

Abstract
Podocyte loss is central to the progression of proteinuric kidney diseases leading to end-stage kidney disease (ESKD), requiring renal replacement therapy, such as dialysis. Despite modern tools and techniques, the 5-year mortality of some patients requiring dialysis remains at about 70% to 80%. Thus, there is a great unmet need for podocyte-specific treatments aimed at preventing podocyte loss and the ensuing development of ESKD. Here, we show that ablation of the podocyte death-promoting protein dendrin delays the onset of ESKD, thereby expanding the life span of mice lacking the adapter protein CD2AP. Ablation of dendrin delays onset and severity of proteinuria and podocyte loss. In addition, dendrin ablation ameliorates mesangial volume expansion and up-regulation of mesangial fibronectin expression, which is mediated by a podocyte-secreted factor. In conclusion, onset of ESKD and death can be markedly delayed by blocking the function of dendrin.
Podocytes are highly specialized cells that constitute a central component of the kidney filtration barrier. Disruption of podocyte function damages the kidney filter, resulting in proteinuria.1 Primary and secondary podocytopathies combined account for about 90% of end-stage kidney disease (ESKD) at a cost of $20 billion per year in the United States alone.2 The health care burden and economic impact is further enhanced by the fact that proteinuria is a strong and independent risk factor for cardiovascular disease.3 Podocytes are terminally differentiated cells that cannot regenerate in response to injury or loss. As a result, podocyte loss causes glomerulosclerosis, in which a reduction in podocyte number below a critical 40% threshold results in high-level sustained proteinuria and decreased renal function.4,5 Clear clinical correlations between podocytopenia and proteinuria have also been established. Podocyte loss contributes to the progression of diabetic nephropathy in patients with type II diabetes mellitus.6 Podocyte cell number is also decreased in patients of all ages with type I diabetes, where the reduction in podocyte number correlates with an increased albumin excretion rate.7,8 In IgA nephropathy, podocyte loss correlates closely with the degree of proteinuria, glomerulosclerosis, and renal dysfunction.9 Podocytopenia is also associated with disease progression in membranous nephropathy, focal segmental glomerulosclerosis (FSGS), amyloidosis, and the aging kidney.10
Despite remarkable advances in clarifying the details of the molecular architecture of podocytes and their interdigitating foot processes, mechanisms underlying podocyte injury and loss remain poorly understood. This results in a significant unmet clinical need. We previously identified dendrin as a dual-compartment signaling molecule anchored at the slit diaphragm under normal conditions.11 Dendrin has no known homologous partners and an unclear role in normal physiology. Under the influence of cell death–inducing high-dose transforming growth factor-β (TGF-β), dendrin relocates to the podocyte nucleus and promotes apoptosis, which is ameliorated after gene silencing of dendrin.11 Of note, dendrin-mediated podocyte apoptosis is inhibited by the prosurvival Yes associated protein (YAP).12 In rodents, nuclear dendrin has been observed in inflammatory glomerulonephritis, in CD2-associated protein (Cd2ap)-deficient (Cd2ap−/−) mice and in adriamycin (ADR) nephropathy.11,13,14 Nuclear dendrin has also been found in biopsies from patients with FSGS, membranous nephropathy, and class V lupus nephritis.14 Conversely, dendrin-null (Ddn−/−) mice display no overt pathology; they have a normal life span and do not develop proteinuria or renal failure.15
Here, we tested the hypothesis that the ablation of dendrin would improve renal survival in progressive glomerulosclerosis. We deleted the dendrin gene in Cd2ap−/− mice that develop podocyte injury with proteinuria at 2 to 3 weeks of age and die of renal failure at 8 to 9 weeks of age.16 The phenotype of Cd2ap−/− mice was completely reversed with podocyte-specific expression of CD2AP,17 confirming podocyte injury as the root cause of kidney failure and death. Clinical evidence for the role of CD2AP as an essential component of the glomerular filtration barrier has been provided by the identification of patients with FSGS-causing Cd2ap−/− gene mutations.18–20 Here, we show that dendrin ablation delays the onset of proteinuria, glomerulosclerosis, renal failure, and death in Cd2ap−/− mice. These results implicate dendrin as a potential podocyte-specific therapeutic target in slowing the progression from proteinuria to ESKD and death.
Materials and Methods
All studies involving mice were performed in accordance with Icahn School of Medicine institutional guidelines under an institutional animal care and use committee–approved protocol.
Mice Strains and Genotyping
DNA for genotyping was extracted using a sodium hydroxide extraction protocol from The Jackson Laboratory (Bar Harbor, ME). Briefly, 2-mm tail was incubated with 25 mmol/L NaOH/0.2 mmol/L EDTA at 98°C for 1 hour in a thermocycler with subsequent temperature reduction to 15°C. After adding 40 mmol/L Tris-HCl (pH 5.5), the mixture was centrifuged at 16,000 × g for 3 minutes and an aliquot removed for PCR. The following primers were used for genotyping Cd2ap in the mice: forward 5′-GTTGACTATATTGTGGAATATGACTATGA-3′ and reverse 5′-CAGCAGGTTGCCAATCACAAATGTACCTATA-3′ for wild type; forward 5′-CAAGTCTGTCATAGGTCACACTCAAGAAG-3′ and reverse 5′-AGGTGCCACTCCCACTGTCCTTTCCTAATAA-3′ for knockout. The expected sizes for wild type is around 300 bp and for knockout is around 400 bp. The following primers were used for genotyping dendrin in the mice: forward 5′-GGAGGATCTCAGCGTCCATA-3′ and reverse 5′-AGGTTCAAGGCCTCTCCATT-3′ for wild type; forward 5′-AATTCCATCAGACCTCGACCT-3′ and reverse 5′-AGGTTCAAGGCCTCTCCATT-3′ for knockout. The expected size for wild type is 574 bp and for knockout is 405 bp.
Measurement of Urine Albumin/Creatinine Ratios and Serum Creatinine
Urine albumin was quantified by enzyme-linked immunosorbent assay according to the manufacturer's protocol (Bethyl Laboratory, Montgomery, TX). Urine creatinine levels were measured in the same samples using the creatinine assay kit (Cayman Chemical Company, Ann Arbor, MI) according to the manufacturer's protocol. Urine albumin excretion rate was expressed as the ratio of albumin to creatinine. Serum creatinine measurements were performed by liquid chromatography-tandem mass spectrometry at the UAB-UCSD O'Brien Core Center for Acute Kidney Injury Research (Birmingham, AL).
Histology and Immunohistochemistry
Mice were perfused with filtered phosphate-buffered saline containing 3% paraformaldehyde (Sigma-Aldrich, St. Louis, MO) at a flow rate of 9 mL per minute for 3 minutes. After perfusion, kidneys were harvested and further fixed in 2.5% glutaraldehyde in 0.1 mol/L sodium cacodylate buffer (pH 7.4; Electron Microscopy Services, Hatfield, PA). Four-micrometer paraffin-processed, formalin-fixed kidney sections were stained with periodic acid-Schiff or, after antigen retrieval [pressure cooker or microwave in citrate buffer (pH 6)], with antibodies against synaptopodin (G1 mouse monoclonal antibody, dilution 1:10; supplied by P.M.), PAX2 (paired box protein Pax-2; rabbit polyclonal antibody, dilution 1:75; Invitrogen/Life Technologies, Carlsbad, CA), WT1 (rabbit polyclonal antibody, dilution 1:50; Santa Cruz Biotechnology, Dallas, TX), cleaved caspase 3 (dilution 1:20; Abcam, Cambridge, UK), cathepsin L (dilution 1:100; Sigma-Aldrich), and dendrin (dilution 1:50; EMD Millipore, Billerica, MA), followed by horseradish peroxidase–conjugated secondary antibodies (Dako, Carpinteria, CA), and counterstained with hematoxylin. One-micrometer sections of resin-embedded tissue were stained with toluidine blue. Images were taken with an Olympus BX53 microscope with a DP72 camera (Olympus, Tokyo, Japan) and processed using Adobe Photoshop software (Adobe Systems, San Jose, CA). Histology of three to six animals in each group was assessed. Periodic acid-Schiff– and toluidine blue–stained sections were analyzed in a blinded fashion (A.W.).
Electron Microscopy
Ultra-thin 80-nm sections of resin-embedded kidney tissue were mounted on copper grids, treated with uranyl acetate and lead citrate, and examined by a pathologist (A.W.) in a blinded fashion using a JEOL 1010 transmission electron microscope (JEOL, Tokyo, Japan). Electron micrographs were assessed in a blinded fashion (A.W.).
Morphometry
One-millimeter cubes of kidney cortex were fixed in glutaraldehyde and embedded in EPON resin. One-micrometer-thick sections were cut using an ultramicrotome fitted with a Histo Jumbo Diamond Knife (Diatome US, Hatfield, PA). Serial sections were cut, and every tenth section and the adjacent section were saved to a microscope slide and stained with toluidine blue.21 A total of 20 pairs of sections were saved per kidney. Images were obtained using the 100× objective lens (numerical aperture = 1.40) of an Olympus BX51 microscope fitted with a DP71 digital camera and DP Controller software DP-BSW-Ver3.2 (Olympus America, Center Valley, PA). Images were observed using Photoshop (Adobe Systems) on a 24-inch monitor at a window magnification of 100%. Ten glomeruli per kidney were analyzed for all morphometric parameters.
Podocyte Number
Podocytes were counted using the fractionator/disector principle.22,23 Briefly, images of adjacent sections from throughout a glomerulus were observed, and the number of profiles from podocyte nuclei present in the second section of each pair, but not present in the first section, was counted. The number of podocyte nuclei was used as a surrogate for the number of podocytes. It was assumed there was only one nucleus per podocyte. The number of podocytes per glomerulus was calculated using the equation , where 10 is the reciprocal of the fraction of the glomerulus sampled and ΣQ− is the sum of nuclei counted in the second section, but not present in the first section, of each section pair. An average of 137 Q− (range, 48 to 212) was counted per animal.
Glomerular Volume
Glomerular volume was measured using the Cavalieri principle.24 Briefly, using the Layers function of Photoshop, a counting grid was randomly placed over the image of the first section of each section pair from a glomerulus, and the number of grid point falling on the glomerulus was counted.21 Glomerular volume was calculated using the equation: , where 10 is the distance in micrometers between the measured sections, ΣP is the sum of grid points falling on all of the measured images from the glomerulus, d is the distance in micrometers between points on the grid, and mag is the image magnification. An average of 726 (range, 337 to 2721) grid points on glomeruli were counted per animal.
Podocyte Numerical Density per Glomerulus
Podocyte numerical density was calculated by dividing podocyte number by glomerular volume and expressed as number per cubic micrometer.
Glomerular Component Volumes
First, glomerular component volume densities [Vv(Component X/glomerulus)] were measured using electron microscopy.25 Briefly, silver-gold sections were cut with an Ultra Diamond Knife (Diatome US) placed on single-slot grids and stained with uranyl acetate and lead citrate. Images were obtained using a JEM-1200EX electron microscope (JEOL). Images were used at a final magnification of ×3200 and were taken for the 3-, 4-, and 5-week-old animals. Measurements were performed on five animals per group and 10 glomeruli per animal. A glomerulus was defined as the area within a minimal string polygon drawn around a glomerulus including all of the podocytes. Only glomeruli with a complete profile were used. Five components within the glomerulus were defined: i) mesangium including mesangial matrix, mesangial cell, and mesangial glomerular basement membrane (GBM); ii) podocytes (excluding cysts); iii) capillary lumen including endothelial cells and other cells within the lumen; iv) cysts within the podocytes; and v) remainder tissues, including Bowman's space, GBM, and nonresolvable tissue. Using the Layer function of Photoshop, a grid of points was randomly placed over an image of the complete glomerular profile, and the number of points falling on each of the five glomerular components was counted. The volume density for component X was calculated using the equation: , where PComponent X is the number of points falling on a component X (ie, mesangium) and ΣPFive components is the number of points falling on all five components. The volume of each component was calculated by multiplying each appropriate volume density by the glomerular volume. For example, for mesangial volume: .
An average of 882 points (range, 388 to 1665) were counted per animal. An average podocyte volume for the mouse was calculated by dividing the total podocyte volume within a glomerulus by the average number of podocytes per glomerulus and was reported as cubic micrometers.
Cell Culture
Conditionally immortalized murine podocytes26 were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies) supplemented with 10% heat-inactivated fetal bovine serum (Life Technologies) and 100 U/mL penicillin–streptomycin (Life Technologies) on type I collagen (Corning, Corning, NY) coated dishes or flasks. Undifferentiated podocytes were maintained at 33°C in the presence of 10 U/mL mouse γ-interferon (Cell Sciences, Canton, MA) to drive T-antigen expression. To induce differentiation, podocytes were thermo-shifted to a 37°C incubator and switched to medium lacking γ-interferon. Mouse mesangial cells (MMCs) were cultured in DMEM containing 10% heat-inactivated fetal bovine serum and 100 U/mL penicillin–streptomycin.
Gene Silencing and Conditioned Media Transfers
pLKO.1 lentiviral shRNA plasmids were purchased from Addgene (Cambridge, MA) for scramble control and from Sigma-Aldrich for CD2AP and dendrin. We used sequence 5′-CCGGCAAACCTAAACCCAGATATTTCTCGAGAAATATCTGGGTTTAGGTTTGTTTTTG-3′ to target CD2AP. We used sequence 5′-CCGGGATTGAAGTGAAGACTATTTCCTCGAGGAAATAGTCTTCACTTCAATCTTTTTG-3′ to target dendrin. The pLKO.1 plasmids, along with the helper plasmids psPAX2 and pMD2.G, were transfected into HEK293T cells at 70% confluence using FuGENE 6 Transfection Reagent (Promega, Madison, WI). Medium was replaced 16 to 18 hours after transfection. Virus was collected at 48 hours and 72 hours post-transfection. Cell debris was removed by passing the supernatant through a 0.45-μ filter. After podocytes were differentiated for 7 days, virus was added to the differentiated podocytes in the presence of 4 μg/mL Polybrene (Sigma-Aldrich) overnight. The medium was changed the next day. After 4 days of infection, cells were serum starved in DMEM medium containing 0.1% heat-inactivated fetal bovine serum. Twenty-four hours after serum starvation, the supernatant was collected and spun down to remove cell debris. The collected supernatants (conditioned podocyte medium) were then used to treat MMCs that were themselves previously made quiescent by serum starvation.
MMC Viability and Proliferation Assay
MMCs (1000) in 100 μL of medium were plated into each well of the opaque-bottom 96-well plate (Corning). Cells were allowed to adhere overnight. The next day, the MMCs were serum starved overnight in DMEM containing 0.1% heat-inactivated fetal bovine serum. The following day, 100 μL of supernatant from serum-starved podocytes was used to treat the MMCs. After 24 hours of incubation, cells were allowed to equilibrate to room temperature for 30 minutes. Cell viability was then measured by adding 100 μL of CellTiter-Glo Luminescent Reagent (Promega) to each well, according to the manufacturer's protocol. After 10 minutes of incubation at room temperature, the luminescence of each sample was measured in a plate-reading luminometer.
Quantification of Fibronectin Production
MMCs (40,000) were plated in each well of a 6-well plate. Cells were then serum starved overnight in DMEM medium containing 0.1% heat-inactivated fetal bovine serum. The following day, serum-starved podocyte medium was added to the MMCs. Conditioned media from experimental cultures were collected 24 hours after treatment, spun down at 2300 × g for 5 minutes to remove cell debris, and analyzed by Western immunoblot for fibronectin. Conditioned media were also diluted 1:10 and quantified in duplicates using a Fibronectin Mouse ELISA Kit (Abcam) according to the manufacturer's protocol.
Assessment of MMC Hypertrophy
MMCs (40,000) were plated in each well of a 6-well plate. Cells were then serum starved overnight in DMEM medium containing 0.1% heat-inactivated fetal bovine serum. The following day, serum-starved podocyte medium was added to the MMCs. MMCs were trypsinized, harvested, and counted after 48 hours of treatment. An equal number of cells were lysed with 100 μL of radioimmunoprecipitation assay buffer by incubating on ice for 30 minutes. After 30 minutes, lysates were spun down at 16,000 × g for 30 minutes. Supernatants were transferred to a new tube and quantified using a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Rockford, IL) according to the manufacturer's protocol.
Statistics
Data are given as means ± SD. Statistical analyses were performed using analysis of variance to compare data among groups and t-tests to determine differences between two groups. P < 0.05 was considered statistically significant.
Results
Dendrin Ablation Prolongs Survival and Preservation of Renal Function in Cd2ap−/− Mice
We reported previously the relocation of dendrin to the podocyte nucleus in murine inflammatory glomerulonephritis.11 Now, we found nuclear redistribution of dendrin in podocytes during disease progression in Cd2ap−/− mice (Supplemental Figure S1), suggesting that nuclear dendrin promotes podocyte injury and renal failure in the absence of CD2AP. To directly test this hypothesis, we generated Cd2ap-null mice with and without dendrin deletion: Cd2ap−/−;Ddn−/− and Cd2ap−/−;Ddn+/+. Wild-type littermates (Cd2ap+/+;Ddn+/+) served as controls (Supplemental Figure S2A). The deletion of dendrin in Cd2ap−/− mice was confirmed by genotyping PCR (Supplemental Figure S2B). Because Cd2ap−/− mice die from renal failure,16 we determined the effect of dendrin ablation on life span and found that Cd2ap−/−;Ddn−/− mice survived significantly longer compared to Cd2ap−/−;Ddn+/+ littermates (mean life span: Cd2ap−/−;Ddn+/+: 8.83 ± 0.28 weeks; Cd2ap−/−;Ddn−/−: 13.52 ± 0.38; P < 0.001, t-test) (Figure 1A). No wild-type littermate controls died during the study period (Figure 1A). To test whether the improved survival of Cd2ap−/−;Ddn−/− mice resulted from prolonged preservation of renal function, we assessed serum creatinine levels. At 3 weeks of age, when proteinuria develops in Cd2ap−/− mice,16 serum creatinine values were comparable between Cd2ap−/−;Ddn+/+ (0.78 ± 0.198 μg/mL), Cd2ap−/−;Ddn−/− (0.72 ± 0.086 μg/mL), and wild-type mice (NS, analysis of variance) (Figure 1B). By contrast, at 6 weeks of age, Cd2ap−/−;Ddn+/+ mice had significantly higher mean serum creatinine levels (4.3 ± 0.552 μg/mL) compared to Cd2ap−/−;Ddn−/− littermates (0.64 ± 0.103 μg/mL; P < 0.001, t-test) (Figure 2B). The simplest interpretation of these results is that dendrin ablation increases the life span of Cd2ap−/− mice by delaying the onset of chronic kidney disease and renal failure.
Figure 1.

Dendrin ablation delays loss of renal function and death of Cd2ap−/− mice. A: The life span of Cd2ap−/−;Ddn−/− mice is significantly greater than Cd2ap−/−;Ddn+/+ and Cd2ap+/+;Ddn+/+ littermates. B: At 3 weeks, serum creatinine levels were not different between the three genotypes. At 6 weeks, Cd2ap−/−;Ddn+/+ mice have significantly greater mean serum creatinine than Cd2ap−/−;Ddn−/− and Cd2ap+/+;Ddn+/+ littermates. C: Proteinuria in Cd2ap−/−;Ddn+/+ mice commences at 3 weeks and increases steadily through week 7. Cd2ap−/−;Ddn−/− mice develop proteinuria at 5 weeks. Data are expressed as means ± SD. ∗∗∗P < 0.001, analysis of variance.
Figure 2.

Genetic ablation of dendrin delays severity of glomerular and tubulointerstitial injury in Cd2ap−/− mice. A: At 3 weeks of age, Cd2ap−/−;Ddn+/+ mice develop mesangial expansion, which is milder in Cd2ap−/−;Ddn−/− mice. By 4 weeks of age, Cd2ap−/−;Ddn+/+ mice develop segmental glomerulosclerosis, and global glomerulosclerosis and focal collapsing lesions at 5 weeks of age. By contrast, hardly any glomerular lesions are found in Cd2ap−/−;Ddn−/− mice at 5 weeks. By 6 weeks of age, Cd2ap−/−;Ddn+/+ mice show extensive chronic changes affecting most glomeruli, whereas Cd2ap−/−;Ddn−/− animals have significantly fewer glomerular lesions. B: Quantitative analysis of histological changes. Cd2ap−/−;Ddn−/− mice (gray bars) show fewer segmental and global glomerulosclerosis than Cd2ap−/−;Ddn+/+ mice (black bars) by 4 and 5 weeks of age; the differences reach statistical significance by 6 weeks. Fewer collapsing lesions are also seen in 5- and 6-week-old Cd2ap−/−;Ddn−/− mice compared to age-matched Cd2ap−/−;Ddn+/+ littermates. No interstitial fibrosis and tubular atrophy (IFTA) is seen in animals at 4 weeks, whereas IFTA is significantly more widespread in Cd2ap−/−;Ddn+/+ mice at 5 and 6 weeks. Data are expressed as means ± SD. ∗P < 0.05, ∗∗P < 0.01. Scale bar = 20 μm.
Dendrin Ablation Delays Onset and Progression of Proteinuria in Cd2ap−/− Mice
In humans, proteinuria is a risk factor for cardiovascular disease, kidney failure, and death.3,27–30 Therefore, we tested whether the beneficial effect of dendrin deletion on renal function and survival was associated with differences in the onset, severity, and progression of proteinuria. Consistent with previous reports,16,31 we first detected proteinuria at 3 weeks of age in Cd2ap−/−;Ddn+/+ mice, but not in Cd2ap−/−;Ddn−/− or wild-type littermate controls [mean albumin/creatinine ratio (ACR): Cd2ap−/−;Ddn+/+, 13.31 ± 7.49 mg/mg; Cd2ap−/−;Ddn−/−, 0.126 ± 0.085 mg/mg; P < 0.001, analysis of variance] (Figure 1C). By contrast, in Cd2ap−/−;Ddn−/− mice, we first detected proteinuria at 5 weeks (mean ACR 6.39 ± 4.7 mg/mg) (Figure 1C). Of note, initial ACR in Cd2ap−/−;Ddn−/− mice was significantly lower when compared to initial ACR in Cd2ap−/−;Ddn+/+ or wild-type littermate control at 3 weeks of age (P < 0.05, analysis of variance). Proteinuria increased steadily in Cd2ap−/−;Ddn+/+ and Cd2ap−/−;Ddn−/− mice over time, but mean ACR levels remained significantly higher at all times in Cd2ap−/−;Ddn+/+ mice (5 weeks: Cd2ap−/−;Ddn+/+, 104.67 ± 21.63 mg/mg; Cd2ap−/−;Ddn−/−, 6.39 ± 4.71 mg/mg; P < 0.001, analysis of variance) (Figure 1C). In Cd2ap−/−;Ddn+/+ mice, proteinuria peaked at 8 weeks (mean ACR, 51.29 ± 15.069 mg/mg) compared to 10 weeks in Cd2ap−/−;Ddn−/− mice (mean ACR, 51.29 ± 15.069 mg/mg) (Supplemental Figure S3A); at this time point, all animals in the Cd2ap−/−;Ddn+/+ cohort had already died. These data reveal that genetic deletion of dendrin delays the onset, severity, and progression of proteinuria in mice lacking CD2AP.
Dendrin Ablation Delays the Development of Glomerulosclerosis in Cd2ap−/− Mice
Because podocyte injury and loss are central to the pathogenesis of renal failure and death in Cd2ap−/− mice,17 we conducted a detailed comparative analysis of renal pathology in Cd2ap−/−;Ddn+/+, Cd2ap−/−;Ddn−/−, and wild-type littermate control mice. To uncover dendrin-dependent changes in glomerular disease progression, we assessed changes in kidney morphology by light microscopy. At 3 weeks of age, glomeruli of Cd2ap−/−;Ddn+/+ and Cd2ap−/−;Ddn−/−, but not of wild-type mice, showed mild mesangial expansion, but no other acute or structural changes (Figure 2A). However, starting at 4 weeks of age, nonsclerosed glomeruli of Cd2ap−/−;Ddn+/+ mice showed more mesangial expansion than those of Cd2ap−/−;Ddn−/− mice, which became even more obvious at 5 weeks of age (Figure 2A). By 6 weeks of age, the majority of glomeruli in Cd2ap−/−;Ddn+/+ kidneys showed chronic glomerular lesions with advanced chronic tubulointerstitial disease, whereas most glomeruli in Cd2ap−/−;Ddn−/− kidneys displayed mesangial expansion, but only a few glomerular lesions (Figure 2A). By 8 to 12 weeks of age, glomeruli of Cd2ap−/−;Ddn−/− mice showed chronic glomerular changes and advanced kidney disease comparable to 6-week-old Cd2ap−/−;Ddn+/+ littermates (Supplemental Figure S3B).
We quantified the light microscopic changes at 4, 5, and 6 weeks of age (Figure 2B). At 4 and 5 weeks, Cd2ap−/−;Ddn−/− mice showed a lower number of glomeruli with segmental glomerulosclerosis when compared to Cd2ap−/− Ddn+/+ mice, (4 weeks: Cd2ap−/−;Ddn−/−, n = 3, 0.27% ± 0.27%; Cd2ap−/−;Ddn+/+, n = 5, 4.20% ± 1.77%, NS; 5 weeks: Cd2ap−/−;Ddn−/−, n = 3, 0.69% ± 0.37%; Cd2ap−/−;Ddn+/+, n = 3, 19.15% ± 6.16%; NS, t-test). The Cd2ap−/−;Ddn−/− mice also showed a lower number of glomeruli with global glomerulosclerosis (4 weeks: Cd2ap−/−;Ddn−/−, n = 3, 0% global glomerulosclerosis; Cd2ap−/−;Ddn+/+, n = 6, 1.97% ± 1.6%; NS; 5 weeks: Cd2ap−/−;Ddn+/+, n = 3, 0%; Cd2ap−/−;Ddn+/+, n = 6, 3.80% ± 1.5%; NS, t-test). The differences reached statistical significance in 6-week-old animals (FSGS: Cd2ap−/−;Ddn−/−, n = 3, 11.95% ± 7.62%; Cd2ap−/−;Ddn+/+, n = 3, 36.05% ± 3.93%; P < 0.05; global glomerulosclerosis: Cd2ap−/−;Ddn−/−, n = 3, 2.6% ± 2.6%; Cd2ap−/−;Ddn−/−, n = 3, 18.96% ± 1.96%; P < 0.01, t-test) (Figure 2B). Although the difference was not statistically significant, we also counted a lower number of glomeruli with collapsing lesions in 5- and 6-week-old Cd2ap−/−;Ddn−/− mice, but not in glomeruli of Cd2ap−/−;Ddn+/+ littermates of the same ages (5 weeks: 0%, n = 3 versus 3.56% ± 1.29%, n = 6; NS; 6 weeks: 2.63% ± 1.31%, n = 3 versus 5.18% ± 0.23%; NS, t-test). Although chronic tubulointerstitial changes were absent in both groups at 4 weeks, interstitial fibrosis and tubular atrophy were significantly more widespread in Cd2ap−/−;Ddn+/+ mice at 5 and 6 weeks (5 weeks: 0%, n = 6 versus 9% ± 2.92%, n = 5; P = 0.0076; 6 weeks: 8.33% ± 3.33%, n = 3 versus 33.33% ± 3.33%; P < 0.01, t-test) (Figure 2B).
Dendrin Deficiency Delays the Onset of Podocyte Injury in Cd2ap−/− Mice
Because podocyte injury is central to the pathogenesis of FSGS,1 we performed a detailed analysis of podocyte morphology by transmission electron microscopy (Figure 3). At 3 weeks, podocytes in Cd2ap−/−;Ddn+/+ mice showed widespread foot process broadening (FPB), segmental foot process effacement (FPE), microvillous transformation of the cell membrane, and focal vacuolization. By contrast, Cd2ap−/−;Ddn−/− mice showed only mild FPB at 3 weeks (Figure 3). At 4 weeks, Cd2ap−/−;Ddn+/+ mice showed widespread FPE, whereas individual foot processes were still mostly preserved in Cd2ap−/−;Ddn−/− littermates (Figure 3). At 5 weeks, podocytes of Cd2ap−/−;Ddn+/+ mice showed diffuse FPE and widespread microvillous transformation of their cell membrane, as well as features of podocyte hypertrophy, which was reflected by the presence of long, thin extensions of the cell bodies (Figure 3). In addition, an expanded mesangial matrix infiltrated the space between the glomerular endothelium and the GBM (Figure 3). The glomerular endothelium showed mild degenerative changes with loss of fenestrations. By contrast, there was only segmental FPE and FPB in 5-week-old Cd2ap−/−;Ddn−/− animals. At 6 weeks, both Cd2ap−/−;Ddn+/+ and Cd2ap−/−;Ddn−/− mice showed widespread FPE, but only in the Cd2ap−/−;Ddn+/+ mice could areas of denuded GBM be frequently identified (Figure 3). Semiquantitative analysis of the degree of FPE on a scale from 0 (no FPE) to 4 (diffuse FPE) (Figure 4A) showed significant differences at all time points analyzed (3 weeks: Cd2ap−/−;Ddn+/+, n = 4, 2.63 ± 0.24; Cd2ap−/−;Ddn−/−, n = 4, 1.625 ± 0.13; P < 0.05; 4 weeks: Cd2ap−/−;Ddn+/+: n = 4, 2.75 ± 0.25; Cd2ap−/−;Ddn−/−, n = 4, 1.38 ± 0.24; P < 0.01; 5 weeks: Cd2ap−/−;Ddn+/+, n = 6, 4 ± 0; Cd2ap−/−;Ddn−/−, n = 4, 1.75 ± 0.43; P < 0.0005; 6 weeks: Cd2ap−/−;Ddn+/+, n = 5, 4 ± 0; Cd2ap−/−;Ddn−/−, n = 5, 2.4 ± 0.4; P < 0.005, t-test) (Figure 4B).
Figure 3.
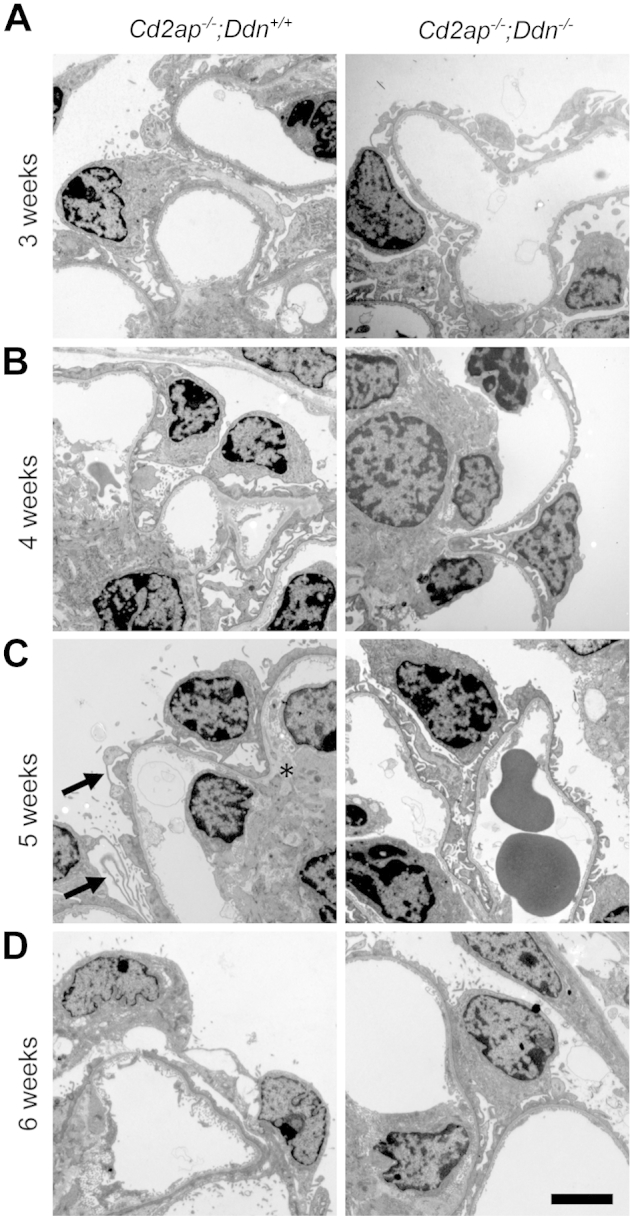
Dendrin ablation ameliorates podocyte injury in Cd2ap−/− mice. A: At 3 weeks, Cd2ap−/−;Ddn+/+ mice show widespread foot process broadening (FPB), segmental foot process effacement (FPE), microvillous transformation of the podocyte cell membrane, and focal vacuolization of podocytes. By contrast, Cd2ap−/−;Ddn−/− mice show only mild FPB at 3 weeks. B: Although Cd2ap−/−;Ddn+/+ mice show widespread FPE at 4 weeks, podocyte foot processes are still mostly preserved with very focal FPE and FPB in Cd2ap−/−;Ddn−/− mice. C: At 5 weeks, podocytes of Cd2ap−/−;Ddn+/+ mice show diffuse FPE, widespread microvillous transformation of their cell membrane, and features of hypertrophy, characterized by thin and long processes (arrows). In addition, an expanded mesangial matrix infiltrates the area between the endothelium and the GBM (asterisk). The glomerular endothelium shows mild loss of fenestrations. By contrast, there is only segmental FPE and extensive FPB in 5-week-old Cd2ap−/−;Ddn−/− animals. D: At 6 weeks, both Cd2ap−/−;Ddn+/+ and Cd2ap−/−;Ddn−/− mice show widespread FPE; however, in the Cd2ap−/−;Ddn+/+ mice, areas of denuded GBM are frequently identified. Scale bar = 2 μm.
Figure 4.

Dendrin ablation protects against filter barrier damage in Cd2ap−/− mice. A: Extensive foot process effacement (FPE) is seen in Cd2ap−/−;Ddn+/+ mice as early as 4 weeks of age, progressing to diffuse FPE by 5 weeks. By contrast, Cd2ap−/−;Ddn−/− mice show extensive foot process broadening, but not diffuse FPE, by 6 weeks of age. B: Semiquantitative analysis of FPE in Cd2ap−/−;Ddn+/+ (black bars) and Cd2ap−/−;Ddn−/− mice (gray bars). 0, no FPE; 1, mild FPE; 2, moderate FPE; 3, extensive FPE; 4, diffuse FPE. Data are expressed as means ± SD. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001. Scale bar = 2 μm.
Podocyte Loss in Cd2ap−/− Mice Triggers a Parietal Epithelial Cell Response, Which Is Delayed in Cd2ap−/−;Ddn−/− Mice
Podocyte loss triggers a parietal epithelial cell (PEC) reaction in ADR nephropathy32 and in membrane-associated guanylate kinase, WW and PDZ domain-containing protein (MAGI)-2–deficient mice.33 Therefore, we tested whether PEC activation also occurs in Cd2ap−/− mice and whether it was delayed in Cd2ap−/−;Ddn−/− mice. We analyzed the expression of the podocyte marker synaptopodin and the PEC marker PAX2 at 4, 5, and 6 weeks in kidneys of wild-type, Cd2ap−/−;Ddn+/+, and Cd2ap−/−;Ddn−/− mice (Figure 5). At 4 weeks, wild-type mice showed typical distribution of synaptopodin staining along the GBM throughout the glomerulus, which appeared weaker but mostly preserved in glomeruli of Cd2ap−/−;Ddn−/− mice. PAX2 staining was limited to the nuclei of PECs lining Bowman's capsule. By contrast, most glomeruli in Cd2ap−/−;Ddn+/+ mice showed a strong reduction in synaptopodin staining, which was confined to the periphery of the glomerulus (Figure 5A); PAX2 staining was exclusively present in the parietal epithelium lining Bowman's capsule (Figure 5A). At 5 weeks, synaptopodin staining was severely diminished in Cd2ap−/−;Ddn+/+ mice, with complete disruption in areas of segmental glomerulosclerosis. By contrast, synaptopodin staining was preserved in Cd2ap−/−;Ddn−/− mice (Figure 5B). Concomitantly, PAX2-positive cells consistent with visceralized PECs (vPECs)32 were present on the tuft in areas of synaptopodin loss in Cd2ap−/−;Ddn+/+ mice, whereas PAX2 staining remained confined to PECs lining Bowman's capsule in Cd2ap−/−;Ddn−/− mice (Figure 5B). At 6 weeks of age, similar to observations in ADR nephropathy,32 affected glomeruli of Cd2ap−/−;Ddn+/+ mice frequently showed severe loss of synaptopodin staining and extensive coverage of the glomerular tuft by PAX2-positive vPECs. By contrast, only a few vPECs were present in focal areas of synaptopodin loss in Cd2ap−/−;Ddn−/− mice (Figure 5C). Similar to the ADR model, a transmission electron micrograph of a segmental collapsing lesion from a Cd2ap−/−;Ddn+/+ mouse demonstrated the attachment of multivacuolated cells, morphologically resembling PECs and originating from the parietal basement membrane, to areas of GBM denudation, whereas the remainder of the GBM was covered by residual, diffusely effaced podocytes with condensed basal actin (Supplemental Figure S4). These results indicate that dendrin-induced loss of synaptopodin-positive podocytes in Cd2ap−/− mice triggers a PEC response reminiscent of that seen in ADR nephropathy,32 suggesting that tuft coverage by activated vPECs is a conserved response to extensive podocyte loss. Of note, genetic ablation of dendrin appeared to slow podocyte loss and, as a result, delay the ensuing PEC activation in Cd2ap−/− mice.
Figure 5.

Dendrin deletion protects against podocyte loss-induced tuft epithelialization by activated parietal cells. A: At 4 weeks, synaptopodin (Synpo) staining is lost in the central part of the glomeruli in Cd2ap−/−;Ddn+/+ mice, but mostly preserved in glomeruli in Cd2ap−/−;Ddn−/− mice, similar to the pattern seen in wild-type mice. PAX2 staining is limited to parietal along Bowman's capsule in all glomeruli shown. B: At 5 weeks, Cd2ap−/−;Ddn+/+ mice show further loss of Synpo staining with complete absence from areas of segmental sclerosis (asterisk). In the sclerotic segments, PAX2-positive epithelial cells [visceralized parietal epithelial cells (vPECs)] populate the tuft (arrows). By contrast, Synpo expression is mostly preserved in Cd2ap−/−;Ddn−/− glomeruli that are largely devoid of sclerosis. Correspondingly, PAX2 staining is limited to the parietal epithelium of Bowman's capsule, indicating that vPECs are absent from the tuft. C: At 6 weeks of age, Synpo staining is severely diminished in glomeruli of Cd2ap−/−;Ddn+/+ mice, with multifocal tuft adhesion and population of the tuft by PAX2-positive vPECs (arrows). Although mesangial expansion becomes more abundant in Cd2ap−/−;Ddn−/− mice, Synpo staining is notably diminished and absent in areas of segmental sclerosis (asterisk). The segmentally sclerosed segment also shows focal population by PAX2-positive vPECs (arrows). Scale bar = 20 μm.
Dendrin Ablation Slows Mesangial Volume Expansion and Podocyte Loss
Dendrin gene silencing reduces podocyte susceptibility to apoptosis in vitro and Cd2ap−/− mice develop podocyte apoptosis at the onset of albuminuria.11,31 We detected podocyte apoptosis in Cd2ap−/−;Ddn+/+ mice at 3 weeks by nuclear blebbing and cleaved caspase 3 colocalization with synaptopodin (Figure 6). No podocyte apoptosis was detected in Cd2ap−/−;Ddn−/− or wild-type littermates (Figure 6). Qualitatively by WT1 labeling, Cd2ap−/−;Ddn+/+ mice displayed a reduction in podocyte number that was particularly noticeable at 5 to 6 weeks of age (Supplemental Figure S5). Podocyte depletion was not detected in Cd2ap−/−;Ddn−/− mice or wild-type littermates at these time points (Supplemental Figure S5). Taken together, these findings suggest that dendrin ablation reduces podocyte loss in Cd2ap−/− mice by the inhibition of podocyte apoptosis.
Figure 6.

Dendrin ablation inhibits podocyte apoptosis in Cd2ap−/− mice. Three-week-old Cd2ap−/−;Ddn+/+ mice show podocyte apoptosis as detected by nuclear blebbing and cleaved caspase 3 staining that colocalizes with synaptopodin. Apoptosis is not detected in the podocytes of wild-type and Cd2ap−/−;Ddn−/− littermates. The insets show the boxed areas at higher magnification. Scale bar = 50 μm.
To more accurately characterize the contribution of dendrin to podocyte loss and glomerular disease progression in an unbiased quantitative fashion, we performed detailed morphometric analyses of glomerular compartment volumes using stereological techniques in Cd2ap−/−;Ddn+/+ and Cd2ap−/−;Ddn−/− mice, as well as wild-type mice (Figure 7). Glomerular volume, mesangial volume, podocyte number, and podocyte volume were measured at 3, 4, 5, and 6 weeks of age. At 3 weeks, we detected a significant increase in mean glomerular volume in Cd2ap−/−;Ddn+/+ mice (mean, 151,804 ± 44,463 μm3) compared to Cd2ap−/−;Ddn−/− mice (107,831 ± 13,648 μm3; P < 0.05, t-test), which showed a similar mean glomerular volume as wild-type littermates (90,307 ± 17,906 μm3; NS, t-test) (Figure 7A). Of note, the difference in glomerular volume coincided with a significant increase in mean mesangial volume in Cd2ap−/−;Ddn+/+ mice (46,120 ± 14,368 μm3) compared to Cd2ap−/−;Ddn−/− mice (27,297 ± 4180 μm3; P < 0.05, t-test) and wild-type littermates (17,035 ± 3453 μm3; P < 0.05, t-test) (Figure 6B). Mean glomerular volume continued to increase in Cd2ap−/−;Ddn+/+ mice through 6 weeks, peaking at 252,704 ± 51,348 μm3 (Figure 7A). Mesangial volume continued to increase in all groups, although the increase in volume for Cd2ap−/−/Ddn+/+ mice was consistently greater than in Cd2ap−/−;Ddn−/− littermates (Figure 7B). We also counted podocyte numbers using the disector/fractionator principle by which nuclei present in the second section of each pair, but not present in the first section, were counted (Supplemental Figure S6). In support of the results in Figure 5 and Supplemental Figure S5, we found a significantly reduced podocyte number at 5 weeks in Cd2ap−/−;Ddn+/+ mice (n ≥ 5: 46 ± 20 podocyte nuclei/glomerulus) compared to Cd2ap−/−;Ddn−/− mice (n ≥ 5: 70 ± 6; P < 0.05, t-test) or wild-type littermates (n ≥ 5: 77 ± 13; P < 0.05, t-test) (Figure 6C). Podocyte depletion continued through the 6-week time point, with Cd2ap−/−;Ddn+/+ mice having a mean podocyte number of 27 ± 6 versus 72 ± 2 for Cd2ap−/−;Ddn−/− mice and 70 ± 4 for wild-type littermates (n ≥ 5; P < 0.01 and 0.004, respectively, t-test) (Figure 7C). The reduction in podocyte number at 5 weeks was associated with an increase in mean podocyte volume for remaining cells in Cd2ap−/−;Ddn+/+ (n ≥ 5, 1072 ± 397 μm3) mice when compared to Cd2ap−/−;Ddn−/− mice (n > 5574 ± 87; P < 0.05, t-test) and wild-type littermates (n ≥ 5, 463 ± 111; P < 0.05, t-test) (Figure 7D). Given the early increase in glomerular volume and subsequent decrease in podocyte number in Cd2ap−/−;Ddn+/+ mice, we also determined podocyte density during disease progression. The increase in mean glomerular volumes at 3 weeks resulted in a lower calculated podocyte density in Cd2ap−/−;Ddn+/+ mice (0.00058 ± 0.00015 podocyte nuclei/μm3) compared to wild-type littermates (0.00080 ± 0.0009; P < 0.05, t-test) and Cd2ap−/− Ddn−/− mice (0.00067 ± 0.0015; NS, t-test) (Figure 7E). At this time point, the decrease in podocyte density was due to the aforementioned increase in mean mesangial and glomerular volumes because mean podocyte numbers were not significantly different (Figure 7, B and C). Overall, Cd2ap−/−;Ddn+/+ mice consistently had a lower podocyte density than Cd2ap−/−;Ddn−/− and wild-type littermates from week 3, with this difference reaching statistical significance compared to Cd2ap−/−;Ddn−/− mice at week 4 (P < 0.005, analysis of variance) (Figure 7E). These findings are important, because a reduction in podocyte density can cause glomerular disease progression.34,35 It was also at 5 weeks that the average podocyte volume for Cd2ap−/− Ddn+/+ mice became significantly greater than that for wild-type (P < 0.05, t-test) and Cd2ap−/−/Ddn−/− (P < 0.05, t-test) littermates (Figure 7D). The simplest interpretation of these results is that mesangial expansion precedes podocyte loss in Cd2ap−/− mice, and that both processes are dendrin dependent. Notably, mesangial and average podocyte volumes could not be measured beyond week 5 for Cd2ap−/−/Ddn+/+ mice because severe sclerosis precluded accurate demarcation of glomerular compartment boundaries.
Figure 7.

Stereological analysis confirms protection of dendrin ablation against glomerular hypertrophy and podocyte loss. A: Glomerular volume is consistently greater in Cd2ap−/−;Ddn+/+ mice compared to Cd2ap−/−;Ddn−/− and Cd2ap+/+;Ddn+/+ littermates, as early as 3 weeks. B: Beginning at 3 weeks, mesangial volume is significantly increased in Cd2ap−/−;Ddn+/+ mice. C: Podocyte number decreases in Cd2ap−/−;Ddn+/+ mice beginning at week 5. D: Average podocyte volume increases significantly in Cd2ap−/−;Ddn+/+ mice at week 5. E: Podocyte density is significantly decreased in Cd2ap−/−;Ddn+/+ mice beginning at week 4. Data are expressed as means ± SD. *P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 comparing Cd2ap−/−;Ddn+/+ versus Cd2ap−/−;Ddn−/− or Cd2ap+/+;Ddn+/+ littermates by analysis of variance.
Cathepsin L Is Not Expressed in Podocytes in Cd2ap−/− Mice
It has been previously postulated that dendrin may be a transcription factor driving cathepsin L expression in Cd2ap−/− mice.13 To test this hypothesis, we analyzed cathepsin L expression by immunohistochemistry in 5-week-old Cd2ap−/−;Ddn+/+, Cd2ap−/−;Ddn−/−, and wild-type littermate control mice. Cathepsin L staining was absent from podocytes in all three phenotypes (Supplemental Figure S7). By contrast, we detected cathepsin L staining in some proximal tubular cells in Cd2ap−/−;Ddn+/+, but not in Cd2ap−/−;Ddn−/− mice or wild-type littermates (Supplemental Figure S7). These data suggest cathepsin L in podocytes does not contribute to glomerular disease progression in Cd2ap−/− mice.
A Dendrin-Regulated Soluble Factor Controls Fibronectin Secretion by Mesangial Cells
Increased expression of the key mesangial matrix component fibronectin has been described in mouse models of glomerulosclerosis, including Cd2ap−/− mice.16,36 Because ablation of dendrin in podocytes reduces mesangial expansion (Figure 7B), we examined whether this was associated with changes in mesangial fibronectin abundance by immunohistochemistry. We found increased fibronectin expression in the mesangial matrix in 4- and 5-week-old Cd2ap−/−;Ddn+/+ mice. This up-regulation of fibronectin was dendrin dependent because it was absent in Cd2ap−/−;Ddn−/− mice (Figure 8A). CD2AP and dendrin are expressed in podocytes, but not in mesangial cells, hinting that the observed effects of dendrin on mesangial volume and fibronectin expression could be regulated by a podocyte-secreted soluble factor. To directly test this hypothesis, we analyzed the effect of conditioned medium derived from CD2AP-depleted, dendrin-depleted, CD2AP and dendrin–co-depleted podocytes, as well as nonsilencing control shRNA-expressing podocytes (Figure 8B), on fibronectin production by cultured MMCs. We found that conditioned medium from dendrin-depleted or CD2AP and dendrin–co-depleted podocytes markedly reduced fibronectin protein abundance in supernatants harvested from MMCs (Figure 8C). Fibronectin expression was not detected in 0.1% fetal calf serum media harvested from untreated MMCs (Figure 8C). The quantification by fold densitometry relative to fetal calf serum media alone confirmed that dendrin depletion or co-depletion of dendrin and CD2AP in podocytes significantly reduced fibronectin production by MMCs (6.36 ± 1.96 and 4.57 ± 0.94, respectively) when compared to CD2AP-depleted (10.17 ± 2.71) or nonsilencing shRNA (9.56 ± 2.21) expressing podocytes (n = 9; P < 0.05 for CD2AP-depleted compared to dendrin-depleted or CD2AP and dendrin–co-depleted studies, t-test) (Figure 8D). We did not detect a significant difference in MMC-derived fibronectin production between conditioned media from dendrin-depleted and dendrin and CD2AP–co-depleted podocytes (n = 9; NS, t-test). To confirm these results with an independent quantitative biochemical approach, we measured fibronectin protein abundance in mesangial cell supernatants using a commercial mouse fibronectin enzyme-linked immunosorbent assay kit (Figure 8E). Consistent with the Western blot results (Figure 8D), media from dendrin-depleted as well as dendrin and CD2AP–co-depleted podocytes induced significantly less fibronectin production by MMCs (3.78 ± 1.50 and 3.96 ± 1.15, respectively) when compared to CD2AP-depleted (5.57 ± 1.51) or nonsilencing shRNA (5.32 ± 1.25) expressing podocytes (n = 4; P < 0.05 for CD2AP-depleted compared to either dendrin-depleted or CD2AP and dendrin–co-depleted studies, t-test) (Figure 8E). Of note, dendrin depletion or co-depletion of dendrin and CD2AP did not affect MMC hypertrophy or proliferation (Supplemental Figure S8, A and B). These results suggest that dendrin drives the release of a soluble factor by podocytes that increases fibronectin production in mesangial cells, thereby altering the composition of the mesangial matrix in Cd2ap−/− mice.
Figure 8.

A dendrin-regulated podocyte-secreted factor increases mesangial fibronectin production. A: Immunohistochemistry reveals increased mesangial fibronectin expression in Cd2ap−/−;Ddn+/+ mice compared to Cd2ap−/−;Ddn−/− and wild-type littermate controls. B: Western blot analysis shows the degree of protein depletion in podocytes expressing shRNAs that target CD2AP or dendrin or both; C: Western blot analysis of fibronectin expression in mesangial cell supernatant after incubation with fetal calf serum (FCS) or the indicated conditioned podocyte supernatants. D: Densitometry quantification of fibronectin expression. E: Enzyme-linked immunosorbent assay quantification of fibronectin expression. Data are expressed as means ± SD. ∗P < 0.05. Scale bar = 20 μm. scr, nonsilencing control shRNA; WT, wild type.
Discussion
Building on seminal studies by Rennke and colleagues,37 work from many groups over the last 25 years has provided compelling evidence that prolonged podocyte injury is the driving force underlying the progression of proteinuric kidney diseases toward ESKD.1 However, to date, little progress has been made toward targeting injurious podocyte signaling to prevent glomerular disease progression. Here, we show that podocyte protection by ablating dendrin signaling is a valid strategy to delay ESKD and death. Importantly, our proof-of-concept study raises the hope that inhibition of pathogenic dendrin signaling in podocytes can be harnessed to delay the loss of kidney function, and the ensuing dialysis-related morbidity and mortality in patients.
More than a decade after the initial description of Cd2ap−/− mice,16 the precise mechanisms of kidney disease progression have not been revealed. Here, we show that similar to ADR nephropathy32 and MAGI-2–deficient mice,33 Cd2ap−/− mice develop light microscopically detectable glomerular lesions by 5 weeks of age, with recruitment of PAX2-positive vPECs to injured tuft segments devoid of synaptopodin expression. Importantly, these changes can be delayed by dendrin ablation. Significant attention has been recently directed toward understanding the pathogenesis and significance of PECs in glomerular disease.38–40 There is evidence that they contribute to the pathogenesis of cellular crescents in crescentic glomerulonephritis,41 pseudocrescents in collapsing glomerulopathy,42 and sclerotic lesions in FSGS.43 Conversely, a subset of podocytes are derived from PECs in juvenile mice, raising the possibility of a podocyte progenitor role under disease conditions, although a functional contribution of PEC-derived epithelial cells to glomerular filtration has not been shown in the adult stage.44 Our results do not clarify whether the PECs we identified are contributing to injury or serve in a regenerative capacity because PAX2 labeling does not distinguish well-described PEC subpopulations.39 Our finding that PAX2 expression in the glomerular tuft coincided with the onset of podocyte depletion in Cd2ap−/− mice suggests that PEC recruitment to the glomerular tuft represents a general response to podocyte loss, independent of the underlying cause of podocyte injury.
Using detailed stereology, we were able to reveal the temporal relationship of three distinct, dendrin-regulated, pathological changes (Figure 9): i) proteinuria with concurrent ultrastructural foot process changes; ii) mesangial volume expansion; and iii) loss of podocytes with ensuing hypertrophy of remaining podocytes. Glomerular volume increase with mesangial expansion represents an early structural change in Cd2ap−/− mice; it can be detected as early as 3 weeks of age and coincides with the onset of proteinuria and early podocyte foot process changes. Importantly, at this stage, podocyte numbers are normal. Significant podocyte depletion does not occur until 2 weeks later (5 weeks of age) and is accompanied by hypertrophy of the remaining podocytes. These pathogenic changes are dendrin mediated because dendrin ablation significantly slowed proteinuria, ultrastructural foot process changes, mesangial volume expansion, podocyte depletion, development of glomerulosclerosis, and renal failure, thereby increasing the life span of Cd2ap−/− mice. A direct versus mediated role for dendrin in these diverse and complex pathogenic consequences will need to be carefully considered in future studies.
Figure 9.

Dual role for dendrin in glomerular disease progression. Genetic deletion of Cd2ap induces nuclear relocation of dendrin, which leads to mesangial volume expansion and increased mesangial fibronectin that is driven by a dendrin-regulated podocyte-secreted factor. Nuclear dendrin also causes direct podocyte injury, leading to proteinuria and foot process effacement (FPE) and, at a later stage, to podocyte loss, thereby furthering the progression of glomerular disease to renal failure and death of the animals.
Nuclear relocation of dendrin promotes apoptosis of podocytes in vitro,11 which can be blocked by dendrin depletion or YAP-mediated inhibition of dendrin signaling.12 Now, we find a beneficial effect of dendrin ablation on podocyte survival in vivo, thereby confirming the previous in vitro data and underscoring the in vivo relevance of studying podocyte cell death mechanisms. Of note, the observed triple effect of dendrin ablation on podocyte loss as a late event, but also on proteinuria/structural foot process changes and mesangial volume expansion as early events preceding podocyte loss, suggests that targeting pathogenic dendrin signaling should offer a superior therapeutic benefit compared to targeting of cell death mechanisms, eg, blocking apoptosis, which would be expected to have undesired systemic side effects. This notion is further supported by the observation that dendrin is not an essential protein; dendrin deletion has no adverse effect at baseline,15 making dendrin blockade an attractive therapeutic target.
The identification of a dendrin-dependent podocyte to mesangial cell signaling axis that increases fibronectin production by mesangial cells offers a novel mechanism for glomerular disease progression through intraglomerular crosstalk between injured podocytes and mesangial cells, similar to recently described podocyte glomerular–endothelial cell crosstalk.45 The observed effect of dendrin on mesangial volume and matrix composition raises the intriguing possibility that mesangial matrix changes seen in other renal diseases, most importantly diabetic nephropathy46 or TGF-β transgenic mice,47 are also a consequence of primary podocyte injury, further supporting the notion that prevention of podocyte injury represents a key event in the prevention of renal disease progression to ESKD. Future studies will be required to test whether dendrin plays a role in diabetic nephropathy. Additional studies will also be necessary to reveal the molecular identity of the podocyte-secreted factor(s) responsible for increasing mesangial fibronectin secretion.
In conclusion, genetic ablation of dendrin significantly prolongs preservation of kidney function, thereby increasing the life span of Cd2ap−/− mice (Figure 9). In addition to mediating proteinuria and podocyte loss, nuclear dendrin promotes the release of podocyte-secreted factor(s) that induce fibronectin secretion from mesangial cells, thereby contributing to mesangial matrix expansion occurring in conjunction with early structural podocyte changes. Our results suggest that inhibition of nuclear dendrin signaling or rerouting of dendrin from the nucleus to the cytoplasm may be a promising starting point for the development of podocyte-protective therapies aimed at delaying the loss of renal function and the need for renal replacement therapy, which is associated with high mortality and tremendous financial costs.
Acknowledgments
We thank Anna Greka (Boston, MA) for helpful discussion and Detlef Schlondorff for providing mouse mesangial cells. Confocal imaging was performed at the Microscopy Shared Resource Facility at the Icahn School of Medicine at Mount Sinai.
Footnotes
See related Commentary on page 2072
Supported by NIH grant DK081617 and a NephCure Foundation Young Investigator grant (K.N.C.), NIH grants DK057683, DK062472, and DK091218 (P.M.), and NIH grant DK093783 (A.W.). Morphometric studies supported by the National Institute on Minority Health and Health Disparities (grant U54MD007598). JEM1200-EX electron microscope at the Electron Imaging Center for NanoMachines supported by NIH (1S10RR23057) and the California NanoSystems Institute at the University of California Los Angeles.
Disclosures: None declared.
A.W. and J.S.W. contributed equally to this work.
Supplemental material for this article can be found at http://dx.doi.org/10.1016/j.ajpath.2015.04.011.
Supplemental Data
Temporal changes in the subcellular distribution of dendrin in Cd2ap−/− mice. Dendrin expression in wild-type control mice is confined to the cytoplasm (upper panels). Partial colocalization with the nuclear protein WT1 reveals nuclear localization of dendrin in a subset of podocytes in 3-week-old Cd2ap−/− mice (middle panels). At 4 weeks of age, dendrin is found in the nuclei of all podocytes in Cd2ap−/− mice (lower panels).
Genetic deletion of dendrin in cd2ap-deficient mice. A: Mating strategy for the generation of wild-type Cd2ap+/+;Ddn+/+, Cd2ap−/−;Ddn+/+ single-null, and Cd2ap−/−;Ddn−/− double-null mice. B: Genotyping PCR for Cd2ap and Ddn.
Glomerular disease progression in Cd2ap−/−;Ddn−/− mice at 8 and 12 weeks of age. A: Proteinuria persisted in Cd2ap−/−;Ddn−/− mice beyond 8 weeks and is consistently greater than in Cd2ap+/+;Ddn+/+ littermates. At these time points, all Cd2ap−/−Ddn+/+ mice had already died. B:Cd2ap−/−;Ddn−/− mice eventually also showed substantial segmental glomerular hyalinosis, podocyte injury, and extensive chronic changes of the tubulointerstitium as early as 8 weeks, however, at a later time point than Cd2ap−/−;Ddn+/+ mice. ∗P < 0.05. Scale bar = 20 μm.
Visceralized parietal epithelial cell (vPEC) attachment to denuded glomerular tuft in Cd2ap−/− mice. Transmission electron microscopy image of a glomerulus in a 6-week-old Cd2ap−/− mouse. The glomerular basement membrane is mostly covered by diffusely effaced podocytes with the typical condensed actin (arrowheads), but shows the presence of several areas of denudation (arrows) where multivacuolated vPECs attach. Scale bar = 500 nm.
Dendrin ablation reduces podocyte loss in Cd2ap−/− mice. WT1 labelling qualitatively shows reduced podocyte number in Cd2ap−/−;Ddn+/+ glomeruli (middle panels) at 5 and 6 weeks compared to glomeruli from Cd2ap−/−;Ddn−/− (lower panels) and wild-type littermates (top panels). Scale bars: 50 μm.
Illustration of disector/fractionator method for measuring podocyte number. Toluidine blue staining shows quantification of podocyte number in adjacent 1-μm-thick EPON sections. Only podocyte nuclei (asterisk) present in the first section, but not present in the second look-up section (arrows), were counted.
Cathepsin L is not induced in podocytes of Cd2ap-deficient mice. Cathepsin L staining is absent from glomeruli in wild-type, Cd2ap−/−;Ddn+/+, and Cd2ap−/−;Ddn−/− mice. Robust staining for cathepsin L is seen in a subset of proximal tubular epithelial cells in Cd2ap−/−;Ddn+/+, serving as positive control. Scale bar = 20 μm.
Conditioned podocyte media do not affect mesangial cell proliferation or hypertrophy. A: Conditioned media from control, CD2AP-depleted, and CD2AP and dendrin–co-depleted podocytes do not affect cell proliferation, as revealed by CellTiter-Glo assay. B: Protein content, as readout of cellular hypertrophy, is not different between mesangial cells treated with conditioned podocyte media from control, CD2AP-depleted, and CD2AP and dendrin–co-depleted podocytes. (n = 7).
References
- 1.Greka A., Mundel P. Cell biology and pathology of podocytes. Annu Rev Physiol. 2012;74:299–323. doi: 10.1146/annurev-physiol-020911-153238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wiggins R.C. The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int. 2007;71:1205–1214. doi: 10.1038/sj.ki.5002222. [DOI] [PubMed] [Google Scholar]
- 3.Agrawal V., Marinescu V., Agarwal M., McCullough P.A. Cardiovascular implications of proteinuria: an indicator of chronic kidney disease. Nat Rev Cardiol. 2009;6:301–311. doi: 10.1038/nrcardio.2009.11. [DOI] [PubMed] [Google Scholar]
- 4.Kim Y.H., Goyal M., Kurnit D., Wharram B., Wiggins J., Holzman L., Kershaw D., Wiggins R. Podocyte depletion and glomerulosclerosis have a direct relationship in the PAN-treated rat. Kidney Int. 2001;60:957–968. doi: 10.1046/j.1523-1755.2001.060003957.x. [DOI] [PubMed] [Google Scholar]
- 5.Wharram B.L., Goyal M., Wiggins J.E., Sanden S.K., Hussain S., Filipiak W.E., Saunders T.L., Dysko R.C., Kohno K., Holzman L.B., Wiggins R.C. Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol. 2005;16:2941–2952. doi: 10.1681/ASN.2005010055. [DOI] [PubMed] [Google Scholar]
- 6.Pagtalunan M.E., Miller P.L., Jumping-Eagle S., Nelson R.G., Myers B.D., Rennke H.G., Coplon N.S., Sun L., Meyer T.W. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest. 1997;99:342–348. doi: 10.1172/JCI119163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steffes M.W., Schmidt D., McCrery R., Basgen J.M. Glomerular cell number in normal subjects and in type 1 diabetic patients. Kidney Int. 2001;59:2104–2113. doi: 10.1046/j.1523-1755.2001.00725.x. [DOI] [PubMed] [Google Scholar]
- 8.White K.E., Bilous R.W., Marshall S.M., El Nahas M., Remuzzi G., Piras G., De Cosmo S., Viberti G. Podocyte number in normotensive type 1 diabetic patients with albuminuria. Diabetes. 2002;51:3083–3089. doi: 10.2337/diabetes.51.10.3083. [DOI] [PubMed] [Google Scholar]
- 9.Lemley K.V., Lafayette R.A., Safai M., Derby G., Blouch K., Squarer A., Myers B.D. Podocytopenia and disease severity in IgA nephropathy. Kidney Int. 2002;61:1475–1485. doi: 10.1046/j.1523-1755.2002.00269.x. [DOI] [PubMed] [Google Scholar]
- 10.Shankland S.J. The podocyte's response to injury: role in proteinuria and glomerulosclerosis. Kidney Int. 2006;69:2131–2147. doi: 10.1038/sj.ki.5000410. [DOI] [PubMed] [Google Scholar]
- 11.Asanuma K., Campbell K.N., Kim K., Faul C., Mundel P. Nuclear relocation of the nephrin and CD2AP-binding protein dendrin promotes apoptosis of podocytes. Proc Natl Acad Sci U S A. 2007;104:10134–10139. doi: 10.1073/pnas.0700917104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Campbell K.N., Wong J.S., Gupta R., Asanuma K., Sudol M., He J.C., Mundel P. Yes-associated protein (YAP) promotes cell survival by inhibiting proapoptotic dendrin signaling. J Biol Chem. 2013;288:17057–17062. doi: 10.1074/jbc.C113.457390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yaddanapudi S., Altintas M.M., Kistler A.D., Fernandez I., Moller C.C., Wei C., Peev V., Flesche J.B., Forst A.L., Li J., Patrakka J., Xiao Z., Grahammer F., Schiffer M., Lohmuller T., Reinheckel T., Gu C., Huber T.B., Ju W., Bitzer M., Rastaldi M.P., Ruiz P., Tryggvason K., Shaw A.S., Faul C., Sever S., Reiser J. CD2AP in mouse and human podocytes controls a proteolytic program that regulates cytoskeletal structure and cellular survival. J Clin Invest. 2011;121:3965–3980. doi: 10.1172/JCI58552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Asanuma K., Akiba-Takagi M., Kodama F., Asao R., Nagai Y., Lydia A., Fukuda H., Tanaka E., Shibata T., Takahara H., Hidaka T., Asanuma E., Kominami E., Ueno T., Tomino Y. Dendrin location in podocytes is associated with disease progression in animal and human glomerulopathy. Am J Nephrol. 2011;33:537–549. doi: 10.1159/000327995. [DOI] [PubMed] [Google Scholar]
- 15.Xiao Z., Rodriguez P.Q., He L., Betsholtz C., Tryggvason K., Patrakka J. Wtip- and gadd45a-interacting protein dendrin is not crucial for the development or maintenance of the glomerular filtration barrier. PLoS One. 2013;8:e83133. doi: 10.1371/journal.pone.0083133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shih N.Y., Li J., Karpitskii V., Nguyen A., Dustin M.L., Kanagawa O., Miner J.H., Shaw A.S. Congenital nephrotic syndrome in mice lacking CD2-associated protein. Science. 1999;286:312–315. doi: 10.1126/science.286.5438.312. [DOI] [PubMed] [Google Scholar]
- 17.Grunkemeyer J.A., Kwoh C., Huber T.B., Shaw A.S. CD2-associated protein (CD2AP) expression in podocytes rescues lethality of CD2AP deficiency. J Biol Chem. 2005;280:29677–29681. doi: 10.1074/jbc.M504004200. [DOI] [PubMed] [Google Scholar]
- 18.Benoit G., Machuca E., Nevo F., Gribouval O., Lepage D., Antignac C. Analysis of recessive CD2AP and ACTN4 mutations in steroid-resistant nephrotic syndrome. Pediatr Nephrol. 2010;25:445–451. doi: 10.1007/s00467-009-1372-x. [DOI] [PubMed] [Google Scholar]
- 19.Gigante M., Pontrelli P., Montemurno E., Roca L., Aucella F., Penza R., Caridi G., Ranieri E., Ghiggeri G.M., Gesualdo L. CD2AP mutations are associated with sporadic nephrotic syndrome and focal segmental glomerulosclerosis (FSGS) Nephrol Dial Transplant. 2009;24:1858–1864. doi: 10.1093/ndt/gfn712. [DOI] [PubMed] [Google Scholar]
- 20.Lowik M.M., Groenen P.J., Pronk I., Lilien M.R., Goldschmeding R., Dijkman H.B., Levtchenko E.N., Monnens L.A., van den Heuvel L.P. Focal segmental glomerulosclerosis in a patient homozygous for a CD2AP mutation. Kidney Int. 2007;72:1198–1203. doi: 10.1038/sj.ki.5002469. [DOI] [PubMed] [Google Scholar]
- 21.Bai X.Y., Basgen J.M. Podocyte number in the maturing rat kidney. Am J Nephrol. 2011;33:91–96. doi: 10.1159/000322701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gundersen H.J. Stereology of arbitrary particles. A review of unbiased number and size estimators and the presentation of some new ones, in memory of William R. Thompson. J Microsc. 1986;143:3–45. [PubMed] [Google Scholar]
- 23.Nyengaard J.R. Stereologic methods and their application in kidney research. J Am Soc Nephrol. 1999;10:1100–1123. doi: 10.1681/ASN.V1051100. [DOI] [PubMed] [Google Scholar]
- 24.Gundersen H.J., Jensen E.B. The efficiency of systematic sampling in stereology and its prediction. J Microsc. 1987;147:229–263. doi: 10.1111/j.1365-2818.1987.tb02837.x. [DOI] [PubMed] [Google Scholar]
- 25.Basgen J.M., Sobin C. Early chronic low-level lead exposure produces glomerular hypertrophy in young C57BL/6J mice. Toxicol Lett. 2014;225:48–56. doi: 10.1016/j.toxlet.2013.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mundel P., Reiser J., Zuniga Mejia Borja A., Pavenstadt H., Davidson G.R., Kriz W., Zeller R. Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp Cell Res. 1997;236:248–258. doi: 10.1006/excr.1997.3739. [DOI] [PubMed] [Google Scholar]
- 27.Tonelli M., Jose P., Curhan G., Sacks F., Braunwald E., Pfeffer M. Proteinuria, impaired kidney function, and adverse outcomes in people with coronary disease: analysis of a previously conducted randomised trial. BMJ. 2006;332:1426. doi: 10.1136/bmj.38814.566019.2F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jefferson J.A., Shankland S.J., Pichler R.H. Proteinuria in diabetic kidney disease: a mechanistic viewpoint. Kidney Int. 2008;74:22–36. doi: 10.1038/ki.2008.128. [DOI] [PubMed] [Google Scholar]
- 29.Asanuma K., Mundel P. The role of podocytes in glomerular pathobiology. Clin Exp Nephrol. 2003;7:255–259. doi: 10.1007/s10157-003-0259-6. [DOI] [PubMed] [Google Scholar]
- 30.Donadio J.V., Bergstralh E.J., Grande J.P., Rademcher D.M. Proteinuria patterns and their association with subsequent end-stage renal disease in IgA nephropathy. Nephrol Dial Transplant. 2002;17:1197–1203. doi: 10.1093/ndt/17.7.1197. [DOI] [PubMed] [Google Scholar]
- 31.Schiffer M., Mundel P., Shaw A.S., Bottinger E.P. A novel role for the adaptor molecule CD2-associated protein in transforming growth factor-beta-induced apoptosis. J Biol Chem. 2004;279:37004–37012. doi: 10.1074/jbc.M403534200. [DOI] [PubMed] [Google Scholar]
- 32.Hakroush S., Cebulla A., Schaldecker T., Behr D., Mundel P., Weins A. Extensive podocyte loss triggers a rapid parietal epithelial cell response. J Am Soc Nephrol. 2014;25:927–938. doi: 10.1681/ASN.2013070687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balbas M.D., Burgess M.R., Murali R., Wongvipat J., Skaggs B.J., Mundel P., Weins A., Sawyers C.L. MAGI-2 scaffold protein is critical for kidney barrier function. Proc Natl Acad Sci U S A. 2014;111:14876–14881. doi: 10.1073/pnas.1417297111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wiggins J.E., Goyal M., Sanden S.K., Wharram B.L., Shedden K.A., Misek D.E., Kuick R.D., Wiggins R.C. Podocyte hypertrophy, “adaptation,” and “decompensation” associated with glomerular enlargement and glomerulosclerosis in the aging rat: prevention by calorie restriction. J Am Soc Nephrol. 2005;16:2953–2966. doi: 10.1681/ASN.2005050488. [DOI] [PubMed] [Google Scholar]
- 35.Fukuda A., Chowdhury M.A., Venkatareddy M.P., Wang S.Q., Nishizono R., Suzuki T., Wickman L.T., Wiggins J.E., Muchayi T., Fingar D., Shedden K.A., Inoki K., Wiggins R.C. Growth-dependent podocyte failure causes glomerulosclerosis. J Am Soc Nephrol. 2012;23:1351–1363. doi: 10.1681/ASN.2012030271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bergijk E.C., Baelde H.J., De Heer E., Killen P.D., Bruijn J.A. Specific accumulation of exogenous fibronectin in experimental glomerulosclerosis. J Pathol. 1995;176:191–199. doi: 10.1002/path.1711760213. [DOI] [PubMed] [Google Scholar]
- 37.Fries J.W., Sandstrom D.J., Meyer T.W., Rennke H.G. Glomerular hypertrophy and epithelial cell injury modulate progressive glomerulosclerosis in the rat. Lab Invest. 1989;60:205–218. [PubMed] [Google Scholar]
- 38.Smeets B., Moeller M.J. Parietal epithelial cells and podocytes in glomerular diseases. Semin Nephrol. 2012;32:357–367. doi: 10.1016/j.semnephrol.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 39.Shankland S.J., Smeets B., Pippin J.W., Moeller M.J. The emergence of the glomerular parietal epithelial cell. Nat Rev Nephrol. 2014;10:158–173. doi: 10.1038/nrneph.2014.1. [DOI] [PubMed] [Google Scholar]
- 40.Romagnani P., Remuzzi G. Renal progenitors in non-diabetic and diabetic nephropathies. Trends Endocrinol Metab. 2013;24:13–20. doi: 10.1016/j.tem.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 41.Smeets B., Uhlig S., Fuss A., Mooren F., Wetzels J.F., Floege J., Moeller M.J. Tracing the origin of glomerular extracapillary lesions from parietal epithelial cells. J Am Soc Nephrol. 2009;20:2604–2615. doi: 10.1681/ASN.2009010122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suzuki T., Matsusaka T., Nakayama M., Asano T., Watanabe T., Ichikawa I., Nagata M. Genetic podocyte lineage reveals progressive podocytopenia with parietal cell hyperplasia in a murine model of cellular/collapsing focal segmental glomerulosclerosis. Am J Pathol. 2009;174:1675–1682. doi: 10.2353/ajpath.2009.080789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smeets B., Kuppe C., Sicking E.M., Fuss A., Jirak P., van Kuppevelt T.H., Endlich K., Wetzels J.F., Grone H.J., Floege J., Moeller M.J. Parietal epithelial cells participate in the formation of sclerotic lesions in focal segmental glomerulosclerosis. J Am Soc Nephrol. 2011;22:1262–1274. doi: 10.1681/ASN.2010090970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Appel D., Kershaw D.B., Smeets B., Yuan G., Fuss A., Frye B., Elger M., Kriz W., Floege J., Moeller M.J. Recruitment of podocytes from glomerular parietal epithelial cells. J Am Soc Nephrol. 2009;20:333–343. doi: 10.1681/ASN.2008070795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Daehn I., Casalena G., Zhang T., Shi S., Fenninger F., Barasch N., Yu L., D'Agati V., Schlondorff D., Kriz W., Haraldsson B., Bottinger E.P. Endothelial mitochondrial oxidative stress determines podocyte depletion in segmental glomerulosclerosis. J Clin Invest. 2014;124:1608–1621. doi: 10.1172/JCI71195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Steffes M.W., Bilous R.W., Sutherland D.E., Mauer S.M. Cell and matrix components of the glomerular mesangium in type I diabetes. Diabetes. 1992;41:679–684. doi: 10.2337/diab.41.6.679. [DOI] [PubMed] [Google Scholar]
- 47.Schiffer M., Bitzer M., Roberts I.S., Kopp J.B., ten Dijke P., Mundel P., Bottinger E.P. Apoptosis in podocytes induced by TGF-beta and Smad7. J Clin Invest. 2001;108:807–816. doi: 10.1172/JCI12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Temporal changes in the subcellular distribution of dendrin in Cd2ap−/− mice. Dendrin expression in wild-type control mice is confined to the cytoplasm (upper panels). Partial colocalization with the nuclear protein WT1 reveals nuclear localization of dendrin in a subset of podocytes in 3-week-old Cd2ap−/− mice (middle panels). At 4 weeks of age, dendrin is found in the nuclei of all podocytes in Cd2ap−/− mice (lower panels).
Genetic deletion of dendrin in cd2ap-deficient mice. A: Mating strategy for the generation of wild-type Cd2ap+/+;Ddn+/+, Cd2ap−/−;Ddn+/+ single-null, and Cd2ap−/−;Ddn−/− double-null mice. B: Genotyping PCR for Cd2ap and Ddn.
Glomerular disease progression in Cd2ap−/−;Ddn−/− mice at 8 and 12 weeks of age. A: Proteinuria persisted in Cd2ap−/−;Ddn−/− mice beyond 8 weeks and is consistently greater than in Cd2ap+/+;Ddn+/+ littermates. At these time points, all Cd2ap−/−Ddn+/+ mice had already died. B:Cd2ap−/−;Ddn−/− mice eventually also showed substantial segmental glomerular hyalinosis, podocyte injury, and extensive chronic changes of the tubulointerstitium as early as 8 weeks, however, at a later time point than Cd2ap−/−;Ddn+/+ mice. ∗P < 0.05. Scale bar = 20 μm.
Visceralized parietal epithelial cell (vPEC) attachment to denuded glomerular tuft in Cd2ap−/− mice. Transmission electron microscopy image of a glomerulus in a 6-week-old Cd2ap−/− mouse. The glomerular basement membrane is mostly covered by diffusely effaced podocytes with the typical condensed actin (arrowheads), but shows the presence of several areas of denudation (arrows) where multivacuolated vPECs attach. Scale bar = 500 nm.
Dendrin ablation reduces podocyte loss in Cd2ap−/− mice. WT1 labelling qualitatively shows reduced podocyte number in Cd2ap−/−;Ddn+/+ glomeruli (middle panels) at 5 and 6 weeks compared to glomeruli from Cd2ap−/−;Ddn−/− (lower panels) and wild-type littermates (top panels). Scale bars: 50 μm.
Illustration of disector/fractionator method for measuring podocyte number. Toluidine blue staining shows quantification of podocyte number in adjacent 1-μm-thick EPON sections. Only podocyte nuclei (asterisk) present in the first section, but not present in the second look-up section (arrows), were counted.
Cathepsin L is not induced in podocytes of Cd2ap-deficient mice. Cathepsin L staining is absent from glomeruli in wild-type, Cd2ap−/−;Ddn+/+, and Cd2ap−/−;Ddn−/− mice. Robust staining for cathepsin L is seen in a subset of proximal tubular epithelial cells in Cd2ap−/−;Ddn+/+, serving as positive control. Scale bar = 20 μm.
Conditioned podocyte media do not affect mesangial cell proliferation or hypertrophy. A: Conditioned media from control, CD2AP-depleted, and CD2AP and dendrin–co-depleted podocytes do not affect cell proliferation, as revealed by CellTiter-Glo assay. B: Protein content, as readout of cellular hypertrophy, is not different between mesangial cells treated with conditioned podocyte media from control, CD2AP-depleted, and CD2AP and dendrin–co-depleted podocytes. (n = 7).