

Abstract
Background
Pigmented purpuric dermatoses (PPD) are a spectrum of disorders characterized by a distinct purpuric rash. Although PPD can be easily diagnosed, the disease entity remains an enigma and a therapeutic challenge.
Objective
The purpose of this study was to investigate the characteristics and clinical manifestations of PPD and to elucidate the relationship between assumed etiologic factors and the clinical manifestations of PPD and treatment responses.
Methods
Retrograde analyses were performed to identify appropriate PPD patients who visited Korea University Medical Center Anam Hospital from 2002 to 2012.
Results
Information on 113 patients with PPD was analyzed, and 38 subjects with skin biopsy were included for this study. Schamberg's disease was the most frequent clinical type (60.5%). Concomitant diseases included hypertension (15.8%), diabetes (10.5%), and others. Associated medication histories included statins (13.2%), beta blockers (10.5%), and others. Possibly associated etiologic factors were recent upper respiratory infection (5.3%), high orthostatic pressure due to prolonged standing (2.6%), and strenuous exercise (2.6%). A total of 36 patients (94.7%) were treated with one or more treatment methods, including oral antihistamines, pentoxifylline, topical steroids, and/or phototherapy. There was no significant difference in disease progress according to underlying diseases, medications, or association factors (p>0.05).
Conclusion
Our overall results were grossly consistent with the existing literature, excluding several findings. Although a possible relationship between PPD and cardiovascular disease or cardiovascular medication was proposed at the beginning of the study, no statistically significant correlations were found according to the specific clinical types and treatment responses (p>0.05).
Keywords: Classification, Etiology, Pigmented purpuric dermatoses, Therapy
INTRODUCTION
Pigmented purpuric dermatosis (PPD) is a general term used to describe a group of chronic and relapsing cutaneous lesions. Such lesions are characterized by common petechiae and pigmentary macules of the skin that are primarily localized to the lower limbs; the histomorphologic features include superficial lymphocytic infiltration and marked hemosiderin deposition with erythrocyte extravasation1,2.
PPD is generally categorized into six groups: progressive PPD (i.e., Schamberg's disease), purpura annularis telangiectodes (i.e., Majocchi's disease), lichen aureus, PPD of Gougerot-Blum, itching purpura, and eczematid-like purpura of Doucas-Kapetanakis. Although the clinical patterns of PPD may represent different features of the same disorder with a similar histopathology, this does not generally influence management or prognosis2,3,4. The cause of PPD is unknown, but several underlying diseases and drugs have been suggested to be associated with PPD, such as chronic venous hypertension, diabetes mellitus, bezafibrate, and others2,4. There are several published case reports of certain clinical types of PPD or possible causes, but few systematic reviews or large-scale studies on PPD have been performed.
Therefore, this study investigated the characteristics and clinical manifestations of PPD and elucidated the relationships among the assumed etiologic factors, clinical manifestations of PPD, and treatment responses in the Korean population.
MATERIALS AND METHODS
Study participants
A total of 113 patients diagnosed with PPD were retrospectively selected from among patients who visited the Dermatology Clinic of Korea University College of Medicine (KUMC), Anam Hospital from January 2002 to December 2012. Among the 113 patients, only 38 underwent skin biopsy and were included. The exclusion criteria were lack of clinical photos or related medical history and ambiguous information about clinical progress. The diagnosis of PPD was made by clinical observation with or without histopathological examination by dermatologists.
Study design
Patient data including sex, age, disease duration, underlying illness and medication, area of involvement, clinical type, histologic findings, treatment methods, and clinical progress were gathered retrospectively from the electronic medical records of KUMC, the archive of clinical photographs, and pathologic slides. This study was initially designed to investigate the growing number of PPD patients who had a history of cardiovascular disease and/or received medications from the Department of Cardiovascular, KUMC. Therefore, we focused on underlying illnesses and medications used to treat cardiovascular diseases to analyze possible associations with PPD. Archived histopathologic slides of the patients were evaluated, and typical findings and atypical features were analyzed according to clinical type. Furthermore, determine whether there were differences in outcome with respect to clinical characteristics or specific treatment methods. The overall progress of patients with PPD was classified into the following four categories: (1) completely resolved, (2) substantially improved, (3) partially improved, and (4) unimproved or aggravated. For multivariate analysis, the favorable categories (1) and (2) were classified as "responsive," and categories (3) and (4) were classified as "resistant." Statistical analysis was performed using Fisher's exact test, and the level of significance was set at p<0.05. All analyses were performed using IBM SPSS Statistics ver. 20.0 (IBM Co., Armonk, NY, USA).
RESULTS
Sex and age
There were a total of 38 patients including 18 men (47.4%) and 20 women (52.6%); the male to female ratio was 1:1.11. The overall mean age at diagnosis was 42.6±19.3 years, ranging from 13~74 years. The mean ages of male and female patients were 37.4±21.2 and 47.3±16.6 years, respectively.
Area of involvement
The most commonly involved area was the lower extremities (78.9%), followed by the upper and lower extremities (13.2%). Upper and lower extremity with trunk involvement (5.3%) and upper extremity and trunk involvement (2.6%) were less common.
Clinical characteristics
On the basis of clinical and histopathological features, the 38 patients were categorized into five clinical groups: Schamberg's disease (n=23, 60.5%) (Fig. 1A), Majocchi's disease (n=8, 21.1%) (Fig. 1B), lichen aureus (n=2, 5.3%) (Fig. 1C), PPD of Gougerot-Blum (n=3, 7.9%) (Fig. 2A) and eczematid-like purpura of Doucas-Kapetanakis (n=2, 5.3%) (Fig. 2B, C). The sex, age, and involved areas of each clinical group are shown in Table 1. There were five adolescent patients (13.2%), and the youngest was 13 years old. Most adolescent patients had Majocchi's disease and not Schamberg's disease.
Fig. 1. (A) Clinical findings of Schamberg's disease. Characteristic "cayenne pepper" pigmentation on the lower leg. (B) Clinical findings of Majocchi's disease. Red to lightbrown annular patches on the lower leg exhibiting peripheral extension. (C) Clinical findings of lichen aureus. Multiple lichenoid purpuric papules on the lower leg.

Fig. 2. (A) Clinical findings of pigmented purpuric dermatosis of Gougerot-Blum. Lichenoid papules that tend to fuse into plaques of redbrown-colored pigmentation. (B, C) Clinical findings of eczematid-like purpura of Doucas-Kapetanakis. Severely pruritic diffusely eczematous purpuric patches are visible on both lower extremities.

Table 1. Numbers, sexual distributions, average ages and involved areas of 38 patients who received skin biopsy according to the clinical characteristics.
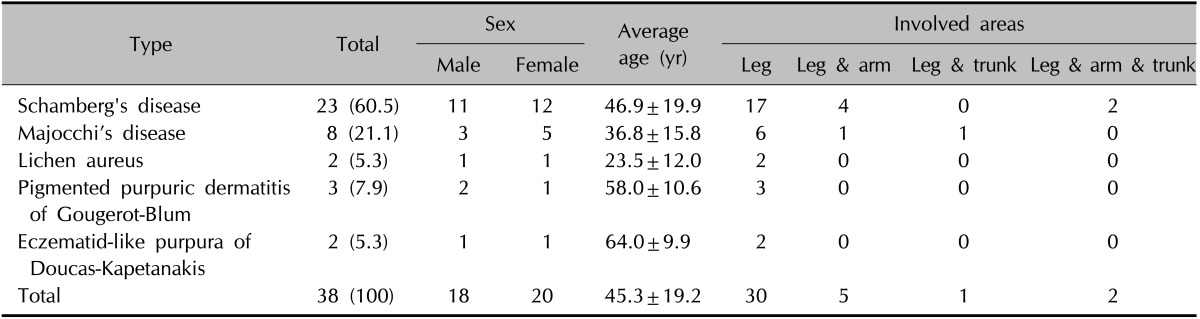
Values are presented as number (%), mean±standard deviation, or number only.
Histopathology
Histologically, PPD showed numerous findings including perivascular lymphocytic infiltration, red blood cell extravasation, hemosiderin deposition, endothelial cell swelling, spongiosis, lymphocyte exocytosis, and lichenoid lymphocytic infiltration (Fig. 3A~D)2,5,6,7. Other findings recorded for further evaluation included interface change and basal hyperpigmentation. The most common pathological characteristic was perivascular lymphocytic infiltration (79.0%), followed by endothelial cell swelling (52.6%), red blood cell extravasation (50.0%), interface changes (42.1%), lichenoid lymphocytic infiltration (39.5%), basal cell hyperpigmentation (29.0%), hemosiderin deposition (26.3%), lymphocyte exocytosis (18.4%), and spongiosis (13.2%) (Table 2).
Fig. 3. (A, B) Histologic findings of Schamberg's disease and (C, D) Majocchi's disease. (A) Dense perivascular lymphocytic infiltrations from the upper to deep dermis (H&E, ×40). (B) Mild spongiosis and vacuolar degeneration, extravasated red blood cells without the presence of leukocytoclastic vasculitis (H&E, ×100). (C) Lichenoid and superficial perivascular lymphocytic infiltrations (H&E, ×40). (D) Marked vacuolar degenerations, extravasated red blood cells, and numerous hemosiderin-laden macrophages without the presence of leukocytoclastic vasculitis (H&E, ×100).

Table 2. Histological findings of the 38 patients with pigmented purpuric dermatoses.
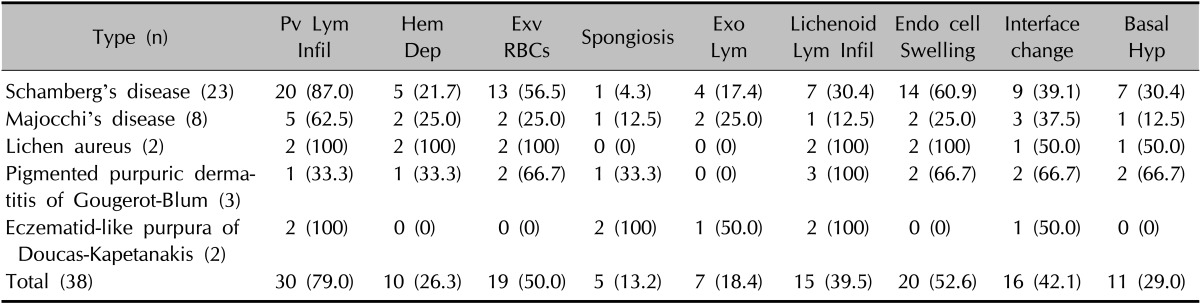
Values are presented as number (%). Pv: perivascular, Lym: lymphocytic or lymphocyte, Infil: infiltration, Hem: hemosiderin, Dep: deposition, Exv: extravasation, RBCs: red blood cells, Exo: exocytosis, Endo: endothelial, Hyp: hyperpigmentation.
Combined underlying diseases, etiologic factors, and medications
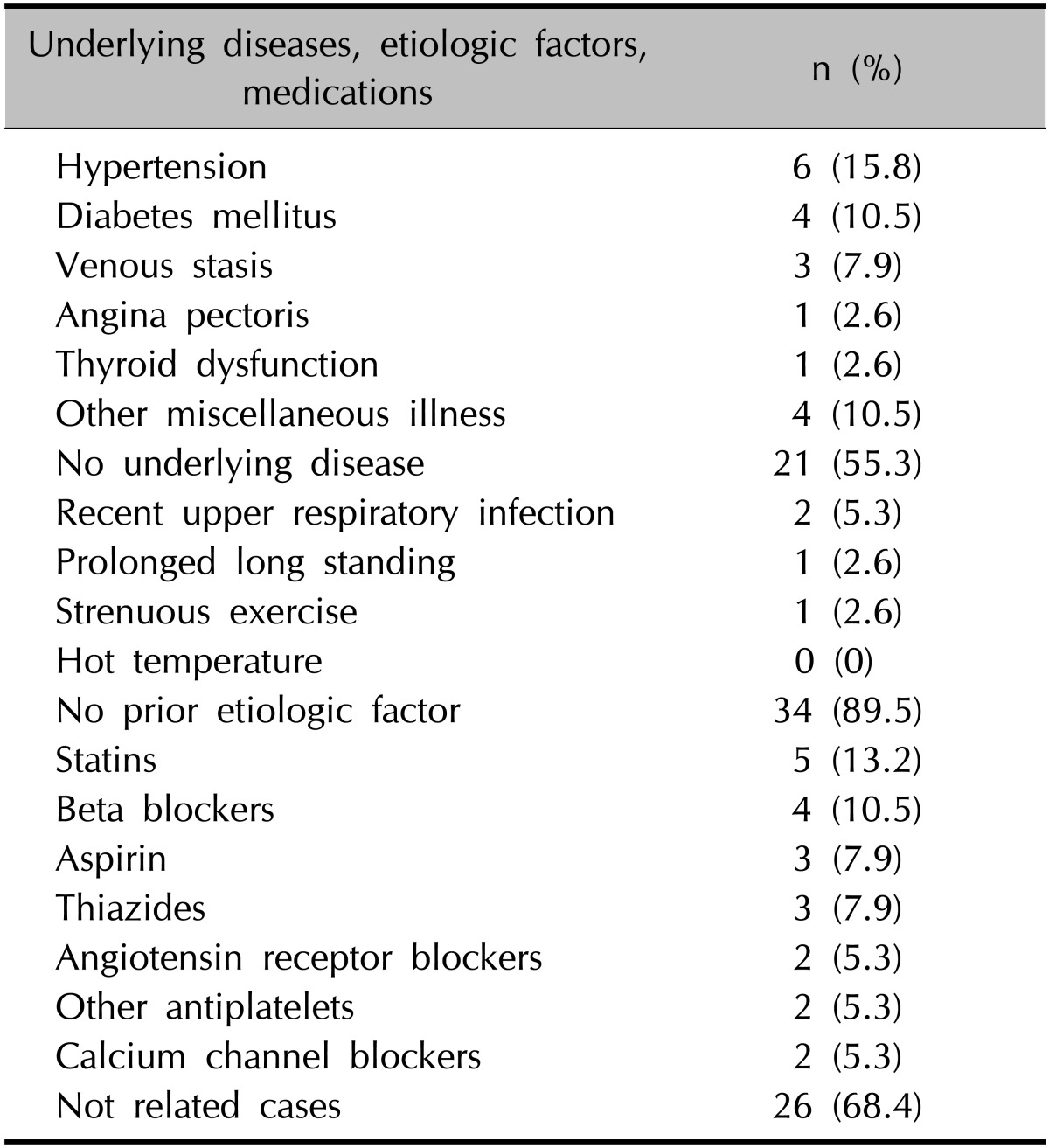
The concomitant underlying diseases were hypertension (15.8%), diabetes mellitus (10.5%), venous stasis (7.9%), angina pectoris (2.6%), thyroid dysfunction (2.6%), and others (10.5%) including hyperlipidemia, atherosclerosis, rheumatoid arthritis, gout, chronic hepatitis, aplastic anemia, and depression. Possible prior etiologic factors were reported by four patients (10.5%), including recent upper-respiratory infection (5.3%), high orthostatic pressure due to prolonged standing (2.6%), and strenuous exercise (2.6%) among others. Other previously reported causes of PPD, such as contact with metal or high temperatures, were not recorded. Associated medication histories were statins (13.2%), beta blockers (10.5%), aspirin (7.9%), thiazides (7.9%), angiotensin receptor blockers (5.3%), antiplatelet agents (5.3%), and calcium channel blockers (5.3%) (Table 3).
Table 3. Combined underlying diseases, etiologic factors, and medications of the 38 patients with pigmented purpuric dermatoses.
Response to and progress of treatments
Thirty-six patients (94.7%) were treated with one or more treatment methods. Thirty-one (81.6%) received oral antihistamines, and 22 (57.9%) were administered oral pentoxifylline. Several medications were administered as adjunct therapies, such as oral tetracycline antibiotics and colchicine (36.8%). Six patients received phototherapy as a monotherapy or as an adjuvant (15.8%). Most patients showed substantial or partial improvement; the average grade of the results was 2.3, indicating "substantial improvement." In detail, the PPD lesions of 4 (10.5%), 22 (57.9%), 10 (26.3%), and 2 (5.3%) patients' lesions were completely resolved, substantially improved, partially improved, and unimproved or aggravated, respectively. No clinical type had a significantly favorable disease progression compared to the others (p=0.287). There was no significant difference in treatment response between patients who had a specific history of medications or underlying diseases and those who did not (p>0.05).
DISCUSSION
PPD disorders are relatively uncommon skin disorders, and their etiology remains incompletely understood. PPD occurs more frequently in men, while Majocchi's disease is more common in women. PPD is infrequently reported in preadolescent children and adolescents, with the most common manifestation being Schamberg's disease; the pigmented purpuric lichenoid dermatoses variant has not been reported in this age group. Lichen aureus and Majocchi's disease predominant affect children and young adults2,8,9,10. Overall, the patients of this study showed a subtle female predominance; this tendency was also observed for not only Majocchi's disease, but Schamberg's disease as well. These results are consistent with those of a previous study of 37 Korean PPD patients3. Although further epidemiologic studies are required for verification, it can be presumed Korean patients with PPD are predominantly female.
As mentioned earlier, PPD can be divided into six distinct categories. Schamberg's disease is mostly observed on the lower legs but can appear anywhere; it is characterized by pinhead-sized reddish puncta resembling grains of cayenne pepper that further form irregular plaques of orange or brown pigmentation. Their localization is sometimes associated with venous insufficiency. Schamberg's disease is generally asymptomatic and persistent2,8,11. The early lesions of Majocchi's disease are bluish-red annular macules in which dark-red telangiectatic puncta appear. A peripheral extension providing an annular configuration fades beyond the central part of the lesion. The eruption begins on the lower extremities and subsequently extends to the upper ones; there may be few or innumerable lesions. The lesions are asymptomatic and usually last several months with relapses and remissions. Unlike Schamberg's disease, Majocchi's disease is unassociated with venous stasis2,8,11. Lichen aureus or lichen purpuricus is a more localized, persistent, more intensely purpuric eruption; the eruption often has a golden-brown color. Lesions may be sufficiently infiltrated clinically and histologically exhibit a lichenoid pattern. The most commonly involved area is the lower limbs, although the trunk and face are occasionally involved. The lesions tend to be chronic, remaining stable or progressing slowly2,9,12,13. PPD of Gougerot-Blum appears as lichenoid papules that tend to fuse into plaques of various hues; they occur on the legs and rarely on the trunk and thighs. This disease occurs more frequently in males and has a chronic course2,7,11. Eczematid-like purpura of Doucas-Kapetanakis are severely pruritic and comparably more extensive than the other types. This disease may be associated with a contact allergy to clothing/rubber and may represent a variant of Schamberg's disease. It spreads rapidly over 15~30 days and fades without treatment over several months to years2,8,11. Most findings of the present study are consistent with those of former reports, except for the presence of PPD of Gougerot-Blum in contrast to the study of 37 Korean PPD patients3 and the fact that only 1 of 23 patients with Schamberg's disease presented with venous insufficiency (4.3%).
The histopathologic findings of PPD include superficial lymphocytic infiltration and macrophages centered on the superficial small blood vessels of the skin, with endothelial cell swelling and luminal narrowing. Variable epitheliotropism and red blood cell extravasation with marked hemosiderin deposition in macrophages are typical findings of PPD. Neutrophils can be found in patients with eczematid-like purpura of Doucas-Kapetanakis. Epidermal spongiosis and exocytosis of lymphocytes are seen in all variants except lichen aureus. Lichenoid lymphocytic infiltration aids the differential diagnosis of PPD of Gougerot-Blum, and marked spongiosis and/or neutrophils are one of the features of eczematid-like purpura of Doucas-Kapetanakis1,5,6. Ackermann emphasizes that PPD is not a capillaritis, because fibrin is not present in the luminal wall and thrombi are not observed in the lumens. All of these disorders essentially share similar pathologic findings with some minor differences that are associated with some of the clinical types14. In the present study, several patients exhibited unusual findings of PPD, such as interface changes and basal hyperpigmentation. Varying degrees of epidermal spongiosis and lymphocyte exocytosis were observed in almost all types of PPD. Although all patients with PPD of Gougerot-Blum exhibited lichenoid lymphocytic infiltration, all patients with lichen aureus and eczematid-like purpura of Doucas-Kapetanakis also exhibited such findings; moreover, all patients with eczematid-like purpura of Doucas-Kapetanakis had marked spongiosis in contrast to others.
The precise cause of PPD remains unknown. However, several cofactors are reported to be associated with the appearance or presentation of PPD, including venous hypertension, strenuous exercise, orthostatic pressure, capillary fragility, focal infections, and chemical ingestion2,3,8,9,12. Several medications appear to provoke PPD, especially Schamberg's disease, including acetaminophen, aspirin, nonsteroidal anti-inflammatory drugs, glipizide, glybuzole, diuretics (i.e., furosemide and thiazides), dipyridamole, bezafibrate, furosemide, hydralazine, etc. Other causes include contact allergy to metals or dyes, clothing, and alcohol ingestion2,3,4,8,9,11,15,16,17,18,19. Many of the abovementioned drugs are used to treat cardiovascular diseases and diabetes. Furthermore, diabetes and dyslipidemia themselves can be associated with PPDs7,8,10,11. Psoriasis is independently associated with increased cardiovascular mortality; PPD of Gougerot-Blum is presumably a variant of psoriasis in which the proliferating epidermis is cut off from the blood supply and the influence of the dermis owing to bleeding in the dermal papillae8,20. Thus, it would be worthwhile to investigate PPD patients who have chronic cardiovascular diseases because of the noticeably higher frequency of departmental consultations and the previously reported etiologic backgrounds.
No single or combination therapy is demonstrably superior for the treatment of PPD. Nevertheless, treatments aiming to improve related etiologic factors might help in some cases. Various therapies are reported to improve outcomes in PPD, including topical steroids, oral antihistamines, pentoxifylline, and ultraviolet light therapy2,21,22. Some of the patients in the present study received oral colchicine or tetracycline antibiotics because of unresponsiveness to initial therapies. However, none of the various treatments administered showed significantly superior outcomes, which is consistent with the litature2,3.
The limitations of this study are the lack of a matched control group comprising patients from the Department of Internal Medicine, the possible omission of etiologic factors from medical records, and potential inaccuracies regarding treatment responses and the consequent distortion of the results. Future studies should also compare diagnostic accuracy before and after skin biopsies.
Overall, the findings of PPD in the present study are generally consistent with the literature, except for the female predominance in Asians, the pattern of age distribution according to clinical type, additional possible histologic features, and potentially associated coexistent medical circumstances. This study was initially designed to verify the relationships between provocations of PPD and coexisting cardiovascular history; however, there were no significant associations with respect to the frequencies or prognosis of PPD. Nevertheless, larger randomized systematic studies encompassing several theoretical backgrounds and clinical experiences could be valuable.
References
- 1.Magro CM, Schaefer JT, Crowson AN, Li J, Morrison C. Pigmented purpuric dermatosis: classification by phenotypic and molecular profiles. Am J Clin Pathol. 2007;128:218–229. doi: 10.1309/AQMU3JFE2A66LC7E. [DOI] [PubMed] [Google Scholar]
- 2.Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482–488. doi: 10.1111/j.1365-4632.2004.02213.x. [DOI] [PubMed] [Google Scholar]
- 3.Cho JH, Lee JD, Kang H, Cho SH. The clinical manifestations and etiologic factors of patients with pigmented purpuric dermatoses. Korean J Dermatol. 2005;43:45–52. [Google Scholar]
- 4.Yung A, Goulden V. Pigmented purpuric dermatosis (capillaritis) induced by bezafibrate. J Am Acad Dermatol. 2005;53:168–169. doi: 10.1016/j.jaad.2005.04.072. [DOI] [PubMed] [Google Scholar]
- 5.Smoller BR, Kamel OW. Pigmented purpuric eruptions: immunopathologic studies supportive of a common immunophenotype. J Cutan Pathol. 1991;18:423–427. doi: 10.1111/j.1600-0560.1991.tb01378.x. [DOI] [PubMed] [Google Scholar]
- 6.Ratnam KV, Su WP, Peters MS. Purpura simplex (inflammatory purpura without vasculitis): a clinicopathologic study of 174 cases. J Am Acad Dermatol. 1991;25:642–647. doi: 10.1016/0190-9622(91)70246-x. [DOI] [PubMed] [Google Scholar]
- 7.Fishman HC. Pigmented purpuric lichenoid dermatitis of Gougerot-Blum. Cutis. 1982;29:260–261. 264. [PubMed] [Google Scholar]
- 8.Newton RC, Raimer SS. Pigmented purpuric eruptions. Dermatol Clin. 1985;3:165–169. [PubMed] [Google Scholar]
- 9.Tristani-Firouzi P, Meadows KP, Vanderhooft S. Pigmented purpuric eruptions of childhood: a series of cases and review of literature. Pediatr Dermatol. 2001;18:299–304. doi: 10.1046/j.1525-1470.2001.01932.x. [DOI] [PubMed] [Google Scholar]
- 10.Saito R, Matsuoka Y. Granulomatous pigmented purpuric dermatosis. J Dermatol. 1996;23:551–555. doi: 10.1111/j.1346-8138.1996.tb02650.x. [DOI] [PubMed] [Google Scholar]
- 11.Devere TS, Patel AB. Pigmented purpuric dermatoses. In: Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ, Wolff K, editors. Fitzpatrick's dermatology in general medicine. 7th ed. New York: McGraw-Hill; 2008. pp. 2049–2054. [Google Scholar]
- 12.Taketuchi Y, Chinen T, Ichikawa Y, Ito M. Two cases of unilateral pigmented purpuric dermatosis. J Dermatol. 2001;28:493–498. doi: 10.1111/j.1346-8138.2001.tb00018.x. [DOI] [PubMed] [Google Scholar]
- 13.Rudolph RI. Lichen aureus. J Am Acad Dermatol. 1983;8:722–724. doi: 10.1016/s0190-9622(83)70087-3. [DOI] [PubMed] [Google Scholar]
- 14.Hercogova J. Persistent pigmented purpuric dermatoses: who are you? J Eur Acad Dermatol Venereol. 2001;15:15. doi: 10.1046/j.1468-3083.2001.00199.x. [DOI] [PubMed] [Google Scholar]
- 15.Filo V, Galbavý S, Filová A, Borecká D, Novotná V. Unilateral progressive pigmented capillaropathy (Schamberg's disease?) of the arm. Br J Dermatol. 2001;144:190–191. doi: 10.1046/j.1365-2133.2001.03976.x. [DOI] [PubMed] [Google Scholar]
- 16.Kwon SJ, Lee CW. Figurate purpuric eruptions on the trunk: acetaminophen-induced rashes. J Dermatol. 1998;25:756–758. doi: 10.1111/j.1346-8138.1998.tb02497.x. [DOI] [PubMed] [Google Scholar]
- 17.Adams BB, Gadenne AS. Glipizide-induced pigmented purpuric dermatosis. J Am Acad Dermatol. 1999;41:827–829. doi: 10.1016/s0190-9622(99)70335-x. [DOI] [PubMed] [Google Scholar]
- 18.Wong WK, Ratnam KV. A report of two cases of pigmented purpuric dermatoses treated with PUVA therapy. Acta Derm Venereol. 1991;71:68–70. [PubMed] [Google Scholar]
- 19.Nishioka K, Katayama I, Masuzawa M, Yokozeki H, Nishiyama S. Drug-induced chronic pigmented purpura. J Dermatol. 1989;16:220–222. doi: 10.1111/j.1346-8138.1989.tb01252.x. [DOI] [PubMed] [Google Scholar]
- 20.Pietrzak A, Brzozowska A, Lotti T, Mosiewicz J, WysokiXMLLink_XYZski A, Mieczkowska J, et al. Future diagnosis, today's treatment -cardiomyopathy in the course of psoriasis: a case report. Dermatol Ther. 2013;26:489–492. doi: 10.1111/dth.12021. [DOI] [PubMed] [Google Scholar]
- 21.Kocaturk E, Kavala M, Zindanci I, Zemheri E, Sarigul S, Sudogan S. Narrowband UVB treatment of pigmented purpuric lichenoid dermatitis (Gougerot-Blum) Photodermatol Photoimmunol Photomed. 2009;25:55–56. doi: 10.1111/j.1600-0781.2009.00398.x. [DOI] [PubMed] [Google Scholar]
- 22.Mun JH, Jwa SW, Song M, Kim HS, Ko HC, Kim BS, et al. Extensive pigmented purpuric dermatosis successfully treated with pentoxifylline. Ann Dermatol. 2012;24:363–365. doi: 10.5021/ad.2012.24.3.363. [DOI] [PMC free article] [PubMed] [Google Scholar]