

Abstract
Autophagy is a catabolic process that has been shown to have a role in many cellular processes including the removal of excessive or damaged proteins and protein aggregates. The salivary glands play a critical role in oral health, and their secretory capacity may be critically intertwined with the autophagic process. This review describes the role of autophagy activation in normal salivary gland homeostasis and during the glandular stress responses of therapeutic radiation, ductal ligation, autoimmunity, and salivary gland adenoid cystic carcinoma.
Keywords: radiation, ductal ligation, cancer, salivary physiology, Sjogren’s syndrome, rapalogues
Overview of Autophagy
Autophagy is a catabolic process that contributes to the maintenance of basal cellular and tissue homeostasis and serves as a survival mechanism in cellular adaptation to stressors. A major function of autophagy is to remove misfolded proteins or damaged organelles from intracellular compartments, and many studies suggest that autophagy is necessary to maintain cellular homeostasis (Klionsky 2007; Qu et al. 2007; Yousefi and Simon 2007; Glick et al. 2010). In addition, autophagy can be induced during metabolic stress, ultimately providing energy and increasing the survival capacity to cells under these conditions (Klionsky 2007; Mathew et al. 2007; Glick et al. 2010). An overview of the autophagic activation pathway and the autophagy-related genes (ATGs) that regulate this process is diagrammed in Figure 1. Key features include the development of double membrane vesicles (autophagosomes) that envelope the cargo, conversion of LC3-I to LC3-II, and reductions in p62 (Martinet et al. 2006; Mizushima and Yoshimori 2007; Tasdemir et al. 2008; Klionsky et al. 2012). Autophagy is a dynamic process with a number of experimental procedures that aid in determining pathway activation or inhibition. Conditional knockout models of autophagy are necessary in vivo as a complete knockout of autophagy is lethal shortly after birth (Kuma et al. 2004; Schiaffino et al. 2008; Wirawan et al. 2012). Previous studies utilizing conditional knockouts of autophagy in a variety of tissues have found developmental and/or functional changes in the target tissue when compared to the tissue of wild-type mice (Komatsu et al. 2005; Hara and Matsui 2006; Komatsu et al. 2006; Nakai et al. 2007; Pua et al. 2007; Jung et al. 2008; Raben et al. 2008; Zhang et al. 2009).
Figure 1.
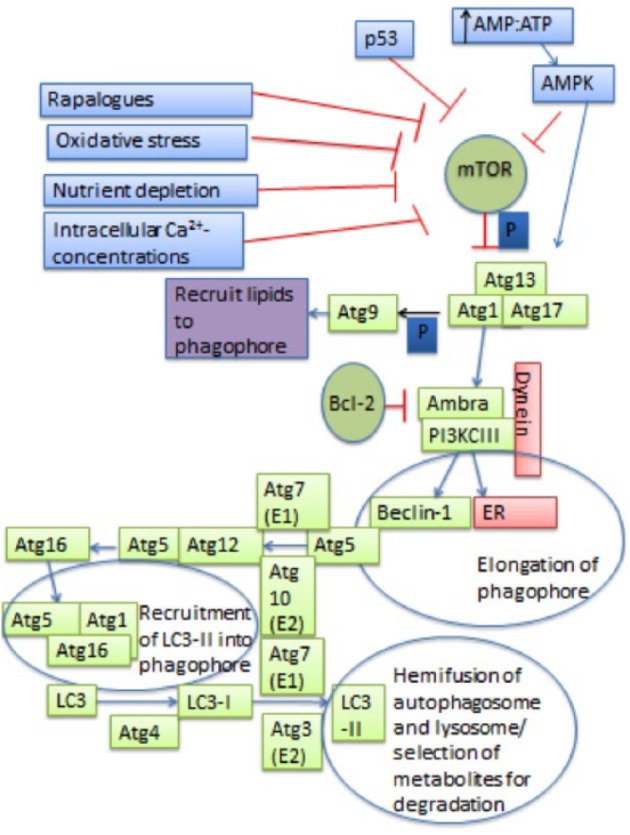
Schematic of the autophagic process. The autophagic process begins with the formation of the Atg1, Atg13, and Atg17 complex that signals Atg9 to bring lipids to the phagophore. This step is regulated by mTOR, which inhibits this step by phosphorylating (represented by the dark blue “P”) Atg13, making it unable to form a complex with Atg1 or Atg17. mTOR can be inhibited by pharmaceuticals such as rapalogues as well as systemic conditions such as nutrient depletion or oxidative stress. Increased intracellular Ca2+ concentrations, decreased ATP levels, and p53 and AMPK signaling pathways also allow for the inhibition of mTOR. Vps34 regulates autophagy by forming a complex with Beclin-1 by increasing levels of PI3P, which is necessary for phagophore elongation. Atg5 activates Atg12 by binding to its carboxy terminal, where it acts like an E1 ubiquitin–activating enzyme. Then, Atg12 is transferred to Atg10, which acts like an E2 ubiquitin carrier protein and allows Atg12 to complex with Atg5. The Atg12/Atg5 complex binds with Atg16. This new complex induces curvature of the phagophore. Then, LC3 is proteolytically cleaved by Atg4 to create LC3-I. Next, the exposed carboxy terminal of LC3-I is activated by Atg7 and transferred to Atg3, where phosphatidylethanolamine is conjugated to carboxyl glycine and LC3-II is formed. The Atg5/Atg12 complex allows for the recruitment and integration of LC3-II into the phagophore. LC3-II is then required for hemifusion of the membranes and selection of metabolites for degradation.
Autophagy and Homeostasis of Normal Salivary Glands
The Atg5f/f;Aqp5-Cre mouse model was developed to study the role of autophagy in normal salivary gland function. This mouse model was developed by crossing Aqp5-Cre mice (Flodby et al. 2010) with Atg5f/f mice (Hara and Matsui 2006). Aquaporin 5 (AQP5) is the primary aquaporin channel expressed on acinar cells and is localized to the apical membrane of acinar cells but not ductal cells. In these mice, the Cre recombinase is knocked-in to exon 1 of 1 copy of endogenous AQP5, and the Atg5 gene is flanked with lox P sites, leading to the preferential loss of Atg5 in salivary acinar cells, thereby rendering these mice autophagy deficient (Morgan-Bathke et al. 2013). Initial characterization of this mouse model focused on homeostasis parameters in young mice (4–6 wk) and found that the Atg5f/f;Aqp5-Cre mice have no differences in apoptosis, proliferation, or carbachol-induced salivary secretion. Total secreted protein profiles as well as amylase levels remained similar in Atg5f/f;Aqp5-Cre mice when compared with wild-type controls (Morgan-Bathke et al. 2013). It has previously been suggested that autophagy could entail a highly specialized step in the maturation of secretory organelles since the Golgi is involved in both autophagy and secretory granule biogenesis (Deretic et al. 2012). Salivary-specific Atg5 inactivation leads to a moderate increase in acinar cell hypertrophy and the accumulation of secretory granules that became more pronounced with aging (6 and 18 mo) (Morgan-Bathke et al. 2013). The treatment of autophagy-deficient mice with isoproterenol resulted in the retention of secretory granules, suggesting that autophagy may regulate mucous and/or serous granule biogenesis and secretion (Morgan-Bathke et al. 2013). In summary, we speculate that autophagy could be a key regulator for the homeostatic regulation of cellular organelles and proteins in salivary acinar cells.
Autophagy and Salivary Stress Responses
Autophagy and Radiation
As stated previously, autophagy can serve as a major survival mechanism during cellular adaptations to stress. One common stressor in salivary glands is the exposure of nondiseased salivary tissues to therapeutic radiation during the treatment of head and neck cancer. Importantly, radiation induces the acute and chronic loss of salivary function, resulting in a severe diminishment in the quality of life for these patients (Hancock et al. 2003; Dirix et al. 2006; Cady 2007). Previous studies using knockdown models of autophagy in a variety of cell types have reported that the loss of autophagic capacity leads to increased DNA damage, cell death, and dysregulation of the cell cycle following treatment with cytotoxic agents and radiation (Levine and Abrams 2008; Bae and Guan 2011). Studies using the salivary-specific conditional knockout model of Atg5, Atg5f/f;Aqp5-Cre, determined that the inactivation of autophagy in the salivary glands leads to a significant decrease in stimulated salivary flow rates following a single 5-Gy dose of targeted head and neck radiation in both males and females when compared with irradiated Atg5+/+;Aqp5-Cre (wild-type) mice at 3, 14, and 30 d after treatment (Morgan-Bathke, Hill, et al. 2014). The administration of insulin-like growth factor 1 (IGF-1) has been previously shown to prevent radiation-induced salivary gland dysfunction (Limesand et al. 2009; Limesand et al. 2010); however, IGF-1 was unable to preserve function in autophagy-deficient mice (Atg5f/f;Aqp5-Cre) (Morgan-Bathke, Hill, et al. 2014). This loss of salivary secretory function is most likely due to increased rates of apoptosis observed at acute time points (24–48 h) (Fig. 2), analogous to other models of radiation-induced salivary gland dysfunction (Avila et al. 2009; Limesand et al. 2009). This elevated apoptosis in the Atg5f/f;Aqp5-Cre mice leads to an early induction of compensatory proliferation, as demonstrated by increased proliferating cell nuclear antigen levels 48 to 72 h following treatment (Morgan-Bathke, Harris, et al. 2014). Radiation treatment has also been shown to decrease microvasculature density in the parotid gland (Xu et al. 2010); however, it is currently unclear if autophagy contributes to microvascular endothelial cell loss. The compensatory proliferation of irradiated salivary glands has been previously shown to occur approximately 1 wk after damage in many studies (Peter et al. 1994; Radfar and Sirois 2003; Muhvic-Urek et al. 2006; Grundmann et al. 2010), and chronically elevated proliferation in the salivary glands has been associated with poor salivary gland function (Grundmann et al. 2010). Atg5f/f;Aqp5-Cre autophagy-deficient mice continue to exhibit dysregulated proliferation 30 d after radiation, resulting in focal areas of hyperplasia (Morgan-Bathke, Harris, et al. 2014). We hypothesize that autophagy may aid in the re-establishment of cellular homeostasis following radiation by balancing apoptosis and proliferation in salivary glands (Fig. 2).
Figure 2.

Autophagy and the radiation response. DNA damage following radiation leads to the stabilization and transcriptional activation of p53, resulting in the induction of apoptosis. High levels of apoptosis are correlated with a significant loss of salivary gland function. The utilization of growth factors, such as insulin-like growth factor 1 (IGF-1) treatment (displayed by the GF receptor at the top of the figure), allows for the inhibition of apoptosis and potentially the induction of autophagy, thereby preventing the loss of salivary gland function after radiation. Autophagy induction after radiation damage may lead to a resolution of excessive oxidative stress and compensatory proliferation, resulting in the restoration of salivary gland function.
Autophagy can be induced pharmacologically through the use of rapamycin and its derivatives CCI-779 and RAD-001 (Ryter et al. 2013). These pharmaceuticals act similarly through the inhibition of mTOR complex 1 (Laplante and Sabatini 2013) (Fig. 1). A few studies have utilized this approach following radiation treatment of noncancerous tissues. Following radiation-induced salivary gland dysfunction, CCI-779 treatment (4–8 d after radiation) resulted in improved stimulated salivary flow rates (30 d after radiation) that are similar to unirradiated controls (Morgan-Bathke, Harris, et al. 2014). Radiotherapy for head and neck cancer also damages the oral epithelium, leading to mucositis in patients. In a rodent model of radiation-induced mucositis, rapamycin was able to promote the repair of this epithelial layer and the resolution of oral sores (Iglesias-Bartolome et al. 2012). Mechanistically, a study conducted with primary human keratinocytes proposed that rapamycin induced increases in mitochondrial superoxide dismutase and therefore a reduction of reactive oxygen species (ROS) (Iglesias-Bartolome et al. 2012). However, this inhibition of ROS may also be due to the induction of autophagy with rapamycin based on the well-defined ability of autophagy to inhibit oxidative stress (Vernon and Tang 2013). In addition, rapamycin has been shown to potently inhibit proliferation (Feng et al. 2005; Oliveira et al. 2008; Laplante and Sabatini 2013) and administration after radiation-induced salivary gland dysfunction restored proliferation indices of the parotid gland to unirradiated levels (Morgan-Bathke, Harris, et al. 2014). Overall, these studies suggest that the loss of autophagy exacerbates the negative response to radiation and that the activation of autophagy could play a beneficial role in re-establishing salivary gland homeostasis.
Autophagy and Ductal Ligation
Ligation of the major excretory duct of the submandibular gland (SMG) prevents the outflow of saliva, and extended obstruction leads to acinar cell and glandular atrophy. Studies using animal models of duct ligation as a model of atrophy regulation have proposed that these findings could extend to obstructive sialadenitis, Sjogrën syndrome, or patients receiving head and neck radiation therapy (Silver et al. 2010; Lin et al. 2014). Autophagy as well as mTOR pathways were both activated in salivary glands as early as 1 d following sustained duct ligation, and thus, both pathways are implicated in salivary gland atrophy and survival, albeit possibly targeting a different population of cells (Silver et al. 2010). Furthermore, daily postligation treatments of the mTOR inhibitor rapamycin delayed ligation-induced salivary gland atrophy in mice (Bozorgi et al. 2014). However, rapamycin is also a potent inducer of autophagy (Sarkar et al. 2009). It is unclear what effect rapamycin has on autophagy signaling in this duct ligation model. Furthermore, using a Atg5f/–;Aqp5-Cre mouse duct ligation model, the effects of ATG5 deficiency in acinar cells and the reduced ATG5 level in duct cells of SMGs were studied simultaneously in mice subjected to sustained ligation for up to 7 d (Lin et al. 2014) (Fig. 3). Ligation-induced acinar cell apoptosis was delayed in autophagy-impaired acinar cells in SMGs. Consistent with the animal study, in vitro analyses showed that the treatment with autophagy inhibitors, chloroquine and bafilomycin A1, or knockout of the Atg7 gene reduced the susceptibility to H2O2-induced cell death in mouse embryonic fibroblasts and salivary cells (Lin et al. 2014). In contrast to ligation-induced atrophy in acinar cells, granular convoluted tubules (GCTs) were dilated at 1 d due to a stoppage in saliva outflow but exhibited premature cellular senescent phenotypes upon continued obstruction at 3 d after ligation. The premature senescence in SMG GCTs with a reduced level of ATG5 remains unresolved at 7 d after ligation, while control wild-type GCTs return toward baseline (Lin et al. 2014) (Fig. 3). Therefore, autophagy functions in both acinar and duct cells, and the manipulation of autophagic capacity can potentially provide intervention in the treatment of oral diseases and conditions involving salivary glands.
Figure 3.

Role of autophagy in tissue injury responses to ductal ligation. Ligation of the main excretory duct of the submandibular gland (SMG) for up to 7 d elicits apoptosis in acinar cells and premature senescence in granular convoluted tubules (GCTs). Compared with cells from duct-ligated Atg5 wild-type (WT) SMGs, apoptosis is delayed in targeted Atg5 knockout (KO) acinar cells, while the reduced expression of Atg5 in GCT cells (knockdown [KD]) renders persisted premature senescence phenotypes after ligation. The cell index is defined as relative changes in cell populations that are apoptag(+) for apoptosis and β-gal(+) for stress-induced premature senescence, respectively.
Autophagy and Autoimmune Responses
It has been postulated that the autophagic process could deliver self-antigens to major histocompatibility complex class II molecules and thereby contribute to an autoimmune disease. In a systemic lupus erythematosus (SLE) mouse model, peripheral T cells had more autophagic vesicles when compared with controls (Gros et al. 2012). Additionally, T cells from patients with SLE had a similar increase in autophagic vesicles. Intriguingly, 1 patient in a comparison nonlupus autoimmune group had primary Sjogrën syndrome and did not display increased autophagosomes in T cells (Gros et al. 2012). This evidence has led to a clinical trial evaluating the efficacy of rapamycin in the treatment of SLE (Monneaux and Muller 2009; Perl 2009). Currently, there is a limited amount of studies directly evaluating autophagy and Sjogrën syndrome. In cultures of salivary human submandibular gland cells, the disruption of autophagy enhances the apoptotic effect of endoplasmic reticulum (ER) stress–inducing agents that deplete Ca2+ stores, such as thapsigargin. ER stress also led to the redistribution of autoantibodies (Ro and La) to the cell membrane (Katsiougiannis et al. 2015). It has been hypothesized that alterations in intracellular Ca2+ lead to autophagy induction in order to promote cell survival (Sukumaran et al. 2015). More studies will be necessary to fully elucidate the role of autophagy in primary Sjogrën syndrome.
Autophagy and Salivary Cancer
The dysregulation of autophagy has been implicated in a variety of human diseases, including intestinal, pulmonary, vascular, infectious, metabolic, and neurodegenerative diseases, as well as in aging and a variety of cancers (Choi et al. 2013; Schneider and Cuervo 2014). The association of autophagy and cancer development may be stage dependent; initially, autophagy may serve a preventative role against cancer and, in later stages of tumor development, serve to protect cancer cells (White 2012; Jiang and Mizushima 2014). Furthermore, autophagy inhibitors and inducers have been exploited as targets for anticancer therapy (Janku et al. 2011) based on their role in promoting tumor suppression or progression, respectively. One of the most common malignancies of the major and minor salivary glands is adenoid cystic carcinoma (ACC). The treatment of salivary ACC cells with anticancer drugs, including cisplatin (Ma et al. 2013; Jiang et al. 2014), mTOR inhibitor temsirolimus (Liu et al. 2014), obatoclax (Liang et al. 2014), YM155 (Wang et al. 2014), isoliquiritigenin (Chen et al. 2012), and zoledronic acid (Ge et al. 2014), reportedly induced autophagy but with different outcomes. Among these agents, cisplatin-based adjuvant chemotherapy is widely used in the treatment of salivary ACC. Autophagy played a protective role in ACC cells treated with cisplatin, such that autophagy inhibition by 3-methyladenine (3-MA), chloroquine, or Beclin-1 siRNA sensitized ACC cells toward cisplatin-induced cytotoxicity (Ma et al. 2013; Jiang et al. 2014). In contrast, chemotherapeutic agents zoledronic acid (Ge et al. 2014) and isoliquiritigenin (Chen et al. 2012) induced autophagic and/or apoptotic cell death that was attenuated by autophagy inhibitor 3-MA or when the expression of autophagy machinery genes (Atg5, Atg7, Beclin-1) was suppressed in ACC cells. Autophagy in these cases thus played a pro-death role by facilitating apoptosis. Therefore, the types of chemotherapeutic agents, treatment intensity, duration, sequence, and heterogeneity of ACC cells could all contribute to differential responses of salivary ACC toward autophagy manipulations. In addition to the aforementioned autophagy induction by chemotherapeutics, many anticancer agents, such as zoledronic acid (Ge et al. 2014), are known to generate ROS in cells. Studies have suggested that autophagy and ROS pathways are mutually linked and that their crosstalk plays an important role in both cancer progression and cellular responses to chemotherapeutics (Dewaele et al. 2010). The clinical impact of autophagy induction or inhibition is yet to be defined in anticancer therapy, and indeed, the function of autophagy in cancer is clearly context dependent.
Future Directions
It is widely reported that autophagy activation is similar to a double-edged sword with beneficial and detrimental properties (Shintani and Klionsky 2004). This also appears to be the case in the salivary glands under basal and stressed conditions. Following radiation or ductal ligation, it is evident that autophagy contributes to the repair response at acute time points, leading to acinar cell homeostasis. However, under sustained stress conditions, autophagy may promote programmed cell death responses. Further characterization of the acinar versus ductal response under various stress conditions would clarify the conditions for rapamycin utilization and possibly expand therapeutic options for patients with xerostomia.
Author Contributions
M. Morgan-Bathke, H.H. Lin, D.K. Ann, K.H. Limesand, contributed to conception, design, data acquisition, and interpretation, drafted and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Footnotes
This work was funded in part by National Institute of Dental and Craniofacial Research (NIDCR) grants RC1 DE020335 (K.H.L. and D.K.A.), R01 DE023534 (K.H.L.), and R01 R01DE10742 and R21 DE023298 (D.K.A.).
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Avila JL, Grundmann O, Burd R, Limesand KH. 2009. Radiation-induced salivary gland dysfunction results from p53-dependent apoptosis. Int J Radiat Oncol Biol Phys. 73(2):523–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae H, Guan JL. 2011. Suppression of autophagy by FIP200 deletion impairs DNA damage repair and increases cell death upon treatments with anticancer agents. Mol Cancer Res. 9(9):1232–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozorgi SS, Proctor GB, Carpenter GH. 2014. Rapamycin delays salivary gland atrophy following ductal ligation. Cell Death Dis. 5:e1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cady J. 2007. Nutritional support during radiotherapy for head and neck cancer: the role of prophylactic feeding tube placement. Clin J Oncol Nurs. 11(6):875–880. [DOI] [PubMed] [Google Scholar]
- Chen G, Hu X, Zhang W, Xu N, Wang FQ, Jia J, Zhang WF, Sun ZJ, Zhao YF. 2012. Mammalian target of rapamycin regulates isoliquiritigenin-induced autophagic and apoptotic cell death in adenoid cystic carcinoma cells. Apoptosis. 17(1):90–101. [DOI] [PubMed] [Google Scholar]
- Choi AM, Ryter SW, Levine B. 2013. Autophagy in human health and disease. N Engl J Med. 368(7):651–662. [DOI] [PubMed] [Google Scholar]
- Deretic V, Jiang S, Dupont N. 2012. Autophagy intersections with conventional and unconventional secretion in tissue development, remodeling and inflammation. Trends Cell Biol. 22(8):397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewaele M, Maes H, Agostinis P. 2010. ROS-mediated mechanisms of autophagy stimulation and their relevance in cancer therapy. Autophagy. 6(7):838–854. [DOI] [PubMed] [Google Scholar]
- Dirix P, Nuyts S, Van den BW. 2006. Radiation-induced xerostomia in patients with head and neck cancer: a literature review. Cancer. 107(11):2525–2534. [DOI] [PubMed] [Google Scholar]
- Feng Z, Zhang H, Levine AJ, Jin S. 2005. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 102(23):8204–8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flodby P, Borok Z, Banfalvi A, Zhou B, Gao D, Minoo P, Ann DK, Morrisey EE, Crandall ED. 2010. Directed expression of Cre in alveolar epithelial type 1 cells. Am J Respir Cell Mol Biol. 43(2):173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge XY, Yang LQ, Jiang Y, Yang WW, Fu J, Li SL. 2014. Reactive oxygen species and autophagy associated apoptosis and limitation of clonogenic survival induced by zoledronic acid in salivary adenoid cystic carcinoma cell line SACC-83. PLoS ONE. 9(6):e101207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick D, Barth S, Macleod KF. 2010. Autophagy: cellular and molecular mechanisms. J Pathol. 221(1):3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gros F, Arnold J, Page N, Décossas M, Korganow AS, Martin T, Muller S. 2012. Macroautophagy is deregulated in murine and human lupus T lymphocytes. Autophagy. 8(7):1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundmann O, Fillinger JL, Victory KR, Burd R, Limesand KH. 2010. Restoration of radiation therapy-induced salivary gland dysfunction in mice by post therapy IGF-1 administration. BMC Cancer. 10(1):417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock PJ, Epstein JB, Sadler GR. 2003. Oral and dental management related to radiation therapy for head and neck cancer. J Can Dent Assoc. 69(9):585–590. [PubMed] [Google Scholar]
- Hara T, Matsui M. 2006. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 441:885–889. [DOI] [PubMed] [Google Scholar]
- Iglesias-Bartolome R, Patel V, Cotrim A, Leelahavanichkul K, Molinolo AA, Mitchell JB, Gutkind JS. 2012. mTOR inhibition prevents epithelial stem cell senescence and protects from radiation-induced mucositis. Cell Stem Cell. 11(3):401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janku F, McConkey DJ, Hong DS, Kurzrock R. 2011. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol. 8(9):528–539. [DOI] [PubMed] [Google Scholar]
- Jiang L, Huang S, Zhang D, Zhang B, Li K, Li W, Zhang S, Zhang W, Zheng P. 2014. Inhibition of autophagy augments chemotherapy in human salivary adenoid cystic carcinoma. J Oral Pathol Med. 43(4):265–272. [DOI] [PubMed] [Google Scholar]
- Jiang P, Mizushima N. 2014. Autophagy and human diseases. Cell Res. 24(1):69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung HS, Chung KW, Won Kim J, Kim J, Komatsu M, Tanaka K, Nguyen YH, Kang TM, Yoon KH, Kim JW, et al. 2008. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 8(4):318–324. [DOI] [PubMed] [Google Scholar]
- Katsiougiannis S, Tenta R, Skopouli FN. 2015. Endoplasmic reticulum stress causes autophagy and apoptosis leading to cellular redistribution of the autoantigens Ro/SSA and La/SSB in salivary gland epithelial cells. Clin Exp Immunol. [epub ahead of print 2015 Apr 4] doi: 10.1111/cei.12638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ. 2007. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 8(11):931–937. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. 2012. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 8(4):445–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al. 2006. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 441(7095):880–884. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. 2005. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 169(3):425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. 2004. The role of autophagy during the early neonatal starvation period. Nature. 432(7020):1032–1036. [DOI] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. 2013. Regulation of mTORC1 and its impact on gene expression at a glance. J Cell Sci. 126(Pt 8):1713–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Abrams J. 2008. p53: the Janus of autophagy? Nat Cell Biol. 10(6):637–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang LZ, Ma B, Liang YJ, Liu HC, Zhang TH, Zheng GS, Su YX, Liao GQ. 2014. Obatoclax induces Beclin 1 and ATG5-dependent apoptosis and autophagy in adenoid cystic carcinoma cells. Oral Dis. 21(4):470–477. [DOI] [PubMed] [Google Scholar]
- Limesand KH, Avila JL, Victory K, Chang HH, Shin YJ, Grundmann O, Klein RR. 2010. IGF-1 preserves salivary gland function following fractionated radiation. Int J Radiat Oncol Biol Phys. 78(2):579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limesand KH, Said S, Anderson SM. 2009. Suppression of radiation-induced salivary gland dysfunction by IGF-1. PLoS ONE. 4(3):e4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HH, Lin SM, Chung Y, Vonderfecht S, Camden JM, Flodby P, Borok Z, Limesand KH, Mizushima N, Ann DK. 2014. Dynamic involvement of ATG5 in cellular stress responses. Cell Death Dis. 5:e1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Huang S, Chen Z, Wang H, Wu H, Zhang D. 2014. Temsirolimus, the mTOR inhibitor, induces autophagy in adenoid cystic carcinoma: in vitro and in vivo. Pathol Res Pract. 210(11):764–769. [DOI] [PubMed] [Google Scholar]
- Ma B, Liang LZ, Liao GQ, Liang YJ, Liu HC, Zheng GS, Su YX. 2013. Inhibition of autophagy enhances cisplatin cytotoxicity in human adenoid cystic carcinoma cells of salivary glands. J Oral Pathol Med. 42(10):774–780. [DOI] [PubMed] [Google Scholar]
- Martinet W, De Meyer GR, Andries L, Herman AG, Kockx MM. 2006. Detection of autophagy in tissue by standard immunohistochemistry: possibilities and limitations. Autophagy. 2(1):55–57. [DOI] [PubMed] [Google Scholar]
- Mathew R, Karantza-Wadsworth V, White E. 2007. Role of autophagy in cancer. Nat Rev Cancer. 7(12):961–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T. 2007. How to interpret LC3 immunoblotting. Autophagy. 3(6):542–545. [DOI] [PubMed] [Google Scholar]
- Monneaux F, Muller S. 2009. Molecular therapies for systemic lupus erythematosus: clinical trials and future prospects. Arthritis Res Ther. 11(3):234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan-Bathke M, Harris ZI, Arnett DG, Klein RR, Burd R, Ann DK, Limesand KH. 2014. The rapalogue, CCI-779, improves salivary gland function following radiation. PLoS ONE. 9(12):e113183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan-Bathke M, Hill GA, Harris ZI, Lin HH, Chibly AM, Klein RR, Burd R, Ann DK, Limesand KH. 2014. Autophagy correlates with maintenance of salivary gland function following radiation. Sci Rep. 4:5206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan-Bathke M, Lin HH, Chibly AM, Zhang W, Sun X, Chen CH, Flodby P, Borok Z, Wu R, Arnett D, et al. 2013. Deletion of ATG5 shows a role of autophagy in salivary homeostatic control. J Dent Res. 92(10):911–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhvic-Urek M, Bralic M, Curic S, Pezelj-Ribaric S, Borcic J, Tomac J. 2006. Imbalance between apoptosis and proliferation causes late radiation damage of salivary gland in mouse. Physiol Res. 55(1):89–95. [DOI] [PubMed] [Google Scholar]
- Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, et al. 2007. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 13(5):619–624. [DOI] [PubMed] [Google Scholar]
- Oliveira JC, Souza KK, Dias MM, Faria MC, Ropelle ER, Flores MB, Ueno M, Velloso LA, Saad ST, Saad MJ, et al. 2008. Antineoplastic effect of rapamycin is potentiated by inhibition of IRS-1 signaling in prostate cancer cells xenografts. J Cancer Res Clin Oncol. 134(8):833–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perl A. 2009. Emerging new pathways of pathogenesis and targets for treatment in systemic lupus erythematosus and Sjogren’s syndrome. Curr Opin Rheumatol. 21(5):443–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter B, Van Waarde MA, Vissink A, s-Gravenmade EJ, Konings AW. 1994. Radiation-induced cell proliferation in the parotid and submandibular glands of the rat. Radiat Res. 140(2):257–265. [PubMed] [Google Scholar]
- Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. 2007. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med. 204(1):25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Zou Z, Sun Q, Luby-Phelps K, Cheng P, Hogan RN, Gilpin C, Levine B. 2007. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell. 128(5):931–946. [DOI] [PubMed] [Google Scholar]
- Raben N, Hill V, Shea L, Takikita S, Baum R, Mizushima N, Ralston E, Plotz P. 2008. Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Hum Mol Genet. 17(24):3897–3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radfar L, Sirois DA. 2003. Structural and functional injury in minipig salivary glands following fractionated exposure to 70 Gy of ionizing radiation: an animal model for human radiation-induced salivary gland injury. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 96(3):267–274. [DOI] [PubMed] [Google Scholar]
- Ryter SW, Cloonan SM, Choi AM. 2013. Autophagy: a critical regulator of cellular metabolism and homeostasis. Mol Cells. 36(1):7–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC. 2009. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 16(1):46–56. [DOI] [PubMed] [Google Scholar]
- Schiaffino S, Mammucari C, Sandri M. 2008. The role of autophagy in neonatal tissues: just a response to amino acid starvation? Autophagy. 4(5):727–730. [DOI] [PubMed] [Google Scholar]
- Schneider JL, Cuervo AM. 2014. Autophagy and human disease: emerging themes. Curr Opin Genet Dev. 26:16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani T, Klionsky DJ. 2004. Autophagy in health and disease: a double-edged sword. Science. 306(5698):990–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver N, Proctor GB, Arno M, Carpenter GH. 2010. Activation of mTOR coincides with autophagy during ligation-induced atrophy in the rat submandibular gland. Cell Death Dis. 1:e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumaran P, Sun Y, Vyas M, Singh BB. 2015. TRPC1-mediated Ca(2)(+) entry is essential for the regulation of hypoxia and nutrient depletion-dependent autophagy. Cell Death Dis. 6:e1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasdemir E, Galluzzi L, Maiuri MC, Criollo A, Vitale I, Hangen E, Modjtahedi N, Kroemer G. 2008. Methods for assessing autophagy and autophagic cell death. Methods Mol Biol. 445:29–76. [DOI] [PubMed] [Google Scholar]
- Vernon P, Tang D. 2013. Eat-me: autophagy, phagocytosis, and reactive oxygen species signaling. Antioxid Redox Signal. 18(6):677–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YF, Zhang W, He KF, Liu B, Zhang L, Zhang WF, Kulkarni AB, Zhao YF, Sun ZJ. 2014. Induction of autophagy-dependent cell death by the survivin suppressant YM155 in salivary adenoid cystic carcinoma. Apoptosis. 19(4):748–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E. 2012. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 12(6):401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirawan E, Vanden Berghe T, Lippens S, Agostinis P, Vandenabeele P. 2012. Autophagy: for better or for worse. Cell Res. 22(1):43–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Yan X, Gao R, Mao L, Cotrim AP, Zheng C, Zhang C, Baum BJ, Wang S. 2010. Effect of irradiation on microvascular endothelial cells of parotid glands in the miniature pig. Int J Radiat Oncol Biol Phys. 78(3):897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousefi S, Simon HU. 2007. Apoptosis regulation by autophagy gene 5. C rit Rev Oncol Hematol. 63(3):241–244. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Goldman S, Baerga R, Zhao Y, Komatsu M, Jin S. 2009. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc Natl Acad Sci U S A. 106(47):19860–19865. [DOI] [PMC free article] [PubMed] [Google Scholar]