

Abstract
Complement activation plays a major role in several renal pathophysiological conditions. The three pathways of complement lead to C3 activation, followed by the formation of the anaphylatoxin C5a and the terminal membrane attack complex (MAC) in blood and at complement activating surfaces, lead to a cascade of events responsible for inflammation and for the induction of cell lysis. In case of ongoing uncontrolled complement activation, endothelial cells activation takes place, leading to events in which at the end thrombotic microangiopathy can occur. Atypical haemolytic uremic syndrome (aHUS) is a thrombotic microangiopathy characterized by excessive complement activation on the surface of the microcirculation. It is a severe, rare disease which leads to end-stage renal failure (ESRF) and/or to death in more than 50% of patients without treatment. In the first decade of the second millennium, huge progress in understanding the aetiology of this disease was made, which paved the way to better treatment. First, protocols of plasma therapy for treatment, prevention of relapses and for renal transplantation in those patients were set up. Secondly, in some severe cases, combined kidney and liver transplantation was reported. Finally, at the end of this decade, the era of complement inhibitors, as anti-C5 monoclonal antibody (anti-C5 mAb) began. The past five years have seen growing evidence of the favourable effect of anti-C5 mAb in aHUS which has made this drug the first-line treatment in this disease. The possible complication of meningococcal infection needs appropriate vaccination before its use. Unfortunately, the worldwide use of anti-C5 mAb is limited by its very high price. In the future, extension of indications for anti-C5 mAb use, the elaboration of generics and of mAbs directed towards other complement factors of the terminal pathway of the complement system might succeed in reducing the cost of this new valuable therapeutic approach and render it available worldwide for patients from all social classes.
Keywords: atypical haemolytic uremic syndrome, combined kidney–liver transplantation, complement-related HUS, eculizumab, mutations, plasma exchange
Introduction
Haemolytic uremic syndrome (HUS) is a thrombotic microangiopathy (TMA) which is clinically recognized by the simultaneous occurrence of microangiopathic haemolytic anaemia, thrombocytopenia and acute renal failure. Another TMA manifestation is thrombotic thrombocytopenic purpura (TTP) which is characterized, aside from haemolytic anaemia and thrombocytopenia, mainly by neurological involvement and generally by moderate renal impairment.
The clinical and the pathogenic distinctions between HUS and TTP remained poorly defined for decades. The terminology atypical HUS (aHUS) was initially used for HUS not related to diarrhoea by opposition to typical diarrhoea-related HUS, which is the main form of HUS in children. When the latter was found to be caused by infection with Shiga toxin producing Escherichia coli (STEC), the denomination of typical HUS was replaced by STEC HUS. TMA can result from different causes as classified in a report of the European Paediatric Study Group for HUS [Besbas et al. 2006]. The following aetiologies and clinical associations have been reported in the literature: (a) infection induced: STEC, Shigella dysenteriae type 1, Streptococcus pneumoniae, HIV; (b) complement-related; (c) ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) deficiency; (d) diacylglycerol kinase epsilon (DGKE) mutations; (e) defective cobalamine metabolism (hyperhomocysteinemia); (f) drug-induced; (g) transplantation; (h) malignancy; (i) pregnancy- and contraceptive pill-associated; (j) systemic lupus erythematosus; (k) glomerulopathy; and (l) malignant hypertension. In some cases, however, no aetiology or disease association can be found.
This paper describes the clinical, pathophysiological and genetic aspects and the actual treatment options of aHUS, in particular the use of an anti-complement factor 5 monoclonal antibody (anti-C5 mAb) that specifically binds to the complement protein C5 with high affinity, which has been used with success since several years for the treatment of paroxysmal nocturnal haemoglobinuria (PNH) [McKeage, 2011] and more recently for aHUS.
Complement system in aHUS: pathophysiology, genetics and clinical features
The activation of the alternative pathway of the complement system in HUS has been known for several decades [Cameron and Vick, 1973; Monnens et al. 1974; Thompson and Winterborn, 1981].
The mechanisms of activation of the complement system are well known and have been summarized in several reviews [Berger et al. 2005; Loirat and Frémeaux-Bacchi, 2011; Verhave et al. 2014]. The complement system is an important component of the adaptive and innate immune system and consists of more than 40 plasma and membrane-associated proteins. The innate most important roles of the complement system are the recognition of pathogens, the activation and chemotaxis of leucocytes, and the induction of cell lysis by incorporation of the membrane attack complex (MAC). The three pathways of complement activation (classical, lectin and alternative) lead to complement factor 3 (C3) activation followed by MAC formation on complement-activating surfaces (Figure 1). Uncontrolled complement activation at the cell surface can lead to endothelial cells dysfunction and a cascade of events responsible for TMA.
Figure 1.
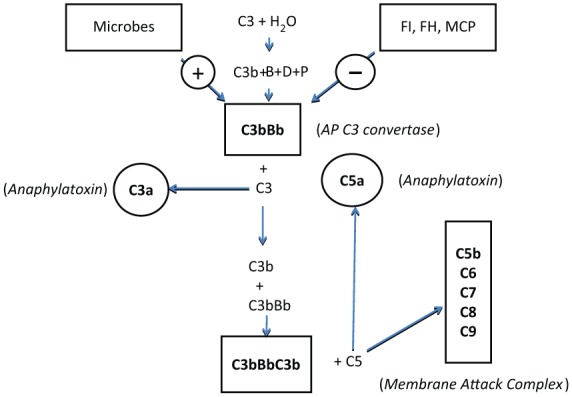
Alternative pathway of the complement system.
The activation of the alternative pathway of the complement system depends on spontaneous hydrolysis of complement factor 3 (C3) in plasma leading to the formation of C3(H2O) [Berger et al. 2005]. This molecule binds to factor B. Subsequent activation by factor D results in the formation of C3(H2O)Bb. This complex cleaves additional C3 to C3a and C3b (split products of C3) constantly and at a low rate. In the presence of an activating surface (e.g. a bacterial wall), C3b is protected from inactivation by circulating regulatory proteins like factor I and factor H or by membrane co-factor protein (MCP) which is a protein anchored to the cell’s membrane. As a result the more active alternative pathway C3 convertase C3bBb is formed, which is further stabilized by properdin. The binding of C3b to the C3 convertase results in the formation of the C5 convertases (C3bBbC3b) for the alternative pathway [Berger et al. 2005]. The C5 convertases initiate the formation of the membrane attack complex (MAC) by cleavage of C5 to C5a (anaphylatoxin, split product of complement factor 5) and C5b. C5b binds to respectively C6, C7, C8 and multiple molecules of C9 to form MAC. MAC is able to incorporate to cell membranes and to provoke cell activation or even cell lysis [Berger et al. 2005].
Mutations
Several extensive reports of patient series and reviews of pathophysiology on this topic have been published [Constantinescu et al. 2004; Esparza-Gordillo et al. 2005; Noris and Remuzzi, 2005, 2009, 2010; Caprioli et al. 2006; Sellier-Leclerc et al. 2007; Johnson and Taylor, 2008; Rodrigues de Cordoba and de Jorge, 2008; Coppo et al. 2010; Dragon-Durey et al. 2010; Kavanagh and Goodship, 2010; Sánchez-Corral and Melgosa, 2010; Scheiring et al. 2010; Westra et al. 2010; Loirat and Frémeaux-Bacchi, 2011; Keir and Coward, 2011; Zuber et al. 2011; Geerdink et al. 2012; Bresin et al. 2013; Kavanagh et al. 2013; Skerka et al. 2013; Verhave et al. 2014). The prevalence of mutations and of associated low serum concentrations in aHUS are listed in Table 1 [Cameron and Vick, 1973; Thompson and Winterborn, 1981; Warwicker et al. 1998; Neumann et al. 2003; Frémeaux-Bacchi et al. 2008; Loirat et al. 2008; Lhotta et al. 2009; Roumenina et al. 2009; Tawadrous et al. 2010; Maga et al. 2010; Kavanagh and Goodship, 2010; Noris and Remizzi, 2010; Bresin et al. 2013; Verhave et al. 2014].
Table 1.
Prevalence of mutations for genes coding for proteins affecting the inhibition of the AP convertase, anti-CFH antibodies and low C3 serum concentration in aHUS.
| Mutations and anti-CFH antibodies | Prevalence (%) | Low C3 serum concentration (%) |
|---|---|---|
| CFH* | 20–30 | 30–50 |
| MCP$ | 5–15 | 25 |
| CFI‡ | 4–10 | 20–30 |
| CFB‖ | 1–4 | 100 |
| C3¶ | 2–10 | 70–80 |
| THBD# | 0–5 | 50 |
| Combined mutations.** | 2–12 | 30 |
References
[Caprioli et al. 2006; Sellier-Leclerc et al. 2007; Neumann et al. 2003; Rodrigues de Cordoba and de Jorge, 2008; Noris and Remuzzi, 2010; Kavanagh and Goodship, 2010; Loirat and Frémeaux-Bacchi, 2011; Bresin et al. 2013; Verhave et al. 2014].
[Sellier-Leclerc et al. 2007; Fang et al. 2007; Maga et al. 2010; Noris and Remuzzi, 2010; Sullivan et al. 2010; Frémeaux-Bacchi et al. 2013; Verhave et al. 2014].
[Caprioli et al. 2006; Noris and Remuzzi, 2010; Loirat and Frémeaux-Bacchi, 2011; Kavanagh et al. 2013; Verhave et al. 2014].
[Goicoechea de Jorge et al. 2007; Roumenina et al. 2009; Maga et al. 2010; Tawadrous et al. 2010; Loirat and Frémeaux-Bacchi, 2011; Frémeaux-Bacchi et al. 2013; Verhave et al. 2014].
[Frémeaux-Bacchi et al. 2008; Lhotta et al. 2009; Noris and Remuzzi, 2010; Kavanagh and Goodship, 2010; Bresin et al. 2013; Loirat and Frémeaux-Bacchi, 2011; Verhave et al. 2014].
[Delvaeye et al. 2009; Loirat and Frémeaux-Bacchi, 2011; Kavanagh et al. 2013; Frémeaux-Bacchi et al. 2013; Verhave et al. 2014].
[Caprioli et al. 2006; Sellier-Leclerc et al. 2007; Neumann et al. 2003; Rodrigues de Cordoba and de Jorge, 2008; Noris and Remuzzi, 2010; Kavanagh and Goodship, 2010; Loirat and Frémeaux-Bacchi, 2011; Bresin et al. 2013; Frémeaux-Bacchi et al. 2013; Verhave et al. 2014].
aHUS, atypical haemolytic uremic syndrome; AP, alternative pathway; C3, complement factor 3; CFH, complement factor H.
In aHUS, the hyperactivity of the alternative pathway (AP) results from a stabilization of the AP C3 convertase (C3 convertase of the alternate pathway of the complement system) (Figure 1). This stabilization may be caused by loss of function mutations of the genes coding for AP C3 convertase inhibitors [complement factor H (CFH), complement factor I (CFI) and membrane cofactor protein (MCP)] which prevent those molecules from playing their physiological role. Other mutations coding for C3 and complement factor B (CFB) called gain of function mutations increase the solidity of the binding of C3b and Bb (split product of complement factor B) in the AP C3 convertase. Another cause of AP C3 convertase stabilization resides in antibodies directed against CFH. The latter are frequently associated with a homozygous deletion of the genes coding for two CFH-related proteins, CFHR1 and CFHR3 [Dragon-Durey et al. 2010; Lee et al. 2009; Moore et al. 2010; Noris and Remuzzi, 2010; Westra et al. 2010; Frémeaux-Bacchi et al. 2013]. Some cases result also from loss of function mutations of the membrane-bound protein thrombomodulin (THBD), involved in the coagulation process [Delvaeye et al. 2009]. The latter also binds to C3b and CFH, and increases the CFI-mediated inactivation of C3b (split product of C3) in the presence of CFH.
Up to 12% of aHUS patients have various combinations of 2 or more mutations of CFH, CFI, MCP, C3, CFB, THBD and/or antibodies against CFH [Esparza-Gordillo et al. 2006; Goicoechea de Jorge et al. 2007; Loirat et al. 2008; Rodrigues de Cordoba and Jorge, 2008; Delvaeye et al. 2009; Bienaimé et al. 2009; Maga et al. 2010; Noris and Remuzzi, 2010; Geerdink et al. 2012; Bresin et al. 2013; Frémeaux-Bacchi et al. 2013]. Not only combined mutations of gene coding for complement factors but also other genetic susceptibility factors such as the risk haplotypes (polymorphisms) in the CFH and MCP genes and mutations of genes coding for factors of the cyanocobalamin system [Bouts et al. 2010] or for DGKE (a protein involved in the coagulation process) [Lemaire et al. 2013] might increase the risk of TMA and as such contribute to the development of aHUS.
One or several abnormalities of the complement system are presently demonstrated today in 50–70% of children and adults with aHUS, leaving 30–50% of aHUS remaining unexplained. Not unexpectedly, the percentage of patients within the various subgroups shows some variations over the years according to countries and registries.
Familial aHUS, incomplete penetrance and genetic variability
A familial clustering of the disease is observed in approximately 20% of all aHUS cases [Sellier-Leclerc et al. 2007; Noris and Remuzzi, 2010]. The disease inheritance is in an autosomal recessive or dominant trait. The majority of complement mutations are heterozygous in aHUS patients. The penetrance of complement mediated-aHUS has been found to be present in only approximately 50% for all mutations, as half of the family members who carry the mutation do not present the disease by age 45 [Esparza-Gordillo et al. 2006; Goicoechea de Jorge et al. 2007; Frémeaux-Bacchi et al. 2008; Rodrigues de Cordoba and de Jorge, 2008; Lhotta et al. 2009; Noris and Remuzzi, 2010; Kavanagh and Goodship, 2010; Sullivan et al. 2010]. The expression of the disease not only depends on the presence of mutations or at-risk polymorphisms in the complement system, but can also be triggered by infection, pregnancy or medications [Fang et al. 2007; Fakhouri et al. 2010; Noris and Remuzzi, 2010]. In practice, apart from adequate information about the disease and its presenting symptoms, it is impossible to foreseen the risk of occurrence of HUS in family members presenting the same mutation.
Clinical features
Upper respiratory tract infection or diarrhoea/gastroenteritis triggers onset of aHUS in at least half of patients [Noris and Remuzzi, 2010] and up to 80% in paediatric patients [Sellier-Leclerc et al. 2007; Loirat et al. 2008; Geerdink et al. 2012]. Interestingly, diarrhoea preceded aHUS in 20–30% of paediatric patients [Sellier-Leclerc et al. 2007; Noris and Remuzzi, 2010], which complicates the differential diagnosis with STEC-HUS in children. Pregnancy is a frequent triggering event [Fang et al. 2007, Rodrigues de Cordoba and de Jorge, 2008; Fakhouri et al. 2010; Noris and Remuzzi, 2010; Noris et al. 2010]: 20% of women who develop aHUS experience their first presentation in the context of gravidity [Fakhouri et al. 2010]. It takes place mainly at the occasion of the first pregnancy and during the postpartum period [Noris et al. 2010]. In a large cohort of HUS neither associated with STEC infection nor ADAMST13 abnormalities, but attributed to other various TMA causes (such as glomerulonephritis and cancer), a few cases presented with genetic abnormalities in complement system factors [Besbas et al. 2006; Noris et al. 2010].
The onset of aHUS is generally sudden. Symptoms in young children are pallor, general distress, poor feeding, vomiting, fatigue and sometimes oedema. Adults mention fatigue and general distress. Often symptoms of hypertension are present. Most patients have the complete biologic features of HUS at first investigation and have to be dialysed directly on admission due to the severity of the acute renal failure.
Extra renal manifestations are observed in 20% of patients. Most often reported is the central nervous system (CNS) involvement, ranging from mild orientation disorders to alterations in consciousness, apraxia and epileptic seizures and even coma. But myocardial infarct and skin lesions are also mentioned [Sellier-Leclerc et al. 2007; Noris and Remuzzi, 2010].
Some patients have a less abrupt onset with subclinical anaemia and thrombocytopenia, while renal function is preserved with variable evolution [Sellier-Leclerc et al. 2007].
The outcome of untreated patients is mostly severe [Berger et al. 2005]. At 5 years after onset, the percentage of patients with end-stage renal failure (ESRF) was 73, 50 and 38% in the CFH, CFI and MCP groups, respectively. Relapsing aHUS may occur whatever the genotype. Relapse with complete recovery is characteristic of patients with MCP. Some observations suggest that complement dysregulation may be responsible for atheroma-like vascular complications [Loirat et al. 2010; Davin et al. 2011b], especially when patients are submitted to prolonged periods of dialysis.
After renal transplantation, aHUS may occur either as a recurrent or de novo form [Loirat and Frémeaux-Bacchi, 2008; Noris and Remuzzi, 2010; Zuber et al. 2011]. The risk of post-transplant recurrence of aHUS depends on the genetic abnormality involved, and ranges from 15 to 20% in patients with MCP mutations and from 50 to 100% in patients with mutations in the genes that encode circulating regulators of complement. In de novo aHUS after renal transplantation, the genetic abnormalities in complement regulators are reported in 30% of recipients.
Differences between children and adults
Frémeaux-Bacchi and colleagues recently compared the genetic and clinical features of aHUS initiated during childhood and adulthood [Frémeaux-Bacchi et al. 2013]. Onset of aHUS occurred as frequently during adulthood (58.4%) as during childhood (41.6%). The percentages of patients who developed the disease were 23, 40, 70, and 98% by age 2, 18, 40 and 60 years, respectively. Mortality was higher in children than in adults (6.7% versus 0.8% at 1 year; p = 0.02), but progression to ESRF after the first aHUS episode was more frequent in adults (46% versus 16%; p = 0.001). The renal outcome was not significantly different in adults regardless of genetic background. Only MCP and undetermined aHUS were less severe in children than adults. The frequency of relapse after 1 year was 92% in children with MCP-associated HUS and approximately 30% in all other subgroups. The onset occurred as frequently in adulthood (58.4%) as in childhood (41.6%).
Differential diagnosis
It has been proposed recently to define aHUS as a TMA characterized by excessive complement activation at the surface of the renal microvasculature [Scully and Goodship, 2014]. This definition has a major therapeutic implication since plasma therapy and overall anti-C5 mAb have been shown to be successful in the treatment of aHUS, particularly when they are initiated promptly. However, several issues exist for diagnosing aHUS [Scully and Goodship, 2014]. Indeed, a systemic activation of the complement system is not always present. Furthermore, patients with unexplained HUS do not always present with inherited or acquired abnormality affecting components of the complement pathway, whereas such abnormalities are detected in some patients presenting with HUS apparently secondary to other causes [Scully and Goodship, 2014]. For this reason, exclusion and inclusion criteria have been recently established in the UK by the aHUS Rare Disease Group (RDG) (http://rare-renal.org).
TTP and STEC-HUS are TMAs at highest risk to be confounded with aHUS. Indeed, as stated above, 20–30% of aHUS are presenting with diarrhoea. Activation of the AP of the complement resulting from the binding of STEC toxin to CFH has been shown in STEC-HUS [Orth et al. 2009]. Furthermore, the renal function can be reduced in TTP [Wada et al. 2014]. The diagnosis of TTP requires, in the majority of cases, that ADAMST13 activity is less than 5–10% [Wada et al. 2014]. STEC- HUS can be diagnosed in the stool by a positive culture of STEC and the presence of Shiga toxin (Shiga toxin polymerase chain reaction) or by the presence of serological antibodies to the O-antigen of the STEC such as O-157 antigen [Espié et al. 2008]. Serological and genetic abnormalities of the complement system should be looked for in any patient with HUS when TTP and STEC- HUS have been definitely excluded. As any HUS at-tributed to other causes can mask aHUS [Fakhouri et al. 2010], the same parameters should also been screened in those cases. The same remark also concerns STEC-HUS with an uncommon course (familial with different time period at presentation, relapses, late onset, atypical presentation).
It can be questioned why screening for genetic abnormalities is important. Indeed the results are generally not known for several weeks. Therefore they cannot be used for acute indication of plasma therapy or of anti-C5 mAb. Moreover, no genetic abnormalities are found in 30–50% of patients in whom no other cause of HUS is found. However, knowing the genetic features of the patient is important for the prognosis and for the treatment. aHUS related to MCP mutations is often characterized by multiple relapses healing spon-taneously with less risk of ESRF than other mutations.
Another issue concerns the reimbursement of the cost of anti-C5 mAb by health insurance companies. In some countries (e.g. Belgium), the presence of an abnormality of the complement system is required for reimbursement.
Treatment of aHUS
Plasma therapy
The success obtained with plasma replacement in TTP decades ago led to the early use of the latter in aHUS [Rock et al. 1991; Loirat 1992].
Fresh frozen plasma (FFP) brings normal complement proteins to the patient (CFH, CFI, CFB and C3). Plasmapheresis (PE) using plasma as exchange adds to the quantitative and qualitative amounts of ‘good’ complement proteins, the removal of mutant CFH, CFI, CFB and C3, as well as anti-CFH antibodies and inflammatory products inducing endothelial dysfunction and platelet aggregation. Preventing hyperproteinemia and volume overload eventually resulting in cardiac failure represents another advantage of PE over plasma infusion (PI).
The efficacy of plasma therapy can only be determined on individual reports since no prospective trial has been performed. Because spontaneous recovery is possible with all types of mutations and because patients are treated with different protocols, it is impossible to evaluate correctly the efficacy of plasma therapy in reported series of aHUS patients.
A retrospective attempt was recently made by the members of the European Paediatric Study Group for Haemolytic Uraemic Syndrome. They investigated their clinical practice guidelines for the investigation and initial therapy of aHUS [Ariceta et al. 2009] by a questionnaire among paediatric nephrologists across Europe in the months prior to the use of anti-C5 mAb [Johnson et al. 2014]. A total of 59 out of 71 (83%) patients received plasma therapy within the first 33 days after diagnosis, of whom 10 received PI only. There were no deaths during the study period of the first 33 days after diagnosis. However, 17% were still dialysis-dependent, somehow comparable with published paediatric studies and better than the adult outcome [Geerdink et al. 2012; Bresin et al. 2013]. In this study, 31% had complications related to central venous access by day 33.
Differences of efficacy can also be expected from the type of mutation implicated. As MCP is not a circulating protein, a beneficial effect of plasma therapy is unlikely to be expected in MCP mutated patients. Indeed at least 90% of patients with a MCP mutation enter remission after acute episodes, whether or not they receive plasma therapy [Caprioli et al. 2006; Sellier-Leclerc et al. 2007; Noris and Remuzzi, 2010].
Case reports show clearly that intensive and prophylactic PE is the only plasma therapy modality to recover and maintain a normal renal function at long term in difficult cases associated with CFH mutations [Loirat and Frémeaux-Bacchi, 2011]. In a family of three sisters presenting with the disease and the same CFH mutation, two of them presented with ESRF in the follow up. The only one who still presented a normal renal function after 11 years follow up despite 2 relapses, received initially daily PE sessions until plasma creatinine normalization and prophylactic sessions every 2 weeks indefinitely with return to daily sessions in case of relapse [Davin et al. 2008; Davin et al. 2011b]. This is confirmed by the paediatric cases of aHUS with CFH mutation reported in the literature and reviewed by Loirat and Frémeaux-Bacchi [Loirat and Frémeaux-Bacchi, 2011]. The latter showed that intensive followed by maintenance PE in 10 out 12 aHUS patients resulted in preserved renal function after 1–6 years, whereas all patients treated with plasma therapy during acute episodes only all died or were in ESRF within less than 1 year.
Although case reports suggest that plasma therapy could be useful in patients with CFI, C3, CFB or THBD mutations [Goicoechea de Jorge et al. 2007; Fang et al. 2007; Roumenina et al. 2009; Lhotta et al. 2009; Noris and Remuzzi, 2010; Tawadrous et al. 2010; Loirat and Frémeaux-Bacchi, 2011; Johnson et al. 2014], larger series with a better documentation of the protocol used are necessary to better evaluate this therapy in those cases.
In case of anti-CFH antibodies, prompt PE sessions to lower antibodies, associated with immunosuppression to prevent the level of antibodies increasing again after PE interruption have been shown to be efficacious and safe at long term to prevent relapses [Dragon-Durey et al. 2010; Sana et al. 2014; Sinha et al. 2014]. A later occurrence and a lower frequency of relapses (16.6%) were reported in patients receiving during and after PE sessions, maintenance immunosuppression by prednisone with either mycophenolate mofetil or azathioprine (Sinha et al. 2014). Prospective studies are needed to confirm these findings.
Many patients of historical series have received some form of plasma therapy at the time of recurrence after kidney transplantation [Sellier-Leclerc et al. 2007; Loirat and Frémeaux-Bacchi, 2008; Noris and Remuzzi, 2010]. Most studies suggested that plasma therapy initiated at the time of recurrence often fails to rescue kidney func-tion and to prevent graft loss [Loirat and Frémeaux-Bacchi, 2008; Noris and Remuzzi, 2010]. The high risk of recurrence and graft lost led some authors to contra-indicate transplantation in aHUS [Ruggenenti, 2002]. The first successful kidney transplantation using prophylactic PE, started before the transplantation and continued indefinitely in a case of familial severe aHUS due to a factor H mutation, was reported in 2005 [Olie et al. 2005; Davin et al. 2011a]. Thereafter, preventive protocolled plasma therapy started before and peri-transplantation was recommended until the era of anti-C5 mAb [Zuber et al. 2011].
Unfortunately PE presents numerous disadvantages. It is not always efficacious, certainly when it is not given in time. Adverse reaction to plasma still occurs despite prevention procedures. Resistance to plasma can be present from the start or can develop within time. It requires regular hours during visits to hospital and can induce several complications associated with the vascular access, infections from contaminated plasma or allergic reactions to plasma intolerance. Since the demonstration of the superiority of anti-C5 mAb, the remaining indications of plasma therapy in aHUS, in particular in children, reside in the window phase of the first 4 days of diagnosis of the underlying disease in an unknown patient, and the impossibility of access to anti-C5 mAb and in anti-CFH mediated aHUS.
Combined liver–kidney transplantation to cure aHUS
As CFH, CFI, CFB and C3 are synthesized in the liver, liver or combined liver–kidney transplantation was logically proposed. Several successful cases on condition of intensive plasma therapy during the procedure have been reported [Saland et al. 2009; Loirat and Frémeaux-Bacchi, 2011]. However, due to 18% operative deaths and the new era of complement inhibition, this procedure should be considered only on an individual base.
Use of anti-C5 mAb in aHUS
Eculizumab (Soliris; Alexion Pharmaceutical) is actually the only anti-C5 mAb on the market for clinical purpose. Eculizumab is a humanized, mouse anti-C5 mAb with high affinity for human C5, effectively blocking its cleavage into C5a (anaphylatoxin, split product of complement factor 5) and C5b, and as such the inhibition of the subsequent generation of the terminal complement complex C5b-9 and downstream pro-inflammatory and cell lytic properties. To minimize immunogenicity, murine complementarity-determining regions are grafted into human heavy and light chain germline antibody framework sequences. Additionally, human immunoglobulin G 2 (IgG2) and IgG4 heavy chain sequences were combined to form a hybrid constant region that is unable to bind Fc receptors or to activate the complement cascade (www.ema.europa.eu; www.solirisrems.com).
As eculizumab does not interfere with the proximal activation of the complement system, the splits products of C3 which are necessary for opsonisation of microorganisms and clearance of immune complexes, remain intact (www.ema.europa.eu; www.solirisrems.com). Eculizumab was first used with success in patients with PNH. Nowadays several dozen retrospective observations of children and adults treated with eculizumab for aHUS in native or transplanted kidneys have been published [Gruppo and Rother, 2009; Nürnberger et al. 2009; Chatelet et al. 2009; Davin et al. 2010; Zimmerhackl et al. 2010; Heyne et al. 2011; Nester et al. 2011; Weitz et al. 2011; Wilson et al. 2011; Ariceta et al. 2012; Dorresteijn et al. 2012; Ardissino et al. 2013; Cañigral et al. 2013; Gilbert et al. 2013; Gulleroglu et al. 2013; Legendre et al. 2013b; Pelicano et al. 2013; Povey et al. 2013; Reuter et al. 2013; Sinibaldi et al. 2013; Thajudeen et al. 2013; Vaisbich et al. 2013; Belingheri et al. 2014; Chiodini et al. 2014; Christmann et al. 2014; Fakhouri et al. 2014; Green et al. 2014; Román-Ortiz et al. 2014; Tran et al. 2014]. All described success on renal, neurological, cutaneous and myocardial lesions, in all kind of mutations and also when mutations were not identified, and in anti-CFH antibodies mediated HUS. A relapse can be observed when eculizumab is stopped [Nürnberger et al. 2009]. Use during pregnancy did not induce any complication in the foetus [Ardissino et al. 2013]. The recommended dosage of eculizumab is reported in Table 2 (www.ema.europa.eu; www.solirisrems.com).
Table 2.
Dosing recommendations for eculizumab in patients with aHUS according to patient body weight (www.solirisrems.com).
| • 40 kg and over: |
| ○ 900 mg weekly × 4 doses; 1200 mg at week 5; then 1200 mg every 2 weeks |
| • 30 kg to less than 40 kg: |
| ○ 600 mg weekly × 2 doses; 900 mg at week 3; then 900 mg every 2 weeks |
| • 20 kg to less than 30 kg |
| ○ 600 mg weekly × 2 doses; 600 mg at week 3; then 600 mg every 2 weeks |
| • 10 kg to less than 20 kg |
| ○ 600 mg weekly × 1 dose; 300 mg at week 2; then 300 mg every 2 weeks |
| • 5 kg to less than 10 kg |
| ○ 300 mg weekly × 1 dose; 300 mg at week 2; then 300 mg every 3 weeks |
aHUS, atypical haemolytic uremic syndrome.
Eculizumab efficacy
Eculizumab has been used in renal transplantation for aHUS, as replacement of PE because of intolerance to plasma, as treatment of relapse or as pre-emptive treatment with or without PE and finally as replacement of PE in case of renal–liver transplantation in a patient intolerant for plasma. In all patients, all haematological signs normalized and renal function improved markedly, and the renal transplantations were successful.
International multicentre prospective studies (controlled, open-label, single-arm phase II, duration 26 weeks) were conducted in 2009–2010 in adults and adolescents (⩾12 years) with aHUS (primary disease or post transplantation recurrence [Legendre et al. 2013a].
Patients ⩾12 years-old with newly diagnosed aHUS and persistent TMA despite ⩾4 PE/PI sessions (plasma-resistant aHUS) treated with eculizumab demonstrated a highly significant change in platelet count; 15/17 patients (88%) achieved TMA event-free status and 4/5 patients on dialysis permanently discontinued dialysis [Legendre et al. 2013a]. Of the 13 patients who entered the extension study, all of them continued to maintain normal levels of thrombocytes and renal function improved [Legendre et al. 2013a]. Eculizumab was well tolerated; 10 patients presented with adverse events deemed related to eculizumab which were graded as generally mild/moderate [Legendre et al. 2013a].
Amongst the 20 ⩾ 12 year-old patients with plasma-dependent aHUS sensitive to PE/PI and still receiving plasma therapy at the time of screening, 19 continued into an extension [Legendre et al. 2013a]. All patients maintained normal platelets count during the whole follow up and none of them necessitated any TMA intervention (no re-entry of plasma therapy). Quality of life was improved. Only six patients had adverse events deemed related to the drug. In both studies, eculizumab was similarly effective in patients with or without identified complement mutations and was well tolerated.
In a retrospective study in paediatric patients treated with eculizumab, normalization of thrombocytes was seen in 14/15 patients [Rathbone et al. 2013]. The data from the prospective clinical trial for eculizumab in paediatric patients with aHUS are currently being analysed.
In conclusion, those prospective and retrospective studies confirm the analysis of case reports of the use of eculizumab in aHUS. Eculizumab treats TMA caused by dysregulation of complement, prevents the recurrence of TMA during treatment, improves renal function, allows discontinuation of chronic PE/PI and is well tolerated.
Eculizumab safety
The use of eculizumab increases a patient’s susceptibility to serious meningococcal infections (septicaemia and/or meningitis). Life-threatening and fatal meningococcal infections have occurred. Administration of a tetravalent conjugated meningococcal vaccine (serogroups A, C, Y, W 135) at least 2 weeks before starting eculizumab is recommended. Re-vaccination of patients may be necessary depending on the duration of eculizumab therapy. If urgent therapy is indicated in an unvaccinated patient, it is recommended to administer the meningococcal vaccine as soon as possible and to start antibiotic prophylaxis (www.ema.europa.eu; www.solirisrems.com).
It has been shown that vaccination reduces, but does not eliminate, the risk of meningococcal infection [Struijk et al. 2013]. The reasons of this lack of efficacy can be multiple. Patients can mount insufficient Ab titres. Another reason is that no available vaccines contained a serogroup B antigen, which is the most prevalent in the rest of Europe and other parts of the world [Bouts et al. 2011]. Based on this risk of meningococcal infection by serogroup B that cannot yet be prevented by most used vaccines, penicillin prophylaxis should be strongly considered according some authors [Bouts et al. 2011]. The best strategy depends on the distribution of meningococcal serogroups and the availability of vaccines in different countries. A new vaccine including serogroup B became available recently, but some uncertainty remains about the breadth of the conferred protection [Andrews and Pollard, 2014].
Children treated with anti-C5 mAb may also be at increased risk of developing serious infections due to Streptococcus pneumoniae and Haemophilus influenza type b (Hib). Vaccinations for the prevention of S. pneumoniae and Hib are therefore indicated (www.ema.europa.eu; www.solirisrems.com).
Other side effects reported are hypertension, infections, diarrhoea, vomiting, cough, nasal congestion and tachycardia. Reactions during infusion are possible as for each protein (www.ema.europa.eu; www.solirisrems.com; Legendre et al. 2013a).
As with all proteins there is a potential for immunogenicity. Low titres of antibodies to eculizumab were detected in 1/37 (2.7%) patients with aHUS treated, but no apparent correlation of antibody development to clinical response was observed in both indications [Parker, 2009]. Eculizumab is a relative young medicine used in humans and no data on serious adverse effects are yet available on the long-term use. These data are needed and should be collected to provide information about the safety profile in the long term.
Anti-C5 mAb treatment issues
In the prospective trials on eculizumab in aHUS [Legendre et al. 2013a], the diagnostic criteria were limited to the triad of symptoms (haemolytic anaemia, progressive thrombopenia and impaired renal function) in absence of TTP and STEC infection. Abnormalities of the complement system (presence of mutations or reduce concentration of circulating proteins) were not required. In those trials, even if the use of eculizumab resulted in a significant mean increase from baseline of blood platelet count, lactate dehydrogenase (LDH) concentration and glomerular filtration rate (GFR) in the whole cohort of patients, those improvements were not observed in all patients (12% of them were not TMA event-free during the treatment). The lack of efficacy of anti-C5 mAb in those studies has been attributed to a late treatment. However, it does not exclude the fact that some patients included in the trials did not present a HUS related to complement abnormalities. The absence of response to anti-C5 mAb in HUS without complement abnormalities has been confirmed in a young child presenting with HUS associated with a mutation in the gene DGKE, coding for a protein involved in the coagulation process [Lemaire et al. 2013]. The latter results in a prothrombotic state leading to aHUS in a select group of children less than 1 year old. This underscores the need for extensive diagnostic investigation in non-STEC/non TTP HUS to avoid unnecessary anti-C5 mAb use. For this reason, as mentioned above (see differential diagnosis section), some authors have proposed restricting the terminology of aHUS to a TMA characterized by excessive complement activation at the surface of the renal microvasculature [Scully and Goodship, 2014]. In the UK, criteria for allowance of anti-C5 mAb exclude patients presenting with STEC-HUS, other infection-associated HUS (HIV, Streptococcus pneumonia), ADAMTS 13 deficiency or antibodies, drug-associated HUS, transplantation of any type excepted de novo renal, cobalamin deficiency, systemic lupus erythematosus, antiphospholipid syndrome and scleroderma (http://rare-renal.org).
Because of its very high price (around 400,000 euros per year of non-interrupted treatment of an adult), eculizumab is not available for all aHUS patients in the world. When this is the case, plasma therapy remains the best option. Another concern for the long term is the possibility of developing anti-C5 mAb with the risk of resistance to treatment or of serum sickness (although both complications have not yet been reported in a large cohort of patients treated with eculizumab for PNH [Parker, 2009]. Those issues led to the development of anti-C5 mAb sparing strategies.
The possibility of reducing the frequency of eculizumab injections by monitoring the ability of patient’s serum to raise the formation of C5b-9 in functional assays has been recently shown [Cugno et al. 2014]. A safe eculizumab interruption under strict surveillance by frequent dipstick for haematuria with immediate re-treatment if necessary has been reported in patients presenting mutations with a lower risk of recurrence [Ardissino et al. 2014]. The possible benefit of minimalizing endothelial stress as increasing risk of recurrence has been reported. Four adult aHUS patients at moderate or even high risk for recurrence received a living donor transplant while minimalizing endothelial damage (aggressive treatment of high blood pressure and cholesterol serum levels, low dose of calcineurin inhibitors and quadruple immunosuppression therapy to limit the risk of rejection). In this study neither anti-C5 mAb nor PE were given prophylactically. No recurrence was seen after more than 2 years’ follow up [Verhave et al. 2013].
Conclusion
It is clear that, with the use of anti-C5 mAb for the treatment of aHUS, a new era for patients with this disease has begun. Anti-C5 mAb has been shown to stop TMA, whether the cause of the dysregulation of the complement is known or not. The registration of this drug for aHUS has completely changed the treatment strategy, the prognosis and the quality of life of patients presenting with this illness.
It now remains to develop prospective multi-centre international studies in order to develop guidelines to define what conditions injections might be safely stopped or less frequently given, and to propose protocols for use in case of transplantation.
In the elaboration of anti-C5 mAb sparing strategies, it is important to keep in mind that any delay of treatment of relapse can result in irreversible lesions [Olie et al. 2004] and that any episode of acute renal failure, even of short duration and not needing dialysis, may lead to chronic kidney disease in the long term [Lameire et al. 2013].
In the future, the extension of indications for anti-C5 mAb use, the elaboration of generics and of mAbs directed towards other complement factors of the terminal pathway of the complement system might succeed in reducing the cost of this new valuable therapeutic approach and render it available worldwide for patients from all social classes.
Footnotes
Funding: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement: N.v.d.K. is member of the International Advisory Board of aHUS, Alexion Pharmaceuticals. J.C.D. is a member of the Belgian group of experts for aHUS supported by Alexion Pharmaceuticals.
Contributor Information
Jean-Claude Davin, Paediatric Nephrology Department, Emma Children’s Hospital-Academic Medical Centre, Meibergdreef 9, 1105 AZ Amsterdam Z-O, The Netherlands.
Nicole C. A. J. van de Kar, Department of Paediatric Nephrology, Radboud University Medical Centre, Amalia Children’s Hospital, Nijmegen, The Netherlands
References
- Andrews S., Pollard A. (2014) A vaccine against serogroup B Neisseria meningitidis: dealing with uncertainty. Lancet Infect Dis 14: 426–434. [DOI] [PubMed] [Google Scholar]
- Ardissino G., Ossola M., Baffero G., Rigotti A., Cugno M. (2013) Eculizumab for atypical hemolytic uremic syndrome in pregnancy. Obstet Gynecol 122: 487–489. [DOI] [PubMed] [Google Scholar]
- Ardissino G., Testa S., Possenti I., Tel F., Paglialonga F., Salardi S., Tedeschi S., et al. (2014) Discontinuation of eculizumab maintenance treatment for atypical hemolytic uremic syndrome: a report of 10 cases. Am J Kidney Dis 64: 633–637. [DOI] [PubMed] [Google Scholar]
- Ariceta G., Arrizabalaga B., Aguirre M., Morteruel E., Lopez-Trascasa M. (2012) Eculizumab in the treatment of atypical hemolytic uremic syndrome in infants. Am J Kidney Dis 59: 707–710. [DOI] [PubMed] [Google Scholar]
- Ariceta G., Besbas N., Johnson S., Karpman D., Landau D., Licht C., et al. (2009) Guideline for the investigation and initial therapy of diarrhea-negative hemolytic uremic syndrome. Pediatr Nephrol 24: 687–696. [DOI] [PubMed] [Google Scholar]
- Belingheri M., Possenti I., Tel F., Paglialonga F., Testa S., Salardi S., et al. (2014) Cryptic activity of atypical hemolytic uremic syndrome and eculizumab treatment. Pediatrics 133: e1769–e1771. [DOI] [PubMed] [Google Scholar]
- Berger S., Roos A., Daha M. (2005) Complement and the kidney: what the nephrologist needs to know in 2006? Nephrol Dial Transplant 20: 2613–2619. [DOI] [PubMed] [Google Scholar]
- Besbas N., Karpman D., Landau D., Loirat C., Proesmans W., Remuzzi G., et al. (2006) A classification of hemolytic uremic syndrome and thrombotic thrombocytopenic purpura and related disorders. Kidney Int 70: 423–431. [DOI] [PubMed] [Google Scholar]
- Bienaimé F., Dragon-Durey M., Regnier C., Nillson S., Kwan W., Blouin J., et al. (2009) Mutations in components of complement influences the outcome of Factor I associated atypical hemolytic uremic syndrome. Kidney Int 77: 339–349. [DOI] [PubMed] [Google Scholar]
- Bouts A., Monnens L., Davin J., Struijk G., Spanjaard L. (2011) Insufficient protection by Neisseria meningitidis vaccination alone during eculizumab therapy. Pediatr Nephrol 26: 1919–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouts A., Roofthooft M., Salomons G., Davin J. (2010) CD46-associated atypical haemolytic uremic syndrome with uncommon course caused by cblC deficiency. Pediatr Nephrol 25: 2547–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresin E., Rurali E., Caprioli J., Sanchez-Corral P., Frémeaux-Bacchi V., Rodriguez de, Cordoba S., et al. (2013) Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype. J Am Soc Nephrol 24: 475–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron J., Vick R. (1973) Plasma-C3 in haemolytic-uraemic syndrome and thrombotic thrombocytopenic purpura. Lancet 2: 975. [DOI] [PubMed] [Google Scholar]
- Cañigral C., Moscardó F., Castro C., Pajares A., Lancharro A., Solves P., de la, Rubia J., et al. (2013) Eculizumab for the treatment of pregnancy-related atypical hemolytic uremic syndrome. Ann Hematol 93: 1421–1422. [DOI] [PubMed] [Google Scholar]
- Caprioli J., Noris M., Brioschi S., Pianetti G., Castelleti F., Bettinaglio P., et al. (2006) Genetics of HUS: the impact of MCP, CFH and IF mutations on clinical presentation, response to treatment, and outcome. Blood 108: 1267–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatelet V., Frémeaux-Bacchi V., Lobbedez T., Ficheux M., de Ligny B. (2009) Safety and long-term efficacy of eculizumab in a renal transplant patient with recurrent atypical hemolytic-uremic syndrome. Am J Transplant 9: 2644–2645. [DOI] [PubMed] [Google Scholar]
- Châtelet V., Lobbedez T., Frémeaux-Bacchi V., Ficheux M., Ryckelynck J., Hurault de Ligny B. (2010) Eculizumab: safety and efficacy after 17 months of treatment in a renal transplant patient with recurrent atypical hemolytic-uremic syndrome: case report. Transplant Proc 42: 4353–4355. [DOI] [PubMed] [Google Scholar]
- Chiodini B., Davin J., Corazza F., Khaldi K., Dahan K., Ismaili K., et al. (2014) Eculizumab in anti-factor H antibodies associated with atypical hemolytic uremic syndrome. Pediatrics 133: e1764–e1768. [DOI] [PubMed] [Google Scholar]
- Christmann M., Hansen M., Bergmann C., Schwabe D., Brand J., Schneider W. (2014) Eculizumab as first-line therapy for atypical hemolytic uremic syndrome. Pediatrics 133: e1759–e1763. . [DOI] [PubMed] [Google Scholar]
- Constantinescu A., Bitzan M., Weiss L., Christen E., Kaplan B., Cnaan A., et al. (2004) Non-enteropathic hemolytic uremic syndrome: causes and short-term course. Am J Kidney Dis 43: 976–982. [DOI] [PubMed] [Google Scholar]
- Coppo P., Schwarzinger M., Buffet M., Wynckel A., Clabault K., Presne C., et al. (2010) Predictive features of severe acquired ADAMTS13 deficiency in idiopathic thrombotic microangiopathies: the French TMA reference center experience. PLoS One 5: e10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cugno M., Gualtierotti R., Possenti I., Testa S., Tel F., Griffini S., et al. (2014) Complement functional tests for monitoring eculizumab treatment in patients with atypical hemolytic uremic syndrome. J Thromb Haemost 12: 1440–1448. [DOI] [PubMed] [Google Scholar]
- Davin J., Gracchi V., Bouts A., Groothoff J., Strain L., Goodship T. (2010) Maintenance of kidney function following treatment with eculizumab and discontinuation of plasma exchange after a third kidney transplant for atypical hemolytic uremic syndrome associated with a CFH mutation. Am J Kidney Dis 55: 708–711. [DOI] [PubMed] [Google Scholar]
- Davin J., Groothoff J., Gracchi V., Bouts A. (2011a) Long-term renal function under plasma exchange in atypical hemolytic uremic syndrome. Pediatr Nephrol 26: 1915–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davin J., Majoie C., Groothoff J., Gracchi V., Bouts A., Goodship T., et al. (2011b) Prevention of large-vessel stenoses in atypical hemolytic uremic syndrome associated with complement dysregulation. Pediatr Nephrol 26: 155–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davin J., Strain L., Goodship T. (2008) Plasma therapy in atypical haemolytic uremic syndrome: lessons from a family with a factor H mutation. Pediatr Nephrol 23: 1517–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delvaeye M., Noris M., De Vriese A., Esmon C., Esmon N., Ferrell G., et al. (2009) Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N Engl J Med 361: 345–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorresteijn E., Van De, Kar N., Cransberg K. (2012) Child with plasma resistant atypical HUS escaped end stage renal failure by eculizumab therapy. Ped Nephrol 27: 1193–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragon-Durey M., Sethi S., Bagga A., Blanc C., Blouin J., Ranchin B., et al. (2010) Clinical features of anti-factor h autoantibody-associated hemolytic uremic syndrome. J Am Soc Nephrol 21: 2180–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esparza-Gordillo J., Goicoechea de, Jorge E., Buil A., Carreras Berges L., López-Trascasa M., Sánchez-Corral P., et al. (2005) Predisposition to atypical hemolytic uremic syndrome involves the concurrence of different susceptibility alleles in the regulators of complement activation gene cluster in 1q32. Hum Mol Genet 14: 703–712. [DOI] [PubMed] [Google Scholar]
- Esparza-Gordillo J., Jorge E., Garrido C., Carreras L., López-Trascasa M., Sánchez-Corral P., et al. (2006) Insights into hemolytic uremic syndrome: segregation of three independent predisposition factors in a large, multiple affected pedigree. Mol Immunol 43: 1769–1775. [DOI] [PubMed] [Google Scholar]
- Espié E., Grimont F., Mariani-Kurkdjian P., Bouvet P., Haeghebaert S., Filliol I., et al. (2008) Surveillance of hemolytic uremic syndrome in children less than 15 years of age, a system to monitor O157 and non-O157 Shiga toxin-producing Escherichia coli infections in France, 1996–2006. Pediatr Infect Dis J 27: 595–601. [DOI] [PubMed] [Google Scholar]
- Fakhouri F., Delmas Y., Provot F., Barbet C., Karras A., Makdassi R., et al. (2014) Insights from the use in clinical practice of eculizumab in adult patients with atypical hemolytic uremic syndrome affecting the native kidneys: an analysis of 19 cases. Am J Kidney Dis 63: 40–48. [DOI] [PubMed] [Google Scholar]
- Fakhouri F., Roumenina L., Provot F., Sallée M., Caillard S., Couzi L., et al. (2010) Pregnancy-associated hemolytic uremic syndrome revisited in the era of complement gene mutations. J Am Soc Nephrol 21: 859–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang C., Frémeaux-Bacchi V., Liszewski M., Pianetti G., Noris M., Goodship T., et al. (2007) Membrane cofactor protein mutations in atypical hemolytic uremic syndrome (aHUS), fatal Stx-HUS, C3 glomerulonephritis and the HELLP syndrome. Blood 111: 624–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frémeaux-Bacchi V., Fakhouri F., Garnier A., Bienaimé F., Dragon-Durey M., Ngo S., et al. (2013) Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol 8: 554–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frémeaux-Bacchi V., Miller E., Liszewski M., Strain L., Blouin J., Brown A., et al. (2008) Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood 112: 4948–4952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geerdink L., Westra D., van Wijk J., Dorresteijn E., Lilien M., Davin J., et al. (2012) Atypical hemolytic uremic syndrome in children: complement mutations and clinical characteristics. Pediatr Nephrol 27: 1283–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert R., Fowler D., Angus E., Hardy S., Stanley L., Goodship T. (2013) Eculizumab therapy for atypical haemolytic uraemic syndrome due to a gain-of-function mutation of complement factor, B. Pediatr Nephrol 28: 1315–1318. [DOI] [PubMed] [Google Scholar]
- Goicoechea de, Jorge E., Harris C., Esparza-Gordillo J., Carreras L., Arranz E., Garrido C., et al. (2007) Gain-of-function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc Natl Acad Sci U S A 104: 240–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green H., Harari E., Davidovits M., Blickstein D., Grossman A., Gafter U., et al. (2014) Atypical HUS due to factor H antibodies in an adult patient successfully treated with eculizumab. Ren Fail 36: 1119–1121. [DOI] [PubMed] [Google Scholar]
- Gruppo R., Rother R. (2009) Eculizumab for congenital atypical hemolyticuremic syndrome. N Engl J Med 360: 544–546. [DOI] [PubMed] [Google Scholar]
- Gulleroglu K., Fidan K., Hançer V., Bayrakci U., Baskin E., Soylemezoglu O. (2013) Neurologic involvement in atypical hemolytic uremic syndrome and successful treatment with eculizumab. Pediatr Nephrol 28: 827–830. [DOI] [PubMed] [Google Scholar]
- Heyne N., Weitz M., Guthoff M., Alscher M., Haering H., Nadalin S. (2011) Terminal complement blockade by eculizumab effectively reverses recurrent atypical hemolytic uremic syndrome after kidney transplantation. J Am Soc Nephrol 22: 952A. [Google Scholar]
- Johnson S., Stojanovic J., Ariceta G., Bitzan M., Besbas N., Frieling M., et al. (2014) An audit analysis of a guideline for the investigation and initial therapy of diarrhea negative (atypical) hemolytic uremic syndrome. Pediatr Nephrol 29: 1967–1978. [DOI] [PubMed] [Google Scholar]
- Johnson S., Taylor C. (2008) What’s new in haemolytic uraemic syndrome? Eur J Pediatr 167: 965–971. [DOI] [PubMed] [Google Scholar]
- Kavanagh D., Goodship T. (2010) Genetics and complement in atypical HUS. Pediatr Nephrol 25: 2431–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanagh D., Goodship T., Richards A. (2013) Atypical hemolytic uremic syndrome. Semin Nephrol 33: 508–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keir L., Coward R. (2011) Advances in our understanding of the pathogenesis of glomerular thrombotic microangiopathy. Pediatr Nephrol 26: 523–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lameire N., Bagga A., Cruz D., De Maeseneer J., Endre Z., Kellum J., et al. (2013) Acute kidney injury: an increasing global concern. Lancet 382: 170–179. [DOI] [PubMed] [Google Scholar]
- Lee B., Kwak S., Shin J., Lee S., Choi H., Kang H., et al. (2009) Atypical hemolytic uremic syndrome associated with complement factor H autoantibodies and CFHR1/CFHR3 deficiency. Pediatr Res 66: 336–340. [DOI] [PubMed] [Google Scholar]
- Legendre C., Licht C., Muus P., Greenbaum L., Babu S., Bedrosian C., et al. (2013a) Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med 368: 2169–2181. [DOI] [PubMed] [Google Scholar]
- Legendre C., Sberro-Soussan R., Zuber J., Rabant M., Loupy A., Timsit M., et al. (2013b) Eculizumab in renal transplantation. Transplant Rev 27: 90–92. [DOI] [PubMed] [Google Scholar]
- Lemaire M., Frémeaux-Bacchi V., Schaefer F., Choi M., Tang W., Le Quintrec M., et al. (2013) Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat Genet 45: 531–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lhotta K., Janecke A., Scheiring J., Petzlberger B., Giner T., Fally V., et al. (2009) A large family with a gain-of- function mutation of complement C3 predisposing to atypical hemolytic uremic syndrome, microhematuria, hypertension and chronic renal failure. Clin J Am Soc Nephrol 4: 1356–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loirat C. (1992) Treatment of hemolytic uremic syndrome with fresh frozen plasma or with plasmapheresis. In: Kaplan B., Trompeter R., Moake J. (eds), Hemolytic Uremic Syndrome and Thrombocytopenic Purpura. New York, Hog Kong and Basel: Marcel Dekker, pp. 431–440. [Google Scholar]
- Loirat C., Frémeaux-Bacchi V. (2008) Hemolytic uremic syndrome recurrence after renal transplantation. Pediatr Transpl 12: 619–629. [DOI] [PubMed] [Google Scholar]
- Loirat C., Frémeaux-Bacchi V. (2011) Atypical hemolytic uremic syndrome. Orphanet J Rare Dis 6: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loirat C., Noris M., Frémeaux-Bacchi V. (2008) Complement and the atypical hemolytic uremic syndrome. Pediatr Nephrol 23: 1957–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loirat C., Macher M., Elmaleh-Berges M., Kwon T., Deschênes G., Goodship T., et al. (2010) Non-atheromatous arterial stenoses in atypical haemolytic uraemic syndrome associated with complement dysregulation. Nephrol Dial Transplant 25: 3421–3425. [DOI] [PubMed] [Google Scholar]
- Maga T., Nishimura C., Weaver A., Frees K., Smith R. (2010) Mutations in alternative pathway complement proteins in American patients with atypical hemolytic uremic syndrome. Hum Mutat 31: E1445–E1460. [DOI] [PubMed] [Google Scholar]
- McKeage K. (2011) Eculizumab: a review of its use in paroxysmal nocturnal haemoglobinuria. Drugs 71: 2327–2345. [DOI] [PubMed] [Google Scholar]
- Monnens L., Hendrickx G., van Wieringen P., van Munster P. (1974) Letter: serum complement levels in haemolytic–uraemic syndrome. Lancet 2: 294. [DOI] [PubMed] [Google Scholar]
- Moore I., Strain L., Pappworth I., Kavanagh D., Barlow P., Herbert A., et al. (2010) Association of factor H autoantibodies with deletions of CFHR1, CFHR3, CFHR4 and with mutations in CFH, CFI, CD46, and C3 in patients with atypical haemolytic uraemic syndrome. Blood 115: 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nester C., Stewart Z., Myers D., Jetton J., Nair R., Reed A., et al. (2011) Pre-emptive eculizumab and plasmapheresis for renal transplant in atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol 6: 1488–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann H., Salzmann M., Bohnert-Iwan B., Mannuelian T., Skerka C., Lenk D., et al. (2003) Haemolytic uraemic syndrome and mutations of the factor H gene: a registry-based study of German speaking countries. J Med Genet 40: 676–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noris M., Caprioli J., Bresin E., Mossali C., Pianetti G., Gamba S., et al. (2010) Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol 5: 1844–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noris M., Remuzzi G. (2005) Hemolytic uremic syndrome. J Am Soc Nephrol 16: 1035–1050. [DOI] [PubMed] [Google Scholar]
- Noris M., Remuzzi G. (2009) Atypical hemolytic-uremic syndrome. N Engl J Med 361: 1676–1687. [DOI] [PubMed] [Google Scholar]
- Noris M., Remuzzi G. (2010) Thrombotic microangiopathy after kidney transplantation. Am J Transplant 10: 1517–1523. [DOI] [PubMed] [Google Scholar]
- Nürnberger J., Philipp T., Witzke O., Opazo Saez A., Vester U., Baba H., et al. (2009) Eculizumab for atypical haemolytic–uremic syndrome. N Engl J Med 360: 542–544. [DOI] [PubMed] [Google Scholar]
- Olie K., Florquin S., Groothoff J., Verlaak R., Strain L., Goodship T., et al. (2004) Atypical relapse of hemolytic uremic syndrome after transplantation. Pediatr Nephrol 19: 1173–1176. [DOI] [PubMed] [Google Scholar]
- Olie K., Goodship T., Verlaak R., Florquin S., Groothoff J., Strain L., et al. (2005) Posttransplantation cytomegalovirus-induced recurrence of atypical hemolytic uremic syndrome associated with a factor H mutation: successful treatment with intensive plasma exchanges and ganciclovir. Am J Kidney Dis 45: e12–e15. [DOI] [PubMed] [Google Scholar]
- Orth D., Khan A., Naim A., Grif K., Brockmeyer J., Karch H., et al. (2009) Shiga toxin activates complement and binds factor H: evidence for an active role of complement in hemolytic uremic syndrome. J Immunol 182: 6394–6400. [DOI] [PubMed] [Google Scholar]
- Parker C. (2009) Eculizumab for paroxysmal nocturnal haemoglobinuria. Lancet 373: 759–767. [DOI] [PubMed] [Google Scholar]
- Pelicano M., de Córdoba S., Diekmann F., Saiz M., Herrero S., Oppenheimer F., et al. (2013) Anti-C5 as prophylactic therapy in atypical hemolytic uremic syndrome in living-related kidney transplantation. Transplantation 96: e26–e29. [DOI] [PubMed] [Google Scholar]
- Povey H., Vundru R., Junglee N., Jibani M. (2013) Renal recovery with eculizumab in atypical hemolytic uremic syndrome following prolonged dialysis. Clin Nephrol 82: 326–331. [DOI] [PubMed] [Google Scholar]
- Rathbone J., Kaltenthaler E., Richards A., Tappenden P., Bessey A., Cantrell A. (2013) A systematic review of eculizumab for atypical haemolytic uraemic syndrome (aHUS). BMJ Open 3: e003573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter S., Heitplatz B., Pavenstädt H., Suwelack B. (2013) Successful long-term treatment of TMA with eculizumab in a transplanted patient with atypical hemolytic uremic syndrome due to MCP mutation. Transplantation 27: e74–e76. [DOI] [PubMed] [Google Scholar]
- Rock G., Shumak K., Buskard N., Blanchette V., Kelton J., Nair R., et al. (1991) Comparison of plasma exchange with plasma infusion in the treatment of thrombotic hrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med 325: 393–397. [DOI] [PubMed] [Google Scholar]
- Rodrigues de, Cordoba S., de Jorge E. (2008) Translational mini-review series on complement factor H: genetics and disease associations of human complement factor, H. Clin Exp Immunol 151: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Román-Ortiz E., Mendizabal Oteiza S., Pinto S., López-Trascasa M., Sánchez-Corral P., Rodríguez de Cordoba S. (2014) Eculizumab long-term therapy for pediatric renal transplant in aHUS with CFH/CFHR1 hybrid gene. Pediatr Nephrol 29: 149–153. [DOI] [PubMed] [Google Scholar]
- Roumenina L., Jablonski M., Hue C., Blouin J., Dimitrov J., Dragon-Durey M., Cayla M., et al. (2009) Hyperfunctional C3 convertase leads to complement deposition on endothelial cells and contributes to atypical hemolytic uremic syndrome. Blood 114: 2837–2845. [DOI] [PubMed] [Google Scholar]
- Ruggenenti P. (2002) Post-transplant hemolytic-uremic syndrome. Kidney Int 62: 1093–1104. [DOI] [PubMed] [Google Scholar]
- Saland J., Ruggenenti P., Remuzzi G. and Consensus Study Group. (2009) Liver–kidney transplantation to cure atypical hemolytic uremic syndrome. J Am Soc Nephrol 20: 940–949. [DOI] [PubMed] [Google Scholar]
- Sana G., Dragon-Durey M., Charbit M., Bouchireb K., Rousset-Rouvière C., Bérard E., et al. (2014) Long-term remission of atypical HUS with anti-factor H antibodies after cyclophosphamide pulses. Pediatr Nephrol 29: 75–83. [DOI] [PubMed] [Google Scholar]
- Sánchez-Corral P., Melgosa M. (2010) Advances in understanding the aetiology of atypical Haemolytic Uraemic Syndrome. Br J Haematol 150: 529–542. [DOI] [PubMed] [Google Scholar]
- Scheiring J., Rosales A., Zimmerhackl L. (2010) Clinical practice. Today’s understanding of the haemolytic uraemic syndrome. Eur J Pediatr 169: 7–13. [DOI] [PubMed] [Google Scholar]
- Scully M., Goodship T. (2014). How I treat thrombotic thrombocytopenic purpura and atypical haemolytic uraemic syndrome. Br J Haematol 164: 759–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellier-Leclerc A., Frémeaux-Bacchi V., Macher M., Niaudet P., Guest G., Boudailliez B., et al. (2007) Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome. J Am Soc Nephrol 18: 2392–2400. [DOI] [PubMed] [Google Scholar]
- Sinha A., Gulati A., Saini S., Blanc C., Gupta A., Gurjar B., et al. (2014) Prompt plasma exchanges and immunosuppressive treatment improves the outcomes of anti-factor H autoantibody-associated hemolytic uremic syndrome in children. Kidney Int 85: 1151–1160. [DOI] [PubMed] [Google Scholar]
- Sinibaldi S., Guzzo I., Piras R., Bresin E., Emma F., Dello Strologo L. (2013) Post-transplant recurrence of atypical hemolytic uremic syndrome in a patient with thrombomodulin mutation. Pediatr Transplant 17: E177–E181. [DOI] [PubMed] [Google Scholar]
- Skerka C., Chen Q., Frémeaux-Bacchi V., Roumenina L. (2013) Complement factor H related proteins (CFHRs). Mol Immunol 56: 170–180. [DOI] [PubMed] [Google Scholar]
- Struijk G., Bouts A., Rijkers G., Kuin E., ten Berge I., Bemelman F. (2013) Meningococcal sepsis complicating eculizumab treatment despite prior vaccination. Am J Transplant 13: 819–820. [DOI] [PubMed] [Google Scholar]
- Sullivan M., Erlic Z., Hoffmann M., Arbeiter K., Patzer L., Budde K., et al. (2010) Epidemiological approach to identifying genetic predispositions for atypical hemolytic uremic syndrome. Ann Hum Genet 74: 17–26. [DOI] [PubMed] [Google Scholar]
- Tawadrous H., Maga T., Sharma J., Kupferman J., Smith R., Schoeneman M. (2010) A novel mutation in the complement factor B gene (CFB) and atypical hemolytic uremic syndrome. Pediatr Nephrol 25: 947–951. [DOI] [PubMed] [Google Scholar]
- Thajudeen B., Sussman A., Bracamonte E. (2013) A case of atypical hemolytic uremic syndrome successfully treated with eculizumab. Case Rep Nephrol Urol 3: 139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson R., Winterborn M. (1981) Hypocomplementaemia due to a genetic deficiency of beta 1H globulin. Clin Exp Immunol 46: 110–119. [PMC free article] [PubMed] [Google Scholar]
- Tran H., Chaudhuri A., Concepcion W., Grimm P. (2014) Use of eculizumab and plasma exchange in successful combined liver-kidney transplantation in a case of atypical HUS associated with complement factor H mutation. Pediatr Nephrol 29: 477–480. [DOI] [PubMed] [Google Scholar]
- Vaisbich M., Henriques Ldos S., Watanabe A., Pereira L., Metran C., Malheiros D., et al. (2013) [Eculizumab for the treatment of atypical hemolytic uremic syndrome: case report and revision of the literature]. J Bras Nefrol 35: 237–241. [DOI] [PubMed] [Google Scholar]
- Verhave J., Westra D., van Hamersvelt H., van Helden M., van de, Kar N., Wetzels J. (2013) Living kidney transplantation in adult patients with atypical haemolytic uraemic syndrome. Neth J Med 71: 342–347. [PubMed] [Google Scholar]
- Verhave J., Wetzels J., van de Kar N. (2014) Novel aspects of atypical haemolytic uraemic syndrome and the role of eculizumab. Nephrol Dial Transplant 29(Suppl. 4): iv131–iv141. [DOI] [PubMed] [Google Scholar]
- Wada H., Matsumoto T., Yamashita Y. (2014) Natural history of thrombotic thrombocytopenic purpura and hemolytic uremic syndrome. Semin Thromb Hemost 40: 866–873. [DOI] [PubMed] [Google Scholar]
- Warwicker P., Goodship T., Donne R., Pirson Y., Nicholls A., Ward R., et al. (1998) Genetic studies into inherited and sporadic haemolytic uremic syndrome. Kidney Int 53: 836–844. [DOI] [PubMed] [Google Scholar]
- Weitz M., Amon O., Bassler D., Koenigsrainer A., Nadalin S. (2011) Prophylactic eculizumab prior to kidney transplantation for atypical hemolytic uremic syndrome. Pediatr Nephrol 26: 1325–1329. [DOI] [PubMed] [Google Scholar]
- Westra D., Volokhina E., van der Heijden E., Vos A., Huigen M., Jansen J., et al. (2010) Genetic disorders in complement (regulating) genes in patients with atypical haemolytic uraemic syndrome (aHUS). Nephrol Dial Transplant 25: 2195–2202. [DOI] [PubMed] [Google Scholar]
- Wilson C., Brown A., White S., Goodship T., Sheerin N., Manas D. (2011) Successful treatment of de novo posttransplant thrombotic microangiopathy with eculizumab. Transplantation 92: e42–e43. [DOI] [PubMed] [Google Scholar]
- Zimmerhackl L., Hofer J., Cortina G., Mark W., Wurzner R., Jungraithmayr T., et al. (2010) Prophylactic eculizumab after renal transplantation in atypical hemolytic-uremic syndrome. N Engl J Med 362: 1746–1748. [DOI] [PubMed] [Google Scholar]
- Zuber J., Le Quintrec M., Sberro-Soussan R., Loirat C., Frémeaux-Bacchi V., Legendre C. (2011) New insights into postrenal transplant hemolytic uremic syndrome. Nat Rev Nephrol 7: 23–35. [DOI] [PubMed] [Google Scholar]