

Abstract
Annexin A2 is a multicompartmental protein that orchestrates a spectrum of dynamic membrane-related events. At cell surfaces, A2 forms the (A2•S100A10)2 complex which accelerates tissue plasminogen activator−dependent activation of the fibrinolytic protease, plasmin. Anti-A2 antibodies are associated with clinical thrombosis in antiphospholipid syndrome, whereas overexpression of A2 promotes hyperfibrinolytic bleeding in acute promyelocytic leukemia. A2 is upregulated in hypoxic tissues, and mice deficient in A2 are resistant to hypoxia-related retinal neovascularization in a model of diabetic retinopathy. Within the cell, A2 regulates membrane fusion processes involved in the secretion of pre-packaged, ultra-large molecules. In stimulated dendritic cells, A2 maintains lysosomal membrane integrity, thereby modulating inflammasome activation and cytokine secretion. Together, these findings suggest an emerging, multifaceted role for annexin A2 in human health and disease. The author's work has been inspired by numerous colleagues and mentors, and by the author's grandfather, and former ACCA member, Dr. J. Burns Amberson.
INTRODUCTION
The annexins constitute a family of more than 60 highly conserved, Ca2+-regulated, phospholipid-binding proteins that have existed for more than 500 million years (1). Humans express 12 annexins (annexins A1-A11 and A13), and, among these, annexin A2 (A2) is arguably the most extensively investigated with respect to health and disease (2,3). Typical annexins consist of a 30- to 35-kilodalton “core” domain containing four alpha helical, Ca2+-binding “annexin” repeats, and a more hydrophilic amino-terminal “tail” domain, which is essentially unique to each family member. Through their capacity for Ca2+-dependent membrane binding, annexins add or “annex” proteins to membrane surfaces and also facilitate membrane fusion events. These properties allow the annexins to fulfill a wide variety of intra- and extracellular functions, and the term “annexinopathy” has come to reflect their newly recognized roles in human pathophysiology (4).
ANNEXIN A2 AND ITS PARTNER PROTEINS
In the last 20 years, it has become increasingly clear that the cell surface is a major site for protease assembly and activity (5,6). In the 1980s, however, the concept that human endothelial cells could assemble components of the fibrinolytic system was novel. Our research began with the observation that human endothelial cells reacted specifically with antibodies directed against plasminogen and its tissue activator (tissue plasminogen activator, tPA) (7,8). Ligand blotting of a plasma membrane fraction isolated from human endothelial cell homogenates revealed that both plasminogen and tPA interacted specifically with a 36-kilodalton protein expressed on the cell surface (9). The purified protein bound both plasminogen and tPA in a dose-dependent and high-affinity manner, and amino acid sequencing identified this cell surface protein as annexin A2 (10). We now know that A2 is synthesized by endothelial cells, monocytes, macrophages, dendritic cells, trophoblast cells, epithelial cells, and some tumor cells, and can exist either as a soluble monomer in the cytoplasm, or as a complex associated with cellular membranes (11,12).
The S100 family consists of low molecular weight (9- to 14-kilodalton) dimeric proteins that undergo structural shifts in response to changes in Ca++ concentration, and often interact with annexins (13). By forming a heterotetrameric complex with protein S100A10, A2 increases its sensitivity to Ca++ and its ability to bind to cellular membranes at resting intracellular Ca++ concentrations (14). S100A10 is unique among the family of S100 proteins in that it exists in a permanent “calcium-on” state, and does not require a Ca++-induced conformational change to associate with annexin A2 (15). Crystallographic studies have revealed that, in the tetrameric (A2•S100A10)2 complex, two copies of S100A10 are linked non-covalently to create a molecular groove, which is occupied by the α-helical N-terminal 14 amino acids of A2. In endothelial cells, S100A10 is stabilized by this interaction with A2, which masks a polyubiquitination site that would otherwise destine unpartnered S100A10 for degradation within the proteasome (16). Three additional family members, S100A4, S100A6, and S100A11, have been reported to bind A2 in vitro, but the potential physiologic consequences of these interactions are unknown (17).
THE ANNEXIN A2 COMPLEX ON THE CELL SURFACE
It is now well-established that the (A2•S100A10)2 tetramer serves as an assembly site for two fibrinolytic proteins, plasminogen and tPA, on the endothelial cell surface (2,3) (Figure 1). This assembly promotes plasmin generation (18–21). Upon hydrolysis of its R560-V561 peptide bond by tPA, the zymogen plasminogen is transformed into the principal fibrinolytic protease, plasmin (22–24). The catalytic efficiency of tPA-dependent plasminogen activation increases by 10- to 100-fold in the presence of A2; although significant, this increment is less dramatic than the 500-fold acceleration provided in the presence of fibrin (22). It is hypothesized that, whereas classical fibrinolysis serves to dissolve established intravascular thrombi, the A2-based system provides constitutive surveillance that allows for clearance of fibrin forming on the blood vessel surface in response to more subtle forms of vascular injury.
Fig. 1.
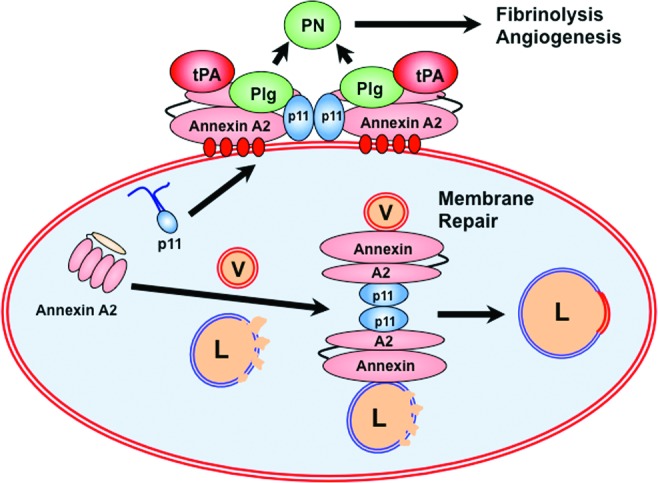
Some biologic functions of annexin A2. At the cell surface, annexin A2 binds to protein S100A10 (p11). The heterotetrameric complex associates with the plasma membrane through calcium linkages (shown in red) with membrane phospholipid, and supports the assembly of plasminogen (Plg) and tissue plasminogen activator (tPA), leading to the efficient generation of the fibrinolytic serine protease, plasmin (PN). Plasmin enables fibrinolysis and angiogenesis. Within the cell, the (annexin A2-p11)2 complex participates in repair of injured lysosomal membranes (L) by “annexing” membrane from cytoplasmic vesicles (V).
Expression of the (A2•S100A10)2 complex on the endothelial cell surface is a dynamic process. Translocation of the complex from the cytoplasm to the outer leaflet of the endothelial cell membrane appears to be a key regulatory step in vascular fibrinolysis, but the precise translocation mechanism is unknown (25,26). Increased cell surface expression of the heterotetramer occurs within minutes to hours of heat stress, receptor-mediated thrombin stimulation, or hypoxia (27–29), and requires both src kinase-mediated phosphorylation of Y23 and expression of S100A10 (28). In addition, the level of expression of the complex on the cell surface is regulated by intracellular protein kinase C, which phosphorylates S11 or S25 residues within the N-terminal tail domain of A2, thereby dissociating the (A2•S100A10)2 complex, and preventing further translocation to the cell surface (30,31). Serine phosphorylation of A2 by protein kinase C (PKC) appears to be triggered by cell surface plasmin, which cleaves A2 and activates toll-like receptor 4. This negative feedback mechanism may allow plasmin to limit its own activation.
THE ANNEXIN A2 COMPLEX IN HEMOSTASIS
Several animal studies support the hypothesis that annexin A2 regulates hemostasis in vivo. First, Anxa2−/− mice, while displaying uncompromised development, fertility, and lifespan, accumulate fibrin in both intra- and extravascular locations within the lungs, spleen, small intestine, liver, and kidney (32) Second, experimental injury to the carotid artery leads to a significant increase in thrombotic occlusion in Anxa2−/− versus Anxa2+/+ mice (32), and S100a10−/− mice also display increased vascular fibrin and reduced clearance of thrombi (33). Third, Anxa2−/− microvascular endothelial cells fail to support tPA-dependent plasmin generation (32). Fourth, in mice with diet-induced hyperhomocysteinemia, homocysteine derivatizes A2 and blocks its ability to bind tPA and generate plasmin, leading to fibrin accumulation and deficits in angiogenic potential (34,35). Fifth, A2 alone or in combination with tPA enhances vascular patency and reduces infarct size in several rodent models of stroke (36–40).
These results are reflected in recent observations in humans. In antiphospholipid syndrome and in a cohort of patients with cerebral venous thrombosis, high-titer anti-A2 autoantibodies are prevalent and correlate with major thrombosis (41–45), suggesting that cell surface A2 represents a prominent auto-antibody target associated with a thrombosis (46). In children with sickle cell disease, in addition, single nucleotide polymorphisms in the ANXA2 gene are associated with increased risk of stroke (47,48), whereas additional ANXA2 SNPs have been associated with elevated risk of avascular necrosis of bone (osteonecrosis) (49).
In acute promyelocytic leukemia, which, conversely, is associated with life-threatening hemorrhage at the time of presentation (50), there is typically robust expression of A2 in leukemic blast cells (51). The resulting coagulopathy appears to reflect a combination of disseminated intravascular coagulation and hyperfibrinolysis, the latter evidenced by elevated fibrin degradation products, depletion of plasma fibrinogen, and consumption of alpha2-antiplasmin (51). In cultured acute promyelocytic leukemia−like cells, elevated steady state levels of A2 mRNA returned to normal after treatment with the therapeutic differentiating agent all-trans retinoic acid (51). Follow-up studies have confirmed these findings and shown that S100A10 is also elevated in these cells (52,53).
ANNEXIN A2 IN PROLIFERATIVE RETINAL ANGIOPATHY
In several models of stimulated postnatal angiogenesis, Anxa2−/− mice have shown a diminished ability to form new blood vessels (32). In addition, wild-type mice with diet-induced hyperhomocysteinemia display impaired corneal neoangiogenesis due to covalent modification of annexin A2 by homocysteine; in this case, angiogenesis can be restored to normal with intravenous infusion of recombinant A2 (34). In the oxygen-induced retinopathy model, which mimics many aspects of human proliferative diabetic retinopathy (54), the typical vascular proliferative response is blunted by approximately 50% in Anxa2−/− mice (29). The data suggest that angiogenic impairment in the Anxa2−/− mouse may reflect reduced vascular fibrinolysis and fibrin accumulation around blood vessels.
INTRACELLULAR ANNEXIN A2
As a multicompartmental protein, annexin A2 is poised to fulfill a range of intracellular membrane-related functions, including organization of specialized membrane microdomains, recruitment of peripheral membrane proteins, and regulation of membrane fusion and repair events (11). Whereas heterotetrameric (A2•S100A10)2 resides on the plasma membrane, monomeric annexin A2 is distributed throughout the cytoplasm, but may transition to intracellular membranes in response to signals, such as changes in Ca2+ concentration, pH, or membrane phospholipid composition, and the availability of ancillary S100 proteins, such as S100A10. How these multiple activities may relate to human health and disease, however, is not yet clear.
Annexin A2 mediates a number of intracellular vesicular remodeling events. Within its N-terminal domain, annexin A2 possesses a single isoleucine-leucine pair motif (amino acids 6 and 7) that may function as an endosomal targeting sequence (55), thus allowing A2 to bind to endosomes and possibly mediate their fusion (56,57). In addition, A2 is required for the biogenesis of multivesicular bodies, and is also a constituent of exosomes that is frequently cited in proteomic studies (58,59). Through interactions with soluble NSF (N-ethylmaleimide-sensitive factor) attachment receptor (SNARE) proteins, A2 participates in the regulated exocytosis of chromaffin granules (60,61), von Willebrand factor−containing Weibel-Palade bodies (62,63), lamellar body−containing surfactant (64,65), and collagen VI multimers (66) from chromaffin, endothelial, type II alveolar, and bronchial epithelial cells, respectively.
Through its ability to associate with lysosomal membranes (Figure 1), A2 dysfunction is implicated in the inflammatory response associated with aseptic arthritis, which occurs in 10% to 15% of patients undergoing the several million joint replacement procedures performed each year in the United States (67). In aseptic arthritis, wear debris particles are shed into the joint space upon articulation of prosthetic joint surfaces. These particles are endocytosed by inflammatory macrophages and dendritic cells, and can induce lysosomal and endosomal membrane damage, which is normally associated with recruitment of cytoplasmic annexin A2 to the lyso-endosomal membrane. In A2-deficient cells, lysosomal injury leads to leakage of lysosomal cathepsins into the cytosol. Through an as yet unknown mechanism, cytosolic cathepsins activate the nucleotide-binding, leucine-rich, pyrin-containing-3 (NLRP3) inflammasome, leading to secretion of interleukin-1 and accelerated inflammation (68).
SUMMARY
The annexin A2 system serves a growing spectrum of biologic functions both atop and beneath the plasma membrane. At the cell surface, the (A2•S100A10)2 heterotetrameric system localizes plasmin activity and promotes fibrinolysis, angiogenesis, and cell migration. Within intracellular compartments, A2 appears to facilitate membrane organization, fusion, and repair in an array of activities including endocytosis, exocytosis, and lysosomal membrane repair. The physiologic consequences of these activities are under active investigation, and the next several years of annexin A2 research should offer exciting insights into human health and disease.
TRIBUTE
In the work described herein, I have been inspired by numerous colleagues and mentors, including James Burns Amberson, Jr., MD (Figure 2). He was born in 1890 in Waynesboro, Pennsylvania, attended Lafayette College, and graduated from the Johns Hopkins University School of Medicine in 1917. In 1918, while working with Dr. E. W. Goodpasture in the pathology lab at Hopkins, Dr. Amberson experienced an episode of hemoptysis, and the diagnosis of pulmonary tuberculosis was made. He was admitted to Loomis Sanatorium in upstate New York, where, after a year of recuperation, he became a staff physician, and ultimately Physician-in-Chief. In 1929, he accepted a faculty position at Columbia's College of Physicians and Surgeons, and began working at the Bellevue Chest Service. There, he devoted the rest of his career to the study and treatment of tuberculosis, serving as Physician-in-Charge.
Fig. 2.

James Burns Amberson, Jr., MD. Reprinted from Richards, D. L. “Presentation of the Academy Plaque to James Burns Amberson, M.D.” Bull NY Acad Med 1970;46:663−5. Courtesy of the New York Academy of Medicine.
Dr. Amberson was elected to the American Clinical and Climatological Association (ACCA) in 1922 at the age of 32, and served as its Vice President in 1940 (69). His first publication in the ACCA Transactions was entitled “Clinical Studies of the Healing of Tuberculosis: I. Absorption of Pulmonary Deposits,” and demonstrated the importance of correlating serial clinical and radiologic findings in the treatment of tuberculosis (70). This paper revealed that pulmonary tuberculosis can heal by resolution, in addition to fibrosis and calcification, the more commonly recognized modes of healing. In his ACCA Memorial to J. Burns Amberson, Dr. George W. Wright described him as “a scholarly, gentle person…a teacher par excellence, a superb clinician, and a compassionate physician” (71). This is an apt description of someone well-known to me, as Dr. Amberson was, after all, my grandfather.
ACKNOWLEDGEMENTS
This work was supported by NIH grants HL42493, HL046403 and HL090895, and by a grant from the March of Dimes 6-FY12-356.
Footnotes
Potential Conflicts of Interest: None disclosed.
DISCUSSION
Boxer, Ann Arbor: What is the mechanism by which annexin induces fusion? And my other question, does annexin II play a role in the periodic fever syndromes?
Hajjar, New York: So, Larry we don't know whether it plays a role in periodic fever syndromes. I think that might be a very interesting thing to investigate. There are actually a number of questions that have come to mind, and we have to prioritize them. But I think that would be very interesting. And your other question was about the mechanism by which it induces inflammation. What we think is that its normal function is to maintain the integrity of the lysosome. And when the lysosomal contents leak into the cytoplasm, that induces assembly of the inflammasome, and we get all these downstream events and secretion of cytokines.
Schuster, New York: Have you had a chance to try blocking antibodies in animal models of diabetic retinopathy. And secondly, not all endothelia are the same, so what is distribution of annexin across the arterial and then the venous endothelium?
Hajjar, New York: The distribution of annexin II; we find it in every endothelial cell that we have looked at throughout the body. So we think that it's fairly ubiquitously distributed. We have a panel of antibodies that we have developed with some collaborators and we are getting ready to test those in mice.
Hochberg, Baltimore: Is there any role for cyclo-oxygenase enzymes in the expression of annexin on the cell membrane and its intracellular location to the lysosomal membrane?
Hajjar, New York: The answer is we don't know.
Wenzel, Richmond: Have you looked at drugs that might block the inflammasome specifically and see what happens to the activity or to the levels of annexin in either sepsis or the periodic fever or something related?
Hajjar, New York: We have another model of inflammatory bowel disease that we have been working on, and I didn't think I could fit that into the 12 minutes. But essentially we have used two inhibitors of inflammasome activation in DSS-induced inflammatory bowel disease in the mouse, and in both cases the increased severity that we see in the knockout reverts to the level of disease we see in the wild type. So that would confirm your hypothesis.
REFERENCES
- 1.Moss SE, Morgan RO. The annexins. Genome Biol. 2004;5:1–8. doi: 10.1186/gb-2004-5-4-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hedhli N, Falcone DJ, Huang B, et al. The annexin A2/S100A10 system in health and disease: Emerging paradigms. J Biomed Biotechnol. 2012:406273–406286. doi: 10.1155/2012/406273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luo M, Hajjar KA. Annexin A2 system in human biology: cell surface and beyond. Semin Thromb Hemost. 2013;39:338–46. doi: 10.1055/s-0033-1334143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rand JH. “Annexinopathies” — a new class of diseases. N Engl J Med. 1999;340:1035–6. doi: 10.1056/NEJM199904013401310. [DOI] [PubMed] [Google Scholar]
- 5.Puente XS, Sanchez LM, Overall CM, Lopez-Otin C. Human and mouse proteases: a comparative genomic approach. Nat Rev Gen. 2003;4:544–58. doi: 10.1038/nrg1111. [DOI] [PubMed] [Google Scholar]
- 6.Szabo R, Bugge T. Membrane-anchored serine proteases in vertebrate cell and developmental biology. Ann Rev Cell Develop Biol. 2011;27:213–35. doi: 10.1146/annurev-cellbio-092910-154247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hajjar KA, Harpel PC, Jaffe EA, Nachman RL. Binding of plasminogen to cultured human endothelial cells. J Biol Chem. 1986;261:11656–62. [PubMed] [Google Scholar]
- 8.Hajjar KA, Hamel NM, Harpel PC, Nachman RL. Binding of tissue plasminogen activator to cultured human endothelial cells. J Clin Invest. 1987;80:1712–9. doi: 10.1172/JCI113262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hajjar KA, Hamel NM. Identification and characterization of human endothelial cell membrane binding sites for tissue plasminogen activator and urokinase. J Biol Chem. 1990;265:2908–16. [PubMed] [Google Scholar]
- 10.Hajjar KA. The endothelial cell tissue plasminogen activator receptor: specific interaction with plasminogen. J Biol Chem. 1991;266:21962–70. [PubMed] [Google Scholar]
- 11.Gerke V, Creutz CE, Moss SE. Annexins: linking Ca++ signalling to membrane dynamics. Nat Rev Mol Cell Biol. 2005;6:449–61. doi: 10.1038/nrm1661. [DOI] [PubMed] [Google Scholar]
- 12.Gerke V, Moss SE. Annexins: from structure to function. Physiol Rev. 2002;82:331–71. doi: 10.1152/physrev.00030.2001. [DOI] [PubMed] [Google Scholar]
- 13.Donato R. S100: a multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. Int J Biochem Cell Biol. 2001;33:637–68. doi: 10.1016/s1357-2725(01)00046-2. [DOI] [PubMed] [Google Scholar]
- 14.Rescher U, Gerke V. S100A10/p11: family, friends, and functions. Pflugers Arch. 2007;455:575–82. doi: 10.1007/s00424-007-0313-4. [DOI] [PubMed] [Google Scholar]
- 15.Rety S, Sopkova J, Renouard M, et al. The crystal structure of a complex of p11 with the annexin II N-terminal peptide. Nat Struct Biol. 1999;6:85–9. doi: 10.1038/4965. [DOI] [PubMed] [Google Scholar]
- 16.He KL, Deora AB, Xiong H, et al. Endothelial cell annexin A2 regulates polyubiquitination and degradation of its binding partner S100A10/p11. J Biol Chem. 2008;283:19192–200. doi: 10.1074/jbc.M800100200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y, Myrvang HK, Dekker LV. Annexin A2 complexes with S100 proteins: structure, function, and pharmacological manipulation. Br J Pharmacol. 2015;172:1664–1676. doi: 10.1111/bph.12978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hajjar KA, Jacovina AT, Chacko J. An endothelial cell receptor for plasminogen/tissue plasminogen activator: I. Identity with annexin II. J Biol Chem. 1994;269:21191–7. [PubMed] [Google Scholar]
- 19.Cesarman GM, Guevara CA, Hajjar KA. An endothelial cell receptor for plasminogen/tissue plasminogen activator: II. Annexin II-mediated enhancement of t-PA–dependent plasminogen activation. J Biol Chem. 1994;269:21198–203. [PubMed] [Google Scholar]
- 20.O'Connell PA, Surette AP, Liwski RS, Svenningsson P, Waisman DM. S100A10 regulates plasminogen-dependent macrophage invasion. Blood. 2010;116:1136–46. doi: 10.1182/blood-2010-01-264754. [DOI] [PubMed] [Google Scholar]
- 21.Das R, Burke T, Plow EF. Histone H2B as a functionally important plasminogen receptor on macrophages. Blood. 2007;110:3763–72. doi: 10.1182/blood-2007-03-079392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cesarman-Maus G, Hajjar KA. The molecular basis of fibrinolysis. Br J Haematol. 2005;129:307–21. doi: 10.1111/j.1365-2141.2005.05444.x. [DOI] [PubMed] [Google Scholar]
- 23.Hajjar KA. The Molecular Basis of Fibrinolysis. In: Orkin SH, et al., editors. Nathan and Oski's Hematology of Infancy and Childhood. Philadelphia, PA: Saunders; 2008. pp. 1425–47. [Google Scholar]
- 24.Hajjar KA, Ruan J. Fibrinolysis and Thrombolysis. In: Kaushansky K, et al., editors. Williams Hematology. New York, NY: McGraw-Hill; 2010. pp. 2219–46. [Google Scholar]
- 25.Dassah M, Deora A, He K, Hajjar KA. The endothelial cell annexin A2 system and vascular fibrinolysis. Gen Physiol Biophys. 2009;28:F20–8. [PMC free article] [PubMed] [Google Scholar]
- 26.Flood EC, Hajjar KA. The annexin A2 system and vascular homeostasis. Vasc Pharmacol. 2011;54:59–67. doi: 10.1016/j.vph.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petersen EA, Sutherland MR, Nesheim ME, Pryzdial EL. Thrombin induces endothelial cell-surface exposure of the plasminogen receptor annexin 2. J Cell Sci. 2003;116:2399–408. doi: 10.1242/jcs.00434. [DOI] [PubMed] [Google Scholar]
- 28.Deora AB, Kreitzer G, Jacovina AT, Hajjar KA. An annexin 2 phosphorylation switch mediates its p11-dependent translocation to the cell surface. J Biol Chem. 2004;279:43411–18. doi: 10.1074/jbc.M408078200. [DOI] [PubMed] [Google Scholar]
- 29.Huang B., Deora A.B., He K., et al. Hypoxia-inducible factor-1 drives annexin A2 system-mediated perivascular fibrin clearance in oxygen-induced retinopathy in mice. Blood. 2011;118:2918–29. doi: 10.1182/blood-2011-03-341214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He K, Sui G, Xiong H, et al. Feedback regulation of endothelial cell surface plasmin generation by PKC dependent phosphorylation of annexin A2. J Biol Chem. 2011;286:15428–39. doi: 10.1074/jbc.M110.185058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jost M, Gerke V. Mapping of a regulatory important site for protein kinase C phosphorylation in the N-terminal domain of annexin II. Biochim Biophys Acta. 1996;1313:283–9. doi: 10.1016/0167-4889(96)00101-2. [DOI] [PubMed] [Google Scholar]
- 32.Ling Q, Jacovina AT, Deora A. Annexin II regulates fibrin homeostasis and neoangiogenesis in vivo. J Clin Invest. 2004;113:38–48. doi: 10.1172/JCI200419684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Surette AP, Madureira PA, Phipps KD, Miller VA, Svenningsson P, Waisman DM. Regulation of fibrinolysis by S100A10 in vivo. Blood. 2011;118:3172–81. doi: 10.1182/blood-2011-05-353482. [DOI] [PubMed] [Google Scholar]
- 34.Jacovina AT, Deora AB, Ling Q, et al. Homocysteine inhibits neoangiogenesis in mice through blockade of annexin A2-dependent fibrinolysis. J Clin Invest. 2009;119:3384–94. doi: 10.1172/JCI39591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hajjar KA, Mauri L, Jacovina AT, et al. Tissue plasminogen activator binding to the annexin II tail domain: direct modulation by homocysteine. J Biol Chem. 1998;273:9987–93. doi: 10.1074/jbc.273.16.9987. [DOI] [PubMed] [Google Scholar]
- 36.Zhu H, Fan J, Liu J, et al. Annexin A2 combined with low dose tPA improves thrombolytic therapy in a rat model of focal embolic stroke. J Cerebral Blood Flow Metab. 2010;30:1137–46. doi: 10.1038/jcbfm.2009.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tanaka Y, Ishii H, Hiraoka M, et al. Efficacy of recombinant annexin 2 for fibrinolytic therapy in a rat embolic stroke model: a magnetic resonance imaging study. Brain Res. 2007;1165:135–43. doi: 10.1016/j.brainres.2007.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walvick RP, Bratane BT, Henninger N, et al. Visualization of clot lysis in a rat embolic stroke model: application to comparative lytic efficacy. Stroke. 2011;42:1110–5. doi: 10.1161/STROKEAHA.110.602102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ishii H, Yoshida M, Hiraoka M, et al. Recombinant annexin II modulates impaired fibrinolytic activity in vitro and in rat carotid artery. Circ Res. 2001;89:1240–5. doi: 10.1161/hh2401.101066. [DOI] [PubMed] [Google Scholar]
- 40.Fan X, Zhanyang Y, Liu J, et al. Annexin A2: a tissue plasminogen activator amplifier for thrombolytic stroke therapy. Stroke. 2010;41:S54–8. doi: 10.1161/STROKEAHA.110.596106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cockrell E, Espinola RG, McCrae KR. Annexin A2: biology and relevance to the antiphospholipid syndrome. Lupus. 2008;17:943–51. doi: 10.1177/0961203308095329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cohen D, Berger SP, Steup-Beekman GM, Bloemenkamp KWM, Bajema IM. Diagnosis and management of the antiphosphlipid syndrome. Br Med J. 2010;340:1125–32. doi: 10.1136/bmj.c2541. [DOI] [PubMed] [Google Scholar]
- 43.Cesarman-Maus G, Rios-Luna NP, Deora AB, et al. Autoantibodies against the fibrinolytic receptor, annexin 2, in antiphospholipid syndrome. Blood. 2006;107:4375–82. doi: 10.1182/blood-2005-07-2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wen A, Zheng H, Chen XW, Shen Y, Yang CD. Anti-annexin II antibody is associated with thrombosis and/or pregnancy morbidity in antiphospholipid syndrome and systemic lupus erythematosus with thrombosis. Rheumatol Int. 2011;31:865–9. doi: 10.1007/s00296-010-1379-4. [DOI] [PubMed] [Google Scholar]
- 45.Cesarman-Maus G, Cantu-Brito C, Barinagarrementeria F, et al. Autoantibodies against the fibrinolytic receptor, annexin A2, in cerebral venous thrombosis. Stroke. 2011;42:501–3. doi: 10.1161/STROKEAHA.110.592121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krone KA, Allen KL, McCrae KR. Impaired fibrinolysis in the antiphospholipid syndrome. Curr Rheumatol Rep. 2010;12:53–7. doi: 10.1007/s11926-009-0075-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sebastiani P, Ramoni MF, Nolan V, Baldwin CT, Steinberg MH. Genetic dissection and prognostic modeling of overt stroke in sickle cell anemia. Nat Genet. 2005;37:435–40. doi: 10.1038/ng1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Flanagan JM, Frohlich DM, Howard TA, et al. Genetic predictors for stroke in children with sickle cell anemia. Blood. 2011;117:6681–4. doi: 10.1182/blood-2011-01-332205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baldwin CT, Nolan VG, Wyszynski DF, et al. Association of klotho, bone morphogenetic protein 6, and annexin A2 polymorphisms with sickle cell disease. Blood. 2005;106:372–5. doi: 10.1182/blood-2005-02-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stein E, McMahon B, Kwaan H, Altman JK, Frankfurt O, Tallman MS. The coagulopathy of acute promyelocytic leukaemia revisited. Best Pract Res Clin Haematol. 2009;22:152–63. doi: 10.1016/j.beha.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 51.Menell JS, Cesarman GM, Jacovina AT, McLaughlin MA, Lev EA, Hajjar KA. Annexin II and bleeding in acute promyelocytic leukemia. N Engl J Med. 1999;340:994–1004. doi: 10.1056/NEJM199904013401303. [DOI] [PubMed] [Google Scholar]
- 52.Liu Y, Wang Z, Jiang M, Dai L, Zhang W, Wu D, Ruan C. The expression of annexin II and its role in the fibrinolytic activity in acute promyelocytic leukemia. Leuk Res. 2011;35:879–84. doi: 10.1016/j.leukres.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 53.O'Connell PA, Madureira PA, Berman JN, Liwski RS, Waisman DM. Regulation of S100A10 by the PML-RARalpha oncoprotein. Blood. 2011;117:4095–4105. doi: 10.1182/blood-2010-07-298851. [DOI] [PubMed] [Google Scholar]
- 54.Stahl A, Connor KM, Sapieha P, et al. The mouse retina as an angiogenesis model. Invest Ophthalmol Vis Sci. 2010;51:2813–26. doi: 10.1167/iovs.10-5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jost M, Zeusschner D, Seemann J, Weber K, Gerke V. Identification and characterization of a novel type of annexin-membrane interaction: Ca2+ is not required for the association of annexin II with early enodsomes. J Cell Sci. 1997;110:221–28. doi: 10.1242/jcs.110.2.221. [DOI] [PubMed] [Google Scholar]
- 56.Emans N, Gorvel JP, Walter C, et al. Annexin II is a major component of fusogenic endosomal vesicles. J Cell Biol. 1993;120:1357–69. doi: 10.1083/jcb.120.6.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Harder T, Gerke V. The subcellular distribution of early endosomes is affected by the annexin II-p11 complex. J Cell Biol. 1993;123:1119–32. doi: 10.1083/jcb.123.5.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gruenberg J, Stenmark H. The biogenesis of multivesicular endosomes. Nat Rev Mol Cell Biol. 2004;5:317–23. doi: 10.1038/nrm1360. [DOI] [PubMed] [Google Scholar]
- 59.Mathivanan S, Hong J, Simpson RJ. Exosomes: extracellular organelles important in intercellular communication. J Proteomics. 2010;73:1907–20. doi: 10.1016/j.jprot.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 60.Umbrecht-Jenck E, Demais V, Calco V, Bailly Y, Bader MF, Chasserot-Golaz S. S100A10-mediated translocation of annexin-A2 to SNARE proteins in adrenergic chromaffin cells undergoing exocytosis. Traffic. 2010;11:958–71. doi: 10.1111/j.1600-0854.2010.01065.x. [DOI] [PubMed] [Google Scholar]
- 61.Creutz CE, Pazoles CJ, Pollard HB. Identification and purification of an adrenal medullary protein (synexin) that causes calcium-dependent aggregation of isolated chromaffin granules. J Biol Chem. 1978;253:2858–66. [PubMed] [Google Scholar]
- 62.Rojo Pulido I, Nightengale TD, Darchen F, Seabra MC, Cutler DF, Gerke V. Myosin Va acts in concert with Rab27a and MyRIP to regulate acute von-Willebrand factor release from endothelial cells. Traffic. 2011;12:1371–82. doi: 10.1111/j.1600-0854.2011.01248.x. [DOI] [PubMed] [Google Scholar]
- 63.Knop J, Aareskjold E, Bode G, Gerke V. Rab3D and annexin A2 play a role in regulated secretion of vWF, but not tPA, from endothelial cells. EMBO J. 2004;23:2982–92. doi: 10.1038/sj.emboj.7600319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dietl P, Haller T, Frick M. Spatio-temporal aspects, pathways, and actions of Ca++ in surfactant secreting pulmonary alveolar type II pneumocytes. Cell Calcium. 2012;52:296–302. doi: 10.1016/j.ceca.2012.04.010. [DOI] [PubMed] [Google Scholar]
- 65.Wang P, Chintagari NR, Gou D, Su L, Liu L. Physical and functional interactions of SNAP-23 with annexin A2. Am J Respir Cell Mol Biol. 2007;37:465–76. doi: 10.1165/rcmb.2006-0447OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dassah M, Almeida D, Hahn R, Bonaldo P, Worgall S, Hajjar KA. Annexin A2 mediates collagen VI secretion, pulmonary elasticity, and bronchial epithelial cell apoptosis. J Cell Sci. 2014;127:828–44. doi: 10.1242/jcs.137802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cobelli N, Scharf B, Crisi GM, Hardin J, Santambrogio L. Mediators of the inflammatory response to joint replacement devices. Nat Rev Rheumatol. 2011;7:600–18. doi: 10.1038/nrrheum.2011.128. [DOI] [PubMed] [Google Scholar]
- 68.Scharf B, Clement CC, Wu XX, et al. Annexin A2 binds to endosomes following organelle destabilization by particulate wear debris. Nature Comm. 2012;3:755–65. doi: 10.1038/ncomms1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Former Officers and Deceased Members. Trans Am Climatol Clin Assoc. 2014;125:viii–xviii. [Google Scholar]
- 70.Amberson JB. Clinical studies of the healing of tuberculosis, I. Absorption of pulmonary deposits. Trans Am Climatol Clin Assoc. 1924;40:174–85. [PMC free article] [PubMed] [Google Scholar]
- 71.Wright GW. James Burns Amberson. Trans Am Climatol Clin Assoc. 1982;93:xxxiii–xxxv. [Google Scholar]