

Abstract
AIM: To examine DNA methylation profiles in a longitudinal comparison of pre-diabetes mellitus (Pre-DM) subjects who transitioned to type 2 diabetes mellitus (T2DM).
METHODS: We performed DNA methylation study in bisulphite converted DNA from Pre-DM (n = 11) at baseline and at their transition to T2DM using Illumina Infinium HumanMethylation27 BeadChip, that enables the query of 27578 individual cytosines at CpG loci throughout the genome, which are focused on the promoter regions of 14495 genes.
RESULTS: There were 694 CpG sites hypomethylated and 174 CpG sites hypermethylated in progression from Pre-DM to T2DM, representing putative genes involved in glucose and fructose metabolism, inflammation, oxidative and mitochondrial stress, and fatty acid metabolism. These results suggest that this high throughput platform is able to identify hundreds of prospective CpG sites associated with diverse genes that may reflect differences in Pre-DM compared with T2DM. In addition, there were CpG hypomethylation changes associated with a number of genes that may be associated with development of complications of diabetes, such as nephropathy. These hypomethylation changes were observed in all of the subjects.
CONCLUSION: These data suggest that some epigenomic changes that may be involved in the progression of diabetes and/or the development of complications may be apparent at the Pre-DM state or during the transition to diabetes. Hypomethylation of a number of genes related to kidney function may be an early marker for developing diabetic nephropathy.
Keywords: Epigenetic changes, Pre-diabetes, Diabetes, Nephropathy
Core tip: Many independent predictors of diabetes including markers of metabolic dysfunction (high body mass index, hypertension, low HDL and smoking) were significantly increased early on in pre-diabetes mellitus (Pre-DM) and sustained in diabetes groups. The innovation in high-throughput epigenome of DNA methylation studies suggests that some epigenomic changes that may be involved in the progression of diabetes and/or the development of complications may be apparent at the Pre-DM state or during the transition to diabetes.
INTRODUCTION
Pre-diabetes mellitus (Pre-DM) is a condition characterized by elevated blood glucose concentrations that denote the incipient development of type 2 diabetes mellitus (T2DM), along with its co-morbid conditions of cardiovascular disease and renal disease. The most common definitions of Pre-DM refer to impaired glucose tolerance (IGT) and impaired fasting glucose (IFG). IGT and IFG are assumed to define categories of glycemia associated with an increased risk of developing diabetes[1-3]. It is estimated that roughly 86 million individuals in the United States aged 21 years and older have Pre-DM. People with Pre-DM are 5-15 times more likely to develop T2DM than are people with normal glucose values[1,4]. The risk of people with IGT and/or IFG developing T2DM is not uniform. An analysis of several prospective studies showed that the incidence rates of developing T2DM in people with IGT ranged from 35.8 to 87.3 per 1000 person-years[5]. Environmental exposures, sedentary lifestyle, and high calorie, high-fat diets correlate with the development of metabolic syndrome including obesity and insulin resistance. All of these factors influence the rate of progression of Pre-DM. Progression to T2DM among those with Pre-DM is not inevitable and also is variable in terms of development of complications related to T2DM. People with Pre-DM who lose weight and engage in moderate physical activity can prevent or delay T2DM and may even return their blood glucose levels to normal[6]. Similar to the prevalence of Pre-DM and T2DM in the United States, estimates suggest that more than 31 million people in the United States are affected by kidney disease, with less than 500000 (< 0.2%) having kidney failure treated by dialysis or transplantation. T2DM is an important independent risk factor for kidney disease in the United States and almost half of all new cases of End Stage Renal Disease (ESRD) are due to diabetic nephropathy. A tremendous amount of work has been done to better understand diabetic nephropathy and the risk of progression to ESRD and much of the work in renoprotection has focused on this population.
The etiological origins of T2DM are complex. A data-mining approach, which analyzed over 12 million Medline records to identify factors associated with the pathology of T2DM, identified epigenetic changes as among the most important causal factors in the pathogenesis of T2DM[7]. The epigenome is increasingly gaining acceptance as playing an important role in diabetes and obesity, and the role of both nutritional status and endocrine disruptors would appear to be major factors in these conditions[8-10]. Initial observations indicating a role for environmental cues in establishing epigenetic patterns came from studies of the agouti mouse model with offspring suffering from obesity, hyperinsulinaemia and diabetes[11]. In human studies it has been shown that trans-generational effects of nutrition may be passed on to future generations. In a study of historical records from Överkalix, Sweden, the grandsons of men who were well-nourished prior to puberty had an increased risk of developing T2DM[12].
A general defect in DNA methylation in T2DM is suggested by the observation that S-adenosylmethionine (SAM), the main physiologic donor of methyl groups, is decreased in erythrocytes of diabetic patients. In addition, decreased erythrocyte concentrations of SAM and other alterations were found to be associated with disease progression[13]. Methylation plays an important role in regulating gene expression, most likely including the expression of those genes essential for the strict maintenance of normal blood glucose levels. Expression patterns that develop in response to changes in diet or in response to environmental factors are likely to become locked by DNA methylation early in development[14]. Methylation of DNA on specific cytosine residues in CpG islands, especially in promoter regions, leads to DNA hypermethylation, which generally is associated with lowering gene expression, while removal of methyl groups, leading to DNA hypomethylation, is generally associated with increasing gene expression. Methylation patterns have been suggested to be involved in the propagation of insulin resistance in insulin target tissues and, being a reversible modification, might also confer the adaptability of metabolism to loss of body weight.
In this longitudinal study, we used archived biological samples to examine the methylation patterns in DNA obtained from subjects at the time they were classified as Pre-DM and were also later obtained from the same subjects after they had transitioned to T2DM. All subjects in this cohort eventually developed diabetic nephropathy. This allowed for a longitudinal comparison of changes associated with transitioning from Pre-DM to T2DM. Our aims were two-fold: first, to obtain a global comparison of hyper- and hypomethylation patterns between the Pre-DM and T2DM states and analysis of these differences in terms of altered metabolic pathways; and second, to examine for methylation changes at the T2DM stage that might suggest early markers to future development of diabetic complications, specifically diabetic nephropathy.
MATERIALS AND METHODS
The study protocol was approved by the Human Subject Research Review Committee of the University of New Mexico Health Sciences Center. Genome-wide screening for DNA methylation was carried out with the Infinium 27K methylation array (Illumina Infinium® HumanMethylation27 BeadChip, Illumina, San Diego, CA). Quantitative measurements of DNA methylation were determined for 27578 CpG dinucleotide spanning 14495 genes. This methodology combines bisulfite conversion of genomic DNA and whole-genome amplification with direct, array-based capture and enzymatic scoring of the CpG loci. Allele-specific single-base extension of the oligos on the BeadChip, using the captured DNA as a template, incorporates detectable labels on the BeadChip and determines the methylation profile for the sample. One microgram of DNA was treated with sodium bisulfite using the Zymo EZ DNA Methylation Kit to convert un-methylated cytosines to uracil, while methylated cytosines remain unchanged. The DNA was purified and quantified in preparation for whole genome amplification, followed by fragmentation and ethanol precipitation. The DNA was re-suspended in hybridization buffer and applied to the bead chip array for an overnight incubation. Following hybridization, the arrays were washed to eliminate un-hybridized and non-specifically hybridized DNA. The samples then underwent single base extension and staining followed by more washing. The arrays were allowed to dry and then scanned using the Illumina iScan system. Analysis of the scanned results is achieved using Illumina’s GenomeStudio software in conjunction with the GenomeStudio methylation module. GenomeStudio Software is a modular analysis tool for genotyping, gene expression, and methylation applications. The Methylation Module allows users to combine Infinium methylation assay data with mRNA data, enabling convergence of data across gene expression and epigenetic analyses.
To evaluate the genes identified by the Infinium methylation assay for groupings that may identify metabolic pathways, the data were analyzed with ingenuity pathway analysis (IPA) (Ingenuity Systems, Redwood City, CA). Differentially expressed transcripts satisfying the statistical conditions were exported to IPA. This software determines the top canonical pathways by using the ratio of the number of genes in a given pathway that meet cutoff criteria divided by the total number of genes that constitute that pathway. The significance of a pathway for the data set reflects the likelihood that the pathway is associated with the dataset by random chance. The methylation assay results were analyzed and scored for significance of hyper and hypomethylation compared to controls. Output (Beta) was used in computations creating a P-value from a Diff score; DiffScore = 10*sgn(Beta Condition - Beta Reference)*log10p. Level of significance related to DiffScore are as follows; P-value of 0.05, DiffScore = ± 13; For a P-value of 0.01, DiffScore = ± 22; For a P-value of 0.001, DiffScore = ± 33.
This procedure was carried out using DNA isolated from 11 Pre-DM non-Hispanic white male subjects when they were diagnosed with Pre-DM and repeated after transitioning to T2DM, and from two reference subjects used to establish a baseline. The Pre-DM samples and the T2DM samples were normalized to this baseline in order to determine expression changes that occur over the extended time period between Pre-DM and T2DM. The cohort was limited to 11 non-Hispanic white males to minimize confounding variables of ethnicity and gender. Blood leukocyte samples were taken at each stage and phenotype for clinical parameters and anthropomorphic measurements. Every patient’s transition from Pre-DM to T2DM involved a higher body mass index (BMI), weight gain as well as higher levels of blood glucose and HbA1c. Clinical standards set by the American Diabetes Association were used to classify the subjects: fasting plasma glucose levels of 99 mg/dL or below are considered normal; plasma glucose levels between 100 to 125 mg/dL indicate Pre-DM; and plasma glucose levels of 126 mg/dL and higher indicate T2DM. Subjects with a normal plasma glucose but elevated HbA1c (5.7%-6.0%) were also classified as Pre-DM.
RESULTS
The mean age at Pre-DM diagnosis was 40.27 ± 5.46, the mean transition time was 7.09 ± 1.97 years, and the mean age at T2DM diagnosis was 47.36 ± 5.97 years of age. In the Pre-DM state the BMI = 32.2 ± 6.9, HbA1c = 5.5 ± 0.31, glucose = 99.1 ± 15.9 (mg/dL), and weight = 226.6 ± 68.9 (lbs). In the diabetic state, the BMI = 37.2 ± 6.9, HbA1c = 9.6 ± 2.15, glucose = 225.9 ± 78.8 (mg/dL), and weight = 261.4 ± 65.1 (lbs).
Global changes in methylation patterns
Comparisons of the epigenetic profiles of methylated CpG loci in DNA from 11 non-Hispanic white male revealed that 694 CpG sites were consistently hypomethylated and 174 were hypermethylated in the DNA obtained at the time of transition to T2DM compared to the DNA obtained at Pre-DM (Figure 1). Analysis of the genes with IPA identified numerous putative genes associated with carbohydrate and lipid metabolism, inflammation, immune cell function and cell signaling, suggesting increased activities in these pathways at the T2DM state compared to the Pre-DM state. The five Top Associated Networks identified in the Ingenuity Pathway Analysis are summarized in Table 1.
Figure 1.
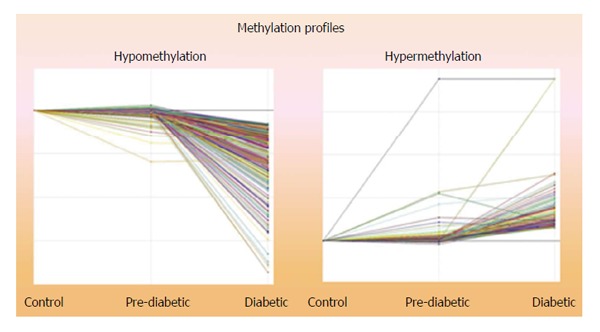
Global methylation profile depicting a total of 868 genes. Total of 694 were hypomethylated, 174 were hypermethylated.
Table 1.
The top associated networks from ingenuity pathway analysis
| Carbohydrate metabolism, small molecule biochemistry, cell signaling |
| Lipid metabolism, small molecule biochemistry, drug metabolism |
| Inflammatory response, cardiovascular system development and function, lymphoid tissue structure and development |
| Cellular function and maintenance, inflammatory response, cell-to-cell signaling and interaction |
| Cellular movement, hematological system development and function, immune cell trafficking |
The Top Biological Functions identified by IPA and the number of genes involved are summarized in Table 2. These include numerous genes that suggest changes that may be directly associated with or are early markers for diseases and disorders, molecular and cellular function and physiological system development and function, possibly related to the Pre-DM state transitioning to the T2DM state and related nephropathy.
Table 2.
The top biological functions identified by ingenuity pathway analysis and the number of genes involved
| # of genes | |
| Diseases and disorders | |
| Inflammatory disease | 218 |
| Inflammatory response | 202 |
| Immunological disease | 195 |
| Respiratory disease | 126 |
| Hematological disease | 112 |
| Molecular and cellular functions | |
| Cellular growth and proliferation | 230 |
| Cell death | 224 |
| Cell-to-cell signaling and interaction | 188 |
| Cellular development | 152 |
| Physiological system development and function | |
| Hematological system development/function | 214 |
| Immune cell trafficking | 137 |
| Hematopoiesis | 126 |
| Tissue morphology | 105 |
| Cell-mediated immune response | 85 |
Genes associated with kidney disease
The entire ensemble of genes was evaluated by literature search to identify key candidate genes involved in kidney disease. Sixteen genes were selected: SLC22A12, Transient Receptor Potential Melastatin subtype 6 (TRPM6), aquaporin 9 (AQP9), HP, HPR, ABCC2, alanine-glyoxylate aminotransferase (AGXT), UGT2A3, HAL, HYAL2, SLC13A1, SERPINF1, CD22, SIGLEC5, NEU4, and NOX1. These genes may be directly related to risk factors or biomarkers that signify kidney damage progression or vulnerability. Hypomethylation is seen in all sixteen of the genes. The methylation patterns are shown in Figure 2.
Figure 2.
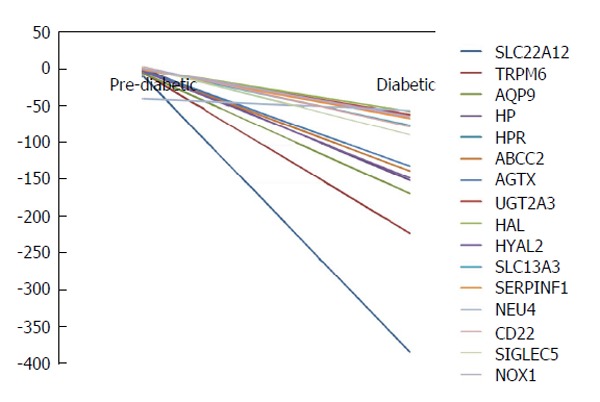
A total of 92 genes were hypomethylated and 54 were hypermethylated in all subjects using Illumina bead studio. Significantly hypomethylated genes were compared to putative genes identified in literature review forming of a sub-group of sixteen genes related to kidney disease. These sixteen genes are represented here as cumulative average DiffScore deviations from control comparisons. Scores of ± 13 P-value of 0.05, ± 22 P-value of 0.01.
A total of 92 genes were hypomethylated and 54 were hypermethylated in all subjects. Analysis of the sub-group of sixteen genes (Figure 2) associated with kidney disease for genes which were hypomethylated in all subjects identified six genes. The data for these six genes are shown in Figure 3. These genes and their associated products or functions are: SLC22A12 (a urate transporter on the proximal tubule); TRPM6 (a cation channel in the kidney); AQP9 (an aquaporin); HP (haptoglobin, which binds plasma hemoglobin); AGXT (which is involved in oxalic acid secretion); HYAL2 (a hyaluronidase).
Figure 3.
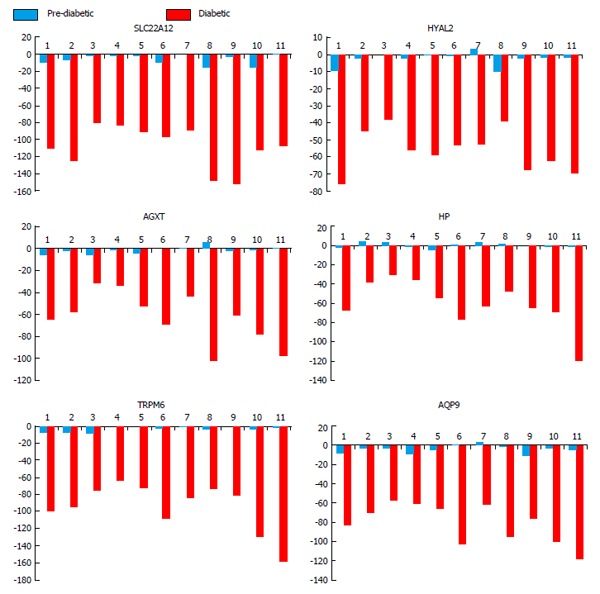
Hypo-methylated kidney disease associated gene loci. Six putative genes related to kidney disease were hypomethylated across patients (numbered 1-11) compared to controls (n = 2). Red bars indicate diabetic time point and blue bars representing prediabetic time point. Data represented as DiffScore with ± 13 equating to P-value 0.05, ± 22 equating to P-value of 0.01, and ± 33 equating to P-value of 0.001. SLC22A12 (a urate transporter on the proximal tubule); TRPM6 (a cation channel in the kidney); AQP9 (an aquaporin); HP (haptoglobin, which binds plasma hemoglobin); AGXT (alanine-glyoxylate aminotransferase which is involved in oxalic acid secretion); HYAL2 (a hyaluronidase).
DISCUSSION
This study demonstrated that there are a large number of methylation changes in the progression of Pre-DM to T2DM in a homogeneous longitudinal cohort of white males of which IPA of the associated genes identified numerous cellular pathways that potentially can be altered, leading to development and/or prediction of diabetes-related complications. Of particular interest, this study identified six genes that may be associated with/or predict the development of diabetic nephropathy. These six genes were hypomethylated in all subjects in the progression from Pre-DM to T2DM. The sample size was limited by the longitudinal observation of nearly a decade time period (approximately 7 years) required for the mean transition time from Pre-DM to T2DM. Although this sample size is small, the study identified a limited set of markers in this cohort that were hypomethylated in all subjects. Longitudinal studies similar to the present study but with larger numbers of subjects are especially difficult owing to the rare availability of DNA samples. However, the results from this study will aid in the design of future studies. For example, DNA from a range of subjects with diabetic nephropathy can be analyzed to confirm whether these changes in expression are observed in other ethnic groups during transition from Pre-DM to T2DM and also to compare with subjects who are resistant to developing T2DM related nephropathy.
Below is a brief discussion of the six genes
SLC22A12: Uric acid, which is the metabolic end product of purine metabolism in humans, has protective antioxidant properties but can also be pro-oxidant. Urate, the ionized form of uric acid, scavenges potentially harmful radicals. Defective renal handling of urate is a frequent pathophysiologic factor in hyperuricemia. In response to genetic or environmental factors, such as diet, hyperuricemia may cause gout, nephrolithiasis, hypertension, and vascular disease. However, hypouricemia may also have pathological consequences. Humans have higher serum uric acid levels compared to other mammalian species; this is the result of genetic silencing of hepatic uricase, an enzyme that metabolizes uric acid into allantoin. Uric acid homeostasis is maintained by balance between production, intestinal secretion, and renal excretion. The kidney is important in the regulation of circulating uric acid levels through control of re-absorption of filtered urate and through uric acid excretion. In humans, urate transporters URAT1, MRP4, OAT1, and OAT3 play central roles in homeostasis. SLC22A12, a member of the organic anion transport family, encodes for the protein URAT1, which is a kidney-specific urate transporter that transports urate across the apical membrane of the proximal tubule various mutations in SLC22A12 have been associated with renal disease. Given the importance of urate homeostasis, and the critical role of URAT1 activity in determining whether urate absorption vs secretion in balanced, epigenetic hypomethylation may be a determinant in the activity of URAT1[15-21] (Figure 3).
TRPM6: TRPM6 is a member of the Transient Receptor Potential superfamily of cation channels, which are widely expressed and function in the regulation of absorption and secretion of cations. Many TRPs are expressed in kidney along the nephron. These channels are involved in hereditary as well as acquired kidney disorders. Increased expression of TRPM6 transporters is associated with diabetes mellitus and kidney damage in experimental animal models[22,23]. TRPM6 channels are primarily located in the renal distal convolution, the main site of active transcellular Ca(2+) and Mg(2+) transport in the kidney. The channels are regulated by many factors and hormones to maintain systemic concentrations of Ca(2+) and Mg(2+). Loss-of-function mutations in TRPM6 are a molecular cause of hypomagnesemia with secondary hypocalcemia. TRPM6 may be viewed as the gatekeeper of the body’s Mg2+ balance although, even in the distal convolutions, multiple proteins involved in Mg2+ transport have been identified (TRPM6, proEGF, and FXYD2 which is the Na+/K+-ATPase gamma-subunit). Drug treatment, acid-base status, and several hormones have been shown to regulate TRPM6 expression[24-27].
AQP9: AQP are integral membrane channels for the transfer of water, and in some cases, small solutes across the membrane. Aquaporins are conserved in bacteria, plants, and animals. There are more than 10 isoforms of AQP. Several of the mammalian aquaporins (e.g., AQP1, AQP2, AQP4, and AQP5) are selective for the passage of water; others also transport glycerol (e.g., AQP3 and AQP8) and even larger solutes (AQP9). The human aquaporins, AQP3, AQP7, AQP8 and AQP9 are also permeable to ammonia. AQP9 is an aquaporin which stimulates urea transport and allows passage of a wide variety of non-charged solutes. AQP9 is expressed in numerous tissues and is especially abundant in liver. AQP9 as well as other AQPs also are expressed in kidney. The ammonia-transporting AQPs, including AQP9, supplement the ammonia transport of the Rhesus proteins; AQP9 also supplements the urea transporters. AQP9 can also transport arsenic trioxide[28-32]. Given the wide distribution of the AQPs, including AQP9, in is unclear whether epigenetic hypomethylation of APQ9 in diabetes would contribute to development of diabetic nephropathy. One possibility is that the special properties of AQP9 in urea transport may require highly controlled expression in tissues, such as kidney, which have important roles in urea transport.
HP: The HP gene encodes for haptoglobin (Hp) which functions to bind free plasma hemoglobin, thereby helping to prevent loss of iron through the kidney and protecting the kidneys from damage by hemoglobin. Iron status is influenced by environmental and genetic factors. The genetic polymorphism of Hp has been shown to affect iron turnover. Hp captures hemoglobin in plasma to allow hepatic recycling of heme iron, which helps to prevent kidney damage during hemolysis. Hp acts as an anti-oxidant by binding hemoglobin. Two common alleles for Hp (1 and 2) produce three common Hp genotypes: Hp1-1, Hp2-1, and Hp2-2. The protein encoded by Hp1-1 provides superior antioxidant protection compared with that encoded by Hp2-2. Hp genotype is an independent risk factor for complications; individuals with Hp2-2 are more likely to develop nephropathy, retinopathy, and cardiovascular disease as compared with those with Hp2-1 or Hp1-1. In diabetic patients, urinary Hp levels and genotype predict renal functional decline. Aged animals are especially sensitive to the nephrotoxicity of hemoglobin. Hp synthesis is primarily a function of liver where Hp up regulation is a major stress response. However, in acute kidney injury, Hp synthesis in the proximal tubules is a major stress response[33-39].
AGXT: The AGXT gene codes for the peroxisomal enzyme AGXT, which converts glyoxylate into glycine using L-alanine as the amino-group donor. Mutations in the AGXT are responsible for primary hyperoxaluria type 1 (PH1), which is a rare disease characterized by excessive hepatic oxalate production. When AGXT activity is absent, glyoxylate is converted to oxalate. Oxalate forms insoluble calcium salts that accumulate in the kidney. PH1 patients are at risk for recurrent deposition of calcium oxalate in the renal pelvis/urinary tract, deposition of calcium oxalate in the renal parenchyma, or ESRD. The PH1 is mostly due to single point mutations on the AGXT gene; more than 150 so far been identified[40-42]. The epigenetic hypomethylation of AGXT (Figure 3), where hypomethylation generally is associated with enhanced expression, would seem counter to a role for AGXT where PH1 is associated with diminished activity. However, in a recent cluster analysis of microarray expression data for genes associated with T2DM and nephropathy, AGXT was identified as one of the more highly expressed genes[43].
HYAL2: Hyaluronidases degrade hyaluronan, one of the major glycosaminoglcans of the extracellular matrix. The human genome contains six hyaluronidase-like genes. HYAL2 and HYAL1 are the major mammalian hyaluronidases in somatic tissues. They work together to degrade high molecular weight hyaluronan to tetrasaccharides. Initially large hyaluronan fragments (20 kD) are generated at the cell surface from digestion by the glycosylphosphatidyl-inositol-anchored HYAL2. These fragments are internalized and further digested by HYAL1. Alterations in hyaluronan have been reported in numerous renal diseases. The accumulation of hyaluronan in the renal cortex is observed in inflammatory renal diseases. In addition, the large fragments of hyaluronan produced by HYAL2 display inflammatory effects in vitro and may contribute to immune renal injury. Increased activity of renal hyaluronidase occurs in streptozotocin-induced diabetic rats; this activity increases in multiple areas of the kidney during the progression of diabetic nephropathy[44-47].
There are a number of limitations to this study. The sample size was small, reflecting the challenges in obtaining DNA samples from subjects at the Pre-DM and T2DM stages of patients who eventually developed diabetic nephropathy. The six selected kidney disease-associated gene based on literature evaluation, which were hypomethylated in all of the subjects, suggests but does not prove that the expression levels of these genes were up regulated during the progression to T2DM. In addition, the hypomethylation of these genes does not predict the interval of time before the development of nephropathy. Nevertheless, the fact that all of the subjects exhibited hypomethylation of these genes raises the question whether these changes might be predictive of diabetic nephropathy.
COMMENTS
Background
Type 2 diabetes mellitus (T2DM) affects more than 29 million in United States and about 79 million adults have pre-diabetes mellitus (Pre-DM). Environmental exposures, sedentary lifestyle, and high calorie, high-fat diets correlate with the development of metabolic syndrome including obesity and insulin resistance. All of these factors influence the rate of progression of Pre-DM. Recent studies suggest that gene-environment interactions relevant for T2DM are at least partly regulated by epigenomic mechanisms.
Research frontiers
The epigenome is increasingly gaining acceptance as playing an important role in diabetes and obesity, and the role of both nutritional status and endocrine disruptors would appear to be major factors in these conditions. A general defect in DNA methylation in T2DM is suggested by the observation that S-adenosylmethionine (SAM), the main physiologic donor of methyl groups, is decreased in erythrocytes of diabetic patients. In addition, decreased erythrocyte concentrations of SAM and other alterations were found to be associated with disease progression.
Innovations and breakthroughs
The study demonstrated that there are a large number of methylation changes in the progression of Pre-DM to T2DM. The study results revealed that 694 CpG sites were consistently hypomethylated and 174 were hypermethylated in the DNA obtained at the time of transition to T2DM compared to the DNA obtained at Pre-DM. The putative genes identified are associated with carbohydrate and lipid metabolism, inflammation, immune cell function and cell signaling, suggesting increased activities in these pathways at the T2DM state compared to the Pre-DM state. The authors further observed methylation changes in six candidate genes in all patients at the T2DM stage with nephropathy suggesting future development of diabetic complications.
Applications
Characterizing the epigenomic components that may regulate the transcriptional potential of a cell and contribute to the etiology, severity and progression of Pre-DM to T2DM and to complications including kidney disease will provide novel insights into disease pathogenesis and therapeutic approaches. This knowledge will enhance our ability to investigate, diagnose and ameliorate T2DM and kidney disease with a significant epigenomic component.
Peer-review
It is an interesting prospective study, analyzing DNA methylation profiling in 11 pre diabetic and 2 control individuals. In addition hypomethylation may be associated to difference genes in the nephropaty progression.
Footnotes
P- Reviewer: Hwu CM, Sanlioglu AD, Sanchez R, Wu CS S- Editor: Ji FF L- Editor: A E- Editor: Liu SQ
Supported by The grants from the National Center for Research Resources, No. 5P20RR016480-12; The National Institute of General Medical Sciences of the NIH, No. 8P20GM103451-12; the partial support from the National Center for Advancing Translational Sciences of the National Institutes of Health, No. 8UL1TR000041; the University of New Mexico Clinical and Translational Science Center; the cost for clinical phenotyping and payments to participants was supported under a UNM Health Sciences Center-based Cardiovascular and Metabolic Diseases Signature Program.
Institutional review board statement: The study protocol was approved by the Human Subject Research Review Committee of the University of New Mexico Health Sciences Center.
Clinical trial registration statement: The Clinical trial registration is not applicable for our study.
Informed consent statement: All participants rendered written informed consent.
Conflict-of-interest statement: No potential conflicts of interest relevant to this original article were reported by authors.
Data sharing statement: We will coordinate data sharing and make available both the raw data and analyzed data upon request.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: February 27, 2015
First decision: May 14, 2015
Article in press: July 14, 2015
References
- 1.Center for Disease Control and Prevention. National Diabetes Fact Sheet. 2011. Available from: http://www.cdc.gov/diabetes/pubs/factsheet11.htm. [Google Scholar]
- 2.International Diabetes Federation. IDF Diabetes Atlas. 6th ed. Available from: http://www.idf.org/diabetesatlas/
- 3.Nathan DM, Davidson MB, DeFronzo RA, Heine RJ, Henry RR, Pratley R, Zinman B. Impaired fasting glucose and impaired glucose tolerance: implications for care. Diabetes Care. 2007;30:753–759. doi: 10.2337/dc07-9920. [DOI] [PubMed] [Google Scholar]
- 4.DeFronzo RA, Abdul-Ghani M. Assessment and treatment of cardiovascular risk in prediabetes: impaired glucose tolerance and impaired fasting glucose. Am J Cardiol. 2011;108:3B–24B. doi: 10.1016/j.amjcard.2011.03.013. [DOI] [PubMed] [Google Scholar]
- 5.Edelstein SL, Knowler WC, Bain RP, Andres R, Barrett-Connor EL, Dowse GK, Haffner SM, Pettitt DJ, Sorkin JD, Muller DC, et al. Predictors of progression from impaired glucose tolerance to NIDDM: an analysis of six prospective studies. Diabetes. 1997;46:701–710. doi: 10.2337/diab.46.4.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Colberg SR, Sigal RJ, Fernhall B, Regensteiner JG, Blissmer BJ, Rubin RR, Chasan-Taber L, Albright AL, Braun B. Exercise and type 2 diabetes: the American College of Sports Medicine and the American Diabetes Association: joint position statement. Diabetes Care. 2010;33:e147–e167. doi: 10.2337/dc10-9990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wren JD, Garner HR. Data-mining analysis suggests an epigenetic pathogenesis for type 2 diabetes. J Biomed Biotechnol. 2005;2005:104–112. doi: 10.1155/JBB.2005.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gallou-Kabani C, Junien C. Nutritional epigenomics of metabolic syndrome: new perspective against the epidemic. Diabetes. 2005;54:1899–1906. doi: 10.2337/diabetes.54.7.1899. [DOI] [PubMed] [Google Scholar]
- 9.Gallou-Kabani C, Vigé A, Junien C. Lifelong circadian and epigenetic drifts in metabolic syndrome. Epigenetics. 2007;2:137–146. doi: 10.4161/epi.2.3.4897. [DOI] [PubMed] [Google Scholar]
- 10.Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007;8:253–262. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miltenberger RJ, Mynatt RL, Wilkinson JE, Woychik RP. The role of the agouti gene in the yellow obese syndrome. J Nutr. 1997;127:1902S–1907S. doi: 10.1093/jn/127.9.1902S. [DOI] [PubMed] [Google Scholar]
- 12.Kaati G, Bygren LO, Edvinsson S. Cardiovascular and diabetes mortality determined by nutrition during parents’ and grandparents’ slow growth period. Eur J Hum Genet. 2002;10:682–688. doi: 10.1038/sj.ejhg.5200859. [DOI] [PubMed] [Google Scholar]
- 13.Poirier LA, Brown AT, Fink LM, Wise CK, Randolph CJ, Delongchamp RR, Fonseca VA. Blood S-adenosylmethionine concentrations and lymphocyte methylenetetrahydrofolate reductase activity in diabetes mellitus and diabetic nephropathy. Metabolism. 2001;50:1014–1018. doi: 10.1053/meta.2001.25655. [DOI] [PubMed] [Google Scholar]
- 14.Gluckman PD, Lillycrop KA, Vickers MH, Pleasants AB, Phillips ES, Beedle AS, Burdge GC, Hanson MA. Metabolic plasticity during mammalian development is directionally dependent on early nutritional status. Proc Natl Acad Sci USA. 2007;104:12796–12800. doi: 10.1073/pnas.0705667104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hediger MA, Johnson RJ, Miyazaki H, Endou H. Molecular physiology of urate transport. Physiology (Bethesda) 2005;20:125–133. doi: 10.1152/physiol.00039.2004. [DOI] [PubMed] [Google Scholar]
- 16.Sakurai H. Urate transporters in the genomic era. Curr Opin Nephrol Hypertens. 2013;22:545–550. doi: 10.1097/MNH.0b013e328363ffc8. [DOI] [PubMed] [Google Scholar]
- 17.Bobulescu IA, Moe OW. Renal transport of uric acid: evolving concepts and uncertainties. Adv Chronic Kidney Dis. 2012;19:358–371. doi: 10.1053/j.ackd.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anzai N, Jutabha P, Amonpatumrat-Takahashi S, Sakurai H. Recent advances in renal urate transport: characterization of candidate transporters indicated by genome-wide association studies. Clin Exp Nephrol. 2012;16:89–95. doi: 10.1007/s10157-011-0532-z. [DOI] [PubMed] [Google Scholar]
- 19.So A, Thorens B. Uric acid transport and disease. J Clin Invest. 2010;120:1791–1799. doi: 10.1172/JCI42344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tasic V, Hynes AM, Kitamura K, Cheong HI, Lozanovski VJ, Gucev Z, Jutabha P, Anzai N, Sayer JA. Clinical and functional characterization of URAT1 variants. PLoS One. 2011;6:e28641. doi: 10.1371/journal.pone.0028641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Enomoto A, Endou H. Roles of organic anion transporters (OATs) and a urate transporter (URAT1) in the pathophysiology of human disease. Clin Exp Nephrol. 2005;9:195–205. doi: 10.1007/s10157-005-0368-5. [DOI] [PubMed] [Google Scholar]
- 22.Lee CT, Lien YH, Lai LW, Chen JB, Lin CR, Chen HC. Increased renal calcium and magnesium transporter abundance in streptozotocin-induced diabetes mellitus. Kidney Int. 2006;69:1786–1791. doi: 10.1038/sj.ki.5000344. [DOI] [PubMed] [Google Scholar]
- 23.Menè P, Punzo G, Pirozzi N. TRP channels as therapeutic targets in kidney disease and hypertension. Curr Top Med Chem. 2013;13:386–397. doi: 10.2174/1568026611313030013. [DOI] [PubMed] [Google Scholar]
- 24.Dietrich A, Chubanov V, Gudermann T. Renal TRPathies. J Am Soc Nephrol. 2010;21:736–744. doi: 10.1681/ASN.2009090948. [DOI] [PubMed] [Google Scholar]
- 25.Dimke H, Hoenderop JG, Bindels RJ. Hereditary tubular transport disorders: implications for renal handling of Ca2+ and Mg2+ Clin Sci (Lond) 2010;118:1–18. doi: 10.1042/CS20090086. [DOI] [PubMed] [Google Scholar]
- 26.Woudenberg-Vrenken TE, Bindels RJ, Hoenderop JG. The role of transient receptor potential channels in kidney disease. Nat Rev Nephrol. 2009;5:441–449. doi: 10.1038/nrneph.2009.100. [DOI] [PubMed] [Google Scholar]
- 27.Hsu YJ, Hoenderop JG, Bindels RJ. TRP channels in kidney disease. Biochim Biophys Acta. 2007;1772:928–936. doi: 10.1016/j.bbadis.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 28.Litman T, Søgaard R, Zeuthen T. Ammonia and urea permeability of mammalian aquaporins. Handb Exp Pharmacol. 2009;(190):327–358. doi: 10.1007/978-3-540-79885-9_17. [DOI] [PubMed] [Google Scholar]
- 29.Verkman AS, Mitra AK. Structure and function of aquaporin water channels. Am J Physiol Renal Physiol. 2000;278:F13–F28. doi: 10.1152/ajprenal.2000.278.1.F13. [DOI] [PubMed] [Google Scholar]
- 30.Takata K, Matsuzaki T, Tajika Y. Aquaporins: water channel proteins of the cell membrane. Prog Histochem Cytochem. 2004;39:1–83. doi: 10.1016/j.proghi.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 31.Sugiura K, Aste N, Fujii M, Shimada K, Saito N. Effect of hyperosmotic stimulation on aquaporins gene expression in chick kidney. Comp Biochem Physiol A Mol Integr Physiol. 2008;151:173–179. doi: 10.1016/j.cbpa.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 32.Liu Z. Roles of vertebrate aquaglyceroporins in arsenic transport and detoxification. Adv Exp Med Biol. 2010;679:71–81. doi: 10.1007/978-1-4419-6315-4_6. [DOI] [PubMed] [Google Scholar]
- 33.Delanghe JR, Langlois MR. Haptoglobin polymorphism and body iron stores. Clin Chem Lab Med. 2002;40:212–216. doi: 10.1515/CCLM.2002.035. [DOI] [PubMed] [Google Scholar]
- 34.Nakhoul FM, Miller-Lotan R, Awaad H, Asleh R, Levy AP. Hypothesis--haptoglobin genotype and diabetic nephropathy. Nat Clin Pract Nephrol. 2007;3:339–344. doi: 10.1038/ncpneph0467. [DOI] [PubMed] [Google Scholar]
- 35.Asleh R, Levy AP. In vivo and in vitro studies establishing haptoglobin as a major susceptibility gene for diabetic vascular disease. Vasc Health Risk Manag. 2005;1:19–28. doi: 10.2147/vhrm.1.1.19.58930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Orchard TJ, Sun W, Cleary PA, Genuth SM, Lachin JM, McGee P, Paterson AD, Raskin P, Anbinder Y, Levy AP. Haptoglobin genotype and the rate of renal function decline in the diabetes control and complications trial/epidemiology of diabetes interventions and complications study. Diabetes. 2013;62:3218–3223. doi: 10.2337/db13-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bhensdadia NM, Hunt KJ, Lopes-Virella MF, Michael Tucker J, Mataria MR, Alge JL, Neely BA, Janech MG, Arthur JM. Urine haptoglobin levels predict early renal functional decline in patients with type 2 diabetes. Kidney Int. 2013;83:1136–1143. doi: 10.1038/ki.2013.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nath KA, Grande JP, Farrugia G, Croatt AJ, Belcher JD, Hebbel RP, Vercellotti GM, Katusic ZS. Age sensitizes the kidney to heme protein-induced acute kidney injury. Am J Physiol Renal Physiol. 2013;304:F317–F325. doi: 10.1152/ajprenal.00606.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zager RA, Vijayan A, Johnson AC. Proximal tubule haptoglobin gene activation is an integral component of the acute kidney injury “stress response”. Am J Physiol Renal Physiol. 2012;303:F139–F148. doi: 10.1152/ajprenal.00168.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hernández-Fernaud JR, Salido E. Differential expression of liver and kidney proteins in a mouse model for primary hyperoxaluria type I. FEBS J. 2010;277:4766–4774. doi: 10.1111/j.1742-4658.2010.07882.x. [DOI] [PubMed] [Google Scholar]
- 41.Cellini B, Oppici E, Paiardini A, Montioli R. Molecular insights into primary hyperoxaluria type 1 pathogenesis. Front Biosci (Landmark Ed) 2012;17:621–634. doi: 10.2741/3948. [DOI] [PubMed] [Google Scholar]
- 42.Danpure CJ. Molecular aetiology of primary hyperoxaluria type 1. Nephron Exp Nephrol. 2004;98:e39–e44. doi: 10.1159/000080254. [DOI] [PubMed] [Google Scholar]
- 43.Guttula SV, Rao AA, Sridhar GR, Chakravarthy MS, Nageshwararo K, Rao PV. Cluster analysis and phylogenetic relationship in biomarker identification of type 2 diabetes and nephropathy. Int J Diabetes Dev Ctries. 2010;30:52–56. doi: 10.4103/0973-3930.60003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun L, Feusi E, Sibalic A, Beck-Schimmer B, Wüthrich RP. Expression profile of hyaluronidase mRNA transcripts in the kidney and in renal cells. Kidney Blood Press Res. 1998;21:413–418. doi: 10.1159/000025893. [DOI] [PubMed] [Google Scholar]
- 45.Declèves AE, Caron N, Voisin V, Legrand A, Bouby N, Kultti A, Tammi MI, Flamion B. Synthesis and fragmentation of hyaluronan in renal ischaemia. Nephrol Dial Transplant. 2012;27:3771–3781. doi: 10.1093/ndt/gfs098. [DOI] [PubMed] [Google Scholar]
- 46.Andre B, Duterme C, Van Moer K, Mertens-Strijthagen J, Jadot M, Flamion B. Hyal2 is a glycosylphosphatidylinositol-anchored, lipid raft-associated hyaluronidase. Biochem Biophys Res Commun. 2011;411:175–179. doi: 10.1016/j.bbrc.2011.06.125. [DOI] [PubMed] [Google Scholar]
- 47.Ikegami-Kawai M, Suzuki A, Karita I, Takahashi T. Increased hyaluronidase activity in the kidney of streptozotocin-induced diabetic rats. J Biochem. 2003;134:875–880. doi: 10.1093/jb/mvg214. [DOI] [PubMed] [Google Scholar]