

Abstract
Brain-derived neurotrophic factor (BDNF) is a member of a family of neurotrophins which include nerve growth factor, neurotrophin 3, and neurotrophin 4. Studies over the last three decades have identified mature BDNF as a key regulator of neuronal differentiation, structure, and function; actions mediated by the TrkB receptor. More recently identified isoforms which are translated from the bdnf gene, including the uncleaved precursor, pro-BDNF, and the cleaved prodomain, have been found to elicit opposing functions in neurons through the activation of distinct receptors. This work emphasizes the critical roles for all three isoforms of BDNF in modulating neuronal activity that impact complex human behaviors including memory, anxiety, depression, and hyperphagia.
DISCOVERY OF BRAIN-DERIVED NEUROTROPHIC FACTOR AND IDENTIFICATION OF MATURE BDNF ACTIVITIES
Brain-derived neurotrophic factor (BDNF) was first isolated in the 1980s (1), nearly three decades after the identification of the related factor, nerve growth factor (NGF). NGF, identified by Rita Levi-Montalcini, Victor Hamberger, and Stanley Cohen (2–4) was recognized to support the survival of subpopulations of peripheral neurons, prompting Yves-Alain Barde and Hans Thoenen to search for a growth factor that exhibited similar properties for other populations of neurons. BDNF was initially purified from pig brain (1), and was found to be expressed at low concentrations; 1.5 kg of starting material was required to purify 1 μg of BDNF. Similar to many other growth factors, BDNF is highly charged (pI [isoelectric point] = ∼9–10), stable during biochemical purification, and supports the survival and differentiation of distinct populations of neurons (1). Once the amino acid sequence of BDNF was deduced (5), it was recognized that BDNF and NGF were highly related, and two additional family members, neurotrophin 3 and neurotrophin 4, were identified by molecular cloning. These four related growth factors comprise the highly conserved neurotrophin family.
Similar to all neurotrophins, BDNF is initially synthesized as a precursor form (pro-BDNF), consisting of a prodomain of 129 amino acids and a mature domain of 118 amino acids. The mature domain forms a cysteine knot structure, leading to non-covalent dimerization of the mature domains (6). When the prodomain is cleaved from intact pro-BDNF, through the actions of proconvertase at a conserved RVRR sequence, the dimeric mature domains are released, and are called mature BDNF, or simply BDNF.
Thousands of publications during the past 25 years have characterized the biological actions of mature BDNF and have been well summarized (7–10). It has been established that BDNF mediates survival and differentiative activities on neurons by binding and activating the tropomycin receptor kinase B or TrkB, a member of the larger family of Trk receptors (reviewed in [10]). Although BDNF promotes survival of subclasses of neurons in vitro, genetic loss of BDNF in the central nervous system does not cause a significant reduction in neuronal cell number, but rather leads to a reduction in spine density and dendritic complexity (11,12). These results suggest that BDNF acts primarily as a differentiation factor rather than a survival factor in vivo.
BDNF also plays a critical role in modulating the synaptic function of neurons. Following biosynthesis, BDNF is axonally transported in dense core vesicles to axon terminals, and is secreted into the synaptic cleft following membrane depolarization (13,14). Delivery of recombinant BDNF to hippocampal slices enhances the efficacy of excitatory synapses (15,16) but depresses the firing of inhibitory synapses of GABAergic neurons (17). These effects are mediated presynaptically by altering the efficacy of presynaptic neurotransmitter release (18), or post-synaptically by enhancing the magnitude of neurotransmitter effects, primarily through alterations in NMDA receptor function (reviewed in [9]). Both mechanisms require the binding of BDNF to TrkB, which is present both on the presynaptic and post-synaptic membranes. In addition to these effects on basal synaptic transmission, BDNF facilitates long-term potentiation (19–21), and can promote the structural enlargement of dendritic spines (22) to regulate the function of mature neural circuits. In the periphery, BDNF promotes the development of neuromuscular synapses (23). As such, even moderate changes in BDNF levels in rodents results in significant alterations in hippocampal function in rodents, including impaired memory, anxiety, aggression, and hyperphagia (24) (reviewed in [25]). Some of these behavioral characteristics are phenocopied in humans with genetic abnormalities leading to impaired BDNF-TrkB function (25).
REGULATION OF BDNF EXPRESSION
Because BDNF plays such a crucial role in the maintenance and refinement of neural circuits and behaviors, multiple mechanisms are used to tightly regulate BDNF levels. Although the bdnf gene comprises nine exons, the coding sequence resides in exon nine, with eight upstream exons that encode promoters that regulate regional and cell type specific expression (reviewed in [26]). Among these, exon IV has been most extensively characterized, as this exon, containing promotor elements, regulates activity-dependent bdnf expression. Specifically, induced mutations in this promoter affect the function of inhibitory circuits in the hippocampus and prefrontal cortex (27,28). The bdnf gene also encodes two different 3'UTRs, which may facilitate localization to axons or dendrites, although the function of dendritically targeted BDNF has not been examined in detail (29). Epigenetic regulation of chromatin structure may also play an important role in altering the efficacy of activity-dependent BDNF transcription. For example, electroconvulsive treatment or chronic social defeat stress in mice can alter histone methylation and promoter IV activity (30,31). However, it is unclear whether normal physiologic neuronal activity is sufficient to induce epigenetic changes that regulate BDNF levels in vivo.
ACTIONS of Pro-BDNF
As noted above, BDNF is initially synthesized as a precursor, pro-BDNF in the endoplasmic reticulum, and the N-terminal prodomain can be cleaved from the C-terminal mature domain by the actions of furin or proconvertases in the trans-Golgi network or in secretory granules. The efficiency of cleavage, and hence the ratio of pro-BDNF to mature BDNF, is different at distinct developmental stages and during postnatal life; specifically, in the neonatal and adolescent stages in mice, both pro-BDNF and mature BDNF are detectable, whereas in adulthood, mature BDNF predominates (32). The precursor forms or proneurotrophins, including pro-BDNF, were initially thought to be inactive precursors; however, studies in 2001 described proneurotrophins as independent ligands that activate the p75 receptor, rather than TrkB that is activated by mature BDNF (33). The p75 receptor is a member of the tumor necrosis factor family of receptors that encode a cytoplasmic death domain and can initiate apoptosis following ligand binding. The p75 receptor binds to the mature domain region of pro-BDNF, whereas the prodomain region binds to sortilin (a Vps10p-domain sorting receptor) or SorCS2, a related sortilin family member (34,35). Indeed, treatment of neurons that express p75 with recombinant pro-BDNF induces cell death (34). Additional activities of recombinant pro-BDNF were rapidly uncovered, and include retraction of growth cones (35), similar to that observed with pro-NGF (36) pruning of axonal processes (37), facilitation of long-term depression at mature hippocampal synapses (32,38,39), and synaptic elimination of the neuromuscular junction (40). These effects are often opposite of those elicited by mature BDNF (i.e., enhancement of outgrowth of growth cones or mature spine, and long-term potentiation), leading to the “yin-yang” hypothesis of neurotrophin action in which mature BDNF and pro-BDNF exhibit opposing functions (7).
In studies using cultured neurons, some investigators detect secretion of both pro-BDNF and mature BDNF upon depolarization (40–42) whereas other investigators detect predominantly mature BDNF (43). To clarify these results and to identify the biological actions of pro-BDNF in vivo, a proBDNF knockin mouse was generated to examine the effects of pro-BDNF under the control of its endogenous promoter and other regulatory elements (32). In this in vivo model, pro-BDNF expression negatively regulates hippocampal dendritic complexity and spine density, effects that are mediated by the p75 receptor. Most importantly, hippocampal slices from these pro-BDNF−expressing mice exhibit depressed synaptic activity and enhanced long-term depression (32). These finding suggest that pro-BDNF acts in vivo as a biologically active factor that regulates hippocampal structure, synaptic transmission, and synaptic plasticity, effects that are distinctive from mature BDNF. These results, coupled with the higher levels of expression of pro-BDNF in the developing post-natal brain, suggest that this ligand may be a key regulator in shaping neural circuitry and synaptic plasticity in adolescence, effects that may be maintained through adulthood (32). Indeed, abnormalities in pro-BDNF/mature BDNF levels are observed in the brains of individuals with autism, as well as in individuals with HIV-associated cognitive impairment, suggesting that the balance of these two BDNF isoforms may be dysregulated in disease states (44,45).
THE PRODOMAIN OF BDNF
For several decades after the identification of BDNF, the fate of the cleaved prodomain region has been unknown. Based on predictions that the prodomain was an intrinsically disordered protein, similar to the prodomain region of pro-NGF (46), most investigators believed that it was subject to rapid proteolytic degradation after cleavage from pro-BDNF. However, two recent studies (14,35) describe the detection of the prodomain in the hippocampus and other brain regions upon fixation of protein from hippocampal lysates to western blot membranes that tether the prodomain and permit detection. Indeed, the levels of the prodomain are very low in the neonatal rodent brain, but rapidly rise during adolescence and adulthood, paralleling the increase in mature BDNF at these same times. In addition, the prodomain is released from neurons upon depolarization, suggesting that it may act as an independent ligand.
THE Val66Met BDNF POLYMORPHISM AND IDENTIFICATION OF THE Met PRODOMAIN AS AN INDEPENDENT LIGAND
Clues to potential functions of the prodomain arose from the recently recognized single nucleotide polymorphism (SNP) within the bdnf gene, at position 66, near the middle of the prodomain (47). In humans, but not in any of the other 70 species studied to date, a methionine (Met) to valine (Val) substitution at position 66 has been identified in approximately 20% of the Caucasion population, and at varying percentages in individuals of other ethnicities. This polymorphism is associated with numerous human phenotypes, including alterations in episodic memory and with enhanced risk to develop depression and anxiety disorders in humans (47–52). More detailed study of the function of this Val66Met SNP was facilitated by the development of a murine model, which demonstrates reduced hippocampal size and an anxiety phenotype (53). The prevailing hypothesis regarding the molecular mechanism by which the Met66 allele contributed to these phenotypes was that the Met66 prodomain region of intact pro-BDNF exhibited impaired interaction with sortilin, resulting in diminished sorting of pro-BDNF to the axon and impaired release of mature BDNF upon neuronal depolarization (53,54). Thus the proposed mechanism was indirect, with the Met substitution resulting in altered intracellular trafficking, and diminished levels of BDNF in the synaptic cleft, leading to impaired synaptic plasticity and synaptic structure.
However, the high sequence conservation in the prodomain and the recent evolutionary emergence of the Val66Met polymorphism prompted inquiry as to whether the prodomain might function as an independent ligand (35). Indeed, structural studies using nuclear magnetic resonance spectroscopy to elucidate the disordered structure of the prodomain demonstrated that the Met and Val prodomains adopt distinct conformations, leading to a differential interaction with the SorCS2 receptor. Neither the Val nor the Met prodomain interact directly with the p75 receptor. However, application of the Val or Met prodomains to cultured neurons that expressed both p75 and SorCS2 resulted in differential responses. Application of the Val prodomain induced no acute structural changes to neuronal growth cones, whereas treatment with the Met prodomain led to acute growth cone retraction which was dependent upon the expression of both SorCS2 and p75. These results suggested that the Met prodomain can function as a third BDNF ligand to promote acute negative remodeling of neuronal architecture using the p75 and SorCS2 receptors (35) (Figure 1).
Fig. 1.
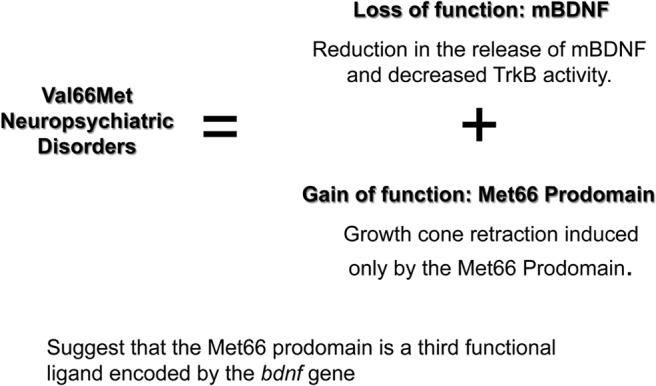
Proposed mechanisms that contribute to the differential actions of Val66BDNF and Met66BDNF are shown. In addition to the well described function of the Met66 allele, to decrease the amount of BDNF that is delivered to synapses, the Met66 prodomain exhibits independent activities utilizing p75/SorCS2 receptors to negatively regulate neuronal morphology.
Collectively, the phenotypes exhibited by humans related to neuropsychiatric disorders associated with the Val66Met polymorphism have been linked to impaired sorting and secretion of BDNF, leading to impaired neuronal differentiation and plasticity due to reduced TrkB activation. These more recent studies, however, suggest that the Met66 prodomain is a newly identified ligand that can selectively activate SorCS2 to alter neuronal morphology, actions that are similar to those of pro-BDNF (Figure 2). Whereas expression of pro-BDNF is largely restricted to the early post-natal brain, the prodomain and SorCS2 are highly expressed in adulthood. Thus, these studies suggest that the Met prodomain may continue to mediate modulate the yin-yang effects of mature BDNF, and may contribute to the altered neural plasticity in humans with this SNP.
Fig. 2.
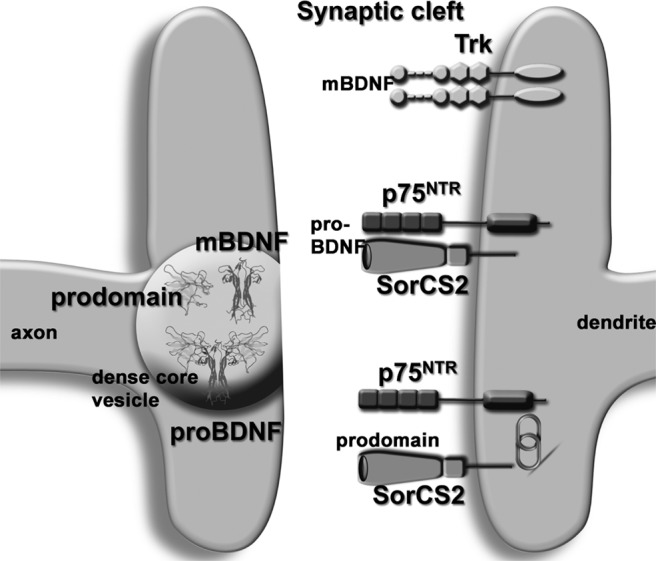
Schematic representation of the BDNF isofoms that are released at the synapse and mediate distinct actions are shown. All three BDNF isoforms are transported anterogradely in dense core vesicles within axons, and are released in an activity-dependent manner into the synaptic cleft. The specificity of BDNF action reflects both the BDNF isoforms that are released (i.e., pro-BDNF vs. mature BDNF, or the Val66prodomain vs. the Met66 prodomain), as well as the receptors that are expressed on the post-synaptic membrane. When p75 and SorCS2 are expressed, pro-BDNF and the Met66 prodomain can mediate negative effects on morphology and potentially synaptic plasticity (synaptic elimination, long term depression), whereas when TrkB is expressed, mature BDNF can mediate positive effects on morphology and plasticity (synaptic growth, long term potentiation).
ACKNOWLEDGMENTS
The author would like to acknowledge the numerous collaborators and laboratory members who have made this project possible. In particular, she would like to thank Agustin Anastasia, W. Clay Bracken, Katrin Deinhart, Jianmin Yang, Lauren Harte, Helen Scharhman, Jean Siao, Qian Ma, and Francis Lee. The author would also like to thank Moses Chao for his many years of mentorship and introduction to the neurotrophin field, and to Ralph Nachman and Andrew Schafer for their continued academic mentorship.
Footnotes
Support for these studies came from the NIH (NS 030687, NS 064114).
Potential Conflicts of Interest: None disclosed.
DISCUSSION
Tweardy, Houston: I enjoyed your presentation and was curious to know if any members of the BNDF family of ligands are upregulated in neurodegenerative diseases. As you indicated, the portion of pro-BNDF that is cleaved off to form BNDF is very unstable; therefore, either pro-BNDF or the portion cleaved off might accumulate in diseases characterized by impaired proteostasis such as neurodegenerative diseases.
Hempstead, New York: We don't know all of the mechanisms by which these proteins are induced following injury or even during neurodegeneration. We know that BDNF isoforms are transcriptionally regulated. They do have hypoxia response elements in some of the promoter regions. Also, in some very interesting recent work, we and others have found that BDNF is regulated by micro-RNAs. I think that as we begin to dig deeper, we will see that nature is re-expressing some of these proteins that normally prune circuits, as it establishes appropriate neuron targeting during development, and reutilizes those same types of strategies in a not particularly good fashion to re-sculpt and actually damage the CNS following injury or in neurodegenerative disease.
Hochberg, Baltimore: Does BDNF or the prodomain have any effect on pain sensation in your preclinical studies?
Hempstead, New York: Of the neurotrophins, the pain molecule is NGF. As a clinician, you could inject NGF into a human once. It activates pain fibers and they'll probably never let you do it again. NGF is the moiety that really regulates pain, via TrkA expressing neurons. BDNF has modest effects in that regard. Probably if you give high doses, there could be cross-talk with the TrkA receptor. But it doesn't activate pain fibers directly.
Fagin, New York: We are now identifying mutations, particularly fusion mutations in various cancers that activate NTRK 1, NTRK3, etcetera, in thyroid cancers and in others. So there is a lot of interest in developing compounds that block the activity of these receptor tyrosine kinases. Could you predict what type of theoretical consequences we might expect if it would cross the blood brain barrier?
Hempstead, New York: I think the companies that are really involved in this are well aware that they are going to need to identify molecules that do not cross the blood brain barrier, or, if so, do so in such a way that these effects can be modulated. There are many tumors that take advantage of Trk receptor tyrosine kinase survival signaling and hence upregulate the Trk receptors. They are an attractive target. But one of the major limitations to date has been CNS effects. There are new generation molecules that cross the blood brain barrier poorly. Some of them are actually in phase 1 trials.
Castle, Ann Arbor: Have you had a chance to look at the expression of these moieties during early neonatal development with modeling of the neuro circuitry?
Hempstead, New York: Pro-BDNF is expressed at the highest level, in the perinatal period, really at about P0 in a mouse, which is comparable to the third trimester of humans, and it rapidly decreases during adolescence. We know that one of the things it is doing is remodeling circuits. Neurons have to figure out where to go, and they are pruned away when they are projecting to the wrong area. An attractive hypothesis is that pro-BDNF may be one of a whole host of factors that is important in mediating this process.
REFERENCES
- 1.Barde YA, Edgar D, Thoenen H. Purification of a new neurotrophic factor from mammalian brain. EMBO J. 1982;1:549–53. doi: 10.1002/j.1460-2075.1982.tb01207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hamburger V, Levi-Montalcini R. Proliferation, differentiation and degeneration in the spinal ganglia of the chick embryo under normal and experimental conditions. J Exp Zool. 1949;111:457–502. doi: 10.1002/jez.1401110308. [DOI] [PubMed] [Google Scholar]
- 3.Levi-Montalcini R, Hamburger V. A diffusible agent of mouse sarcoma producing hyperplasia of sympathetic ganglia and hyperneurotization of viscera in the chick embyro. J Exp Zool. 1953;123:233–87. [Google Scholar]
- 4.Cohen S. Purification of a nerve-growth promoting protein from the mouse salivary gland and its neuro-cytotoxic antiserum. Proc Natl Acad Sci U S A. 1960;46:302–11. doi: 10.1073/pnas.46.3.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leibrock, Lottspeich F, Hohn A, et al. Molecular cloning and expression of brain-derived neurotrophic factor. Nature. 1989;341:149–52. doi: 10.1038/341149a0. [DOI] [PubMed] [Google Scholar]
- 6.Radziejewski C, Robinson RC, DeStefano PS, et al. Dimeric structure and conformational stability of brain-derived neurotrophic factor and neurotrophin-3. Biochemistry. 1992;31:4431–6. doi: 10.1021/bi00133a007. [DOI] [PubMed] [Google Scholar]
- 7.Lu B, Pang PT, Woo NT. The yin and yang of neurotrophin action. Nat Rev Neurosci. 2005;6:604–14. doi: 10.1038/nrn1726. [DOI] [PubMed] [Google Scholar]
- 8.Minichiello L. TrkB signalling pathways in LTP and learning. Nat Rev Neurosci. 2009;10:850–60. doi: 10.1038/nrn2738. [DOI] [PubMed] [Google Scholar]
- 9.Park H, Poo MM. Neurotrophin regulation of neural circuit development and function. Nat Rey Neurosci. 2013;14:7–23. doi: 10.1038/nrn3379. [DOI] [PubMed] [Google Scholar]
- 10.Chao MV. Neurotrophins and their receptors: a convergence point for many signaling pathways. Nat Rev Neurosci. 2003;4:299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- 11.Gao X, Smith GM, Chen J. Impaired dendric development and synaptic formation of postnatal-born dentate gyrus granular neurons in the absence of brain-derived neurotrophic factor signaling. Exp Neurol. 2009;215:178–190. doi: 10.1016/j.expneurol.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 12.Rauskolb S, Zagrebelsky M, Dreznjak A, et al. Global deprivation of brain-derived neurotrophic factor in the CNS reveals an area-specific requirement for dendritic growth. J Neurosci. 2010;30:1739–49. doi: 10.1523/JNEUROSCI.5100-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Conner JM, Lauterborn JC, Yan Q, et al. Distribution of brain-derived neutrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J Neurosci. 1997;17:2295–313. doi: 10.1523/JNEUROSCI.17-07-02295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dieni S, Matsumoto T, Dekkers M, et al. BDNF and its pro-peptide are stored in presynaptic dense core vesicles in brain neurons. J Cell Biol. 2012;196:775–88. doi: 10.1083/jcb.201201038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Korte M, Carroll P, Wolf E, et al. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci U S A. 1995;92:8856–60. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patterson SL, et al. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–45. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- 17.Tanaka T, Saito H, Matsuki N. Inhibition of GABAA synaptic responses by brain-derived neurotrophic factor (BDNF) in rat hippocampus. J Neurosci. 1997;17:2959–66. doi: 10.1523/JNEUROSCI.17-09-02959.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boulanger LM, Poo MM. Presynaptic depolarization facilitates neurotrophin-induced synaptic potentiation. Nat Neurosci. 1999;4:346–51. doi: 10.1038/7258. [DOI] [PubMed] [Google Scholar]
- 19.Kang H, Welcher AA, Shelton D, et al. Neurotrophins and time: different roles for TrkB signaling in hippocampal long-term potentiation. Neuron. 1997;19:653–64. doi: 10.1016/s0896-6273(00)80378-5. [DOI] [PubMed] [Google Scholar]
- 20.Korte M, Kang H, Bonhoeffer T, et al. A role for BDNF in the late-phase of hippocampal long-term potentiation. Neuropharmacology. 1998;37:553–9. doi: 10.1016/s0028-3908(98)00035-5. [DOI] [PubMed] [Google Scholar]
- 21.Cowansage KK, LeDoux JE, Monfils MH. Brain-derived neurotrophic factor; a dynamic gatekeeper of neural plasticity. Curr Mol Pharmacol. 2010;3:12–29. doi: 10.2174/1874467211003010012. [DOI] [PubMed] [Google Scholar]
- 22.Rex CS, Lin CY, Kramar EA, et al. Brain-derived neurotrophic factor promotes long-term potentiation-related cytoskeletal changes in adult hippocampus. J Neurosci. 2007;27:3017–29. doi: 10.1523/JNEUROSCI.4037-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lehof AM, Ip NY, Poo MM. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature. 1993;363:350–3. doi: 10.1038/363350a0. [DOI] [PubMed] [Google Scholar]
- 24.Lyons WE, et al. Brain-derived neurotrophic factor-deficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormalities. Proc Natl Acad Sci U S A. 1999;96:15236–44. doi: 10.1073/pnas.96.26.15239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rios M. Neurotrophins and the regulation of energy balance and body weight. In: Lewin GR, Carter BD, editors. Handbook of Experimental Pharmacology 220. Berlin, Germany: Springer-Verlag; 2014. pp. 283–307. [DOI] [PubMed] [Google Scholar]
- 26.West AE, Pruunsild, Timmusk T. (2014) Neurotrophins: transcription and translation. In: Lewin GR, Carter BD, editors. Handbook of Experimental Pharmacology 220. Berlin, Germany: Springer-Verlag; 2014. pp. 67–100. [DOI] [PubMed] [Google Scholar]
- 27.Hong EJ, McCord AE, Greenberg ME. A biological function for the neuronal activity-dependent component of Bdnf transcription in the development of cortical inhibition. Neuron. 2008;60:610–24. doi: 10.1016/j.neuron.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sakata K, et al. Critical role of promoter-IV driven BDNF transcription in GABAergic transmission and synaptic plasticity in the prefrontal cortex. Proc Natl Acad Sci U S A. 2009;106:5942–7. doi: 10.1073/pnas.0811431106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaneko M, et al. Dendritic BDNF synthesis is required for late phase spine maturation and recovery of cortical responses following sensory deprivation. J Neurosci. 2012;32:4790–802. doi: 10.1523/JNEUROSCI.4462-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsankova NM, et al. Sustained hippocampal chromatin regulation in a mouse model of depression and anti-depressant action. Nature Neurosci. 2006;9:519–25. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- 31.Ma DK, et al. Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science. 2009;323:1074–7. doi: 10.1126/science.1166859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang J, Harte-Hargrove LC, Siao CJ, et al. proBDNF negatively regulates neuronal remodeling, synaptic transmission, and synaptic plasticity in hippocampus. Cell Rep. 2014;7:796–806. doi: 10.1016/j.celrep.2014.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee R, Kermani P, Teng KK, et al. Regulation of cell survival by secreted proneurotrophins. Science. 2001;294:1945–8. doi: 10.1126/science.1065057. [DOI] [PubMed] [Google Scholar]
- 34.Teng HK, Teng KK, Lee R, et al. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J Neurosci. 2005;25:5455–63. doi: 10.1523/JNEUROSCI.5123-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anastasia A, Deinhardt K, Chao MV, et al. Val66Met polymorphism of BDNF alters prodomain structure to induce neuronal growth cone retraction. Nat Commun. 2013;4:2490. doi: 10.1038/ncomms3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deinhardt K, Kim T, Spellman DS, et al. Neuronal growth cone retraction relies on proneurotrophin receptor signaling through Rac. Sci Signal. 2011;4(202):ra82. doi: 10.1126/scisignal.2002060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun Y, Lim Y, Li F, et al. ProBDNF collapses neurite outgrowth of primary neurons by activating RhoA. PLoS One. 2012;7:e35883. doi: 10.1371/journal.pone.0035883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Woo NH, Teng HK, Siao CJ, et al. Activation of p75NTR by proBDNF facilitates hippocampal long-term depression. Nat Neurosci. 2005;8:1069–77. doi: 10.1038/nn1510. [DOI] [PubMed] [Google Scholar]
- 39.Lu Y, Christian K, Lu B. BDNF: a key regulator for protein synthesis-dependent LTP and long-term memory? Neurobiol Learn Mem. 2008;89:312–23. doi: 10.1016/j.nlm.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Je HS, Yang F, Ji Y, et al. Role of pro-brain-derived neurotrophic factor (proBDNF) to mature BDNF conversion in activity-dependent competition at developing neuromuscular synapses. Proc Natl Acad Sci U S A. 2012;109:15924–9. doi: 10.1073/pnas.1207767109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang J, Siao CJ, Nagappan G, et al. Neuronal release of proBDNF. Nat Neurosci. 2009b;12:113–5. doi: 10.1038/nn.2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nagappan G, Zaitsev E, Senatorov VV, Jr, et al. Control of extracellular cleavage of ProBDNF by high frequency neuronal activity. Proc Natl Acad Sci U S A. 2009;106:1267–72. doi: 10.1073/pnas.0807322106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matsumoto T, Rauskolb S, Polack M, et al. Biosynthesis and processing of endogenous BDNF: CNS neurons store and secrete BDNF, not pro-BDNF. Nat Neurosci. 2008;11:131–3. doi: 10.1038/nn2038. [DOI] [PubMed] [Google Scholar]
- 44.Bachis A, Avdoshina V, Zecca L, et al. Human immunodeficiency virus type 1 alters brain-derived neurotrophic factor processing in neurons. J Neurosci. 2012;32:9477–84. doi: 10.1523/JNEUROSCI.0865-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garcia KL, Yu G, Nicolini C, et al. Altered balance of proteolytic isoforms of pro-brain-derived neurotrophic factor in autism. J Neuropathol Exp Neurol. 2012;71:289–97. doi: 10.1097/NEN.0b013e31824b27e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feng D, Kim T, Ozkan E, et al. Molecular and structural insight into proNGF engagement of p75NTR and sortilin. J Mol Biol. 2010;396:967–84. doi: 10.1016/j.jmb.2009.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Egan MF, Kojima M, Callicott JH, et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. 2003;112:256–69. doi: 10.1016/s0092-8674(03)00035-7. [DOI] [PubMed] [Google Scholar]
- 48.Frielingsdorg H, et al. Variant brain-derived neurotrophic factor Val66Met endophenotypes: implications for posttraumatic stress discorder. Ann NY Acad Sci. 2010;1208:157–75. doi: 10.1111/j.1749-6632.2010.05722.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Verhagen M, et al. Meta-analysis of the BDNF Val66Met polymorphism in major depressive disorder: effects of gender and ethnicity. Mol Psychiatry. 2010;15:260–71. doi: 10.1038/mp.2008.109. [DOI] [PubMed] [Google Scholar]
- 50.Dincheva I, Glatt CE, Lee FS. Impact of the BDNF Val66Met polymorphism on cognition: implications for behavior genetics. Neuroscientist. 2012;18:439–51. doi: 10.1177/1073858411431646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hajek T, Kopeck M, Hoschl C. Reduced hippocampal volumes in healthy carriers of brain-derived neutrophic factor Val66Met polymorphi8sm: meta-analysis. World J Biol Psychiatry. 2012;13:178–87. doi: 10.3109/15622975.2011.580005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Soliman F, et al. A genetic variant BDNF polymorphism alters extinction learning in both mouse and human. Science. 2010;327:863–6. doi: 10.1126/science.1181886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen ZY, Patel PD, Sant G, et al. Variant brain-derived neurotrophic factor (BDNF) (Met66) alters the intracellular trafficking and activity-dependent secretion of wild-type BDNF in neurosecretory cells and cortical neurons. J Neurosci. 2004;24:4401–11. doi: 10.1523/JNEUROSCI.0348-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen ZY, Ieraci A, Teng H, et al. Sortilin controls the intracellular sorting of brain-derived neurotrophic factor to the regulated secretory pathway. J Neurosci. 2005;25:6156–66. doi: 10.1523/JNEUROSCI.1017-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]