

Abstract
Islet non-β-cells, the α- δ- and pancreatic polypeptide cells (PP-cells), are important components of islet architecture and intercellular communication. In α-cells, glucagon is found in electron-dense granules; granule exocytosis is calcium-dependent via P/Q-type Ca2+-channels, which may be clustered at designated cell membrane sites. Somatostatin-containing δ-cells are neuron-like, creating a network for intra-islet communication. Somatostatin 1-28 and 1-14 have a short bioactive half-life, suggesting inhibitory action via paracrine signaling. PP-cells are the most infrequent islet cell type. The embryologically separate ventral pancreas anlage contains PP-rich islets that are morphologically diffuse and α-cell deficient. Tissue samples taken from the head region are unlikely to be representative of the whole pancreas. PP has anorexic effects on gastro-intestinal function and alters insulin and glucagon secretion. Islet architecture is disrupted in rodent diabetic models, diabetic primates and human Type 1 and Type 2 diabetes, with an increased α-cell population and relocation of non-β-cells to central areas of the islet. In diabetes, the transdifferentiation of non-β-cells, with changes in hormone content, suggests plasticity of islet cells but cellular function may be compromised. Understanding how diabetes-related disordered islet structure influences intra-islet cellular communication could clarify how non-β-cells contribute to the control of islet function.
Keywords: communication, exocytosis, glucagon, granule, insulin, intra-islet signaling, non-β-cell, paracrine, PP, somatostatin
Introduction
Although β-cells form the largest cellular component of islets in most species—60% to 80% in rodents and 50% to 70% in humans (Cabrera et al. 2006; Clark et al. 1988; Elayat et al. 1995; Rahier et al. 1983a; Steiner et al. 2010)—the non-β-cells have important roles to play in intra-islet coordination and thus in the control of glucose homeostasis. It has been known for many years that the balance between insulin and the counter-regulatory hormone glucagon is of major importance in the fine control of glucose homeostasis and its disruption in diabetes (Unger et al. 1970; Unger and Orci 1975). The observations made with a glucagon receptor knockout mouse demonstrating the prevention of diabetes when glucagon signaling is impaired (Lee et al. 2011) highlighted the important role of α-cell secretion in vivo. The roles of δ-cells and pancreatic polypeptide (PP) cells and their respective hormones in islet function have been largely ignored until recently. The recent studies demonstrating plasticity in adult islets have brought the non-β-cells to the forefront of islet research once again (Brereton et al. 2014; Courtney et al. 2013; Gao et al. 2014; Piran et al. 2014; Talchai et al. 2012; Thorel et al. 2010). Therefore, the non-β-cells have an important regulatory role in facilitating communication between islet cells, controlling glucose homeostasis and metabolism, and maintaining the islet architecture.
Islet Architecture and Cellular Communication
The pancreatic islet functions as a single organ with tightly coordinated signaling between the different cell types. This network allows the islet to respond to changes in blood glucose and to intra-islet signals (via gap junctions or paracrine signaling) and extrinsic nerve impulses in a rapid and sensitive manner. The islet cells communicate via gap junctions or via paracrine secretion and signaling. The architecture of the islet and spatial arrangements of the different cell types are therefore important for this cell-to-cell communication (Figs. 1, 2).
Figure 1.
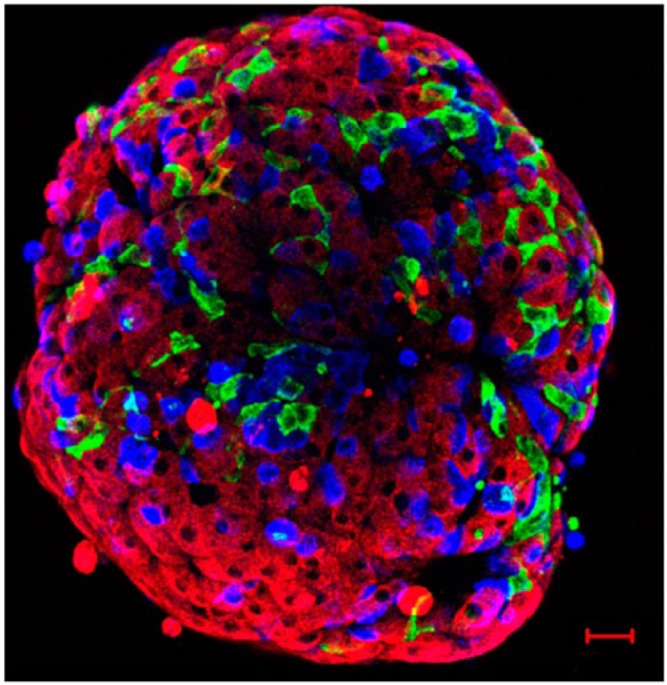
Mouse islet immunolabelled for insulin (red), glucagon (blue), and somatostatin (green). This confocal image reconstruction of the cells at the exterior of the islet demonstrates the network of δ-cells and their proximity to α- and β-cells. Scale, 20 µm.
Figure 2.
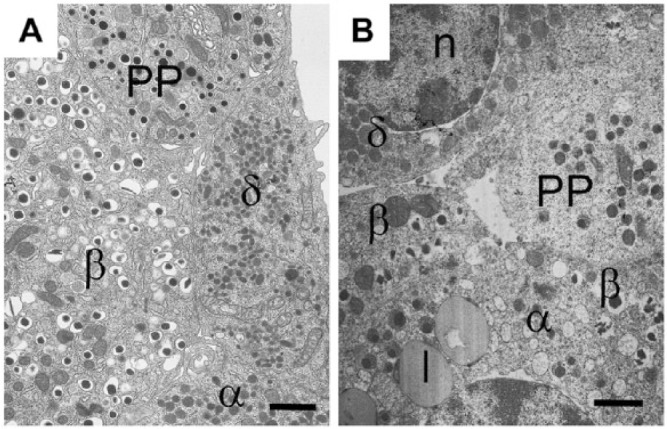
Granule morphologies and islet cell network in an islet from (A) a mouse and (B) a human islet. β-, α-, δ-, and PP-cells viewed by electron microscopy. Insulin secretory granules are similar in both species with an electron-dense core and clear halo. However, human insulin granules sometimes appear crystalline, with angular shaped cores compared to the smooth spherical cores of the mouse islet. Glucagon secretory granules are electron-dense without a clear halo; in human α-cells, some secretory granules have a grey halo surrounding the dense core, whereas others are without a halo, as in the mouse. PP-cells contain spherical smaller granules, which are very heterogeneous in size in both species; some PP granules are similar to those found in α-cells and others have a small halo. Somatostatin-containing granule morphology is very different in mouse and human: in rodents, the granules are small, lozenge-shaped structures; in humans, the granules are larger, slightly electron-opaque but spherical and of similar size to that of glucagon granules. l, lipofuscin body; n, nucleus. Scale, 1.0 µm.
The islet architecture differs amongst species and has puzzled anatomists for many years (Fig. 3) (Falkmer and Ostberg 1977; Steiner et al. 2010). These differences likely relate to the different species-specific functional requirements for hormonal regulation, the islet vascular supply, and the requirement for other intrinsic secreted factors (like ATP, GABA or Zn2+) for islet function.
Figure 3.
Pancreatic islets demonstrating the species-specific differences in cellular architecture. Immunofluorescent labelling of pancreatic sections for insulin (green), glucagon (pink), and somatostatin (yellow). In mouse islets (A), the non-β-cells are situated at the periphery of the islet whereas, in non-human primates (B) and humans (C), the α- and δ-cells are found both at the periphery of the islet cross-section and towards the islet center. This reflects the location of these cells at the perivascular border of both circumferential capillaries (rodents and humans) and those penetrating the islet interior (non-human primates and humans). Scale, 200 μm.
The localization of the non β-cells predominantly determines the islet architecture (Falkmer and Ostberg 1977; Orci and Unger 1975; Steiner et al. 2010). The non-β-cells in all mammalian species are located immediately adjacent to the islet capillaries. Small islets in rodents have a peripheral capillary and so, in tissue sections, the non-β-cells surround the central core of β-cells. In larger islets (and in humans), the vascular supply penetrates the islet center and non-β-cells appear within the core of the islet (Cabrera et al. 2006). This difference in vascular organization is not random but divides the more central β-cell clusters into units, which have ready access to capillaries (Fig. 4). However, it is unclear if every β-cell directly contacts a capillary. Originally, it was suggested that each cell was adjacent to afferent and efferent capillaries (Bonner-Weir and Orci 1982). Polarised insulin secretion towards capillary remnants in isolated islets has been demonstrated (Low et al. 2014). However, recent observations with three-dimensional (3D) electron microscopy (EM) have suggested that many β-cells in rodent islets are located several cell diameters distant from capillaries; these cells would release granules into extracellular spaces, allowing insulin to diffuse to the capillaries (Pfeifer et al. 2014). This “core-mantle” structure has led to suggestions that the direction of blood flow, and, therefore, the sequential delivery of changes in blood glucose to different cell types, is an important coordinator of islet function; either non-β-cells receive the blood-borne signals first and, in turn, influence insulin secretion from β-cells, or vice versa (Bonner-Weir and Orci 1982; Brunicardi et al. 1996; Liu et al. 1993; Nyman et al. 2008; Ohtani et al. 1986; Samols et al. 1986).
Figure 4.
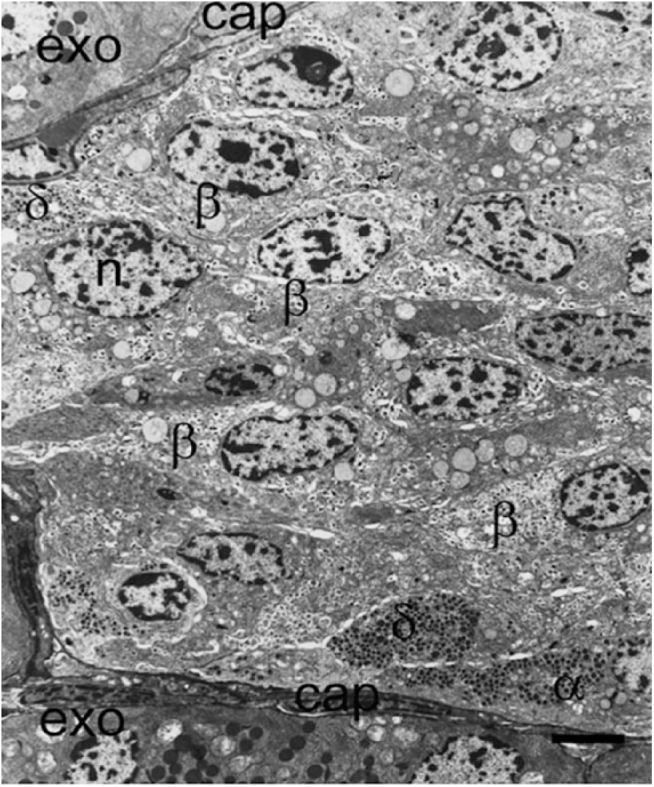
Part of an islet from a non-diabetic patient viewed with electron microscopy demonstrating the presence of insulin-containing β-cells (β), α-cells (α), and δ-cells (δ) all close to the peripheral capillary (cap). A large proportion of β-cells are not situated adjacent or near to a capillary in this thin section. l, lipofuscin body; n, nucleus. Scale, 5 μm.
Although the vascular system is a distribution pathway for islet endocrine secretion and the control of extra-pancreatic metabolic function, intra-islet regulation is likely to be more independent of blood borne factors. Cellular communication in a single islet can be regulated by “paracrine” hormone actions and other factors secreted into the extracellular space. This is an intimate system for the delivery of peptides with a short half-life (e.g., somatostatin and possibly GLP-1) to elicit inhibitory or excitatory responses on hormone secretion. Low concentrations of either agonists or surface receptors will be very effective in the restricted extracellular space. Therefore, the adjacencies of the different cells (and the architecture of the islet) are crucial to ensure correct delivery and receipt of paracrine signals. The development of methods for physiological and biophysical studies on intact islets in vitro (rather than isolated cells) have allowed identification of intercellular regulatory mechanisms involving ions and hormones, particularly involving communications between α-, δ-, and β-cells (Rorsman et al. 2011; Rorsman et al. 2008).
However, the permanency of islet architecture and cellular identity of hormone-producing cells in adult mammals is now being challenged. Islet architecture alters in animal models of diabetes, and the transformation/re-differentiation of islet cells from a specific hormone type to another has been demonstrated in diabetic animals and humans (Brereton et al. 2014; Piran et al. 2014; Spijker et al. 2013; Talchai et al. 2012; Thorel et al. 2010). This suggests that not only is islet cell function compromised in diabetes, but also intra-islet cellular regulation may be disrupted.
α-Cells
Glucagon Production and Glucagon Receptors
Glucagon was discovered in 1923 and named as a gluc-ose agon-ist (Murlin et al. 1923) due to its glucose elevating effect. In the early crude insulin treatment of diabetic animals and humans, whole pancreatic extract was administered and transient hyperglycemia was common; contaminating glucagon in these extracts underlies this paradoxical effect (Murlin et al. 1923).
The expression of glucagon in α-cells was only revealed in 1948 (Sutherland and de Duve 1948) and glucagon receptors have been localized to both hepatic and non-hepatic tissues, including the islet (Dunphy et al. 1998). The peptide glucagon (29 amino acids) is processed from a large precursor preproglucagon (178 amino acids). Although glucagon is exclusively produced by islet α-cells, the gene that encodes preproglucagon is expressed in many different cells and tissues, including L-cells in the gut, and the hypothalamus and thalamus in the brain (Kieffer and Habener 1999). The bioactive hormone product(s) of this precursor protein is determined by the post-translational modification of the prohormone by prohormone convertase enzymes. Pancreatic α-cells express prohormone convertase 2 (PC2), which cleaves the propeptide to produce glucagon and a major proglucagon fragment. L-cells in the intestine express prohormone convertase 1 (also known as PC3; PC1/3); this enzyme cleavage results in production of the incretin, glucagon-like peptide 1 (GLP-1), GLP-2 and glicentin as well as oxyntomodulin (Furuta et al. 1997; Furuta et al. 2001; Pocai 2012).
Recently, it has been reported that PC1/3 and GLP-1 are co-localized in some human α-cells, suggesting that GLP-1 is produced in human islets (Marchetti et al. 2012). Exogenous and circulating GLP-1 has dual effects on pancreatic hormone release: stimulating insulin secretion from β-cells (Gromada et al. 1998) and inhibiting glucagon secretion from α-cells (De Marinis et al. 2010). Whether locally produced and released GLP-1 contributes to these effects has yet to be proven.
Regulation of Glucagon Secretion
Glucagon secretion from α-cells is regulated by nutrients (glucose and amino acids), hormones and neurotransmitters (Gromada et al. 2007). Unlike other islet hormones, glucagon secretion is stimulated at low glucose concentrations (hypoglycemia) and normally suppressed by hyperglycemia when insulin secretion is stimulated. The exact cellular regulation of glucagon secretion remains hotly debated. Mechanisms proposed include paracrine regulation (by factors released from neighboring β- and δ-cells) (Unger and Orci 2010) and neuronal regulation (Taborsky et al. 1998). Since glucagon secretion is inhibited at glucose concentrations below the threshold required to evoke insulin and somatostatin release (3–5 mM glucose) a paracrine signal from these cells is unlikely (e.g., insulin, Zn2+, SST) and a cell-specific glucose or nutrient-dependent regulation of glucagon secretion must operate intrinsically in α-cells. Some evidence implicates ATP-sensitive K+ channels (KATP channel) of the same type as those found in β-cells in this intrinsic regulation (Zhang et al. 2013). Further elevation of glucose (10–15 mM) has been shown to stimulate glucagon secretion (Salehi et al. 2006), a feature characteristic of islets from Type 2 diabetic (T2D) donors (Zhang et al. 2013).
Somatostatin is a strong inhibitor of glucagon secretion (Zhang et al. 2007). α-cells express somatostatin receptor 2 (SSTR2) in both human and rodent islets (Braun 2014). When activated, this leads to inhibition of glucagon secretion by induction of hyperpolarization and reduced firing of action potentials. In addition, somatostatin inhibits depolarization-evoked exocytosis in rat α-cells (Gromada et al. 2001).
Islets are innervated by sympathetic, adrenergic (Lindsay et al. 2006), and parasympathetic cholinergic nerves (Havel and Taborsky 1989; Rossi et al. 2005). Surprisingly, although circulating adrenaline is a strong stimulus for α-cell secretion, adrenaline added in vitro has a relatively mild effect on α-cell electrical activity (De Marinis et al. 2010). Most of the stimulatory effect instead results from adrenaline-induced mobilization of Ca2+ release from intracellular Ca2+ stores (Gylfe and Gilon 2014; Vieira et al. 2004).
Role of Glucagon
The liver is the main site of glucagon action. Upon binding to the glucagon receptor, (a 7-transmembrane domain G protein-coupled receptor), glucagon activates stimulatory G-proteins which, in turn, stimulate adenylyl cyclase. This leads to the production of cAMP and activation of protein kinase A, which phosphorylates glycogen phosphorylase kinase. The activated phosphorylase breaks down glycogen and thus increases hepatic glucose output (Jiang and Zhang 2003). In addition, glucagon is an important regulator of lipid metabolism, reducing plasma triglycerides and stimulating hepatic fatty acid oxidation (Longuet et al. 2008; Vuguin and Charron 2011).
In pancreatic islets, the glucagon receptor has been identified in most β-cells and a subset of α- and δ-cells. Thus, glucagon is likely to function as a paracrine regulator of hormone secretion within the islet (Kieffer et al. 1996). Activation of glucagon receptors in β- and δ-cells stimulates insulin and somatostatin secretion via elevation of intracellular cAMP. The impact of glucagon receptor signaling back on α-cells is difficult to evaluate; glucagon receptor knockout mice are hyperglucagonemic and have hyperplasia of α-cells; this could indicate that glucagon is a negative regulator of its own secretion (Vuguin and Charron 2011).
Morphological Characteristics of α-Cells
Glucagon granules are large, dense core vesicles, some of which have a pale electron-opaque halo (Figs. 2, 5) (Deconinck et al. 1971). Although the core of the granule is similar to that of the insulin secretory granule, the glucagon granule has a smaller diameter (200 nm vs 350 nm) (Gopel et al. 2004; Pfeifer et al. 2014). In mouse islets, the overall cellular density of glucagon granules is 9 granules/µm3, with approximately 7000 granules per cell in total (Barg et al. 2000). High-resolution capacitance measurements have revealed that α-cell exocytosis is dependent on a subset of voltage-gated Ca2+ channels (the P/Q type Ca2+ channel). This suggests a close localization between the release competent granules and the P/Q Ca2+-channels. If these channels are localized to specific sites in the membrane, exocytosis would be restricted to these areas. High-resolution microscopy will be required to identify these sites.
Figure 5.
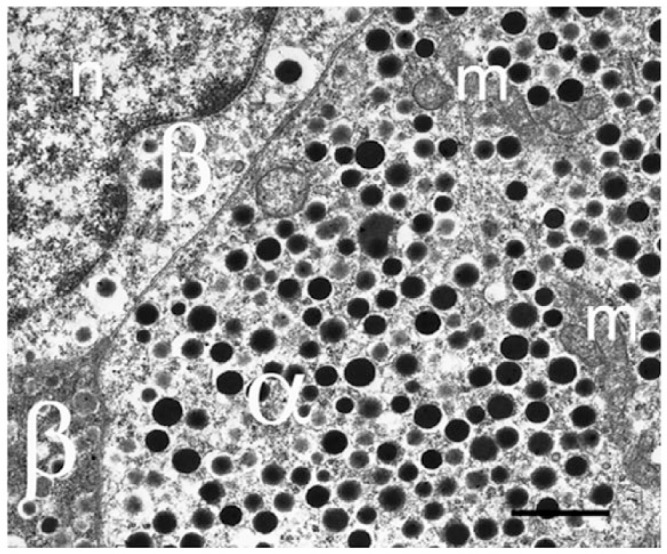
Electron microscopy image of mouse α-cell to demonstrate the dense granulation. α-Cells rarely show degranulation, even in diabetic human or mouse models. m, mitochondrion; n, nucleus; β, β-cell. Scale, 1 μm.
The location of α-cells adjacent to the capillary and in close proximity to the β-cells permits intercellular signaling to create a local, very glucose-sensitive system and immediate adjustment of both insulin and glucagon production for small changes in ambient glucose concentrations. It is now evident that diabetes is a bi-hormonal disorder and α-cells will receive more attention than before in understanding islet dysfunction in disease.
δ-Cells
Pancreatic δ–cells secrete the hormone somatostatin (SST). δ–cells are also present in the hypothalamus, central nervous system (CNS), peripheral neurons and the gastrointestinal tract (Arimura et al. 1975; Hökfelt et al. 1975). Somatostatin, also known as growth hormone inhibiting hormone (GHIH) or somatotropin release-inhibiting hormone (SRIF), inhibits most cellular secretions and was discovered in ovine hypothalamus by Brazeau and colleagues in 1973 (Brazeau et al. 1973).
SST Production and SST Receptors
SST is synthesized as part of a precursor molecule, preprosomatostatin (PPSST, 116 amino acids), which is rapidly cleaved into pro-somatostatin (PSST, 92 amino acids). PSST is processed enzymatically at its C-terminal site in the secretory granule by a protein convertase, probably PC2 (Marcinkiewicz et al. 1994), to yield several mature products. This includes two bioactive forms. Both SST-14 (14 amino acids) and SST-28 (28 amino acids) are very short-lived; somatostatin peptides have a half-life of <1 min in the circulation. In islets, δ-cells release mainly SST-14, whereas SST-28 is the product of the intestinal cells (Francis et al. 1990). Both SST-14 and SST-28 bind to five somatostatin receptor subtypes (SSTR1–SSTR5) (Kumar et al. 1999). SSTRs are G-protein-coupled receptors, linked to the inhibition of adenylyl cyclase (Patel and Srikant 1997) or activation of inwardly rectifying K+ channels (Kreienkamp et al. 1997). Using SST-14-selective and SST-28-selective radioligands, it has been shown that β-cells possess SST-28-preferred binding sites whereas α-cells display SST-14-preferred receptors (Kumar et al. 1999). In rodent islets, α-cells contain SSTR2, whereas β-cells possess SSTR5. In human islets, SSTR2 is the functionally dominant receptor present in both α- and β-cells (Kailey et al. 2012). This suggests that in rodents, SST-14 exerts an inhibitory effect mainly on α-cells whereas, in humans, the peptide can modulate both glucagon and insulin secretion. Further work with specific isoforms and assays are required to answer this question.
Role of SST
The role of SST in the pancreas is poorly understood. SST can inhibit glucagon, insulin and PP release as well as its own secretion through an auto feedback mechanism (Ipp et al. 1979). The insulin and glucagon secretory response to nutrient stimuli was enhanced in vivo in SST knock-out mice; in addition, the glucose-induced suppression of glucagon secretion was absent although no defective insulin and glucagon secretion were detected under basal conditions (Hauge-Evans et al. 2009). This suggests that SST negatively regulates α- and β-cell function only under conditions of nutrient stimulus. Exogenous SST inhibits granule exocytosis from isolated rodent and human α- and β-cells (Gromada et al. 2001; Kailey et al. 2012; Renström et al. 1996). Although SST receptor antagonists increased glucose-induced insulin secretion in isolated islets (Zhang et al. 2014), there was no effect of the same antagonist in the perfused pancreas; this difference could reflect the difficulty of transferring experimental stimulatory and inhibitory effects efficiently from a single islet in vitro to the intact perfused multi-islet organ. Nevertheless, these observations emphasize the importance of coordinated communication within the islet network in controlling hormone secretion.
In rodents, like in humans, SST is secreted in a glucose- and Ca2+-dependent manner (Berts et al. 1996; Braun et al. 2009; Grill and Efendić 1984; Zhang et al. 2007). Given that SST is a strong inhibitor of insulin secretion, why is it released at glucose concentrations at which insulin is required to lower the blood glucose levels? Considering that SST begins to be released at glucose levels that diminish glucagon release (Vieira et al. 2007), it can be hypothesized that SST acts as a “buffering hormone”, preventing the over-secretion of insulin or glucagon. An alternative hypothesis is that at high glucose concentrations, SST primarily inhibits glucagon secretion rather than insulin production.
In mouse islets, δ-cells, like α- and β-, are innervated by parasympathetic and sympathetic axons whereas, in humans, innervation is less clear (Rodriguez-Diaz et al. 2011a). In fact, human α-cells secrete acetylcholine, which can act as a paracrine signal to regulate insulin and SST release (Molina et al. 2014; Rodriguez-Diaz et al. 2011b). The effects of acetylcholine (or carbachol) on somatostatin secretion are controversial. Whereas stimulation of somatostatin release was observed in some studies (Zhang et al. 2007), other studies indicate an inhibitory effect (Hauge-Evans et al. 2009).
Morphological Characteristics of δ-Cells
In most mammals, δ-cells exhibit a neuron-like morphology with cytoplasmic processes extending from the islet capillaries to the central core of an islet (Baskin et al. 1984) (Fig. 6). These dendrite-like extensions would allow paracrine crosstalk with other δ-cells by forming a functional intercellular, syncytial-like arrangement (Grube and Bohn 1983). In addition, the long, thin δ-cells can communicate with many neighboring α- and β-cells to act as efficient regulators of activity. The cellular neighbors of part of one rat δ-cell as it extended from the surface through the islet to a capillary over a depth of 12.5 µm were identified using 3D EM; this small extension was in contact with one α-cell and at least five β-cells (Fig. 7). These data suggest that, although δ-cells are a relatively small in number, they could be regulators of a large proportion of islet cells. In humans, besides δ-cells having a polygonal shape and exhibiting processes of up to 22 µm, a few rounded or pyramidal-shaped δ-cells, whose diameters range between 10 µm and 16 µm, have been described from light microscopic observations (Grube and Bohn 1983). Three-dimensional EM studies will be required to determine if these are a different subset of δ-cells.
Figure 6.
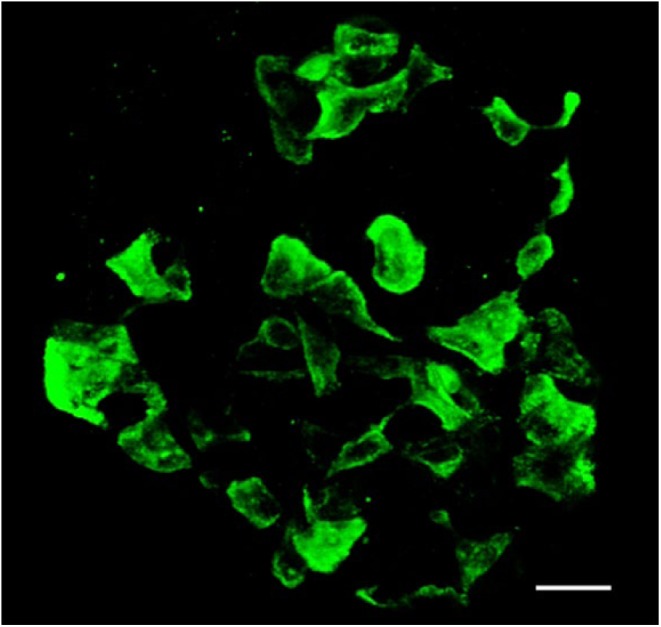
δ-Cells in a mouse islet. Immunofluorescent labelling for somatostatin in a mouse intact islet and reconstructed from confocal images. The cells have long neuronal-like processes that connect with each other to create an interconnecting network of δ-cells in the islet. Scale, 20 µm.
Figure 7.

Sequential electron microscopy images (A–H) to demonstrate the pathway and adjacent cells of a δ-cell in a rat islet. Images were taken using serial block-face scanning electron microscopy of a rat islet. Sections were taken 50 µm apart throughout the islet. This series of sequential images (distance between images in µm listed on the panels) shows a δ-cell (δ) process penetrating the islet from the surface (A) between five adjacent β-cells (β1–5), an α-cell (α1), and an unknown cell type (x1) to a central capillary (c) in (H). Scale, 5 µm.
In mammals, including humans, δ-cells are characterized by a uniform population of moderately electron-opaque secretory granules that tend to be smaller than those found in α- and β-cells. In humans, the diameter of each roughly spherical granule is, on average, 250 nm. In mice, granules are much smaller and are lozenge-shaped (Figs. 2, 8). Differences in granule shape and size are likely to have significant influences on the measurements of granule exocytosis using capacitance changes (Gopel et al. 2004). Because of their complex neuron-like architecture, action potential propagation might actually fulfil a signaling function in the δ-cells. Since there are many close contacts between δ-cells and other cell types, it will be interesting to determine whether there are specialized zones for somatostatin release (analogous to the nerve terminal) at these contact points and whether the ion channel complement in these ‘terminals’ is different from that in the rest of the cell. This could have implications for correct signaling when islet architecture is disrupted in diabetes.
Figure 8.
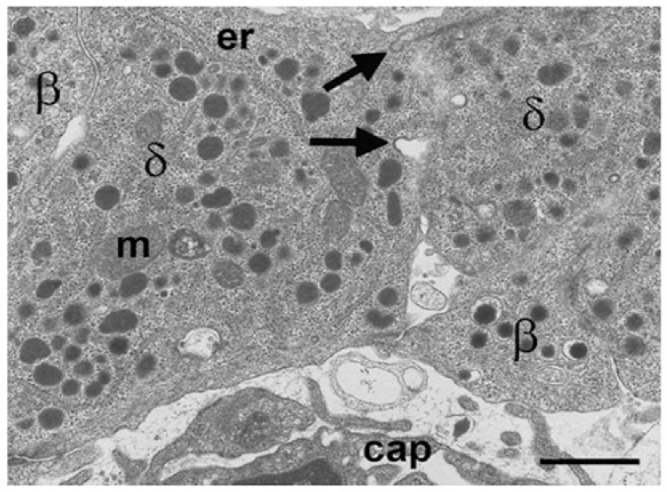
Electron microscopy image of mouse δ-cells (δ) to demonstrate the lozenge-shaped granules and potential intercellular communication between cells. Omega-shaped thickened membrane invaginations (arrows) at the intercellular margin are characteristic of membrane recovery following granule exocytosis. m, mitochondrion; er, endoplasmic reticulum; cap, capillary. Scale, 1 μm.
In conclusion, δ-cells are ideally located in islets with an appropriate neuron-like morphology to act as powerful regulators of islet hormone release. Although the islet has only a relatively small number of δ-cells, they can effectively communicate with each other via elongated projections. This establishes a pan-islet inhibitory network to enable transmission of signals received from both extrinsic nerves and the blood to a larger cellular population in the islet.
PP-Cells
Pancreatic polypeptide (PP) is a 36-amino acid peptide first localized in 1974 in the avian pancreas (Kimmel et al. 1975; Larsson et al. 1975) and later in mammalian islets (Larsson et al. 1975; Lin and Chance 1974) in a specific pancreatic cell type that was different from the originally described “A, B and D” cells. PP-containing cells (also known as F-cells) make up the smallest proportion of all islet cells in most regions of the pancreas (<2% of the islet cell population humans and <5% in rodents) (Clark et al. 1988; Stefan et al. 1982b; Sundler et al. 1977). Despite the relatively low abundance in most of the pancreas, PP-containing cells delineate the embryological origins of the pancreas from two separate pouches of the gut. The highest concentration of PP cells is in the head of the pancreas (known as the PP-rich lobe) where 90% of all PP-cells are located (Rahier et al. 1983b; Stefan et al. 1982a; Wang et al. 2013). In embryonic development in mammals, the pancreas is derived from two primordial pouches from the duodenum—the ventral and dorsal anlage; at the end of development the ventral pancreas forms part of the region of the pancreas known in humans as the uncinate lobe and remains close to the duodenum. In rodents, the ventral pancreas is situated in the region bounded by the bile duct and the duodenum. This small pancreatic region, which forms <10% of the whole pancreas in adult humans (Rahier et al. 1983b; Stefan et al. 1982b), is indistinguishable macroscopically from the lobules of the dorsal lobe but, microscopically, the islets are very different in structure and composition. The islets in the PP-rich lobe in humans and rodents contain very few α-cells (Hellman et al. 1962) and variable amounts of β-cells (Fig. 9) (Clark et al. 1988; Stefan et al. 1982b). Islets in this region appear more diffuse and not the ovoid or spherical structures typical of the major part of the pancreas. In addition, there are scattered, individual PP-containing cells in the exocrine tissue. In the majority of pancreatic islets (embryologically derived from the dorsal anlage), PP-cells are elongated and are located with other non-β-cells towards the periphery of rodent islets and lining the capillaries in human islets. This difference in islet structure and cellular composition is of major importance when taking samples from a pancreas from any species; islets in tissue samples taken from the pancreatic head may include tissue from the ventral pancreas and the cellular content, gene expression and secretory pattern will not necessarily be representative of islets in the major part of the pancreas.
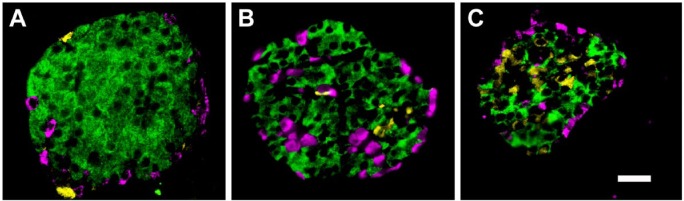
Figure 9.

Tissue from the head region of a human pancreas to demonstrate differences in islet structure in the PP-rich lobule of the ventral pancreas. Immunoperoxidase labelling for glucagon (A) and PP (B). Adjacent sections through the head region show an exocrine lobule (outlined), which has large, diffuse islets containing a high proportion of PP-positive cells. An islet in an adjacent lobule (arrow) has no PP-cells and the usual population of glucagon-positive cells. The lower panel shows immunofluorescent labelling for (C) PP (green), (D) glucagon (red), (E) insulin (blue), and merged images (F). The diffusely structured islet contains mostly PP-positive cells, a single glucagon-positive cell (arrow), and insulin-positive cells distributed randomly throughout. Scale (A, B) 500 μm; (C, D, E, F) 50 μm.
Morphology and Functions of PP-Cells
PP cells contain electron-dense secretory granules bounded by a membrane in both humans and rodents. The size of the PP-cell granules is very heterogeneous in comparison with the similarly electron-dense but larger and more uniform granules of α-cells. PP secretion is an important part of the pancreas–gut–brain axis (Holzer et al. 2012). The peptide is released post-prandially and regulated via the vagus nerve (stimulated by cholinergic agents and blocked by cholinergic inhibitors) and more locally via enteric nerve networks (Field et al. 2010; Schwartz 1983). Although PP secretion is stimulated by arginine, glucose is without effect (Weir et al. 1979). PP has largely inhibitory actions in the gut, reducing gastric emptying and intestinal motor activity (Lin and Chance 1974) through actions on the Y4 receptor (also known as PPYR1). Infused PP has no effect on insulin secretion in vivo (Bastidas et al. 1990) but has been shown to inhibit glucagon secretion at low glucose concentrations via the PPYR1 receptors on α-cells (Aragon et al. 2015). These data suggest that PP could act as an intra-islet regulator of secretion.
Islet Architecture in Diabetes
As we have highlighted, communication within the endocrine cell network of the islet must be fine-tuned to maintain glucose homeostasis (Kanno et al. 2002). Diabetes results not only from dysfunction of one or more of the hormone-expressing cells but also from changes to the islet architecture and a break-down in the communication between the endocrine cells of the pancreatic islet (compare islets in Fig. 3 with those in Fig. 10). Islet structure is altered in most animal models of diabetes and many islets in human diabetes have morphological changes. In Type 1 Diabetes (T1D), an autoimmune attack destroys the β-cells (Foulis et al. 1991; Foulis and Stewart 1984) whereas the non-β-cells remain largely intact. It has been recognized for more than a century that, not only is the β-cell mass affected, but also the α-cell population increases in T2D, and extracellular amyloid deposits disrupt the islet architecture (Butler et al. 2003; Clark et al. 1988; Maclean and Ogilvie 1955; Opie 1901). Although the β-cell is central to these observations, dysfunction in α- or δ-cells can contribute to the disease etiology.
Figure 10.

Immunofluorescent labelling of islet cells rodent and primate diabetes to demonstrate changes in islet architecture in hyperglycemia. Panels show cells immunofluorescently labelled for insulin, glucagon, and somatostatin and the merged signals. Hyperglycemic models: (A) Diabetic Goto-Kakizaki rat; (B) mouse model of diabetes exhibiting beta-cell specific deletion of the Krebs cycle enzyme fumarate hydratase (FH-/-); (C) mouse with impaired insulin secretion due to the expression of an activating KATP channel mutation (βV59M); (D) non-human primate with diabetes; (E) patient with recent-onset T1D; and (F) patient with T2D. In all of these hyperglycemic models, there are marked changes in islet morphology (for comparison see Figure 2 for structure in the absence of diabetes). There was reduced insulin-positive areas and increased proportion of glucagon-positive cells. A typical feature of animal models of diabetes is an increased infiltration of α-cells into the core of the islet. Little change in the expression pattern of somatostatin is apparent in most of the diabetic models. However, somatostatin-expressing cells are increased in βV59M mice and in the patient sample of T1D. Scale, 200 μm.
Pancreatic α-Cells; the Role of Glucagon in Diabetes
The importance of glucagon secretion in the etiology of diabetes was first highlighted nearly 40 years ago by Unger and Orci (1975). They hypothesized that diabetes also results from excessive glucagon secretion, which stimulates hepatic glucose production and contributes to hyperglycemia. Many studies have since demonstrated α-cell dysfunction in diabetes. Patients with uncontrolled T1D, and to a lesser extent, those with T2D, have persistently elevated circulating glucagon levels (Baron et al. 1987; Muller et al. 1973; Reaven et al. 1987; Unger et al. 1970). It is now generally accepted that diabetes is a bi-hormonal disease characterized by insufficient glucagon secretion at low glucose (when it is needed) and excessive release at high glucose (when it is not needed), in addition to impaired insulin secretion and sensitivity (Cryer 2002; Dunning and Gerich 2007; Unger 1985; Unger et al. 1970; Zhang et al. 2013).
Many hypotheses have been generated to explain the underlying mechanism of α-cell dysfunction in diabetes. These include the loss of an intrinsic ability of the α-cell to sense the circulating glucose level (Rorsman et al. 2014; Zhang et al. 2013), and a loss of the paracrine inhibition of glucagon secretion by insulin, zinc, GABA or other β-cell secretory products (Gromada et al. 2007). The importance of targeting and correcting the glucagon secretory defect is exemplified in mice where genetic deletion of the glucagon receptor prevents the development of diet-induced obesity and streptozotocin-induced diabetes (Conarello et al. 2007; Lee et al. 2011).
Alterations in α-cell architecture in diabetes have the potential to result in aberrant glucagon secretion and contribute to the underlying pathophysiology of the disease. An increase in α-cell mass is well recognized in T1D (Fig. 10E) (Rahier et al. 1983a), associated with hyperglucagonaemia (Gromada et al. 2007; Muller et al. 1970). Islets in patients with recent-onset T1D retain their normal population of α-cells but may be devoid of β-cells (Fig. 11) (Foulis and Stewart 1984; Kloppel et al. 1985; Walker et al. 2011a). However, the islets in patients with long-standing T1D are large, no longer ovoid in shape and with a distorted architecture, and contain an expanded population of well-granulated α-cells (Fig. 10E).
Figure 11.
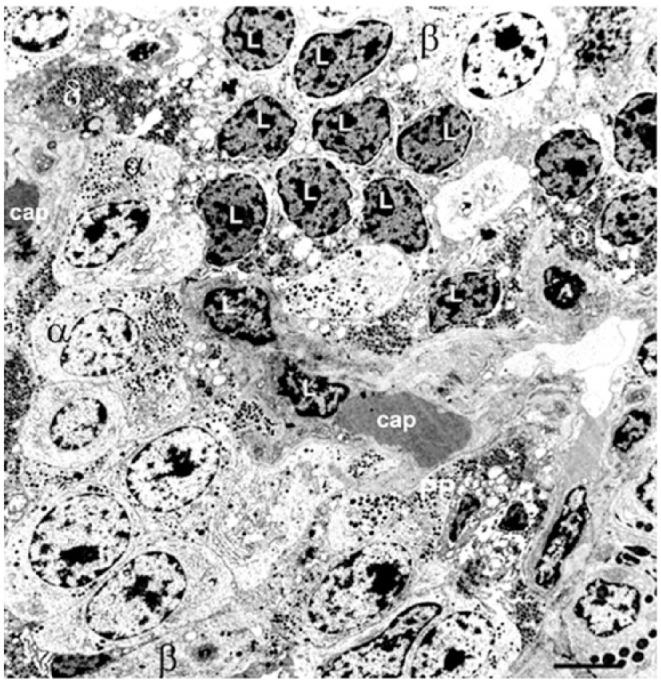
Pancreatic islet structure during the acute, ketotic phase of childhood-onset Type 1 Diabetes (T1D): Electron micrograph shows a few degranulated β-cells (β) but many α- (α) and δ- (δ) cells, most of which are located adjacent to a capillary (cap). Infiltration of lymphocytes (L) was present throughout the islet. Scale, 5 μm.
In T2D, α-cell mass has been reported to be increased (Fig. 10F) (Bosco et al. 2010; Clark et al. 1988; Iki and Pour 2007; Rahier et al. 1983a; Sakuraba et al. 2002; Yoon et al. 2003) or unaltered (Henquin and Rahier 2011; Stefan et al. 1982b). This correlates with a ~3-fold increase in islet glucagon content (Zhang et al. 2013). It has been suggested that an expanded α-cell population results in increased α-cell contacts at the expense of β-to-β contacts, which would affect intra-islet signaling (Kilimnik et al. 2011). However, islet architecture can be disrupted not only by increased non-β-cell populations but also by islet amyloid deposits (Butler et al. 2003; Clark et al. 1988; Jurgens et al. 2011). These extracellular accumulations initially develop in a relatively few islets at perivascular sites and are more likely to be a consequence of hyperglycemia rather than a cause (Clark and Nilsson 2004; Jurgens et al. 2011). Collated electron micrographs covering a large field of view of an islet from a patient with T2D shows numerous granulated β-cells, no amyloidosis and morphologically intact α- and δ-cells (Fig. 12).
Figure 12.
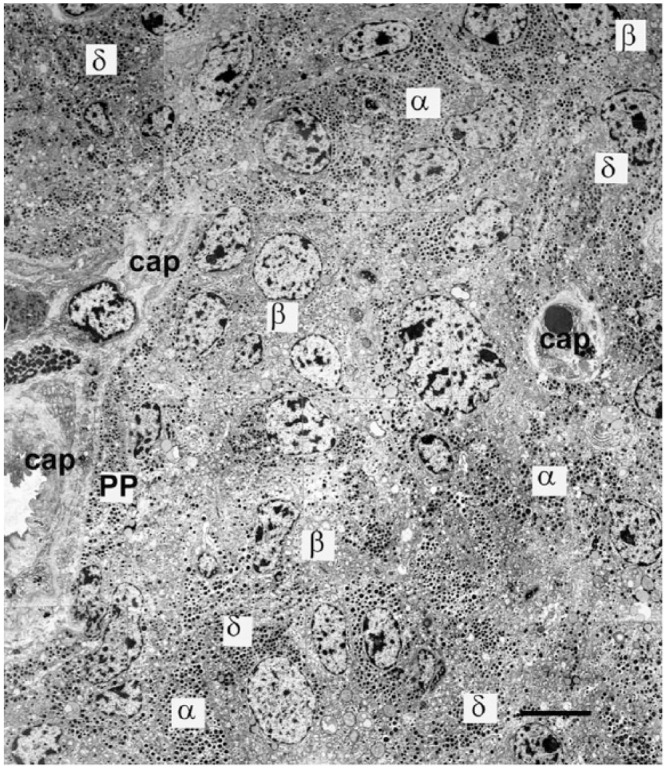
Part of an islet from a patient with Type 2 Diabetes; electron microscopy montage demonstrating the presence of many insulin-containing β-cells (β) and a high proportion of α-cells (α). PP-containing cells (PP) and δ-cells (δ) are close to capillaries (cap). A large proportion of cells are not situated adjacent or near to a capillary in this thin section. Although other islets in this patient contained islet amyloid deposits, there were none in this islet cross section and no cells showing signs of apoptosis. Exo; exocrine tissue. Scale, 5 μm.
α-cell architecture is also disorganized in many animal models of diabetes (Fig. 10A–10D) (Brereton et al. 2014; Guest et al. 2002; Kulkarni et al. 2004). Normally in rodents, the glucagon-expressing cells are located in the periphery of islets. However, in diabetic islets, many α-cells are also found in the islet center. These changes in islet architecture will influence the communication and paracrine signaling within the islet, but the extent to which this underlies the aberrant glucagon secretion in diabetes is poorly understood. It is notable that these changes are reversible once normoglycemia is restored in rodent models (Brereton et al. 2014), suggesting that hyperglycemia can modify the α-cell population and architecture.
Pancreatic δ-Cell; the Role of Somatostatin in Diabetes
Very little attention has been paid to δ-cell structure and function in diabetes. In patients with severe T1D, the proportion of SST-expressing cells increases (Fig. 10E) (Rahier et al. 1983a). The ultrastructure of the non-β-cells remains unaffected by the autoimmune attack and the δ-cells in T1D islets exhibit apparently normal endoplasmic reticulum, granule density and nuclear morphology (Fig. 11). In patients with T2D, there is little change or an increase in the δ-cell population (Figs. 10F, 12) (Clark et al. 1988; Rahier et al. 1983a). It has been reported that the proportion of δ-to-δ and δ-to-α-cell contacts are increased in human T2D (Kilimnik et al. 2011) but if δ-cell mass remains unchanged, this is more likely due to a reduction in the number of β-cells (Rahier et al. 1983a; Saito et al. 1979). In T2D, the expression patterns of SSTRs in the endocrine cells are altered; SSTR1 and SSTR4 subtypes were shown not to be present in α-cells, and SSTR5 was observed to be increased compared to non-diabetic islets (Portela-Gomes et al. 2010), with only the SSTR4 subtype expressed in δ-cells (Portela-Gomes et al. 2010). These changes in receptor expression profiles could contribute to the impaired insulin secretion and elevated glucagon production seen in diabetes, and highlights the importance of intra-islet SST in the regulation of islet function.
Changes in the proportion and location of δ-cells in the islet is also a feature of animal models of diabetes. An increased number of δ-cells in the center of the islet is seen in the hypoinsulinemic and hyperglycemic βV59M mice that express a gain-of-function KATP-channel mutation (Fig. 10C) (Brereton et al. 2014), and it is tempting to attribute these changes to the loss of normal paracrine signaling. Previous studies have shown migration of δ-cells from a “capped” region at the islet periphery adjacent to the vasculature or ductal structures to the center of the islet core in db/db mice, and an apparent increased density of δ-cell projection within the core of β-cells in the center of the islet (Leiter et al. 1979). As with α-cells, δ-cells are rarely degranulated in diabetes, unlike β-cells where this is commonly observed (Figs .10A–10D, 10F, 12).
As the δ-cell serves as the ‘master inhibitory communicator’ within the islet, any changes to the islet architecture in diabetes will have an impact on intra-islet paracrine communication. Deletion of the SST gene or SSTR subtypes in mice resulted in an inverted glucagon secretory profile at high glucose, which is reminiscent of that seen in islets from T2D patients and in vivo (Hauge-Evans et al. 2009; Singh et al. 2007; Walker et al. 2011b). This highlights the complexity of the islet network and suggests that altered paracrine signaling may partly underlie the secretory defects observed in diabetes. It is important to point out, however, that this does not necessarily mean that glucose mediates its inhibitory effect on glucagon secretion via somatostatin. Loss of intra-islet SST signaling may exert long-term effects via altered gene expression, among other effects.
Pancreatic Polypeptide-Cells; the Role of PP in Diabetes
As one of the most poorly understood cells in the pancreatic islet, it is not surprising that the role of PP-cells in diabetes has received little attention. In pancreatitis, the PP-cell population of the affected tissue increases, particularly in the proliferating ductal epithelium. Attempts to quantify the changes in the PP-cell population in patients with T1D and T2D have produced conflicting reports of increased (Gepts et al. 1977) or unaltered (Clark et al. 1988; Rahier et al. 1983b; Stefan et al. 1982b) numbers. Given the propensity for PP-rich islets in the head of the pancreas, these discrepancies may reflect differences in the tissue sampling procedures.
Changes in α- and δ-Cell Identity in Diabetes
The recent emergence of the concept of cellular plasticity in adult islets has led to renewed interest in the non β-cell members of the islet cell family, since cellular transformations have involved both α- and δ-cells. This phenomenon of cellular plasticity challenges the concept that the endocrine cells are terminally differentiated and permanently determined in the adult following fetal/neonatal pancreatic development. Although cells expressing more than one hormone have been identified during pancreatic development (De Krijger et al. 1992; Riedel et al. 2012), it appears that adult islet cells in both humans and animal models can change their hormone-expression profile and seemingly convert from one cell type to another; manipulation of their genetic fingerprint, hyperglycemia or alterations in the intra-islet environment can induce changes in the hormone content (Brereton et al. 2014; Collombat et al. 2009; Courtney et al. 2013; Fernandes et al. 1997; Gao et al. 2014; Piran et al. 2014; Talchai et al. 2012; Thorel et al. 2010; Yang et al. 2011). However, in at least two models of impaired insulin secretion (the βV59M mouse and mice lacking R-type Ca2+ channels), this transformation appears to be largely only in terms of cellular hormone content; cells that had apparently undergone conversion from β- to α-cells contained glucagon granules but had glucose dependence, electrical and transcription factor properties of an insulin-containing cell (Brereton et al. 2014; Jing et al. 2005). This suggests that not all plastic cells may have the cellular fingerprint or be fully functional in their new identity, and raises the question of the role of quantitative immunocytochemistry in the interpretation of changes in islet function. Furthermore, as we have highlighted above, the different islet cell types can be distinguished according to their morphology and ultrastructure. Future studies should aim to investigate the functional and morphological properties of the plastic cells in greater detail as it may provide mechanistic insight into the significance of this phenomenon in diabetes.
Alpha-, Delta- and PP-cells: the Architectural Cornerstones of Islet Structure and Coordination
The intricacies of communication within the islet network depend upon the anatomical localization of the non-β-cells; any change that affects islet architecture can have a profound effect on pancreatic function (Fig. 13). The intimate connection between α- and β-cells fine-tunes the regulation of glucose homeostasis, and the inhibitory properties of the δ-cellular network with the other cells affirm their role as master communicators in the islet. The importance of these minority members of the pancreatic islet structure has become evident, as alterations in their function, structure or identity could contribute to the etiology of diabetes. Therefore, therapeutic strategies should aim to target both cellular function and islet architecture to help restore inter-islet communication and ultimately glucose homeostasis in diabetes.
Figure 13.

Diagrammatic representation of intra-islet connections and regulatory signals in health and diabetes. In normoglycemia, cells are closely packed, with non-β-cells situated more peripherally and adjacent to capillaries. Secretion of insulin (green) and glucagon (purple) exert paracrine effects on α-, δ-(yellow), and β-cells. Somatostatin has inhibitory paracrine effects on all cell types (dotted lines). β-Cell products, Zn2+ and GABA, may affect α-cell function, and acetylcholine from α-cells may influence somatostatin secretion. In the diabetic islet, architecture is disrupted. There are less β-cells, and α-cells are increased and distributed throughout the islet. The influence of insulin on islet function is reduced but the paracrine effects of glucagon and somatostatin are likely to be increased (heavy solid and dotted lines, respectively).
Acknowledgments
We thank Professor Patrik Rorsman for his help and support, Prof Frances M Ashcroft for providing the βV59M mouse samples, Prof Barbara Hansen for the monkey samples, Dr Julie Adams and Prof Patrick Pollard for the FH-/- mouse samples and to Prof C-G Ostenson for the GK rat samples. We are grateful to C Genoud and S Monteith of Gatan’ Pleasantan, CA for their considerable help in preparation and interpretation of the 3D-electron microscopy samples and Dr Juris Galvanovskis for the data analysis of the 3D EM files. Human tissue collection was made according to Local and International human ethical committee recommendations and with appropriate permissions. Animal specimens were collected in accordance with National guidelines (UK Home Office) on animal experimentation.
Footnotes
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: We are grateful to the Wellcome Trust for funding MFB, QZ, EV.
References
- Aragon F, Karaca M, Novials A, Maldonado R, Maechler P, Rubi B. (2015). Pancreatic polypeptide regulates glucagon release through PPYR1 receptors expressed in mouse and human alpha-cells. Biochim Biophys Acta 1850:343-351. [DOI] [PubMed] [Google Scholar]
- Arimura A, Sato H, Dupont A, Nishi N, Schally AV. (1975). Somatostatin: abundance of immunoreactive hormone in rat stomach and pancreas. Science 189:1007-1009. [DOI] [PubMed] [Google Scholar]
- Barg S, Galvanovskis J, Gopel SO, Rorsman P, Eliasson L. (2000). Tight coupling between electrical activity and exocytosis in mouse glucagon-secreting alpha-cells. Diabetes 49:1500-1510. [DOI] [PubMed] [Google Scholar]
- Baron AD, Schaeffer L, Shragg P, Kolterman OG. (1987). Role of hyperglucagonemia in maintenance of increased rates of hepatic glucose output in type II diabetics. Diabetes 36:274-283. [DOI] [PubMed] [Google Scholar]
- Baskin DG, Gorray KC, Fujimoto WY. (1984). Immunocytochemical identification of cells containing insulin, glucagon, somatostatin, and pancreatic polypeptide in the islets of Langerhans of the guinea pig pancreas with light and electron microscopy. Anat Rec 208:567-578. [DOI] [PubMed] [Google Scholar]
- Bastidas JA, Couse NF, Yeo CJ, Schmieg RE, Jr., Andersen DK, Gingerich RL, Zinner MJ. (1990). The effect of pancreatic polypeptide infusion on glucose tolerance and insulin response in longitudinally studied pancreatitis-induced diabetes. Surgery 107:661-668. [PubMed] [Google Scholar]
- Berts A, Ball A, Dryselius G, Gylfe E, Hellman B. (1996). Glucose stimulation of somatostatin-producing islet cells involves oscillatory Ca2+ signaling. Endocrinology 137:693-697. [DOI] [PubMed] [Google Scholar]
- Bonner-Weir S, Orci L. (1982). New perspectives on the microvasculature of the islets of Langerhans in the rat. Diabetes 31:883-889. [DOI] [PubMed] [Google Scholar]
- Bosco D, Armanet M, Morel P, Niclauss N, Sgroi A, Muller YD, Giovannoni L, Parnaud G, Berney T. (2010). Unique arrangement of alpha- and beta-cells in human islets of Langerhans. Diabetes 59:1202-1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun M. (2014). The somatostatin receptor in human pancreatic beta-cells. Vitam Horm 95:165-193. [DOI] [PubMed] [Google Scholar]
- Braun M, Ramracheya R, Amisten S, Bengtsson M, Moritoh Y, Zhang Q, Johnson PR, Rorsman P. (2009). Somatostatin release, electrical activity, membrane currents and exocytosis in human pancreatic delta cells. Diabetologia 52:1566-1578. [DOI] [PubMed] [Google Scholar]
- Brazeau P, Vale W, Burgus R, Ling N, Butcher M, Rivier J, Guillemin R. (1973). Hypothalamic polypeptide that inhibits the secretion of immunoreactive pituitary growth hormone. Science 179:77-79. [DOI] [PubMed] [Google Scholar]
- Brereton MF, Iberl M, Shimomura K, Zhang Q, Adriaenssens AE, Proks P, Spiliotis II, Dace W, Mattis KK, Ramracheya R, Gribble FM, Reimann F, Clark A, Rorsman P, Ashcroft FM. (2014). Reversible changes in pancreatic islet structure and function produced by elevated blood glucose. Nat Commun 5:4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunicardi FC, Stagner J, Bonner-Weir S, Wayland H, Kleinman R, Livingston E, Guth P, Menger M, McCuskey R, Intaglietta M, Charles A, Ashley S, Cheung A, Ipp E, Gilman S, Howard T, Passaro E., Jr. (1996). Microcirculation of the islets of Langerhans. Long Beach Veterans Administration Regional Medical Education Center Symposium. Diabetes 45:385-392. [DOI] [PubMed] [Google Scholar]
- Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. (2003). Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52:102-110. [DOI] [PubMed] [Google Scholar]
- Cabrera O, Berman DM, Kenyon NS, Ricordi C, Berggren PO, Caicedo A. (2006). The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc Natl Acad Sci U S A 103:2334-2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark A, Nilsson MR. (2004). Islet amyloid: a complication of islet dysfunction or an aetiological factor in Type 2 diabetes? Diabetologia 47:157-169. [DOI] [PubMed] [Google Scholar]
- Clark A, Wells CA, Buley ID, Cruickshank JK, Vanhegan RI, Matthews DR, Cooper GJ, Holman RR, Turner RC. (1988). Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis: quantitative changes in the pancreas in type 2 diabetes. Diabetes Res 9:151-159. [PubMed] [Google Scholar]
- Collombat P, Xu X, Ravassard P, Sosa-Pineda B, Dussaud S, Billestrup N, Madsen OD, Serup P, Heimberg H, Mansouri A. (2009). The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into alpha and subsequently beta cells. Cell 138:449-462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conarello SL, Jiang G, Mu J, Li Z, Woods J, Zycband E, Ronan J, Liu F, Roy RS, Zhu L, Charron MJ, Zhang BB. (2007). Glucagon receptor knockout mice are resistant to diet-induced obesity and streptozotocin-mediated beta cell loss and hyperglycaemia. Diabetologia 50:142-150. [DOI] [PubMed] [Google Scholar]
- Courtney M, Gjernes E, Druelle N, Ravaud C, Vieira A, Ben-Othman N, Pfeifer A, Avolio F, Leuckx G, Lacas-Gervais S, Burel-Vandenbos F, Ambrosetti D, Hecksher-Sorensen J, Ravassard P, Heimberg H, Mansouri A, Collombat P. (2013). The inactivation of Arx in pancreatic alpha-cells triggers their neogenesis and conversion into functional beta-like cells. PLoS Genet 9:e1003934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryer PE. (2002). The pathophysiology of hypoglycaemia in diabetes. Diabetes Nutr Metab 15:330-333; discussion 362. [PubMed] [Google Scholar]
- De Krijger RR, Aanstoot HJ, Kranenburg G, Reinhard M, Visser WJ, Bruining GJ. (1992). The midgestational human fetal pancreas contains cells coexpressing islet hormones. Dev Biol 153:368-375. [DOI] [PubMed] [Google Scholar]
- De Marinis YZ, Salehi A, Ward CE, Zhang Q, Abdulkader F, Bengtsson M, Braha O, Braun M, Ramracheya R, Amisten S, Habib AM, Moritoh Y, Zhang E, Reimann F, Rosengren AH, Shibasaki T, Gribble F, Renstrom E, Seino S, Eliasson L, Rorsman P. (2010). GLP-1 inhibits and adrenaline stimulates glucagon release by differential modulation of N- and L-type Ca2+ channel-dependent exocytosis. Cell Metab 11:543-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deconinck JF, Potvliege PR, Gepts W. (1971). The ultrastruture of the human pancreatic islets. I. The islets of adults. Diabetologia 7:266-282. [DOI] [PubMed] [Google Scholar]
- Dunning BE, Gerich JE. (2007). The role of alpha-cell dysregulation in fasting and postprandial hyperglycemia in type 2 diabetes and therapeutic implications. Endocr Rev 28:253-283. [DOI] [PubMed] [Google Scholar]
- Dunphy JL, Taylor RG, Fuller PJ. (1998). Tissue distribution of rat glucagon receptor and GLP-1 receptor gene expression. Mol Cell Endocrinol 141:179-186. [DOI] [PubMed] [Google Scholar]
- Elayat AA, el-Naggar MM, Tahir M. (1995). An immunocytochemical and morphometric study of the rat pancreatic islets. J Anat 186 (Pt 3): 629-637. [PMC free article] [PubMed] [Google Scholar]
- Falkmer S, Ostberg Y. (1977). Comparative morphology of pancreatic islet in animals. In: Volk BW, Wellman KF, eds. The diabetic pancreas. Macmillan, London, pp 15-59. [Google Scholar]
- Fernandes A, King LC, Guz Y, Stein R, Wright CV, Teitelman G. (1997). Differentiation of new insulin-producing cells is induced by injury in adult pancreatic islets. Endocrinology 138:1750-1762. [DOI] [PubMed] [Google Scholar]
- Field BC, Chaudhri OB, Bloom SR. (2010). Bowels control brain: gut hormones and obesity. Nat Rev Endocrinol 6:444-453. [DOI] [PubMed] [Google Scholar]
- Foulis AK, McGill M, Farquharson MA. (1991). Insulitis in type 1 (insulin-dependent) diabetes mellitus in man–macrophages, lymphocytes, and interferon-gamma containing cells. J Pathol 165:97-103. [DOI] [PubMed] [Google Scholar]
- Foulis AK, Stewart JA. (1984). The pancreas in recent-onset type 1 (insulin-dependent) diabetes mellitus: insulin content of islets, insulitis and associated changes in the exocrine acinar tissue. Diabetologia 26:456-461. [DOI] [PubMed] [Google Scholar]
- Francis BH, Baskin DG, Saunders DR, Ensinck JW. (1990). Distribution of somatostatin-14 and somatostatin-28 gastrointestinal-pancreatic cells of rats and humans. Gastroenterology 99:1283-1291. [DOI] [PubMed] [Google Scholar]
- Furuta M, Yano H, Zhou A, Rouille Y, Holst JJ, Carroll R, Ravazzola M, Orci L, Furuta H, Steiner DF. (1997). Defective prohormone processing and altered pancreatic islet morphology in mice lacking active SPC2. Proc Natl Acad Sci U S A 94:6646-6651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta M, Zhou A, Webb G, Carroll R, Ravazzola M, Orci L, Steiner DF. (2001). Severe defect in proglucagon processing in islet A-cells of prohormone convertase 2 null mice. J Biol Chem 276:27197-27202. [DOI] [PubMed] [Google Scholar]
- Gao T, McKenna B, Li C, Reichert M, Nguyen J, Singh T, Yang C, Pannikar A, Doliba N, Zhang T, Stoffers DA, Edlund H, Matschinsky F, Stein R, Stanger BZ. (2014). Pdx1 Maintains beta Cell Identity and Function by Repressing an alpha Cell Program. Cell Metab 19:259-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gepts W, De Mey J, Marichal-Pipeleers M. (1977). Hyperplasia of “pancreatic polypeptide”-cells in the pancreas of juvenile diabetics. Diabetologia 13:27-34. [DOI] [PubMed] [Google Scholar]
- Gopel S, Zhang Q, Eliasson L, Ma XS, Galvanovskis J, Kanno T, Salehi A, Rorsman P. (2004). Capacitance measurements of exocytosis in mouse pancreatic alpha-, beta- and delta-cells within intact islets of Langerhans. J Physiol 556:711-726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grill V, Efendić S. (1984). Stimulation by calcium and barium of somatostatin release. Evidence for lower sensitivity of D- vis-à-vis B- and A-cells. Acta Physiol Scand 122:401-407. [DOI] [PubMed] [Google Scholar]
- Gromada J, Franklin I, Wollheim CB. (2007). Alpha-cells of the endocrine pancreas: 35 years of research but the enigma remains. Endocr Rev 28:84-116. [DOI] [PubMed] [Google Scholar]
- Gromada J, Holst JJ, Rorsman P. (1998). Cellular regulation of islet hormone secretion by the incretin hormone glucagon-like peptide 1. Pflügers Archiv 435:583-594. [DOI] [PubMed] [Google Scholar]
- Gromada J, Høy M, Buschard K, Salehi A, Rorsman P. (2001). Somatostatin inhibits exocytosis in rat pancreatic alpha-cells by G(i2)-dependent activation of calcineurin and depriming of secretory granules. J Physiol 535:519-532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grube D, Bohn R. (1983). The microanatomy of human islets of Langerhans, with special reference to somatostatin (D-) cells. Arch Histol Jpn 46:327-353. [DOI] [PubMed] [Google Scholar]
- Guest PC, Abdel-Halim SM, Gross DJ, Clark A, Poitout V, Amaria R, Ostenson CG, Hutton JC. (2002). Proinsulin processing in the diabetic Goto-Kakizaki rat. J Endocrinol 175:637-647. [DOI] [PubMed] [Google Scholar]
- Gylfe E, Gilon P. (2014). Glucose regulation of glucagon secretion. Diabetes Res Clin Pract 103:1-10. [DOI] [PubMed] [Google Scholar]
- Hauge-Evans AC, King AJ, Carmignac D, Richardson CC, Robinson IC, Low MJ, Christie MR, Persaud SJ, Jones PM. (2009). Somatostatin secreted by islet delta-cells fulfills multiple roles as a paracrine regulator of islet function. Diabetes 58:403-411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havel PJ, Taborsky GJ., Jr. (1989). The contribution of the autonomic nervous system to changes of glucagon and insulin secretion during hypoglycemic stress. Endocr Rev 10:332-350. [DOI] [PubMed] [Google Scholar]
- Hellman B, Wallgren A, Hellerstrom C. (1962). Two types of islet alpha cells in different parts of the pancreas of the dog. Nature 194:1201-1202. [DOI] [PubMed] [Google Scholar]
- Henquin JC, Rahier J. (2011). Pancreatic alpha cell mass in European subjects with type 2 diabetes. Diabetologia 54:1720-1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hökfelt T, Efendić S, Hellerström C, Johansson O, Luft R, Arimura A. (1975). Cellular localization of somatostatin in endocrine-like cells and neurons of the rat with special references to the A1-cells of the pancreatic islets and to the hypothalamus. Acta Endocrinol Suppl (Copenh) 200:5-41. [PubMed] [Google Scholar]
- Holzer P, Reichmann F, Farzi A. (2012). Neuropeptide Y, peptide YY and pancreatic polypeptide in the gut-brain axis. Neuropeptides 46:261-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iki K, Pour PM. (2007). Distribution of pancreatic endocrine cells including IAPP-expressing cells in non-diabetic and type 2 diabetic cases. J Histochem Cytochem 55:111-118. [DOI] [PubMed] [Google Scholar]
- Ipp E, Rivier J, Dobbs RE, Brown M, Vale W, Unger RH. (1979). Somatostatin analogs inhibit somatostatin release. Endocrinology 104:1270-1273. [DOI] [PubMed] [Google Scholar]
- Jiang G, Zhang BB. (2003). Glucagon and regulation of glucose metabolism. Am J Physiol Endocrinol Metab 284:E671-678. [DOI] [PubMed] [Google Scholar]
- Jing X, Li DQ, Olofsson CS, Salehi A, Surve VV, Caballero J, Ivarsson R, Lundquist I, Pereverzev A, Schneider T, Rorsman P, Renstrom E. (2005). CaV2.3 calcium channels control second-phase insulin release. J Clin Invest 115:146-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgens CA, Toukatly MN, Fligner CL, Udayasankar J, Subramanian SL, Zraika S, Aston-Mourney K, Carr DB, Westermark P, Westermark GT, Kahn SE, Hull RL. (2011). beta-cell loss and beta-cell apoptosis in human type 2 diabetes are related to islet amyloid deposition. Am J Pathol 178:2632-2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kailey B, van de, Bunt M, Cheley S, Johnson PR, MacDonald PE, Gloyn AL, Rorsman P, Braun M. (2012). SSTR2 is the functionally dominant somatostatin receptor in human pancreatic beta- and alpha-cells. Am J Physiol Endocrinol Metab 303:E1107-1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno T, Gopel SO, Rorsman P, Wakui M. (2002). Cellular function in multicellular system for hormone-secretion: electrophysiological aspect of studies on alpha-, beta- and delta-cells of the pancreatic islet. Neurosci Res 42:79-90. [DOI] [PubMed] [Google Scholar]
- Kieffer TJ, Habener JF. (1999). The glucagon-like peptides. Endocr Rev 20:876-913. [DOI] [PubMed] [Google Scholar]
- Kieffer TJ, Heller RS, Unson CG, Weir GC, Habener JF. (1996). Distribution of glucagon receptors on hormone-specific endocrine cells of rat pancreatic islets. Endocrinology 137:5119-5125. [DOI] [PubMed] [Google Scholar]
- Kilimnik G, Zhao B, Jo J, Periwal V, Witkowski P, Misawa R, Hara M. (2011). Altered islet composition and disproportionate loss of large islets in patients with type 2 diabetes. PLoS One 6:e27445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel JR, Hayden LJ, Pollock HG. (1975). Isolation and characterization of a new pancreatic polypeptide hormone. J Biol Chem 250:9369-9376. [PubMed] [Google Scholar]
- Kloppel G, Lohr M, Habich K, Oberholzer M, Heitz PU. (1985). Islet pathology and the pathogenesis of type 1 and type 2 diabetes mellitus revisited. Surv Synth Pathol Res 4:110-125. [DOI] [PubMed] [Google Scholar]
- Kreienkamp HJ, Hönck HH, Richter D. (1997). Coupling of rat somatostatin receptor subtypes to a G-protein gated inwardly rectifying potassium channel (GIRK1). FEBS Lett 419:92-94. [DOI] [PubMed] [Google Scholar]
- Kulkarni RN, Roper MG, Dahlgren G, Shih DQ, Kauri LM, Peters JL, Stoffel M, Kennedy RT. (2004). Islet secretory defect in insulin receptor substrate 1 null mice is linked with reduced calcium signaling and expression of sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA)-2b and -3. Diabetes 53:1517-1525. [DOI] [PubMed] [Google Scholar]
- Kumar U, Sasi R, Suresh S, Patel A, Thangaraju M, Metrakos P, Patel SC, Patel YC. (1999). Subtype-selective expression of the five somatostatin receptors (hSSTR1-5) in human pancreatic islet cells: a quantitative double-label immunohistochemical analysis. Diabetes; 48:77-85. [DOI] [PubMed] [Google Scholar]
- Larsson LI, Sundler F, Hakanson R. (1975). Immunohistochemical localization of human pancreatic polypeptide (HPP) to a population of islet cells. Cell Tissue Res 156:167-171. [DOI] [PubMed] [Google Scholar]
- Lee Y, Wang MY, Du XQ, Charron MJ, Unger RH. (2011). Glucagon receptor knockout prevents insulin-deficient type 1 diabetes in mice. Diabetes 60:391-397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiter EH, Gapp DA, Eppig JJ, Coleman DL. (1979). Ultrastructural and morphometric studies of delta cells in pancreatic islets from C57BL/Ks diabetes mice. Diabetologia 17:297-309. [DOI] [PubMed] [Google Scholar]
- Lin TM, Chance RE. (1974). Candidate hormones of the gut. VI. Bovine pancreatic polypeptide (BPP) and avian pancreatic polypeptide (APP). Gastroenterology 67:737-738. [PubMed] [Google Scholar]
- Lindsay TH, Halvorson KG, Peters CM, Ghilardi JR, Kuskowski MA, Wong GY, Mantyh PW. (2006). A quantitative analysis of the sensory and sympathetic innervation of the mouse pancreas. Neuroscience 137:1417-1426. [DOI] [PubMed] [Google Scholar]
- Liu YM, Guth PH, Kaneko K, Livingston EH, Brunicardi FC. (1993). Dynamic in vivo observation of rat islet microcirculation. Pancreas 8:15-21. [DOI] [PubMed] [Google Scholar]
- Longuet C, Sinclair EM, Maida A, Baggio LL, Maziarz M, Charron MJ, Drucker DJ. (2008). The glucagon receptor is required for the adaptive metabolic response to fasting. Cell Metab 8:359-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low JT, Zavortink M, Mitchell JM, Gan WJ, Do OH, Schwiening CJ, Gaisano HY, Thorn P. (2014). Insulin secretion from beta cells in intact mouse islets is targeted towards the vasculature. Diabetologia 57:1655-1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maclean N, Ogilvie RF. (1955). Quantitative estimation of the pancreatic islet tissue in diabetic subjects. Diabetes 4:367-376. [DOI] [PubMed] [Google Scholar]
- Marchetti P, Lupi R, Bugliani M, Kirkpatrick CL, Sebastiani G, Grieco FA, Del Guerra S, D’Aleo V, Piro S, Marselli L, Boggi U, Filipponi F, Tinti L, Salvini L, Wollheim CB, Purrello F, Dotta F. (2012). A local glucagon-like peptide 1 (GLP-1) system in human pancreatic islets. Diabetologia 55:3262-3272. [DOI] [PubMed] [Google Scholar]
- Marcinkiewicz M, Ramla D, Seidah NG, Chrétien M. (1994). Developmental expression of the prohormone convertases PC1 and PC2 in mouse pancreatic islets. Endocrinology 135:1651-1660. [DOI] [PubMed] [Google Scholar]
- Molina J, Rodriguez-Diaz R, Fachado A, Jacques-Silva MC, Berggren PO, Caicedo A. (2014). Control Of Insulin Secretion By Cholinergic Signaling In The Human Pancreatic Islet. Diabetes [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller WA, Faloona GR, Aguilar-Parada E, Unger RH. (1970). Abnormal alpha-cell function in diabetes. Response to carbohydrate and protein ingestion. N Engl J Med 283:109-115. [DOI] [PubMed] [Google Scholar]
- Muller WA, Faloona GR, Unger RH. (1973). Hyperglucagonemia in diabetic ketoacidosis. Its prevalence and significance. Am J Med 54:52-57. [DOI] [PubMed] [Google Scholar]
- Murlin JR, Clough HD, Gibbs CBF, Stokes AM. (1923). Aqueous extracts of pancreas: I. Influence on the carbohydrate metabolism of depancreatized animals. J Biol Chem 56:253-296. [Google Scholar]
- Nyman LR, Wells KS, Head WS, McCaughey M, Ford E, Brissova M, Piston DW, Powers AC. (2008). Real-time, multidimensional in vivo imaging used to investigate blood flow in mouse pancreatic islets. J Clin Invest 118:3790-3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani O, Ushiki T, Kanazawa H, Fujita T. (1986). Microcirculation of the pancreas in the rat and rabbit with special reference to the insulo-acinar portal system and emissary vein of the islet. Arch Histol Jpn 49:45-60. [DOI] [PubMed] [Google Scholar]
- Opie EL. (1901). The Relation Oe Diabetes Mellitus to Lesions of the Pancreas. Hyaline Degeneration of the Islands Oe Langerhans. J Exp Med 5:527-540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orci L, Unger RH. (1975). Functional subdivision of islets of Langerhans and possible role of D cells. Lancet 2:1243-1244. [DOI] [PubMed] [Google Scholar]
- Patel YC, Srikant CB. (1997). Somatostatin receptors. Trends Endocrinol Metab 8:398-405. [DOI] [PubMed] [Google Scholar]
- Pfeifer CR, Shomorony A, Aronova MA, Zhang G, Cai T, Xu H, Notkins AL, Leapman RD. (2014). Quantitative analysis of mouse pancreatic islet architecture by serial block-face SEM. J Struct Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piran R, Lee SH, Li CR, Charbono A, Bradley LM, Levine F. (2014). Pharmacological induction of pancreatic islet cell transdifferentiation: relevance to type I diabetes. Cell Death Dis 5:e1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocai A. (2012). Unraveling oxyntomodulin, GLP1’s enigmatic brother. J Endocrinol 215:335-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portela-Gomes GM, Grimelius L, Westermark P, Stridsberg M. (2010). Somatostatin receptor subtypes in human type 2 diabetic islets. Pancreas 39:836-842. [DOI] [PubMed] [Google Scholar]
- Rahier J, Goebbels RM, Henquin JC. (1983a). Cellular composition of the human diabetic pancreas. Diabetologia 24:366-371. [DOI] [PubMed] [Google Scholar]
- Rahier J, Wallon J, Loozen S, Lefevre A, Gepts W, Haot J. (1983b). The pancreatic polypeptide cells of the human pancreas: the effects of age and diabetes. J Clinical Endocrinology and Metabolism 56:441-444. [DOI] [PubMed] [Google Scholar]
- Reaven GM, Chen YD, Golay A, Swislocki AL, Jaspan JB. (1987). Documentation of hyperglucagonemia throughout the day in nonobese and obese patients with noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab 64:106-110. [DOI] [PubMed] [Google Scholar]
- Renström E, Ding WG, Bokvist K, Rorsman P. (1996). Neurotransmitter-induced inhibition of exocytosis in insulin-secreting beta cells by activation of calcineurin. Neuron 17:513-522. [DOI] [PubMed] [Google Scholar]
- Riedel MJ, Asadi A, Wang R, Ao Z, Warnock GL, Kieffer TJ. (2012). Immunohistochemical characterisation of cells co-producing insulin and glucagon in the developing human pancreas. Diabetologia 55:372-381. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Diaz R, Abdulreda MH, Formoso AL, Gans I, Ricordi C, Berggren PO, Caicedo A. (2011a). Innervation patterns of autonomic axons in the human endocrine pancreas. Cell Metab 14:45-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Diaz R, Dando R, Jacques-Silva MC, Fachado A, Molina J, Abdulreda MH, Ricordi C, Roper SD, Berggren PO, Caicedo A. (2011b). Alpha cells secrete acetylcholine as a non-neuronal paracrine signal priming beta cell function in humans. Nat Med 17:888-892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorsman P, Eliasson L, Kanno T, Zhang Q, Gopel S. (2011). Electrophysiology of pancreatic beta-cells in intact mouse islets of Langerhans. Prog Biophys Mol Biol 107:224-235. [DOI] [PubMed] [Google Scholar]
- Rorsman P, Ramracheya R, Rorsman NJ, Zhang Q. (2014). ATP-regulated potassium channels and voltage-gated calcium channels in pancreatic alpha and beta cells: similar functions but reciprocal effects on secretion. Diabetologia 57:1749-1761. [DOI] [PubMed] [Google Scholar]
- Rorsman P, Salehi SA, Abdulkader F, Braun M, MacDonald PE. (2008). K(ATP)-channels and glucose-regulated glucagon secretion. Trends Endocrinol Metab 19:277-284. [DOI] [PubMed] [Google Scholar]
- Rossi J, Santamaki P, Airaksinen MS, Herzig KH. (2005). Parasympathetic innervation and function of endocrine pancreas requires the glial cell line-derived factor family receptor alpha2 (GFRalpha2). Diabetes 54:1324-1330. [DOI] [PubMed] [Google Scholar]
- Saito K, Yaginuma N, Takahashi T. (1979). Differential volumetry of A, B and D cells in the pancreatic islets of diabetic and nondiabetic subjects. Tohoku J Exp Med 129:273-283. [DOI] [PubMed] [Google Scholar]
- Sakuraba H, Mizukami H, Yagihashi N, Wada R, Hanyu C, Yagihashi S. (2002). Reduced beta-cell mass and expression of oxidative stress-related DNA damage in the islet of Japanese Type II diabetic patients. Diabetologia 45:85-96. [DOI] [PubMed] [Google Scholar]
- Salehi A, Vieira E, Gylfe E. (2006). Paradoxical stimulation of glucagon secretion by high glucose concentrations. Diabetes 55:2318-2323. [DOI] [PubMed] [Google Scholar]
- Samols E, Bonner-Weir S, Weir GC. (1986). Intra-islet insulin-glucagon-somatostatin relationships. Clinical Endocrinology and Metabolism 15:33-58. [DOI] [PubMed] [Google Scholar]
- Schwartz TW. (1983). Pancreatic polypeptide: a hormone under vagal control. Gastroenterology 85:1411-1425. [PubMed] [Google Scholar]
- Singh V, Grotzinger C, Nowak KW, Zacharias S, Goncz E, Pless G, Sauer IM, Eichhorn I, Pfeiffer-Guglielmi B, Hamprecht B, Wiedenmann B, Plockinger U, Strowski MZ. (2007). Somatostatin receptor subtype-2-deficient mice with diet-induced obesity have hyperglycemia, nonfasting hyperglucagonemia, and decreased hepatic glycogen deposition. Endocrinology 148:3887-3899. [DOI] [PubMed] [Google Scholar]
- Spijker HS, Ravelli RB, Mommaas-Kienhuis AM, van Apeldoorn AA, Engelse MA, Zaldumbide A, Bonner-Weir S, Rabelink TJ, Hoeben RC, Clevers H, Mummery CL, Carlotti F, de Koning EJ. (2013). Conversion of mature human beta-cells into glucagon-producing alpha-cells. Diabetes 62:2471-2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefan Y, Grasso S, Perrelet A, Orci L. (1982a). The pancreatic polypeptide-rich lobe of the human pancreas: definitive identification of its derivation from the ventral pancreatic primordium. Diabetologia 23:141-142. [DOI] [PubMed] [Google Scholar]
- Stefan Y, Orci L, Malaisse-Lagae F, Perrelet A, Patel Y, Unger RH. (1982b). Quantitation of endocrine cell content in the pancreas of nondiabetic and diabetic humans. Diabetes 31:694-700. [DOI] [PubMed] [Google Scholar]
- Steiner DJ, Kim A, Miller K, Hara M. (2010). Pancreatic islet plasticity: interspecies comparison of islet architecture and composition. Islets 2:135-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundler F, Hakanson R, Larsson LI. (1977). Ontogeny of rat pancreatic polypeptide (PP) cells. Cell Tissue Res 178:303-306. [DOI] [PubMed] [Google Scholar]
- Sutherland EW, de Duve C. (1948). Origin and Distribution of the hyperglycemic-glycogenolytic factor of the pancreas. J Biol Chem 175:663-674. [PubMed] [Google Scholar]
- Taborsky GJ, Jr., Ahren B, Havel PJ. (1998). Autonomic mediation of glucagon secretion during hypoglycemia: implications for impaired alpha-cell responses in type 1 diabetes. Diabetes 47:995-1005. [DOI] [PubMed] [Google Scholar]
- Talchai C, Xuan S, Lin HV, Sussel L, Accili D. (2012). Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 150:1223-1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorel F, Nepote V, Avril I, Kohno K, Desgraz R, Chera S, Herrera PL. (2010). Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature 464:1149-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger RH. (1985). Glucagon physiology and pathophysiology in the light of new advances. Diabetologia 28:574-578. [DOI] [PubMed] [Google Scholar]
- Unger RH, Aguilar-Parada E, Muller WA, Eisentraut AM. (1970). Studies of pancreatic alpha cell function in normal and diabetic subjects. J Clin Invest 49:837-848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger RH, Orci L. (2010). Paracrinology of islets and the paracrinopathy of diabetes. Proc Natl Acad Sci U S A 107:16009-16012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger RH, Orci L. (1975). The essential role of glucagon in the pathogenesis of diabetes mellitus. Lancet 1:14-16. [DOI] [PubMed] [Google Scholar]
- Vieira E, Liu YJ, Gylfe E. (2004). Involvement of alpha1 and beta-adrenoceptors in adrenaline stimulation of the glucagon-secreting mouse alpha-cell. Naunyn Schmiedebergs Arch Pharmacol 369:179-183. [DOI] [PubMed] [Google Scholar]
- Vieira E, Salehi A, Gylfe E. (2007). Glucose inhibits glucagon secretion by a direct effect on mouse pancreatic alpha cells. Diabetologia 50:370-379. [DOI] [PubMed] [Google Scholar]
- Vuguin PM, Charron MJ. (2011). Novel insight into glucagon receptor action: lessons from knockout and transgenic mouse models. Diabetes Obes Metab 13 Suppl 1:144-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JN, Johnson PR, Shigeto M, Hughes SJ, Clark A, Rorsman P. (2011a). Glucose-responsive beta cells in islets isolated from a patient with long-standing type 1 diabetes mellitus. Diabetologia 54:200-202. [DOI] [PubMed] [Google Scholar]
- Walker JN, Ramracheya R, Zhang Q, Johnson PR, Braun M, Rorsman P. (2011b). Regulation of glucagon secretion by glucose: paracrine, intrinsic or both? Diabetes Obes Metab 13 Suppl 1:95-105. [DOI] [PubMed] [Google Scholar]
- Wang X, Zielinski MC, Misawa R, Wen P, Wang TY, Wang CZ, Witkowski P, Hara M. (2013). Quantitative analysis of pancreatic polypeptide cell distribution in the human pancreas. PLoS One 8:e55501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir GC, Samols E, Loo S, Patel YC, Gabbay KH. (1979). Somatostatin and pancreatic polypeptide secretion: effects of glucagon, insulin, and arginine. Diabetes 28:35-40. [PubMed] [Google Scholar]
- Yang YP, Thorel F, Boyer DF, Herrera PL, Wright CV. (2011). Context-specific alpha- to-beta-cell reprogramming by forced Pdx1 expression. Genes Dev 25:1680-1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon KH, Ko SH, Cho JH, Lee JM, Ahn YB, Song KH, Yoo SJ, Kang MI, Cha BY, Lee KW, Son HY, Kang SK, Kim HS, Lee IK, Bonner-Weir S. (2003). Selective beta-cell loss and alpha-cell expansion in patients with type 2 diabetes mellitus in Korea. J Clin Endocrinol Metab 88:2300-2308. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Bengtsson M, Partridge C, Salehi A, Braun M, Cox R, Eliasson L, Johnson PR, Renström E, Schneider T, Berggren PO, Göpel S, Ashcroft FM, Rorsman P. (2007). R-type Ca(2+)-channel-evoked CICR regulates glucose-induced somatostatin secretion. Nat Cell Biol 9:453-460. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Chibalina MV, Bengtsson M, Groschner LN, Ramracheya R, Rorsman NJ, Leiss V, Nassar MA, Welling A, Gribble FM, Reimann F, Hofmann F, Wood JN, Ashcroft FM, Rorsman P. (2014). Na+ current properties in islet alpha- and beta-cells reflect cell-specific Scn3a and Scn9a expression. J Physiol 592:4677-4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Ramracheya R, Lahmann C, Tarasov A, Bengtsson M, Braha O, Braun M, Brereton M, Collins S, Galvanovskis J, Gonzalez A, Groschner LN, Rorsman NJ, Salehi A, Travers ME, Walker JN, Gloyn AL, Gribble F, Johnson PR, Reimann F, Ashcroft FM, Rorsman P. (2013). Role of KATP channels in glucose-regulated glucagon secretion and impaired counterregulation in type 2 diabetes. Cell Metab 18:871-882. [DOI] [PMC free article] [PubMed] [Google Scholar]