

Abstract
We evaluated the hypothesis that lactate shuttling helps support the nutritive needs of injured brains. To that end, we utilized dual isotope tracer [6,6-2H2]glucose, that is, D2-glucose, and [3-13C]lactate techniques involving arm vein tracer infusion along with simultaneous cerebral (arterial [art] and jugular bulb [JB]) blood sampling. Traumatic brain injury (TBI) patients with nonpenetrating brain injuries (n=12) were entered into the study following consent of patients' legal representatives. Written and informed consent was obtained from control volunteers (n=6). Patients were studied 5.7±2.2 (mean±SD) days post-injury; during periods when arterial glucose concentration tended to be higher in TBI patients. As in previous investigations, the cerebral metabolic rate for glucose (CMRgluc, i.e., net glucose uptake) was significantly suppressed following TBI (p<0.001). However, lactate fractional extraction, an index of cerebral lactate uptake related to systemic lactate supply, approximated 11% in both healthy control subjects and TBI patients. Further, neither the CMR for lactate (CMRlac, i.e., net lactate release), nor the tracer-measured cerebral lactate uptake differed between healthy controls and TBI patients. The percentages of lactate tracer taken up and released as 13CO2 into the JB accounted for 92% and 91% for control and TBI conditions, respectively, suggesting that most cerebral lactate uptake was oxidized following TBI. Comparisons of isotopic enrichments of lactate oxidation from infused [3-13C]lactate tracer and 13C-glucose produced during hepatic and renal gluconeogenesis (GNG) showed that 75–80% of 13CO2 released into the JB was from lactate and that the remainder was from the oxidation of glucose secondarily labeled from lactate. Hence, either directly as lactate uptake, or indirectly via GNG, peripheral lactate production accounted for ∼70% of carbohydrate (direct lactate uptake+uptake of glucose from lactate) consumed by the injured brain. Undiminished cerebral lactate fractional extraction and uptake suggest that arterial lactate supplementation may be used to compensate for decreased CMRgluc following TBI.
Key words: : brain, fuel augmentation, glucose, lactate, mass spectrometry, stable isotopes, TBI
Introduction
Although it was once thought to be the consequence of an inadequate oxygen supply in contracting skeletal muscle, it is now known that lactate is continuously formed and utilized in diverse organs, including the brain. Following traumatic brain injury (TBI), lactate has long been viewed as a waste product of oxygen-limited glycolysis and ischemia. However, both as the product of one metabolic pathway (glycolysis), and as the substrate for a downstream pathway (mitochondrial respiration), lactate can be regarded as the link between glycolytic and oxidative pathways. Importantly, as described in the lactate shuttle hypothesis, this linkage with lactate as the vehicle of exchange can transcend compartment barriers and occur within and between cells, tissues, and organs.1–3
The use of lactate as a preferred fuel over glucose in brain preparations has been observed.4,5 An astrocyte-neuron lactate shuttle has been proposed,6,7 and neurons have a capability for glycolysis and intracellular lactate shuttling.8,9 When blood lactate concentration [lactate] rises in exercising humans, or when exogenous lactate is supplied in the vasculature, lactate is not only the major gluconeogenic precursor,10–12 but also substitutes for glucose as an energy substrate in working skeletal11,13 and cardiac muscle.14–16 Similarly, when blood lactate rises in exercising people, or in response to injury, cerebral lactate uptake increases in a concentration-dependent manner just as it does in working skeletal muscle.11,13 Also, the infusion of Na+-L-lactate has the effect of alkalinizing the blood.17
Results showing improved cognitive function in brain-injured rats given intravenous lactate therapy18 and rapid increases of lactate monocarboxylate transporter (MCT) protein expression in rat brains following TBI19 are a strong inducement for studying lactate metabolism in humans following TBI. Because of the potentially positive role for lactate in cerebral metabolism observed in humans under diverse conditions, we undertook a study of body–brain metabolic interaction in individuals following TBI. In our companion article, we reported massive mobilization of body resources, mainly lactate, to support hepatic and renal gluconeogenesis (GNG); our evidence supports that the body increases lactate production to indirectly supply glucose to the injured brain.24 In this work, by infusing isotopically labeled lactate, we showed how systemic lactate was directly consumed and used by the injured human brain. Further, as opposed to the case in which exogenous lactate infusion could substitute for glucose in working muscles11,13 and in the hearts of healthy men,14–16 our results supported the idea that exogenous lactate infusion could augment cerebral substrate supply when glycolysis was diminished, as it is following TBI.20
Methods
TBI patients
Patients were admitted to the study and medically treated as has been described previously.20 Briefly, eligible patients included all mechanically ventilated moderately or severely brain-injured patients, ≥16 years of age, who were admitted to UCLA Medical Center within 24 h of injury. Moderate or severe brain injury was defined as closed injury with a post-resuscitation Glasgow Coma Score (GCS)≤13 or deterioration to a GCS≤13 within 24 h of admission, and as an injury requiring mechanical ventilation and intracranial pressure (ICP) monitoring. Exclusion criteria included the following: 1) terminal illness (e.g., advanced cancer, AIDS); 2) severe neurological illness (advanced Parkinson's disease, multiple sclerosis, Alzheimer's dementia, disabling cerebrovascular event, mental retardation); and 3) acute complete spinal cord injury. UCLA and UCB institutional review boards approved this protocol, and informed consent was obtained from patients' legal representatives.
General management protocol
Patients were admitted to the intensive care unit (ICU) after initial stabilization or surgical evacuation of an intracranial hematoma, and were treated in accordance with the Brain Trauma Foundation and American Association of Neurological Surgeons (AANS)/Congress of Neurological Surgeons (CNS) TBI Guidelines.21 Management goals included maintenance of ICP<20 mm Hg and cerebral perfusion pressure (CPP)>70 mm Hg. All patients had arterial and JB catheters inserted as soon as possible following patient admission to allow determination of arteriovenous differences (AVD) for glucose, lactate, and oxygen.
For sampling of venous blood from the brain, the dominant jugular vein was selected based on the dominant jugular foramen visualized on admission CT scan. Using standard techniques, a 5 Fr Cordis and a 4 Fr Oxymetric catheter (Baxter Critical Care, Baxter Health Care) were inserted to a depth of ∼15 cm, or until resistance was encountered. Placement of the catheter was confirmed by lateral skull radiodgraphs. The catheter was calibrated in vivo, and repeat calibration was performed every 12 h. Display of light intensity and oxygen saturation was continuously displayed on the monitor.
As a part of patient care, arterial and venous samples and 133Xenon-cerebral blood flow (CBF) studies were scheduled every 24 h during post-injury days 0–5, 7, and 9. It was not possible for all scheduled studies to be completed on all patients because patients' clinical status and treatments took priority, including severely elevated ICP (ICP>30 mm Hg), hemodynamic and/or respiratory instability, or removal of the jugular catheter or extubation following clinical improvement.
For isotope tracer studies, stable, nonradioactive D2-glucose and [3-13C]lactate isotope tracers were infused (discussed subsequently), typically 72–96 h after admission to the ICU when informed consent had been obtained. Intravenous feeding was not used. All glucose-containing intravenous tube feeds were discontinued before isotope infusion. Enteral feeding (Osmolite 1.2 or 1.5; Abbott Laboratories, Abbot Park, IL) was continued during the isotope infusion. The rate of caloric delivery was at the discretion of the attending physician.
Healthy controls
Six healthy, nonsmoking, weight-stable volunteers (28.2±8.2 years) were recruited from the UCLA campus and the surrounding community by posted notices and Internet advertisements. Subjects were admitted into the study if they met the following criteria: 1) had been diet and weight stable for >6 months; 2) were not taking medications; 3) had normal lung function, defined as forced expiratory volume in 1 sec≥70%; and 4) were disease and injury free as determined by a health history questionnaire and physical examination. Control subjects received local anesthetics for catheter placements and experimental procedures. Details for this technique have been previously described.20 Briefly, through use of a femoral vein approach, a JB catheter was inserted under fluoroscopic guidance into proper position in the JB. A radial arterial line was also placed. Again, institutional ethical review boards approved the protocol and subjects provided written informed consent. Dietary records were not recorded for the control group.
Experimental protocol
Control subjects reported to the laboratory and were studied as described. Prior to tracer infusions, a blood sample was collected from the radial artery and JB catheter for baseline isotope enrichment measurements of glucose and lactate. Next, subjects received a primed continuous infusion of D2-glucose and [3-13C]lactate via an arm vein catheter while semisupine for 90 min. The priming bolus for glucose was equal to 125 times the resting glucose infusion rate. Subsequently, D2-glucose was infused at 2 mg/min. The priming bolus for lactate was equal to 23 times the resting lactate infusion rate of 2.5 mg/min.22 As previously, different tracer cocktail infusion rates were managed by means of two separate pumps connected to a common venous infusion port.23 Isotopes were obtained from Cambridge Isotope Laboratories (Tewksbury, MA), diluted in 0.9% sterile saline, and tested for sterility and pyrogenicity prior to use (UCLA Pharmaceutical Services).
Arterial and JB blood samples (1–2 mL) were collected simultaneously every 60 min for a period of 3 h following commencement of tracer infusion. Cannulas were flushed with an equivalent amount of 0.9% saline after each collection.
Processing and analysis of blood
Blood samples were immediately transferred to ice-chilled tubes containing 0.6 M of perchloric acid, shaken and stored on ice until the end of the experiment. Within 1 h of collection, perchloric acid extracts were centrifuged (10 min at 3000 rpm, 2000g, 4°C) and the supernatants transferred to separate tubes for storage at −20°C until further analysis.
Blood lactate was measured in neutralized perchloric extracts using an enzymatic method in addition to mass spectrometry as previously described.13,22,24 Blood lactate isotopic enrichment (IE) was determined using gas chromatography–mass spectrometry (GC-MS) of the N-propylamide heptafluorobutyrate derivative of lactate.25 Briefly, neutralized perchloric extracts were lyophilized, re-suspended in 200 μl of 2,2-dimethoxypropane and 20 μl 10% w/w HCl in methanol, capped and incubated at room temperature for 60 min. Next, 50 μl of N-propylamine was added and the samples were heated at 100°C for 30 min, dried under a stream of N2, re-suspended in ethyl acetate, and transferred to GC-MS vials. Thereafter, the samples were dried under N2, derivatized by adding 20 μl of heptafluorobutyric anhydride (5 min at room temperature), dried again under N2, and re-suspended in ethyl acetate for GC-MS analysis. Methane was used for chemical ionization with selected ion monitoring for mass to charge ratios 328 (unlabeled lactate) and 329 (13C-labeled lactate), respectively.
Blood glucose concentration and IE were determined by GC-MS of the penta-acetate derivative using an Agilent DB-17 GC column and [U-13C]glucose as internal standard. Glucose sample preparation was performed as described previously. Methane was used for chemical ionization and selective ion monitoring was performed for mass-to-charge ratios (M/Z) of 331, 333, and 337 for endogenous glucose, D2-glucose tracer, and [U-13C6]glucose internal standard, respectively. Selected ion abundances were compared against external standard curves for calculation of both concentration and IE, with normalization to internal standard signal for determination of concentration.
Arterial and JB 13CO2 contents were determined by isotope ratio mass spectrometry (IRMS) performed by Metabolic Solutions (Nashua, NH).
Calculations
The arteriovenous differences (AVD) and cerebral metabolic rates (CMR) of glucose and lactate were calculated as follows:
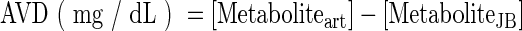 |
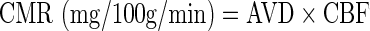 |
where [Metaboliteart] and [MetaboliteJB] are the concentrations of either glucose or lactate from arterial and JB venous sites, respectively, and CBF is the measured cerebral blood flow.20 Positive AVD and CMR reflect net cerebral uptake, whereas negative AVD and CMR indicate net cerebral release.
Tracer-measured cerebral fractional extraction (FExTM) and tracer-measured uptake (TMU) (mg/min) were calculated as follows:
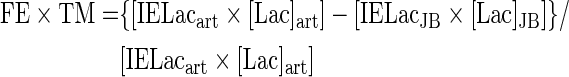 |
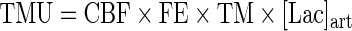 |
where [Lacart] and [LacJB] are arterial and JB lactate concentrations, respectively, and IELacart and IELacJB are the percent enrichment of lactate in arterial and JB blood, respectively. Quantities related to total lactate release include
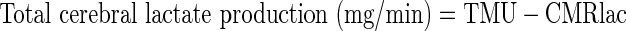 |
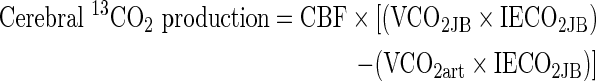 |
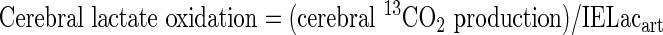 |
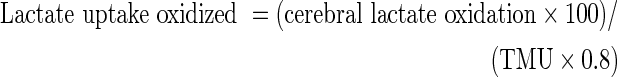 |
where VCO2art and VCO2JB are the volumes of distribution of carbon dioxide in arterial and JB blood, respectively. A correction factor for bicarbonate retention was included and assumed to be 0.8.26, 27
Whole body lactate and glucose rates of appearance (Ra), rates of disappearance (Rd), and metabolic clearance rates (MCR) were calculated using the modified equations of Steele28 for use with stable isotopes,29 and are described in our companion article.24
The percent of whole body lactate Ra that was accounted for by the brain was calculated as 100 × (total cerebral lactate release)/(lactate Ra).
Statistical analysis
Data descriptions and testing were conducted in R version 2.15.1 (R Development Core Team, http://www.R-project.org/; R Foundation for Statistical Computing, Vienna, Austria). Groups were compared both with the conventional t test and its robust analogue, the Yuen test,30 because of the presence of non-normality in the data distributions31; in the text that follows, the larger of the two p values is reported.
Results
Patient status and control subject descriptors
Demographic and anthropometric data on study participants are provided in Table 1 of our companion article.24 Briefly, all TBI patients were male, 35.8±18.8 years of age and weighed 77.6±11.1 kg. Mechanisms of injury for these 12 patients were four motor vehicle accidents, four falls, two motor cycle accidents, one pedestrian versus auto accident and one bicycle versus auto accident. Healthy controls were (5 males and 1 female) aged 28.2±8.2 years of age and weighed 66.2±9.2 kg. Metabolic parameters are summarized in Table 1. Notable significant differences included decreased CBF, AVDO2, and CMRO2 in TBI subjects compared with normal controls. Additionally, over the course of measurement, arterial glucose concentration [glucoseart] was constant in controls and TBI patients; the distributions of [glucoseart] measured were not significantly different between controls and TBI patients (Fig. 1A, Table 1). For both populations, JB glucose concentrations [glucoseJB] were significantly lower (p=0.001) than arterial values, reflecting cerebral net glucose uptake. Despite the tendency for higher arterial glucose in TBI Patients, CMRgluc was depressed compared with normal controls (p<0.001) (Fig. 2A, Table 1).
Table 1.
Metabolic Parameters for Traumatic Brain Injury (TBI) Patients and Healthy Controls (Mean±SE)
| Parameter | TBI patients | Healthy controls | p |
|---|---|---|---|
| CBF (mL/100g/min) | 32.7±10.0 | 43.7±6.4 | <0.02 |
| Sao2 (%) | 98.5±0.6 | 98.1±0.8 | 0.12 |
| SJBo2 (%) | 72.2±3.7 | 66.6±9.4 | 0.12 |
| AVD oxygen (mL/dL) | 3.71±0.62 | 5.60±1.80 | <0.01 |
| CMR oxygen (mL/100g/min) | 1.23±0.35 | 2.45±0.63 | <0.001 |
| Arterial [glucose] (mg/dL) | 113.5±32.6 | 107.6±11.7 | 0.5 |
| Jugular bulb [glucose] (mg/dL) | 104.7±31.8 | 96.2±10.6 | 0.25 |
| Arterial glucose IE | 0.010±0.003 | 0.012±0.002 | 0.3 |
| Jugular bulb glucose IE | 0.010±0.004 | 0.012±0.003 | 0.3 |
| CMR glucose (mg/100g/min) | 2.89±1.83 | 4.89±1.67 | <0.01 |
| FExTM gluc | 11.1±8.2 | 11.5±6.3 | 0.9 |
| Arterial [lactate] (mg/dL) | 8.44±2.9 | 10.3±3.6 | 0.4 |
| Jugular bulb [lactate] (mg/dL) | 8.74±2.2 | 10.59±3.4 | 0.4 |
| Arterial lactate IE | 0.03±0.012 | 0.05±0.013 | <0.02 |
| Jugular bulb lactate IE | 0.025±0.01 | 0.04±0.015 | <0.012 |
| FExTM lac | 9.72±10.6 | 9.7±9.1 | 0.9 |
| CMR lactate (mg/100g/min) | −0.156±.31 | −0.15±0.0.2 | 0.7 |
| TMU lactate (mg/100g/min) | 0.31±0.42 | 0.49±0.57 | 0.3 |
| Total cerebral lactate production (mg/100g/min) | 0.466±.357 | 0.64±0.58 | 0.25 |
| Total cerebral CHO uptake (CMRgluc+TMUlac) (mg/100g/min) |
3.20±2.14 | 5.38±1.37 | <0.001 |
CBF, cerebral blood flow; SJB ; AVD, arteriovenous differences; CMR, cerebral metabolic rates; IE, isotopic enrichment; FExTM, tracer-measured cerebral fractional extraction; TMU, tracer-measured uptake; CHO, total carbohydrate.
FIG. 1.
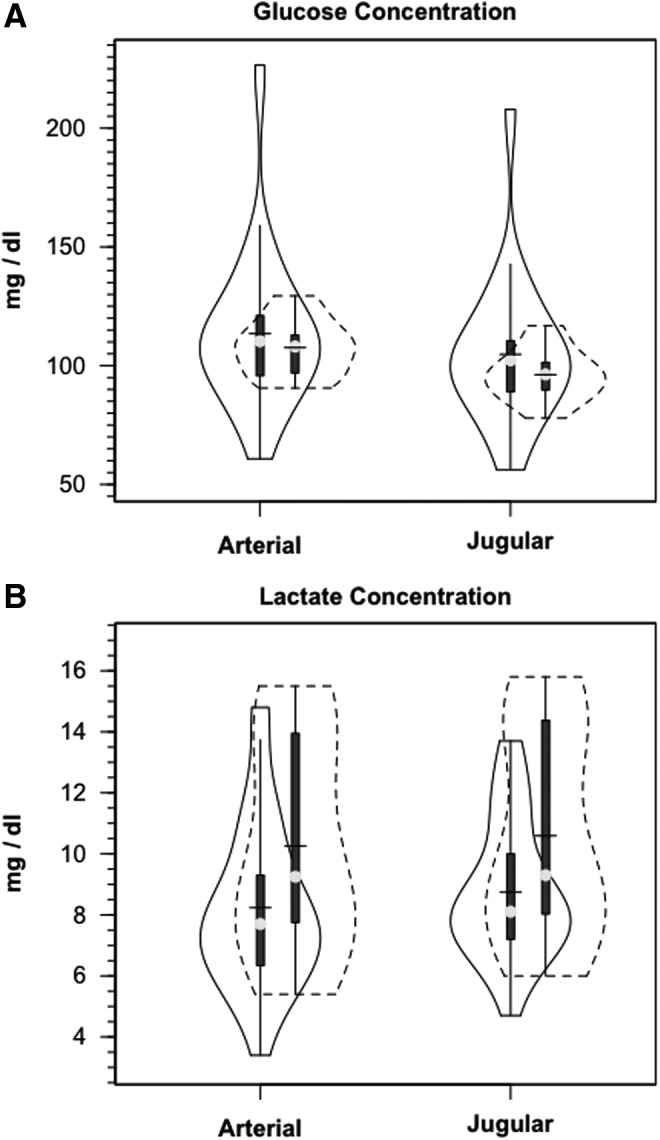
Violin plots of arterial and jugular bulb glucose (A) and lactate concentrations (B). Solid lines represent traumatic brain injury (TBI) patients (n=12) whereas dashed lines are normal control subjects (n=6). This and subsequent figures depict the following components: median (light circle), mean (solid line across), interquartile range (heavy vertical bar), box-plot whisker (thin vertical bar), and a kernel density estimation of the data distribution (replacing the box-plot's rectangular depiction) following Hintze and Nelson56 as visualized by R package “Caroline.” TBI patients are represented by the solid border, controls by dashed lines.
FIG. 2.
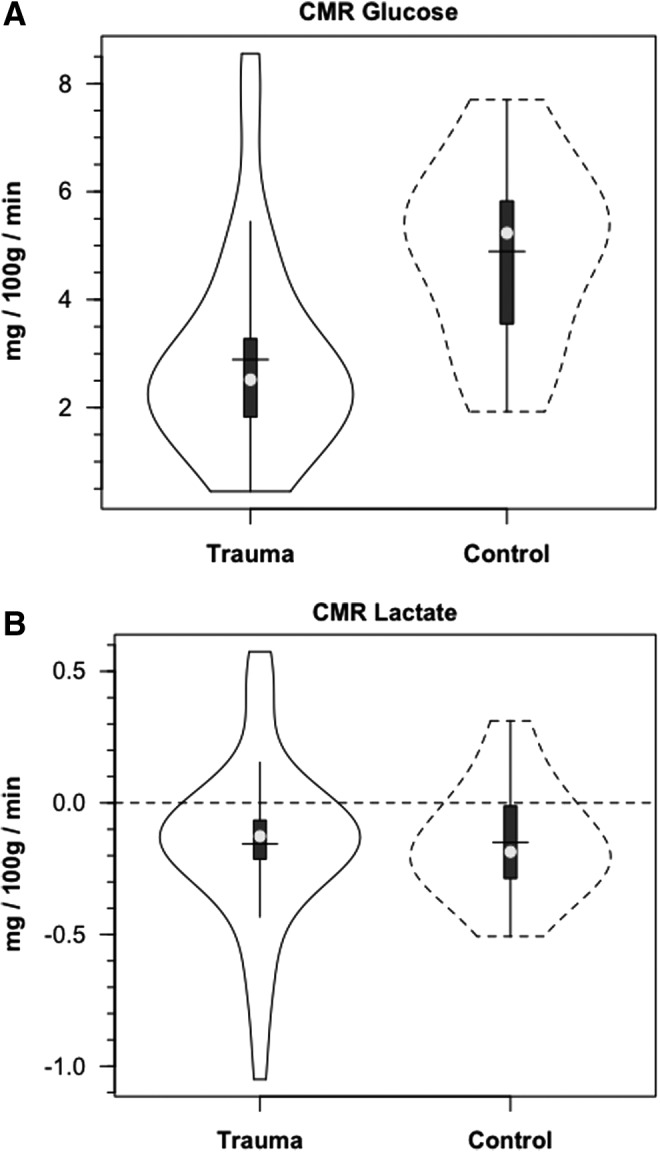
Violin plots of cerebral metabolic rate (CMRgluc, i.e., net uptake) for glucose (A) and lactate (B) (CMRlac i.e., net release). Solid lines represent traumatic brain injury (TBI) patients (n=12) whereas dashed lines are normal control subjects (n=6).
Within 30 min of the commencement of tracer infusion, D2-glucose isotopic enrichments were constant in jugular and arterial blood in control subjects and TBI patients (Fig. 3). Confirming the absence of cerebral glucose production for both populations, there were no significant differences in IEs of simultaneously sampled glucose in arterial and JB blood (Fig. 3). Tracer-measured fractional cerebral glucose extraction (FExTM) (mean≈11%), was not different between control subjects and TBI patients (Fig. 4A, Table 1). Further, TMUgluc did not differ significantly between healthy controls and TBI patients (Fig. 5A).
FIG. 3.

Violin plots of arterial and jugular bulb glucose isotopic enrichments. Glucose isotopic enrichments do not change as blood courses through the brain. Lower blood glucose isotopic enrichments following trauma indicate greater peripheral glucose turnover. Solid lines represent traumatic brain injury (TBI) patients (n=12) whereas dashed lines are normal control subjects (n=6).
FIG. 4.
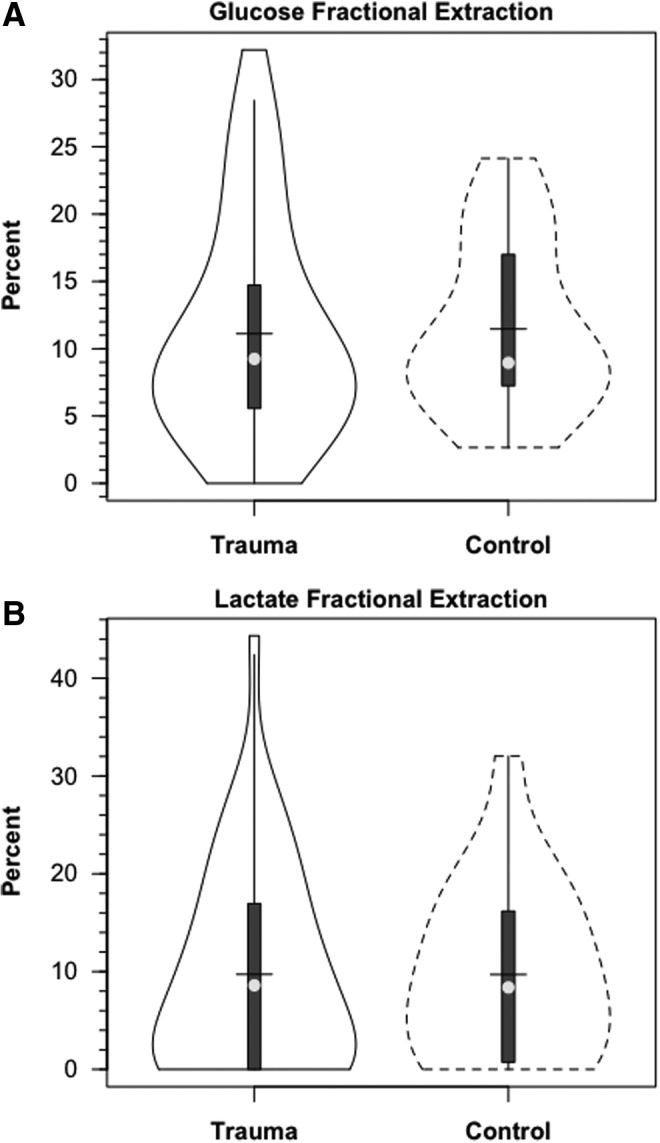
Violin plots of cerebral glucose (A) and lactate (B) fractional extraction in healthy control subjects and traumatic brain injury (TBI) patients. Solid lines represent TBI patients (n=12) whereas dashed lines are normal control subjects (n=6).
FIG. 5.
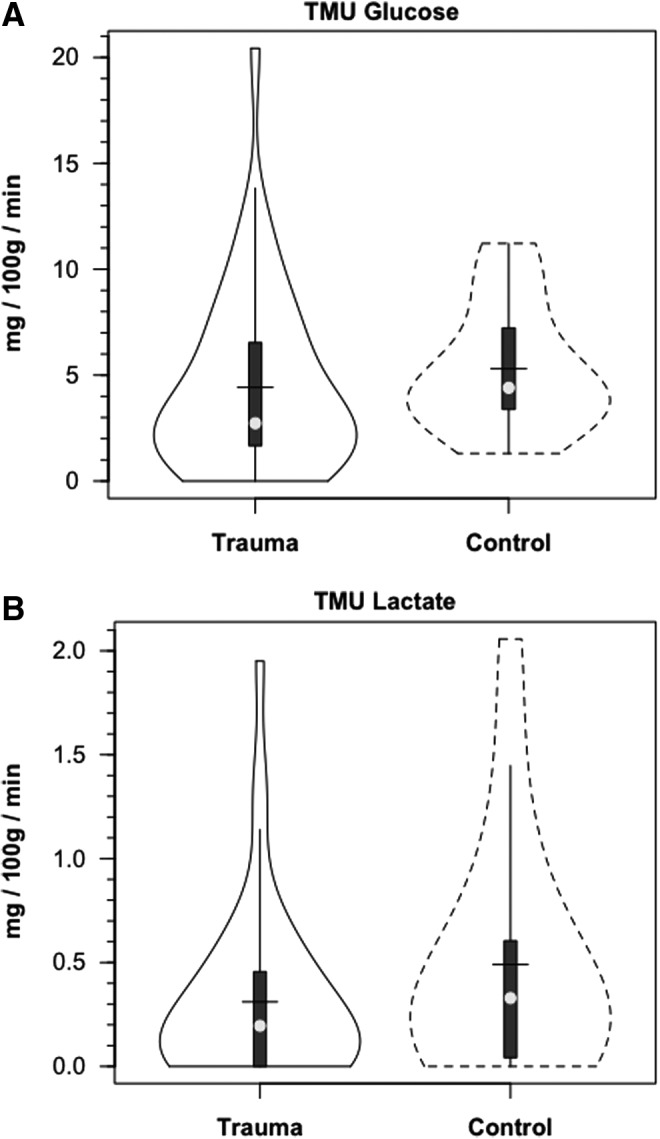
Violin plots of tracer-measured glucose (A) and lactate uptake (B). Solid lines represent traumatic brain injury (TBI) patients (n=12) whereas dashed lines are normal control subjects (n=6).
Arterial lactate [lactateart] was constant in controls, and constant but slightly depressed following TBI (Fig. 1B). The lower blood lactate levels following TBI reflect these patients' status as patients in the ICU 72–96 h post-injury after the initial post-injury stage of cerebral and corporal hyperglycolysis had passed. Based on previous experience, hyperglycolysis immediately after injury raises circulating [lactate].32 Nonetheless, higher lactate concentrations obtained from the JB compared with those obtained from the artery indicated cerebral lactate net release in both control subjects and TBI patients, with no difference between the two groups (Fig. 2B, Table 1). In contrast to CMRgluc, CMRlac was not different between control subjects and TBI patients.
Within 30 min of the commencement of tracer infusion, [3-13C]-lactate isotopic enrichments were constant in jugular and arterial blood in control subjects and TBI patients. For both patient and control populations, [3-13C]-lactate IEs were significantly lower (p<0.02) over the period of study in simultaneously sampled JB blood than in arterial blood, because of the presence of net cerebral lactate release (Fig. 2B) from significant cerebral lactate production. Importantly, although constant over time, arterial lactate IEs were significantly lower in TBI patients, indicating significantly increased systemic lactate production (Ra) and removal (Rd) rates following TBI (Table 1).24
In addition to the presence of net cerebral lactate release in both controls and TBI patients, active cerebral lactate metabolism was indicated by notable (∼ 10%) fractional extraction of lactate tracer (FExTM) from blood traversing the brain (Fig. 4B). Tracer-measured lactate uptake was similar in healthy controls and TBI patients (Fig. 5B). Knowing that cerebral lactate uptake occurred simultaneously with cerebral lactate release allowed calculation of the components of cerebral lactate turnover, which were the sum of uptake and release (Fig. 6). Inherent in calculation of cerebral lactate uptake are components of fractional extraction (Fig. 5) and arterial lactate delivery which, in turn, contains components of arterial [lactate] and CBF (Fig. 1B, Table 1). TMU, and hence total cerebral lactate production rates were not significantly different from control values following TBI (Fig. 6).
FIG. 6.
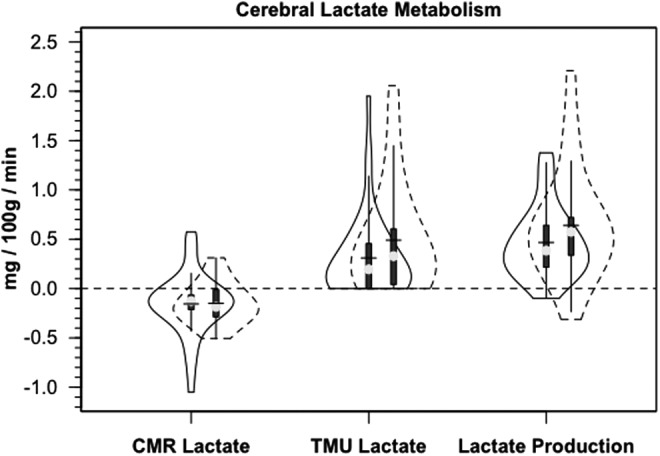
Violin plots of the components of cerebral lactate metabolism. Release (CMRlac), tracer-measured uptake and total cerebral lactate production=TMU-CMRlac. Solid lines represent traumatic brain injury (TBI) patients (n=12) whereas dashed lines are normal control subjects (n=6).
In addition to tracer extraction (TMU), another measure of carbohydrate metabolism is release of 13CO2 (Fig. 7). Cerebral release of 13CO2 did not reach an asymptote over a 3 h course of study. The delayed release of 13CO2 reflects retention in tissue H2CO3 - HCO3- pools.26, 27
FIG. 7.
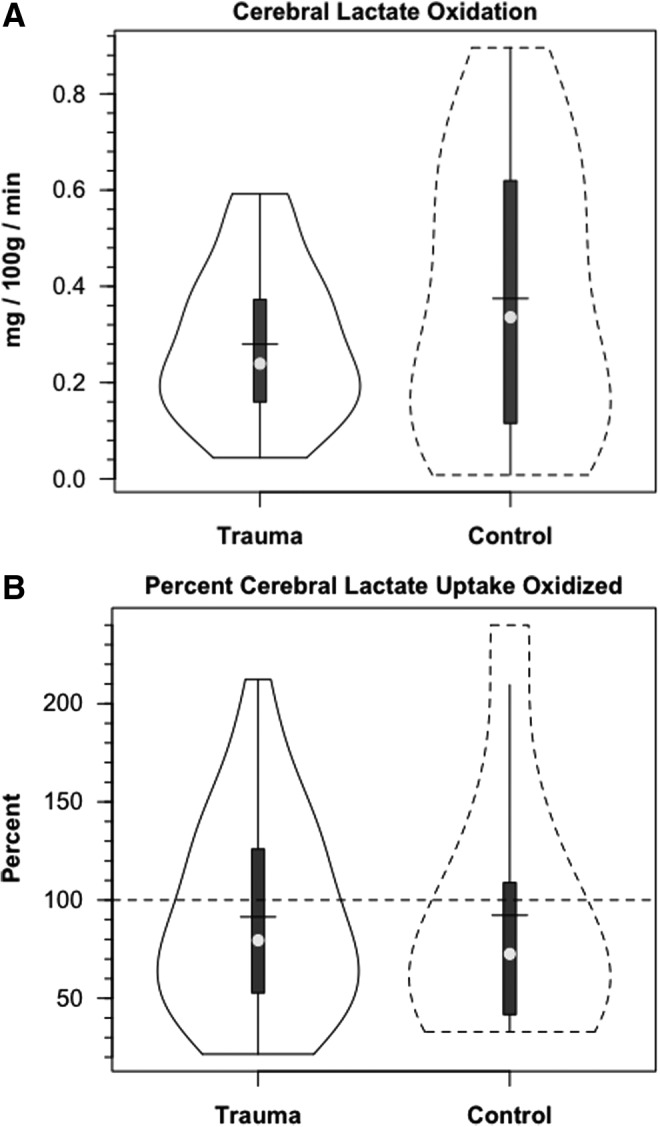
Violin plots of cerebral lactate oxidation in control and traumatic brain injury (TBI) patients. Values given in absolute (mg/100 g/min, panel A), and relative (% lactate uptake oxidized, panel B) terms. In absolute terms, cerebral lactate oxidation declined, but a greater percentage of uptake was oxidized following TBI. Solid lines represent TBI patients (n=12) whereas dashed lines are normal control subjects (n=6).
Discussion
Summary
The major findings of this study were that cerebral lactate uptake is not significantly diminished by TBI. Additionally, lactate that is taken up by the brain is oxidized to CO2; however, TBI did reduce overall cerebral carbohydrate oxidation; that is, the combined contributions of glucose and lactate to brain substrate supply (CMRCHO=TMUgluc+TMUlac). This work proves definitively that the injured human brain can utilize substrates other than glucose, and that non-glucose metabolism may convey an advantage to the recovering brain because glucose uptake and use is diminished following severe injury.20
Lactate and TBI
Lactate has been considered a dead-end waste product of disordered metabolism. This view has changed over the last 30 years, particularly with the introduction of the lactate shuttle concept.1–3 Although no longer assumed to simply be a waste product, cerebral lactate accumulation and lactic acidosis have been considered key secondary insults and have been suggested to be potential causes of delayed cell death after brain trauma and brain ischemia.33,34 Elevated cerebrospinal fluid (CSF) and JB venous lactate have been associated with a poor outcome following TBI.35 High lactate levels are regarded as an important marker of cerebral ischemia in arteriovenous measurements and in magnetic resonance spectroscopy (MRS), and lactate has become a key component of neuromonitoring for metabolic crisis through cerebral microdialysis.
The use of high levels of cerebral lactate as a biomarker for poor outcome are not inconsistent with results of the present investigation showing low [lactateJB], but high levels of whole-body and cerebral lactate flux. High cerebral lactate levels measured by MRS, microdialysis, or other methods may reflect metabolic stasis, and the absence of metabolic flux or high lactate levels may mean the recruitment of glycolysis to supply adenosine triphosphate (ATP) in the face of restricted mitochondrial respiration. Based on the present results, suffice it to say that use of a tracer is necessary to distinguish between lactate accumulation caused by metabolic stasis, and high production complicated by limited disposal.
Maintaining cerebral nutrient delivery is a major physiological priority, especially following brain injury. We now show that support for cerebral nutrient delivery is achieved by mechanisms that operate within the brain. Also, in our companion report, we describe the peripheral support response by which the body serves to fuel the brain. To review results of our companion article, systemic fueling of the brain following TBI is accomplished mainly via the mobilization of total body glycogen reserves and the production of lactate. Lactate is the major gluconeogenic precursor in healthy, post-absorptive individuals,10,12,36,37 and now we have demonstrated that the role of lactate as a gluconeogenic precursor is markedly elevated following TBI. By extension, in this report we describe that fractional lactate uptake is maintained following TBI. Thus, the present results offer insights not only into how the body supports the injured brain, but also into how cerebral lactate metabolism is intact and might be exogenously supported or augmented following TBI.
This article has combined traditional arteriovenous differences of cerebral metabolism with tracer-labeled substrate analyses. For example, and as shown by equivalence of glucose isotopic enrichments in arterial and JB blood (Fig. 3), and parity between CMRgluc and TMUgluc (Table 1), the brain takes up and utilizes but does not produce or release glucose as might result from GNG or the action of debranching enzyme of 1-6 glycosidic linkages. Hence, tracer-measured glucose uptake and CMRgluc are equal because cerebral glucose production is zero. However, the human brain, healthy or traumatized, both takes up and produces lactate. Hence, total cerebral lactate production (alternatively, lactate metabolism or turnover) involves components of uptake (TMUlac) and release (CMRlac).
As with other tissues studied, such as muscle,13,22,38,39 heart,15,16 and lungs,40,41 it is now clear that simultaneous lactate uptake and production are features of cerebral metabolism for both healthy and injured populations. With the technology utilized in this study of global cerebral metabolism, we cannot comment on cerebral compartmentalization of lactate uptake or production. Minimally, our data are consistent with the lactate shuttle concept: we now show that lactate produced in extracerebral tissues is delivered to the brain, where lactate serves as an oxidizable fuel energy source; alternatively, lactate in the systemic circulation may reach the liver for glucose production via GNG,24 with much of the resulting glucose subsequently taken up by the brain from the systemic circulation. And, also as shown here, in the brain, glucose produced from GNG undergoes glycolysis with substrate-level phosphorylation of adenosine diphosphate (ADP) for ATP production or substrate for mitochondrial oxidation. In short, lactate shuttling is a process that serves the brain in health and disease.
Lactate can be oxidized by the human brain
Infusion studies using the stable isotope 13C-labelled sodium-L-lactate in resting volunteers, TBI patients, and exercising humans have shown that lactate can be oxidized to form 13CO2.22,42–44 In resting volunteers, lactate accounted for approximately 9% of the total cerebral energy expenditure (Fig. 8A), with a similar contribution (∼ 10%) after TBI (Fig. 8B). In addition, regional studies have shown that 13C-labelled sodium-L-lactate delivered via microdialysis directly into brain parenchyma can act as an energy source by measuring 13C signals in glutamine.45 Other recent studies have shown indirectly that the brain may oxidize lactate following TBI.46,47
FIG. 8.

Histograms of the absolute and relative contributions to total cerebral carbohydrate (CHO) from lactate, glucose from gluconeogenesis (GNG), and glucose from hepatic glycogenolysis in healthy control subjects (left, panels A and C), and traumatic brain injury (TBI) patients (Right, panels B and D). Compared with panel A, panel B shows the decrease in cerebral metabolic rate for glucose (CMRgluc) following TBI, but also shows increased contributions of lactate and glucose from GNG to total cerebral CHO uptake after TBI. A comparison of panels C (control) and D (TBI) shows the large increase in percentage cerebral CHO uptake contributed by lactate, directly, or indirectly from GNG, following TBI.
In this investigation, glucose and lactate turnover rates are presented in units of mg/min (instead of mMol/min) to facilitate comparisons between the two substrates, the necessity of continuously considering the 2:1 molar ratio between lactate and glucose being obviated. On this basis, it is clear that whereas whole-body glucose turnover increases following TBI, the relative (>71%) increase in whole-body lactate turnover is far greater than is the rise in glucose turnover after TBI. In part, the increased systemic lactate production is disposed of via a quadrupling of the percent GNG following TBI.24 Following TBI, the increase in systemic lactate appearance is far greater than the elevation in glucose disposal; we interpret the results to mean that a carbon source, other than glucose, supports the large increase in lactate production following TBI. That source is presumed to be glycogen that not only exists in skeletal muscle, but also in other large organ systems, such as the integument and liver.2
Fractional lactate extraction (FExTM) is based on an accounting of tracer lactate in JB compared with arterial blood. As blood traverses the cerebral circulation, some isotopic dilution occurs as the result of cerebral lactate production and the resulting net release of lactate (i.e., CMRlac, Fig. 3B). However, what is not apparent in the standard CMRlac calculation is that TMU accounts for most total cerebral lactate production (Fig. 6). Rephrased, both injured and control brains simultaneously produced, consumed, and oxidized lactate. Importantly, because fractional lactate extraction was unaffected (Fig. 4B), tracer-measured lactate uptake was not significantly depressed following TBI (Fig. 5B). One might predict that in our studies the capacity for cerebral lactate uptake was underestimated. Arterial [lactate] was depressed (relative to acute post-injury times) in the TBI patients whom we studied 72–96 hours after injury, and, also, CBF was suppressed (Table 1). Our data predict that augmentations in either or both arterial [lactate] and CBF could increase cerebral lactate uptake, and thus provide nutritive support to the traumatized brain.
In our investigation, we compared 13C-TMU for lactate with the difference between JB and arterial 13CO2 contents. For control subjects, lactate oxidation approximated 0.4 mg/100 g/min, of which excretion as 13CO2 in JB approximated 92% of uptake (Fig. 7A and B, respectively). Following TBI, CMRO2 was decreased (Table 1), as was the 13C-TMU for lactate. For TBI patients, lactate oxidation approximated 0.3 mg/100 g/min (Fig. 7A), of which excretion as 13CO2 in jugular bulb approximated 91% after injury. Therefore, even though CMRO2 was suppressed following TBI (Table 1), cerebral lactate oxidation was nearly complete in both controls and TBI patients (Fig. 7A and B, respectively).
Interpretation of cerebral lactate oxidation data is complicated in this investigation by extreme efforts of the peripheral lactate producing and gluconeogenic organs that, in concert, act to provide nutritive support to the injured brain via lactate shuttling (described previously). Peripheral glycogenolysis raises systemic lactate production by nearly 100%, which provides a precursor supply sufficient to raise hepatic GNG from lactate by>400%. Hence, 13C exists in the arterial circulation as infused 13C-labeled lactate, and also as glucose, secondarily labeled carbon by GNG from lactate.24
In our experiments, two 13C-labeled metabolites circulated; one was the infused 13C-lactate, and the other was 13C-glucose, secondarily labeled from incorporation of 13C from infused tracer lactate. Because both label sources were taken up and oxidized by healthy and injured brains, it was necessary to distinguish their respective contributions to 13CO2 in jugular venous blood. We estimated the relative contributions of 13C-labeled-lactate and secondarily-labeled glucose carbon sources to cerebral CO2 production by comparing the 13C isotopic enrichments of arterial lactate (2.7%) and glucose (1.1%).24 In TBI patients, 69% of 13CO2 appearing in JB blood was from lactate oxidation and the remainder was from the oxidation of glucose secondarily labeled with 13C in GNG from lactate. In the control group, 90% of the 13CO2 in jugular bulb blood was from lactate oxidation, and the remainder was from the oxidation of glucose secondarily labeled. To reiterate, aggregate results such as those in Figure 9 illustrate the role of systemic lactate production in continuously supplying cerebral substrates, in particular under the stress of TBI. In healthy controls as well as TBI patients, both systemic lactate (from peripheral glycogenolysis) and glucose (from GNG of lactate) are taken up by the brain.
FIG. 9.

Violin plots of 13CO2 excretion in jugular bulb (JB) blood. Both infused [3-13C]lactate and glucose secondarily labeled with 13C via gluconeogenesis from lactate contributed to 13CO2 in JB cerebral drainage. The relative isotopic enrichments of precursor pools give the contributions of lactate and glucose to JB 13CO2 in traumatic brain injury (TBI) patients (dotted lines, top) and control subjects (solid lines, below). Solid lines represent TBI patients (n=12) whereas dashed lines are normal control subjects (n=6).
Exigencies to support cerebral functioning following severe injury require immediate and continuous attention by health care professionals. However, it remains that the body corpus and its major organ systems support cerebral functioning always, making it virtually impossible to describe the metabolic needs of the injured brain in the absence of considering responses of the body to support the injured brain.
Of all the responses observed in this and our companion report,24 several interactions are noteworthy. Among those are the relative contributions of hepatic glycogenolysis and GNG from lactate to support brain metabolism (Fig. 10). In terms of CMRgluc (=TMUgluc), the present results (Fig. 10A and C) are consistent with those of others obtained on healthy individuals48–50 in that the brain accounts for 20–25% of whole body glucose disposal. That range sets a firm basis on which to judge our result of 22% of glucose Rd taken up by the healthy brain. As shown in Fig. 10B, following TBI, CMRgluc decreases even though the contribution of glucose from lactate-derived GNG increases, providing 65% of the total net glucose uptake (Fig. 10F). In healthy controls, hepatic glycogenolysis provided most glucose taken up by the brain, with GNG from lactate contributing only 3.3% of net glucose uptake (Fig. 10C). Following TBI, several things happened to cerebral glucose use: the percent of whole body glucose disposal accounted for by the brain decreased because CMRgluc decreased whereas whole body glucose Ra rose, and the contribution of lactate to GNG quadrupled. In healthy controls, hepatic glycogenolysis accounted for 84% of glucose taken up and used by the brain (Fig. 10E), and GNG from lactate contributed the remainder, 16%. In contrast, following TBI, 64% of glucose for brain metabolism was provided by GNG from lactate (Fig. 10F), and hepatic glycogenolysis provided the remainder (36%). This change represented a reversal of the roles of hepatic GNG and glycogenolysis for supplying substrate to the injured brain (Fig. 10E and F).
FIG. 10.

Histograms of the absolute and relative contributions of gluconeogenesis (GNG) from lactate and hepatic glycogenolysis to cerebral glucose uptake in healthy control subjects (left, panels A, C, and E), and traumatic brain injury (TBI) patients (Right, panels B, D and F). Compared with panel A, panel B shows the decrease in cerebral metabolic rate for glucose (CMRgluc) following TBI, but an increase contribution of GNG to CMRgluc after TBI. Compared with panel C, panel D shows the percentage of whole body glucose disposal accounted for by brain metabolism following TBI. The relative decrease after TBI occurs because CMRgluc decreases whereas whole body glucose production (Ra) increases. Panel D also shows the increased role of GNG in supporting CMRgluc. Panels E and F show reversal of the relative roles of GNG from lactate and hepatic glycogenolysis in supporting CMRgluc. More than half (64%) of glucose taken up by the traumatized brain is derived from lactate via GNG.
Glucose and lactate supply carbohydrate energy sources to the brain, and the contributions of each to total body and cerebral energy production vary according to condition. As was developed (Fig. 10), the healthy brain consumes 22% of the whole body glucose disposal (Rd), whereas the brain accounts for only 3% of body lactate disposal in healthy individuals (not shown). However, in terms of total carbohydrate (CHO) uptake in healthy controls, the CMRgluc (4.9 mg/100 g/min from hepatic glycogenolysis+GNG) accounts for 91% of the total CHO (glucose+lactate) uptake because the TMUlac contributes 0.49 mg/100 g/min, or 9% of total CHO uptake (Fig. 8A). Therefore, in terms of the contributions of lactate, GNG from lactate provided 15% and direct lactate uptake contributed 9% of total cerebral CHO use in healthy controls. Alternatively stated, lactate contributed 24% of the CHO energy taken up by the healthy brain, either directly (TMU), or indirectly (the contribution of GNG from lactate to CMRgluc) (Fig. 8C). Glucose from hepatic glycogenolysis contributed the remainder.
Compared with what occurs in healthy controls, the relative role of lactate in fueling the injured brain increases following TBI. This is because TMUlac is not significantly affected by TBI (Fig. 5B), whereas CMRgluc is significantly reduced (Fig. 2A). Therefore, following TBI, the contribution of lactate approximated 10% of the total cerebral carbohydrate uptake, whereas CMRgluc (from hepatic glycogenolysis+GNG) remained at ∼ 90% of total cerebral CHO supply (Fig. 8B). And, within the contribution of glucose, GNG from lactate contributed 58% of cerebral CHO uptake following TBI (Fig. 8B). Following TBI, GNG from lactate accounted for 64% of CMRgluc, or 1.85 mg/100 g/min. Hence, 68% of the carbohydrate energy taken up and used by the injured brain came from lactate, either directly (from TMU), or indirectly (from GNG) (Fig. 8D). With these insights, by simply summing TMUgluc and TMUlac to estimate cerebral CHO uptake, the relative role of lactate is underestimated from controls, and is vastly underrepresented for TBI patients (Fig 8C and D).
Limitations
A limitation of our study imposed by concerns for informed consent was that observations were made several days after injury, when arterial lactate levels were presumably lower than during the immediate post-injury period of hyperglycolysis20,32,51 (Fig. 1B). Therefore, it may be that although cerebral lactate fractional extraction was preserved following TBI (Fig. 3B), perhaps because of increased MCT expression,19 the low glycogen reserves in muscle, skin, liver, and other tissues limited cerebral lactate delivery days after cerebral injury. Studies on the saturability of cerebral lactate uptake as a function of time following injury will be necessary, but for now, the presence of lactate fractional extraction supports the idea of supporting the nutritive requirements of the injured brain by supplying exogenous lactate.18
Another limitation of this study was that only small, tracer amounts of 13C-lactate were given to TBI patients and controls, there having been no attempt to determine the effect of raising arterial lactate and CMRlac by exogenous [lactate] infusion. Hence, we do not know at which blood [lactate] concentration saturation occurred. From our previous efforts, we know that several hours of sodium-lactate infusion at a rate sufficient to raise arterial [lactate] to ∼4 mM does not raise arterial blood glucose or produce untoward effects in healthy young men.10,11,13,23,43 Similarly, Bouzat et al.46 have shown that several hours of sodium-lactate infusion at a rate sufficient to raise arterial lactate to 4–6 mM, a rate sufficient to deliver a majority of the carbohydrate dietary needs without a significant change in systemic glucose, has the additional beneficial effects of lowering intracerebral pressure in patients following severe TBI. However, we do not know how long sodium-lactate infusion can be maintained before untoward effects on blood pH17 or renal function occur to brain-injured persons. Moreover, there is no reason to believe that sodium-lactate would represent the optimal formulation to deliver supplemental fuels and electrolytes to the injured brain; salts or esters of lactate and formulations containing one or more other monocarboxylates delivered continuously, or episodically, to achieve a target [lactate] early or late after injury, may ultimately prove more effective in promoting favorable patient outcomes than sodium-lactate infusion alone.
Conclusions
In summary, we corroborate previous results of depressed net glucose uptake (CMRgluc) by the injured human brain. However, our results obtained using glucose and lactate tracers unmask Herculean attempts of the body to support the energy needs of the brain flowing injury.24 Body glycogen reserves are mobilized, with the result being high rates of systemic lactate production. Although not directly measured in these experiments, we22,52 and others have previously observed that resting skeletal muscles are sites of net lactate release, and, consequently, are sources of circulating lactate. However, it is clear also that in resting, non-exercising individuals, diverse tissues such as the integument, contribute to the circulating lactate load, likely through action of the sympathetic arm of the autonomic nervous system.53–55 Although not apparent in either blood glucose or blood lactate concentration measurements, tracer-measured whole-body glucose and lactate turnover rates are very high following TBI, with lactate giving rise to most glucose production via GNG. Also, we now report that peripherally produced lactate is available as a cerebral energy supply always, including after TBI. Therefore, both directly via cerebral uptake, and indirectly as a gluconeogenic precursor, lactate from the corpus serves the brain's fuel energy needs following injury. Undiminished cerebral lactate fractional extraction and uptake are interpreted to mean that arterial lactate supplementation may be used to compensate for decreased CMRgluc following TBI.
Acknowledgments
Research was supported by the UCLA Brain Injury Research Center award PO1NS058489 from the National Institute of Neurological Disorders and Stroke (NINDS), and a gift from CytoSport, Inc. of Benicia, CA. The authors acknowledge the fine clinical and technical support provided by the staff of the Neurointensive Care Unit at the UCLA Medical Center.
Author Disclosure Statement
George A. Brooks had a financial interest in CytoSport, Inc. Otherwise, no competing financial interests exist.
References
- 1.Brooks G.A. (1984). Glycolytic End Product and Oxidative Substrate During Sustained Exercise in Mammals–The Lactate Shuttle. Comparative Physiology and Biochemistry – Current Topics and Trends, Volume A, Respiration – Metabolism – Circulation. Springer Verlag, Berlin [Google Scholar]
- 2.Brooks G.A. (2009). Cell-cell and intracellular lactate shuttles. J. Physiol. 587, 5591–5600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brooks G.A. (2002). Lactate shuttles in nature. Biochem. Soc. Trans. 30, 258–264 [DOI] [PubMed] [Google Scholar]
- 4.Schurr A. (2006). Lactate: the ultimate cerebral oxidative energy substrate? J. Cereb. Blood Flow Metab. 26, 142–152 [DOI] [PubMed] [Google Scholar]
- 5.Schurr A., and Payne R.S. (2007). Lactate, not pyruvate, is neuronal aerobic glycolysis end product: an in vitro electrophysiological study. Neuroscience 147, 613–619 [DOI] [PubMed] [Google Scholar]
- 6.Pellerin L., Pellegri G., Bittar P.G., Charnay Y., Bouras C., Martin J.L., Stella N., and Magistretti P.J. (1998). Evidence supporting the existence of an activity-dependent astrocyte-neuron lactate shuttle. Dev. Neurosci. 20, 291–299 [DOI] [PubMed] [Google Scholar]
- 7.Pellerin L., and Magistretti P.J. (2011). Sweet sixteen for ANLS. J. Cereb. Blood. Flow. Metab. 32, 1152–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hashimoto T., Hussien R., Cho H.S., Kaufer D., and Brooks G.A. (2008). Evidence for the mitochondrial lactate oxidation complex in rat neurons: demonstration of an essential component of brain lactate shuttles. PLoS One 3, e2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patel A.B., Lai J.C., Chowdhury G.M., Hyder F., Rothman D.L., Shulman R.G., and Behar K.L. (2014). Direct evidence for activity-dependent glucose phosphorylation in neurons with implications for the astrocyte-to-neuron lactate shuttle. Proc. Natl. Acad. Sci. U. S. A. 111, 5385–5390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emhoff C.A., Messonnier L.A., Horning M.A., Fattor J.A., Carlson T.J., and Brooks G.A. (2013). Gluconeogenesis and hepatic glycogenolysis during exercise at the lactate threshold. J. Appl. Physiol. 114, 297–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller B.F., Fattor J.A., Jacobs K.A., Horning M.A., Navazio F., Lindinger M.I., and Brooks G.A. (2002). Lactate and glucose interactions during rest and exercise in men: effect of exogenous lactate infusion. J. Physiol. 544, 963–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bergman B.C., Horning M.A., Casazza G.A., Wolfel E.E., Butterfield G.E., and Brooks G.A. (2000). Endurance training increases gluconeogenesis during rest and exercise in men. Am. J. Physiol. Endocrinol. Metab. 278, E244–251 [DOI] [PubMed] [Google Scholar]
- 13.Miller B.F., Fattor J.A., Jacobs K.A., Horning M.A., Suh S.H., Navazio F., and Brooks G.A. (2002). Metabolic and cardiorespiratory responses to “the lactate clamp”. Am. J. Physiol. Endocrinol. Metab. 283, E889–898 [DOI] [PubMed] [Google Scholar]
- 14.Gertz E.W., Wisneski J.A., Neese R., Bristow J.D., Searle G.L., and Hanlon J.T. (1981). Myocardial lactate metabolism: evidence of lactate release during net chemical extraction in man. Circulation 63, 1273–1279 [DOI] [PubMed] [Google Scholar]
- 15.Gertz E.W., Wisneski J.A., Stanley W.C., and Neese R.A. (1988). Myocardial substrate utilization during exercise in humans. Dual carbon-labeled carbohydrate isotope experiments. J. Clin. Invest. 82, 2017–2025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bergman B.C., Tsvetkova T., Lowes B., and Wolfel E.E. (2009). Myocardial glucose and lactate metabolism during rest and atrial pacing in humans. J. Physiol. 587, 2087–2099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller B.F., Lindinger M.I., Fattor J.A., Jacobs K.A., Leblanc P.J., Duong M., Heigenhauser G.J., and Brooks G.A. (2005). Hematological and acid-base changes in men during prolonged exercise with and without sodium-lactate infusion. J. Appl. Physiol. 98, 856–865 [DOI] [PubMed] [Google Scholar]
- 18.Holloway R., Zhou Z., Harvey H.B., Levasseur J.E., Rice A.C., Sun D., Hamm R.J., and Bullock M.R. (2007). Effect of lactate therapy upon cognitive deficits after traumatic brain injury in the rat. Acta Neurochir. (Wien) 149, 919–927 [DOI] [PubMed] [Google Scholar]
- 19.Prins M.L., and Giza C.C. (2006). Induction of monocarboxylate transporter 2 expression and ketone transport following traumatic brain injury in juvenile and adult rats. Dev. Neurosci. 28, 447–456 [DOI] [PubMed] [Google Scholar]
- 20.Glenn T.C., Kelly D.F., Boscardin W.J., McArthur D.L., Vespa P., Oertel M., Hovda D.A., Bergsneider M., Hillered L., and Martin N.A. (2003). Energy dysfunction as a predictor of outcome after moderate or severe head injury: indices of oxygen, glucose, and lactate metabolism. J. Cereb. Blood Flow Metab. 23, 1239–1250 [DOI] [PubMed] [Google Scholar]
- 21.Bratton S.L., Chestnut R.M., Ghajar J., McConnell Hammond F.F., Harris O.A., Hartl R., Manley G.T., Nemecek A., Newell D.W., Rosenthal G., Schouten J., Shutter L., Timmons S.D., Ullman J.S., Videtta W., Wilberger J.E., and Wright D.W. (2007). Guidelines for the management of severe traumatic brain injury. I. Blood pressure and oxygenation. J. Neurotrauma 24 Suppl. 1, S7–13 [DOI] [PubMed] [Google Scholar]
- 22.Bergman B.C., Wolfel E.E., Butterfield G.E., Lopaschuk G.D., Casazza G.A., Horning M.A., and Brooks G.A. (1999). Active muscle and whole body lactate kinetics after endurance training in men. J. Appl. Physiol. 87, 1684–1696 [DOI] [PubMed] [Google Scholar]
- 23.Messonnier A.L., Emhoff C.W., Fattor J.A., Horning M.A., T.J. C., and Brooks G.A. (2013). Lactate kinetics at the lactate threshold in trained and untrained men. J. Appl. Physiol. 114, 1593–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glenn T.C., Martin N.A., McArthur D.L., Hovda D., Vespa P.M.M., Horning M.A., Johnson M.L., and Brooks G.A. (2014). Endogenous nutritive support following traumatic brain injury: peripheral lactate production for glucose supply via gluconeogenesis. J Neurotrauma. [Epub ahead of print.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tserng K.Y., Gilfillan C.A., and Kalhan S.C. (1984). Determination of carbon-13 labeled lactate in blood by gas chromatography/mass spectrometry. Anal. Chem. 56, 517–523 [DOI] [PubMed] [Google Scholar]
- 26.Henderson G.C., Fattor J.A., Horning M.A., Faghihnia N., Luke–Zeitoun M., and Brooks G.A. (2006). Retention of intravenously infused [13C]bicarbonate is transiently increased during recovery from exercise of hard but not moderate intensity. J. Appl. Physiol. 103, 1604–1612 [DOI] [PubMed] [Google Scholar]
- 27.Trimmer J.K., Casazza G.A., Horning M.A., and Brooks G.A. (2001). Recovery of (13)CO2 during rest and exercise after [1-(13)C]acetate, [2-(13)C]acetate, and NaH(13)CO3 infusions. Am. J. Physiol. Endocrinol. Metab. 281, E683–692 [DOI] [PubMed] [Google Scholar]
- 28.Steele R. (1959). Influences of glucose loading and of injected insulin on hepatic glucose output. Ann. N. Y. Acad. Sci. 82, 420–430 [DOI] [PubMed] [Google Scholar]
- 29.Wolfe R.R. (1982). Radioactive and Stable Isotope Tracers in Biomedicine: Principles and Practice of Kinetic Analysis. Wiley–Liss: New York [Google Scholar]
- 30.Yuen K.F., Lee H., and and Tajuddin I. (1985). Some robust test statistics for the two-sample location problem. J. R. Stat. Soc. D 34, 175-182 [Google Scholar]
- 31.Wilcox R.R. (2012). Introduction to Robust Estimation and Hypothesis Testing, 3rd ed. Academic Press: Waltham, MA [Google Scholar]
- 32.De Salles A.A., Muizelaar J.P., and Young H.F. (1987). Hyperglycemia, cerebrospinal fluid lactic acidosis, and cerebral blood flow in severely head-injured patients. Neurosurgery 21, 45–50 [DOI] [PubMed] [Google Scholar]
- 33.Hakim A.M., and Shoubridge E.A. (1989). Cerebral acidosis in focal ischemia. Cerebrovasc. Brain Metab. Rev. 1, 115–132 [PubMed] [Google Scholar]
- 34.Siesjo B.K., Katsura K., and Kristian T. (1996). Acidosis-related damage. Adv. Neurol. 71, 209–233 [PubMed] [Google Scholar]
- 35.Robertson C.S., Narayan R.K., Gokaslan Z.L., Pahwa R., Grossman R.G., Caram P., Jr., and Allen E. (1989). Cerebral arteriovenous oxygen difference as an estimate of cerebral blood flow in comatose patients. J. Neurosurg. 70, 222–230 [DOI] [PubMed] [Google Scholar]
- 36.Jenssen T., Nurjhan N., Consoli A., and Gerich J.E. (1993). Dose-response effects of lactate infusions on gluconeogenesis from lactate in normal man. Eur. J. Clin. Invest. 23, 448–454 [DOI] [PubMed] [Google Scholar]
- 37.Meyer C., Dostou J.M., Welle S.L., and Gerich J.E. (2002). Role of human liver, kidney, and skeletal muscle in postprandial glucose homeostasis. Am. J. Physiol. Endocrinol. Metab 282, E419–427 [DOI] [PubMed] [Google Scholar]
- 38.Stanley W.C., Gertz E.W., Wisneski J.A., Neese R.A., Morris D.L., and Brooks G.A. (1986). Lactate extraction during net lactate release in legs of humans during exercise. J. Appl. Physiol. 60, 1116–1120 [DOI] [PubMed] [Google Scholar]
- 39.Brooks G.A., Butterfield G.E., Wolfe R.R., Groves B.M., Mazzeo R.S., Sutton J.R., Wolfel E.E., and Reeves J.T. (1991). Decreased reliance on lactate during exercise after acclimatization to 4,300 m. J. Appl. Physiol. 71, 333–341 [DOI] [PubMed] [Google Scholar]
- 40.Johnson M.L., Emhoff C.A., Horning M.A., and Brooks G.A. (2012). Transpulmonary lactate shuttle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 302, R143–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson M.L., Hussien R., Horning M.A., and Brooks G.A. (2011). Transpulmonary pyruvate kinetics. Am. J. Physiol. Regul. Integr. Comp. Physiol. 301, R769–774 [DOI] [PubMed] [Google Scholar]
- 42.Mazzeo R.S., Brooks G.A., Schoeller D.A., and Budinger T.F. (1986). Disposal of blood [1-13C]lactate in humans during rest and exercise. J. Appl. Physiol. 60, 232–241 [DOI] [PubMed] [Google Scholar]
- 43.Emhoff C.A., Messonnier L.A., Horning M.A., Fattor J.A., Carlson T.J., and Brooks G.A. (2013). Direct and indirect lactate oxidation in trained and untrained men. J. Appl. Physiol. 115, 829–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Hall G., Stromstad M., Rasmussen P., Jans O., Zaar M., Gam C., Quistorff B., Secher N.H., and Nielsen H.B. (2009). Blood lactate is an important energy source for the human brain. J. Cereb. Blood Flow Metab. 29, 1121–1129 [DOI] [PubMed] [Google Scholar]
- 45.Gallagher C.N., Carpenter K.L., Grice P., Howe D.J., Mason A., Timofeev I., Menon D.K., Kirkpatrick P.J., Pickard J.D., Sutherland G.R., and Hutchinson P.J. (2009). The human brain utilizes lactate via the tricarboxylic acid cycle: a 13C-labelled microdialysis and high-resolution nuclear magnetic resonance study. Brain 132, 2839–2849 [DOI] [PubMed] [Google Scholar]
- 46.Bouzat P., Sala N., Suys T., Zerlauth J.B., Marques–Vidal P., Feihl F., Bloch J., Messerer M., Levivier M., Meuli R., Magistretti P.J., and Oddo M. (2014). Cerebral metabolic effects of exogenous lactate supplementation on the injured human brain. Intensive Care Med. 40, 412–421 [DOI] [PubMed] [Google Scholar]
- 47.Jalloh I., Helmy A., Shannon R.J., Gallagher C.N., Menon D.K., Carpenter K.L., and Hutchinson P.J. (2013). Lactate uptake by the injured human brain: evidence from an arteriovenous gradient and cerebral microdialysis study. J. Neurotrauma 30, 2031–2037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sokoloff L. (1977). Relation between physiological function and energy metabolism in the central nervous system. J. Neurochem. 29, 13–26 [DOI] [PubMed] [Google Scholar]
- 49.Cahill G.J., Jr., Owen O.E., and Morgan A.P. (1968). The consumption of fuels during prolonged starvation. Adv. Enzyme Regul. 6, 143–150 [DOI] [PubMed] [Google Scholar]
- 50.Institute of Medicine (2005). Dietary Reference Intakes: Energy, Carbohydrate, Fiber, Fat, Fatty Acids, Cholesterol, Protein, and Amino Acids. The National Academies Press: Washington, DC [Google Scholar]
- 51.Bergsneider M., Hovda D.A., Shalmon E., Kelly D.F., Vespa P.M., Martin N.A., Phelps M.E., McArthur D.L., Caron M.J., Kraus J.F., and Becker D.P. (1997). Cerebral hyperglycolysis following severe traumatic brain injury in humans: a positron emission tomography study. J. Neurosurg. 86, 241–251 [DOI] [PubMed] [Google Scholar]
- 52.Brooks G.A., Wolfel E.E., Groves B.M., Bender P.R., Butterfield G.E., Cymerman A., Mazzeo R.S., Sutton J.R., Wolfe R.R., and Reeves J.T. (1992). Muscle accounts for glucose disposal but not blood lactate appearance during exercise after acclimatization to 4,300 m. J. Appl. Physiol. 72, 2435–2445 [DOI] [PubMed] [Google Scholar]
- 53.Ahlborg G., and Felig P. (1982). Lactate and glucose exchange across the forearm, legs, and splanchnic bed during and after prolonged leg exercise. J. Clin. Invest. 69, 45–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mazzeo R.S., Brooks G.A., Butterfield G.E., Podolin D.A., Wolfel E.E., and Reeves J.T. (1995). Acclimatization to high altitude increase muscle sympathetic activity both at rest and during exercise. Am. J. Physiol. 269, R201–207 [DOI] [PubMed] [Google Scholar]
- 55.Brooks G.A., Wolfel E.E., Butterfield G.E., Cymerman A., Roberts A.C., Mazzeo R.S., and Reeves J.T. (1998). Poor relationship between arterial [lactate] and leg net release during exercise at 4,300 m altitude. Am. J. Physiol. 275, R1192–1201 [DOI] [PubMed] [Google Scholar]
- 56.Hintze J.L., and Nelson R.D. (1998). Violin plots: A box plot-density trace synergism. Amer. Stat. 52, 181–184 [Google Scholar]