, IAP antagonist, NF-κB, transcriptome, translation
Introduction
The inhibitor of apoptosis (IAP) proteins are central mediators of a divergent group of cellular signaling pathways, largely involved in immune and inflammatory responses (1). Two of these proteins, cellular inhibitor of apoptosis proteins 1 and 2 (c-IAP1/2), have been implicated as key regulators of NF-κB, a protein family comprised of five different members that dimerize to form active transcription factors (2, 3). The best characterized NF-κB dimers are p65:p50 and p52:RelB, which are often referred to as canonical and non-canonical NF-κB, respectively (4).
The canonical and non-canonical NF-κB pathways are activated by members of the tumor necrosis factor (TNF) receptor superfamily and play important roles in the immune response, inflammation, and cancer (5, 6). While c-IAP1/2 have a demonstrated role in the activation of canonical NF-κB following TNF treatment, their degradation is required to activate the non-canonical NF-κB pathway, a signaling cascade that physiologically occurs following activation of a limited subset of the TNF receptor superfamily that includes CD30, CD40, and TNFR2 (7-9). Prior to stimulation, c-IAP1/2 form a complex with TNF receptor associated factor (TRAF) 2 and TRAF3, and this complex is thought to bind and degrade the constitutively expressed NF-κB-inducing kinase (NIK) (10). Following ligand binding, the receptor recruits the TRAF:c-IAP1/2 complex through the direct binding of the TRAFs. This interaction subsequently triggers the degradation of c-IAP1/2 and results in the gradual accumulation of NIK. NIK, in turn, begins a signaling cascade that leads to the phosphorylation and processing of the NF-κB precursor p100 to the active NF-κB subunit p52. The p52 moiety can dimerize with RelB to form the non-canonical NF-κB transcription factor that subsequently regulates an incompletely defined list of genes involved in multiple cellular processes, including the immune response (3, 4, 10, 11).
The c-IAP1/2 degradation event can be recreated experimentally using a class of synthetic, small molecule compounds known as Smac mimetics (SMs) or IAP antagonists (12-14). These compounds are structurally based on the IAP binding motif (IBM) of a physiological binding partner of the IAPs known as second mitochondria-derived activator of caspase/direct inhibitor of apoptosis-binding protein with low isoelectric point (Smac/DIABLO). These compounds bind to c-IAP1/2 and trigger their autoubiquitination and subsequent degradation, resulting in the processing of p100 to p52 and the activation of non-canonical NF-κB (11-15). Additionally, SM treatment has been shown to activate canonical NF-κB under some circumstances (12, 13). While SMs have been shown to replicate aspects of receptor activation, the extent of the functional overlap between the two classes of stimuli remains unclear.
In the present study, we have taken a systems approach to extensively compare the downstream effects of physiological and synthetic IAP antagonism. We utilized a cellular system in which both receptor activation and SM treatment leads to the degradation of c-IAP1/2 and results in canonical and non-canonical NF-κB activation. While stimulation of the receptor also induced additional signaling pathways, such as JNK and ERK, SM treatment did not, demonstrating that these pathways were independent of c-IAP1/2. To characterize the transcriptional consequences of IAP antagonism-induced NF-κB activation, we used Bru-seq, a technique that we have developed that specifically analyzes newly transcribed RNA, to identify gene expression profiles following each stimulus. While overlapping, the resulting transcriptome profiles revealed differences between the two modes of IAP antagonism. Furthermore, analysis of the transcriptome data revealed novel functions of c-IAP1/2 degradation and sequelae of SM treatment. One novel consequence of c-IAP1/2 degradation was the decreased expression of genes related to the ribosome and translation. In support of this finding, biological assays found that SM treatment resulted in decreased protein synthesis. These findings identify novel functions for the c-IAPs, and provide insight into the mechanism of SMs and their therapeutic potential.
Results
Smac mimetic treatment models CD30-mediated c-IAP1/2 degradation and NF-κB activation
To evaluate the consequences of c-IAP degradation, the effects of a Smac mimetic were compared to that of a cell surface receptor previously shown to induce the degradation of the c-IAPs upon activation. CD30 is a member of the TNF receptor superfamily that binds and degrades c-IAP1/2 as part of a larger complex. CD30 is highly expressed on the surface of certain lymphoma and leukemia cells, including anaplastic large cell lymphoma (ALCL) and Hodgkin's lymphoma cells (16, 17). In healthy individuals, CD30 expression is restricted to a small subset of activated T and B cells, and while its physiological role remains poorly defined, we have previously documented its ability to degrade c-IAP1/2 following activation (11). Using a cell-based system for CD30 stimulation and the ALCL cell lines Karpas 299 and Michel (18), the consequences of Smac mimetic (SM) treatment and CD30 activation were compared. Both stimuli resulted in the degradation of c-IAP1 and c-IAP2 (Fig. 1A, 2A) and induced the processing of p100 to p52, a widely used marker of non-canonical NF-κB activation (Fig. 1B, 2A). Both CD30 stimulation and SM treatment resulted in nuclear translocation of DNA-binding canonical and non-canonical NF-κB, as detected by electrophoretic mobility shift assay (EMSA) and confirmed by supershift analysis (Fig. 1C), indicating that SM treatment models CD30-mediated NF-κB activation. While SM treatment has previously been shown to induce autocrine TNF production (12, 13), pretreatment with the TNF inhibitor Enbrel did not prevent the NF-κB activation (Fig. 1D), indicating that SM-induced NF-κB was independent of TNF. In addition to NF-κB activation, members of the TNFR superfamily can trigger other signaling cascades, including the JNK and ERK pathways (19). To test if SM treatment and CD30 stimulation activated these pathways, Karpas 299 and Michel cells were briefly treated with SM or stimulated with CD30L, and phosphorylation of JNK and ERK was assessed. While receptor stimulation activated these pathways, SM treatment did not (Fig. 1E). Notably, inhibition of JNK and ERK did not affect NF-κB activation, indicating that this occurred independently of CD30-mediated JNK and ERK signaling (Fig. 1F, S1A-C). Collectively, these results demonstrate that the ability of SM treatment to model receptor signaling is limited to activation of NF-κB.
Figure 1. Smac mimetic treatment models CD30-mediated NF-κB activation.

A. Karpas 299 cells were treated with 100 nM SM-164 or exposed to CHO cells expressing CD30L for 3 h. Whole cell lysates were prepared, and c-IAP1/2 degradation was assessed by Western blot. B. Karpas 299 cells were stimulated as in A. Whole cell lysates were prepared, and p100 processing was analyzed by Western blot. Bands marked with an asterisk (*) are non-specific. C. Karpas 299 cells were treated as in A, nuclear extracts were collected, and NF-κB activation was analyzed by EMSA. To identify the NF-κB bands, the CD30L and SM-164 treated samples were incubated with antibodies against the indicated NF-κB subunits or GST as a control and used in a supershift assay. D. Karpas 299 cells were pretreated with the TNF inhibitor Enbrel at 10 μg/mL for 1 h. The cells were then treated with 100 nM SM1-64 for 3 h or 500 U/mL TNF for 30 min. Nuclear extracts were isolated, and NF-κB activation was analyzed by EMSA. E. Karpas 299 and Michel cells were incubated with CHO cells expressing CD30L or treated with 100 nM SM-164 for 15 min. Whole cell lysates were collected and the activation status of the indicated MAP kinase pathways was determined by Western blot. F. Karpas 299 cells were pretreated with 25 μM of the JNK inhibitor SP600125 and 50 nM of the MEK inhibitor Trametinib for 1h. The cells were then exposed to CD30L for the indicated times. Following treatment, nuclear extracts and whole cell lysates were collected from the samples. The nuclear extracts were used to assess NF-κB activation by EMSA. Can. NF-κB, canonical NF-κB; Non-can. NF-κB, non-canonical NF-κB; N.S., non-specific.
Figure 2. Kinetics of NF-κB induction by receptor activation and SM treatment.

A. Karpas 299 and Michel cells were exposed to CD30L-expressing CHO cells (A) or treated with 100 nM SM-164 (B) for the indicated times. Whole cell lysates were collected and used to assess p100 processing and c-IAP1/2 degradation by Western blot. Bands marked with an asterisk (*) are non-specific. B-D. Karpas 299 (B, C) or Michel (D) cells were exposed to CD30L or incubated with 100 nM SM-164 for the indicated times. Nuclear extracts were prepared and NF-κB activation was measured by EMSA.
Kinetics of NF-κB induction by receptor activation and SM treatment
As described above, both CD30 activation and SM treatment were found to trigger the degradation of c-IAP1/2 and to activate canonical and non-canonical NF-κB. However, it was unclear if these processes occurred with similar kinetics. Time course experiments were performed with each stimulus to examine this question. Degradation of c-IAP1/2 occurred rapidly following CD30 stimulation and SM treatment, and substantial loss of protein was observed within 15 min of treatment in both ALCL cell lines (Fig. 2A). Additionally, both stimuli resulted in p100 processing to the active non-canonical NF-κB subunit p52 at 3 h (Fig. 2A), consistent with previous reports describing the delayed kinetics of non-canonical NF-κB activation (10). Notably, CD30 appeared to be a stronger inducer of NF-κB than SM treatment (Fig. 2). It is unclear if this observation is due to the presence of less nuclear NF-κB or weaker DNA-binding activity of NF-κB following SM treatment. A non-canonical NF-κB nuclear protein complex was detected at 3 h by EMSA (Fig. 2B-D), indicating that CD30 stimulation and SM treatment activate non-canonical NF-κB with similar kinetics. Activation of canonical NF-κB, however, differed between the two stimuli. CD30-mediated canonical NF-κB occurred rapidly, being observed 15-30 min following receptor activation, and remained observable throughout the entire time course (Fig. 2B, 2D). Conversely, SM-induced canonical NF-κB was delayed and was initially observed at 1 h (Fig. 2C, 2D). Interestingly, the level of active canonical NF-κB triggered by SM peaked around 6 h before decreasing throughout the remaining time points. This decrease in canonical NF-κB coincided with activation of non-canonical NF-κB (Fig. 2C, 2D). CD30 activation and SM treatment degrade c-IAP1/2 and activate the same NF-κB pathways, albeit with differing rates, suggesting potential mechanistic and functional differences.
Comparison of gene regulation induced by physiological and synthetic IAP antagonism
While the ability of SMs to activate NF-κB has been previously reported (12, 13), it is not known if this resulted in the regulation of a similar gene expression profile as receptor signaling. To address this question, we used a recently developed technique, Bru-seq, that allows transcriptome analysis of newly transcribed RNA (20, 21). As depicted schematically in Figure 3A, Karpas 299 cells were treated with SM or exposed to CD30L, incubated with bromouridine, and the bromouridine-labeled RNA was isolated and converted into cDNA libraries for deep sequencing. In the initial analysis of the Bru-seq data, we compared the expression of two well-characterized NF-κB gene targets: BIRC3, which encodes the c-IAP2 protein, and NFKBIA, the gene encoding IκBα (11, 20, 22). The profiles of the mapped sequencing reads were similar for both stimuli (Fig. 3B, C), indicating that the stimuli produced similarly processed transcripts. Transcription of both genes was induced upon each treatment, with BIRC3 being the most highly transcribed gene in both cases (Table S1, S2). Compared to an unstimulated sample, transcription of BIRC3 was induced 12-fold following CD30 activation, while SM treatment resulted in a 7-fold increase in BIRC3 transcription (Fig. 3B). The Bru-seq results were mirrored by qRT-PCR experiments that also illustrated a more robust expression of genes following CD30 stimulation. Similar to BIRC3, expression of NFKBIA was also markedly higher following CD30L than SM (12-fold increase and 7-fold increase, respectively) (Fig. 3C, Table S1, S2). Since both stimuli degraded c-IAP1/2 to the same degree (Fig. 1A) and with similar rates (Fig. 2A, 2B), these results indicate that the receptor may provide additional signals that strengthen the magnitude of NF-κB activation. Interestingly, IL8, the gene encoding the interleukin-8 cytokine and a canonical NF-κB gene target (20), exhibited the second highest expression following CD30 stimulation (12-fold increase), but was not significantly affected by SM treatment (Fig. 3D, Table S1, S2) indicating different functional consequences of NF-κB activation by the two stimuli. To better visualize and compare the stimuli-induced transcriptomes, the change in transcription of genes following SM treatment was plotted against the change in transcription of genes following CD30 activation (Fig. 3E). While certain genes were affected by both stimuli, such as BIRC3 and NFKBIA, the transcription of other genes, like IL8, were modified by only one of the stimuli. Collectively, these data demonstrate that the transcriptional consequences of IAP antagonism by SM reflect aspects of receptor-induced signaling, while also providing evidence of functional differences between the forms of IAP antagonism.
Figure 3. Comparison of gene regulation induced by physiological and synthetic IAP antagonism.

A. Diagram of the Bru-seq procedure. Karpas 299 cells were exposed to CD30L or treated with 100 nM SM-164 for the indicated times. B-D. Sequencing reads from nascent RNA expressed as reads per thousand base pairs per 1 million reads (RPKM) and mapped to the BIRC3 gene (B), NFKBIA gene (C), and IL8 gene (D) with reference sequence annotation below. The exons and UTRs are denoted as black lines. The CD30L and SM-164 treated samples are shown in blue, and the unstimulated control samples are shown in yellow. For the qRT-PCR, Karpas 299 cells were exposed to CD30L or treated with 100 nM SM-164 for the indicated times. RNA was isolated and converted to cDNA, and the expression of the indicated genes was measured. E. The log2 fold change of genes from the SM-164 treated Bru-seq sample were plotted against the log2 fold change of genes from the CD30L Bru-seq sample. The location of BIRC3, NFKBIA, and IL8 are in blue, and the red dots represent the genes in the KEGG_RIBOSOME gene set.
Gene set analysis reveals novel roles for c-IAP1/2 and IAP antagonists
The initial analysis of the transcriptome profiles generated by each stimulus highlighted the variety of genes regulated by c-IAP1/2 degradation. Due to the initial complexity of classifying these genes with their wide functional diversity, we performed gene set enrichment analysis (GSEA) which allows for the identification of groups of genes that exhibit similar changes in expression using gene sets that have been categorized based on shared, biologically relevant characteristics, such as belonging to a common enzymatic pathway or presence in the same cellular compartment (23). GSEA has advantages over traditional strategies of gene expression analysis, including the ability to detect biologically significant processes involving groups of genes that show only modest changes in expression (23). Analysis of the GSEA data indicated that CD30 activation and SM treatment collectively modulated 256 gene sets (Fig. 4A). There were 119 CD30-specific gene sets identified (Fig. 4A, Table S3), and it was expected that CD30-specific gene sets would be identified since the receptor activated multiple pathways that were not activated by SM (Fig. 1). Examples of CD30-specific gene sets are shown in Figure 4B. Interestingly, 62 SM-specific gene sets were identified (Fig. 4A, Table S3) even though SM treatment was thought to mimic receptor signaling by degrading c-IAP1/2. This suggests that the SM may have additional, uncharacterized effects. These effects may be regulated by additional IAPs, such as X-linked inhibitor of apoptosis (XIAP), which can be antagonized by SMs (12-14). Intriguingly, the majority of the SM-specific gene sets were down-regulated by the compound and several gene sets were functionally related to metabolism and protein synthesis (Fig. 4C, Table S3).
Figure 4. Gene set analysis reveals novel roles for c-IAP1/2 and IAP antagonists.

A. A summary of the results from gene set enrichment analysis (GSEA) performed with the Bru-seq data. The numbers are gene sets modulated by the designated stimulus. The number of gene sets up and down-regulated are noted. B-D. Examples of gene sets with false discovery rates (FDR) < 0.05 that are regulated by CD30 alone (B), SM treatment alone (C), or regulated by both stimuli (D). The bars represent normalized enrichment score for the gene set.
The GSEA data identified 75 gene sets shared between CD30 activation and SM treatment (Fig. 4A). Many of these gene sets were expected, as they were related to functions shared between the two stimuli, such as regulation of NF-κB or involvement in TNFR2 and cell death signaling cascades (Fig. 4D, Table S3). It was unexpected that there would be gene sets significantly down-regulated by both stimuli. Functionally, many of these gene sets involved the regulation of the ribosome and translation, with SM treatment resulting in a more substantial down-regulation of the transcription of these genes and gene sets (Fig. 3E, 4D, Table S1, S2, S3). Experiments with a different Smac mimetic, Birinapant, did not affect the expression of ribosomal genes to the extent of SM-164 (Fig. S2A). However, Birinapant was a weaker inducer of c-IAP2 degradation and NF-κB activation (Fig. S2B), suggesting that the effect on ribosomal gene expression is dependent on efficient degradation of both c-IAP1 and c-IAP2, as well as the subsequent activation of NF-κB. Additionally, treatment with the caspase inhibitor z-VAD-fmk abrogated the SM-induced down-regulation of ribosomal genes (Fig. S3A), indicating a potential regulatory role for caspases in this process. While further work is needed to elucidate the exact mechanism, these analyses indicate that IAP antagonism by receptor activation or synthetic compound has diverse functional consequences, including newly identified potential roles in the regulation of ribosome biogenesis and protein synthesis.
IAP antagonism results in decreased protein synthesis
As described above, the GSEA results indicated novel roles for c-IAP1/2 in the regulation of genes related to the ribosome and translation. To test if the observed transcriptome results had a cellular consequence, protein synthesis was measured following CD30 activation and SM treatment using an assay based on incorporation of a methionine analogue (24, 25). Karpas 299 cells were treated with the designated stimulus and then incubated in L-methionine-free medium that had been supplemented with L-methionine or the methionine analogue L-homopropargylglycine (HPG). The incorporation of HPG was then assessed by flow cytometry. SM treatment resulted in a measurable decrease in protein synthesis by 3 h, and this decrease continued with longer treatment times (Fig. 5A-C, S3D). Importantly, the decrease in protein synthesis was initially observed prior to any substantial SM-induced cell death, but may be regulated by caspases (Fig. S3B, S3C). In contrast, CD30 activation had a minor effect on protein synthesis at early time points and only had a substantial effect by 24 h (Fig. 5D-F, S3E). These findings indicate that IAP antagonism, in the system tested, has an inhibitory impact on global protein synthesis, revealing a novel regulatory consequence for c-IAP1/2 degradation and SM treatment.
Figure 5. IAP antagonism results in decreased protein synthesis.
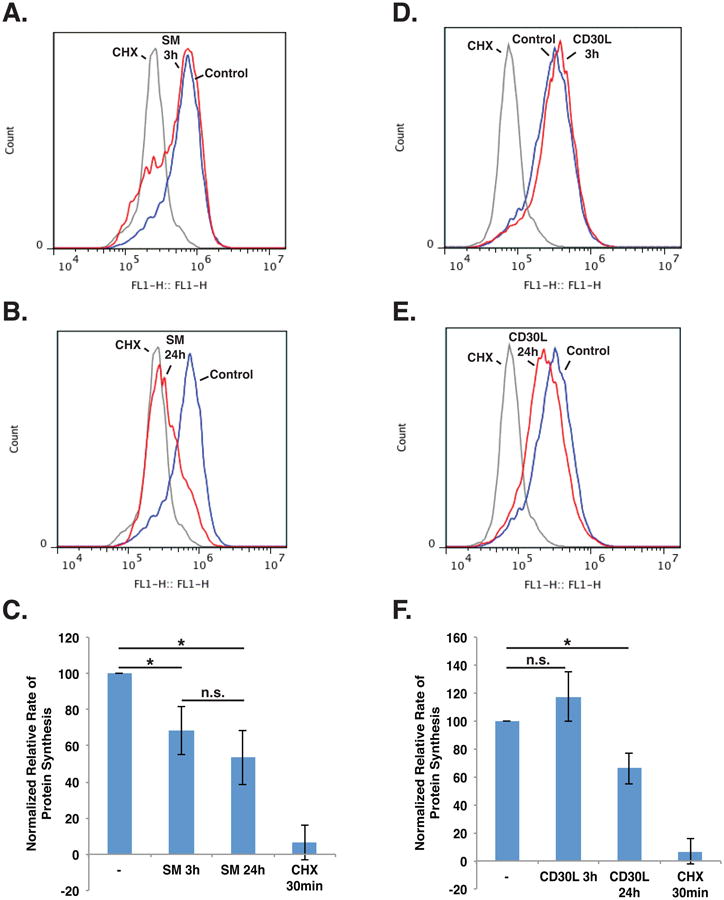
A-C. Karpas 299 cells were incubated with 100 nM SM-164 or 100 μg/mL cycloheximide (CHX) for the indicated times. The medium was then replaced with methionine-free medium supplemented with methionine or the methionine analogue HPG, and the cells were incubated for 1 h. HPG incorporation was measured by flow cytometry. D-F. Karpas 299 cells were exposed to CD30L or 100 μg/mL CHX for the indicated times. The medium was then replace with methionine-free medium supplemented with methionine or HPG, and the cells were incubated for 1 h. HPG incorporation was measured by flow cytometry. Data represent the mean ± standard deviation of at least three independent experiments (*P < 0.01).
Discussion
In this study, we examined the consequences of IAP antagonism on gene regulation by physiological and synthetic stimuli, identifying genes and pathways unique to each stimulus, as well as novel processes affected by both triggers of c-IAP1/2 degradation. One finding of particular interest was the down-regulation of genes related to the ribosome and translation. This observation was identified following SM treatment and CD30 activation, and resulted in decreased protein synthesis following both stimuli, though treatment with the Smac mimetic had an earlier observable effect on protein synthesis than CD30 stimulation, which only became apparent at 24 h. (Fig. 5). It has been previously well established that SMs induce death in certain cells, and that this killing is dependent on TNF (12, 13). However, this study provides evidence that suggests an additional mechanism of SM killing. In this model, SM triggers the degradation of c-IAP1/2 and results in the shutdown of protein synthesis, partially mimicking the effects of cycloheximide. The lethality of TNF would then be due, at least in part, to the inability to synthesize pro-survival proteins, similar to established mechanisms of TNF killing (26-28).
The consequences of CD30-mediated degradation of c-IAP1/2 differed from the IAP antagonism induced by SM treatment. CD30 activation did not exert as rapid an effect on protein synthesis as SM treatment. Furthermore, the decrease in protein synthesis was observed at 24 h, which coincided with previous reports of CD30-mediated cell cycle arrest at that time point (8, 29). Since there is an established connection between cell cycle arrest and decreased protein synthesis (30), this observation raises the intriguing possibility that c-IAP1/2 may regulate cell cycle arrest through their control of protein synthesis.
As noted above, CD30 and SM affected protein synthesis at different rates, and this may be due to several reasons, such as variable expression of CD30 on the cells, additional pathways activated by receptor stimulation such as MAPK pathways (Fig. 1), unique genes regulated by the receptor (Fig. 3, 4), or a combination thereof. Similarly, the unique consequences of SM treatment may contribute to the observed differences. One particular difference may be the mechanism of canonical NF-κB activation by CD30, which appears to differ from SM-induced canonical NF-κB. CD30, like its close TNFR superfamily relatives, is thought to activate NF-κB through its TRAF binding domains (29, 31, 32). However, it has also been shown that CD30 can activate NF-κB in the absence of its TRAF binding domains (32, 33), suggesting that CD30 may activate multiple NF-κB pathways with undefined functions. Less is known, however, about SM-induced canonical NF-κB. Since SM-induced canonical NF-κB appears to be due to c-IAP1/2 degradation, it may be reliant on the accumulation of NIK, a protein normally associated with non-canonical NF-κB activation but also has a reported ability to activate canonical NF-κB (34, 35). The potential mechanistic difference in canonical NF-κB activation is supported by the delayed kinetics of NF-κB activation after SM treatment (Fig. 2), possibly suggesting a reliance on the accumulation of NIK. Furthermore, SM treatment resulted in a weaker NF-κB signal (Fig. 2), potentially indicating additional mechanistic divergence from receptor signaling. Moreover, the canonical NF-κB activated by CD30 may be functionally distinct from the NF-κB activated by SM, potentially explaining the unique gene regulation by each stimulus.
In addition to differences in downstream gene targets, the mechanisms of receptor-induced and pharmacological IAP antagonism are intrinsically different. Receptor-mediated c-IAP1/2 degradation occurs in parallel with TRAF2 degradation (11, 36) and is dependent on translocation of the c-IAP:TRAF2 complex to an insoluble cellular fraction (8). In contrast, SMs directly bind to c-IAP1/2 and selectively trigger their autoubiquitination and subsequent degradation without degrading TRAF2 (11, 15, 37). This highlights another facet of SMs not shared with receptor signaling that may have downstream consequences, as it has been reported that TRAF2 overexpression can trigger p100 processing (11). Furthermore, SMs have been shown to inhibit XIAP. While SM-induced degradation of XIAP is not always observed (14), the SM may still bind and inhibit XIAP, a component in multiple signaling pathways (38, 39), potentially resulting in cellular consequences distinct from CD30 activation since CD30 is not thought to affect XIAP. Notably, caspase inhibition appeared to negate SM-induced ribosomal gene regulation (Fig. S3), potentially supporting a role for XIAP. In addition to providing insight into the functions of c-IAP1/2 in signaling, these findings highlight unique aspects of Smac mimetics that should be useful for defining their therapeutic value, including their regulation of protein synthesis.
Materials and Methods
Cell lines and culture conditions
Karpas 299 and Michel cells were grown in RPMI 1640 (Mediatech, Herndon, VA, USA) medium. The generation of the CD30L+ Chinese hamster ovary (CHO) cells has been previously described (8). CHO cells and CD30L+ CHO cells were cultured in F-12 nutrient medium (Gibco, Carlsbad, CA, USA). All cells were maintained at 37°C in an atmosphere of 5% CO2 and cultured in media supplemented with 10% FBS and 2 mL L-glutamine.
Materials
The following primary antibodies and materials were used in this study: anti-p100/p52 (Millipore); anti-c-IAP1 (Enzo Life Sciences, Farmingdale, NY, USA); anti-c-IAP2 (Cell Signaling, Danvers, MA, USA); anti-GST, anti-p65, and anti-RelB (Santa Cruz Biotechnology, San Diego, CA, USA); anti-phospho-JNK, anti-JNK, anti-phospho-ERK, and anti-ERK (Cell Signaling); anti-β-actin (Sigma-Aldrich, St. Louis, MO, USA); Birinapant (ChemieTek, Indianapolis, IN, USA); Enbrel (University of Michigan Hospital pharmacy); z-VAD-fmk (Cayman Chemical, Ann Arbor, MI, USA); Trametinib (LC Laboratories, Woburn, MA, USA); and SP600125 (SelleckChem, Houston, TX, USA). The Smac mimetic SM-164 (37) was a kind gift from Dr. Shaomeng Wang (University of Michigan).
CD30 stimulation
As described previously (8), Karpas 299 cells were exposed for the times indicated to either CHO cells (negative control) or CD30L+ CHO cells and were collected with gentle pipetting.
Cell lysate and nuclear extract preparation
Cells were treated as described in the figure legends. Following treatment, the cells were collected and washed in PBS. The cell suspension was then divided into tubes, and whole cell lysates and nuclear extracts were subsequently prepared. Whole cell lysates were prepared using RIPA lysis buffer in a process that has been described previously (22). Nuclear extracts were prepared from the cells as described before (39). The nuclear extracts were stored at -80°C.
Immunoblot analysis
Protein concentrations of whole cell lysates were determined using a Pierce BCA Protein Assay Kit (Thermo Scientific, Waltham, MA, USA). Lysates of equal protein concentrations were prepared in LDS sample buffer (Invitrogen, Carlsbad, CA, USA), separated on denaturing NuPAGE 4-12% polyacrylamide gradient gels (Invitrogen), and transferred to nitrocellulose membranes (GE Healthcare, Amersham, UK). Membranes were blocked in a 1:1 mixture of Odyssey blocking buffer (Li-Cor, Lincoln, NE, USA) and Tris-buffered saline (TBS). Membranes were then incubated with primary antibodies in a 1:1 mixture of Odyssey blocking buffer and TBS containing 0.1% Tween 20 (Fisher BioReagents, Waltham, MA, USA) overnight at 4°C. Following washing with TBS with 0.1% Tween 20, membranes were incubated with IRDye secondary antibodies (Li-Cor) for 1 h at room temperature. Membranes were then washed with TBS and analyzed using the Odyssey CLx infrared imaging system (Li-Cor) according to the manufacturer's instructions.
Electrophoretic mobility shift assays
Two complimentary oligonucleotides containing NF-κB consensus binding sites (5′-GATCCAGGGACTTTCCGCTGGGGACTTTCCA-3′ and 5′-GATCTGGAAAGTCCCCAGCGGAAAGTCCCTG-3′) were annealed and radiolabeled using T4 polynucleotide kinase (New England BioLabs, Ipswich, MA, USA) in the presence of [γ-32P] ATP. The radiolabeled probe was then purified using illustra Microspin G-25 Columns (GE Healthcare) according to the manufacturer's instructions. The presence of NF-κB in the nuclear extracts was assessed as described previously (39). Supershift assays were performed by adding 2 μL of antibodies against GST, p65, or RelB (Santa Cruz Biotechnologies) to the reaction mixture. Samples were incubated for 20 min at room temperature prior to running on the gel. For all assays, free probe was run off the gel to obtain maximum resolution. Autoradiography was conducted overnight at -20°C.
Quantitative real-time PCR
Cells were treated as indicated in the figure legends. Following treatment, the cells were washed with PBS, and total RNA was isolated using the RNeasy mini kit (Qiagen, Valencia, CA, USA) according to the manufacture's instructions. 1 μg of total RNA was converted to cDNA using a reverse transcription reaction with random hexamer primers and MultiScribe Reverse Transcriptase (Applied Biosystems, Carlsbad, CA, USA). 1 μL of the resulting cDNA was analyzed for the indicated target genes using the ViiA 7 Real-Time PCR System (Applied Biosystems). Each target assay was normalized to β-actin or 18S levels.
Transcriptome analysis by Bru-seq
Karpas 299 cells were incubated with the treatments indicated in the figure legends. To label nascent RNA, 2 mM bromouridine (Bru) was added to the media for the final 30 min of treatment time. The Bru-seq procedure has been previously described in detail (20, 21). Briefly, total RNA was collected from the treated cells using TRIzol reagent (Invitrogen), and the Bru-labeled, nascent RNA was isolated using anti-BrdU antibodies (BD Biosciences, San Jose, CA, USA) conjugated to magnetic beads (Invitrogen). The isolated RNA was converted into cDNA libraries, which were sequenced at the University of Michigan Sequencing Core using an Illumina HiSeq 2000 sequencer. The sequencing and read mapping were performed as previously described (20, 21). GSEA was used to identify up regulated and down-regulated gene sets by determining which associated genes were significantly enriched in each gene set (23). The log fold change in expression of genes greater than 1kb and expressed above 0.5 RPKM was used as the ranking metric by GSEA. The gene sets were obtained from version 4.0 of the Molecular Signatures Database (http://www.broadinstitute.org/gsea/msigdb/index.jsp). The gene sets used were canonical pathways (KEGG, Reactome, and BioCarta) and gene ontologies (biological processes, molecular functions, and cellular compartment). Gene sets with FDR corrected P-values lower than 0.01 were considered to be significantly enriched and were used in the analysis. The primary sequencing data files from this study will be submitted to the NCBI Gene Expression Omnibus upon acceptance of the manuscript.
Measurement of protein synthesis
Cells were stimulated as indicated in the figure legends. Following the designated times, the cells were collected, and centrifuged at 100 × g for 5 min. The supernatant was removed, and the cells were resuspended in L-methionine free RPMI (Invitrogen) supplemented with 1 mM L-methionine or HPG (50 μM final concentration), and were incubated at 37°C and 5% CO2 for 1 h. The cells were then harvested, washed with PBS, and fixed in 50% ice cold ethanol overnight at -20°C. The samples were then processed using the Click-iT HPG Alexa Fluor 488 Protein Synthesis Assay Kit (Life Technologies, Carlsbad, CA, USA) according to the manufacturer's instructions, and protein synthesis was measured by flow cytometry using an Accuri C6 flow cytometer (BD Biosciences). Relative rates of protein synthesis were calculated by normalizing the treated HPG sample to the control HPG sample after subtracting the auto-fluorescence background.
Viability assays
Following the indicated treatments, cells were harvested, washed with PBS and subsequently resuspended in PBS with 2 μg/mL propidium iodide (PI). The cell viability of the PI-stained cells was assessed by flow cytometry using an Accuri C6 flow cytometer (BD Biosciences).
Supplementary Material
Acknowledgments
We thank Drs. Shaomeng Wang (University of Michigan, Ann Arbor, MI, USA) for the kind gift of SM-164, Judith Sebolt-Leopold (University of Michigan) for the gift of Trametinib, Niall Kenneth (Newcastle University, Newcastle upon Tyne, UK) for his insightful suggestions, Elizabeth Lawlor (University of Michigan, Ann Arbor, MI, USA) for her critical reading of the manuscript, Thomas Wilson (University of Michigan, Ann Arbor, MI, USA) for use of his data analysis software, the members of the Duckett laboratory for their helpful advice, and the personnel at the University of Michigan Molecular and Behavioral Neuroscience Institute computing cluster and the University of Michigan Sequencing Core for their technical assistance. This work was supported in part by NIH R01CA142809 (to C.S.D.), NIH 1R01HG006786 (to M.L.), and NIH T32AI007413 (to A.J.K.).
SM-164 was a kind gift from Dr. Shaomeng Wang (University of Michigan, Ann Arbor, MI, USA).
Footnotes
Disclosure of potential conflicts of interest: The authors declare no conflicts of interest.
Author Contributions: Andrew Kocab and Colin Duckett designed the research. Andrew Kocab and Michelle Paulsen performed the research. Andrew Kocab, Artur Veloso, Mats Ljungman, and Colin Duckett analyzed the data. Andrew Kocab and Colin Duckett wrote the manuscript.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
References
- 1.Salvesen GS, Duckett CS. IAP proteins: blocking the road to death's door. Nat Rev Mol Cell Biol. 2002;3:401–410. doi: 10.1038/nrm830. [DOI] [PubMed] [Google Scholar]
- 2.Hayden MS, Ghosh S. Shared principles in NF-κB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 3.Smale ST. Dimer-specific regulatory mechanisms within the NF-κB family of transcription factors. Immunological Reviews. 2012;246:193–204. doi: 10.1111/j.1600-065X.2011.01091.x. [DOI] [PubMed] [Google Scholar]
- 4.Vallabhapurapu S, Karin M. Regulation and function of NF-κB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 5.DiDonato JA, Mercurio F, Karin M. NF-κB and the link between inflammation and cancer. Immunol Rev. 2012;246:379–400. doi: 10.1111/j.1600-065X.2012.01099.x. [DOI] [PubMed] [Google Scholar]
- 6.Perkins ND. The diverse and complex roles of NF-κB subunits in cancer. Nat Rev Cancer. 2012;12:121–132. doi: 10.1038/nrc3204. [DOI] [PubMed] [Google Scholar]
- 7.Hacker H, Tseng PH, Karin M. Expanding TRAF function: TRAF3 as a tri-faced immune regulator. Nat Rev Immunol. 2011;11:457–468. doi: 10.1038/nri2998. [DOI] [PubMed] [Google Scholar]
- 8.Wright CW, Rumble JM, Duckett CS. CD30 activates both the canonical and alternative NF-κB pathways in anaplastic large cell lymphoma cells. J Biol Chem. 2007;282:10252–10262. doi: 10.1074/jbc.M608817200. [DOI] [PubMed] [Google Scholar]
- 9.Wright CW, Duckett CS. The aryl hydrocarbon nuclear translocator alters CD30-mediated NF-κB-dependent transcription. Science. 2009;323:251–255. doi: 10.1126/science.1162818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vallabhapurapu S, Matsuzawa A, Zhang W, et al. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-κB signaling. Nat Immunol. 2008;9:1364–1370. doi: 10.1038/ni.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Csomos RA, Wright CW, Galban S, Oetjen KA, Duckett CS. Two distinct signalling cascades target the NF-κB regulatory factor c-IAP1 for degradation. Biochem J. 2009;420:83–91. doi: 10.1042/BJ20082140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Varfolomeev E, Blankenship JW, Wayson SM, et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis. Cell. 2007;131:669–681. doi: 10.1016/j.cell.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 13.Vince JE, Wong WW, Khan N, et al. IAP antagonists target cIAP1 to induce TNFα-dependent apoptosis. Cell. 2007;131:682–693. doi: 10.1016/j.cell.2007.10.037. [DOI] [PubMed] [Google Scholar]
- 14.Lu J, Bai L, Sun H, et al. SM-164: a novel, bivalent Smac mimetic that induces apoptosis and tumor regression by concurrent removal of the blockade of cIAP-1/2 and XIAP. Cancer Res. 2008;68:9384–9393. doi: 10.1158/0008-5472.CAN-08-2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Darding M, Feltham R, Tenev T, et al. Molecular determinants of Smac mimetic induced degradation of cIAP1 and cIAP2. Cell Death Differ. 2011;18:1376–1386. doi: 10.1038/cdd.2011.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiarle R, Podda A, Prolla G, Gong J, Thorbecke GJ, Inghirami G. CD30 in normal and neoplastic cells. Clin Immunol. 1999;90:157–164. doi: 10.1006/clim.1998.4636. [DOI] [PubMed] [Google Scholar]
- 17.Mir SS, Richter BW, Duckett CS. Differential effects of CD30 activation in anaplastic large cell lymphoma and Hodgkin disease cells. Blood. 2000;96:4307–4312. [PubMed] [Google Scholar]
- 18.Tian ZG, Longo DL, Funakoshi S, et al. In vivo antitumor effects of unconjugated CD30 monoclonal antibodies on human anaplastic large-cell lymphoma xenografts. Cancer Res. 1995:5335–53341. [PubMed] [Google Scholar]
- 19.Silke J. The regulation of TNF signalling: what a tangled web we weave. Curr Opin Immunol. 2011;23:620–626. doi: 10.1016/j.coi.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 20.Paulsen MT, Veloso A, Prasad J, et al. Coordinated regulation of synthesis and stability of RNA during the acute TNF-induced proinflammatory response. Proc Natl Acad Sci U S A. 2013;110:2240–2245. doi: 10.1073/pnas.1219192110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paulsen MT, Veloso A, Prasad J, et al. Use of Bru-Seq and BruChase-Seq for genome-wide assessment of the synthesis and stability of RNA. Methods. 2014;67:45–54. doi: 10.1016/j.ymeth.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kenneth NS, Hucks GEJ, Kocab AJ, McCollom AL, Duckett CS. Copper is a potent inhibitor of both the canonical and non-canonical NF-κB pathways. Cell Cycle. 2014;13:1006–1014. doi: 10.4161/cc.27922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Signer RA, Magee JA, Salic A, Morrison SJ. Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature. 2014;509:49–54. doi: 10.1038/nature13035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang J, Wang J, Ng S, Lin Q, Shen HM. Development of a novel method for quantification of autophagic protein degradation by AHA labeling. Autophagy. 2014;10:901–912. doi: 10.4161/auto.28267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 27.Darding M, Meier P. IAPs: guardians of RIPK1. Cell Death Differ. 2012;19:58–66. doi: 10.1038/cdd.2011.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-κB signaling pathways. Nat Immunol. 2011;12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 29.Buchan SL, Al-Shamkhani A. Distinct motifs in the intracellular domain of human CD30 differentially activate canonical and alternative transcription factor NF-κB signaling. PLoS One. 2012;7:e45244. doi: 10.1371/journal.pone.0045244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruggero D, Pandolfi PP. Does the ribosome translate cancer? Nat Rev Cancer. 2003;3:179–192. doi: 10.1038/nrc1015. [DOI] [PubMed] [Google Scholar]
- 31.Matsuzawa A, Tseng PH, Vallabhapurapu S, et al. Essential cytoplasmic translocation of a cytokine receptor-assembled signaling complex. Science. 2008;321:663–668. doi: 10.1126/science.1157340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duckett CS, Gedrich RW, Gilfillan MC, Thompson CB. Induction of Nuclear Factor κB by the CD30 Receptor is Mediated by TRAF1 and TRAF2. Mol Cell Biol. 1997;17:1535–1542. doi: 10.1128/mcb.17.3.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buchan SL, Al-Shamkhani A. Distinct motifs in the intracellular domain of human CD30 differentially activate canonical and alternative transcription factor NF-κB signaling. PLoS One. 2012;7:e45244. doi: 10.1371/journal.pone.0045244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malinin NL, Boldin MP, Kovalenko AV, Wallach D. MAP3K-related kinase involved in NF-κB induction by TNF, CD95, and IL-1. Nature. 1997;385:540–544. doi: 10.1038/385540a0. [DOI] [PubMed] [Google Scholar]
- 35.Zarnegar B, Yamazaki S, He JQ, Cheng G. Control of canonical NF-κB activation through the NIK-IKK complex pathway. Proc Natl Acad Sci U S A. 2008;105:3503–3508. doi: 10.1073/pnas.0707959105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duckett CS, Thompson CB. CD30-dependent degradation of TRAF2: implications for negative regulation of TRAF signaling and the control of cell survival. Genes Dev. 1997;11:2810–2821. doi: 10.1101/gad.11.21.2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun H, Nikolovska-Coleska Z, Lu J, et al. Design, synthesis, and characterization of a potent, nonpeptide, cell-permeable, bivalent Smac mimetic that concurrently targets both the BIR2 and BIR3 domains in XIAP. J Am Chem Soc. 2007;129:15279–15294. doi: 10.1021/ja074725f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Galban S, Duckett CS. XIAP as a ubiquitin ligase in cellular signaling. Cell Death Differ. 2010;17:54–60. doi: 10.1038/cdd.2009.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kenneth NS, Duckett CS. IAP proteins: regulators of cell migration and development. Curr Opin Cell Biol. 2012;24:871–875. doi: 10.1016/j.ceb.2012.11.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.