

Abstract
The first 2-pyridylmethyl pendant armed structurally reinforced cyclam ligand has been synthesized and successfully complexed to Mn2+, Fe2+, and Cu2+ cations. X-ray crystal structures were obtained for the diprotonated ligand and its Cu2+ complex demonstrating pentadentate binding of the ligand with trans-II configuration of the side-bridged cyclam ring, leaving a potential labile binding site cis to the pyridine donor for interaction of the complex with oxidants and/or substrates. The electronic properties of these complexes were determined by means of solid state magnetic moment, with a low value of μ = 3.10 μB for the Fe2+ complex suggesting it has a trigonal bipyramidal coordination geometry, matching the crystal structure of the Cu2+ complex, while the μ = 5.52 μB value for the Mn2+ complex suggests it is high spin octahedral. Cyclic voltammetry in acetonitrile revealed reversible redox processes in all three complexes, suggesting catalytic reactivity involving electron transfer processes are possible for these complexes. Screening for oxidation catalysis using hydrogen peroxide as the terminal oxidant identified the Fe2+ complex as the oxidation catalysts most worthy of continued development.
Keywords: structurally reinforced cyclam, side-bridged cyclam, pendant-arm macrocycle, oxidation catalysis, 2-pyridylmethyl
We have studied cross-bridged cyclam complexes of manganese and iron for nearly a decade and a half as oxidation catalysts. [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] The manganese complex of 4,11-dimethyl-1,4,8,11-tetraazabicyclo[6.6.2]hexadecane [15] (Me2EBC, Scheme 1) in particular, has a rich oxidation chemistry. [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] This compound, which we propose to call “the Busch catalyst”, was initially synthesized as an oxidation catalyst because the cross-bridged ligand could rigidly bind the oxygen-reactive manganese metal and stop it from being lost in the form of MnO2. [1] [2] [3] [4] Que has determined that the iron complex of Me2EBC is an efficient olefin epoxidation catalyst with H2O2 oxidant under appropriate conditions as well. [16]
Scheme 1.
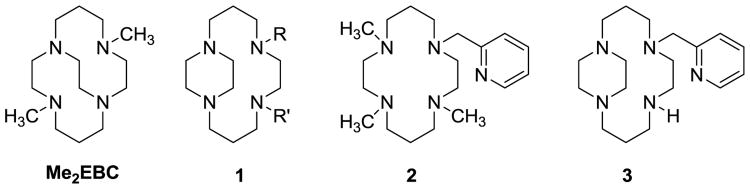
Ligands discussed in this paper.
Cyclam ligands structurally reinforced with an ethylene bridge between nitrogens 1 and 4 (i.e. “side-bridged”) have been known since 1980. [17] [18] [19] [20] [21] [22] [23] [24] [25] [26] The additional bridge provides rigidity, configurational selectivity, and kinetic stability and has been synthetically put in place by several different methods. [17] [18] [19] [20] [21] Like other cyclam ligands, additional utility can be added by addition of pendant arms, which have included in Scheme 1, structure 1: R and/or R′ = H, [17] [21] Me, [19] Et, [20] Bz, [19] [23] [22]Bz-p-NO2, [22] [25]Bz-p-NH2, [22] CH2CO2H, [24] [26]CH2PO3H [25] [26] and others.
An interesting 2-pyridylmethyltrimethylcyclam ligand (Scheme 1, structure 2) was reported by Que and coworkers [27] and stimulated our present work. Its iron complex activated dioxogen and formed an oxoiron(IV)intermediate that was crystallographically characterized, but has not been pursued further as a catalyst. [27] Although 2-pyridylmethyl pendant armed unbridged cyclams are ubiquitous, [28] [29] [30] [31] [32] [33] [34] [35] [36] [37] [38] no 2-pyridylmethyl pendant armed side-bridged or cross-bridged cyclams had been reported prior to our work. Due to our experience with bridged cyclam oxidation catalysts, we set out to synthesize and characterize side- and cross-bridged cyclam ligands containing a 2-pyridylmethyl pendant arm, along with their most biologically relevant oxidation active metal ion complexes: manganese, iron, and copper. Our cross-bridged work has been reported elsewhere. [39] Here, we report on the results of our efforts with the side-bridged cyclam ligand, (Scheme 1, structure 3) and the screening of its complexes as potential oxidation catalysts.
Alkylation of the cyclam-glyoxal bisaminal 4 in typical SN2 solvents like acetonitrile was unsuccessful due to self-reaction of the picolyl chloride. So, we explored the chlorinated solvents [40] to stabilize this reagent in the presence of the bisaminal and found successful, although low- yielding, monoalykylation with picolyl chloride in chloroform at room temperature. [41] We raised the yields of the monoalkyl salt 5 to 66% by addition of KI and increasing the temperature to reflux over 6 days. Typical reduction conditions [23] gave 3 in good yield. Metal complexation with anhydrous metal chlorides in dry solvents under nitrogen gave adequate yields of the desired complexes. [41]
The X-ray crystal structures of the H23Cl2 • 2 H2O and [Cu(3)][PF6]2 • C3H6O were obtained and are depicted in Figure 1. [42] The ligand is protonated at one of the piperazine tertiary nitrogens, and at the secondary nitrogen of the cyclam ring. There is an extensive hydrogen bonding network between the chloride anions, the two waters of crystallization, the cyclam nitrogens, and their protons. The pyridine nitrogen is oriented away from the center of the cyclam ring and is not involved in the hydrogen bonding network.
Figure 1.
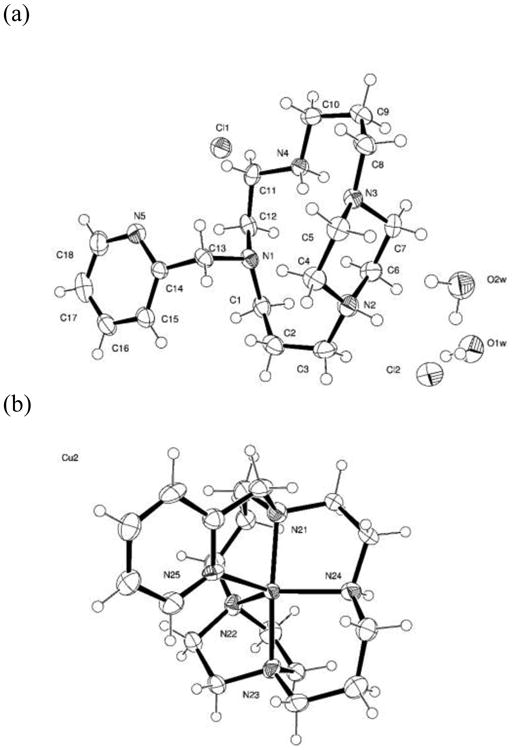
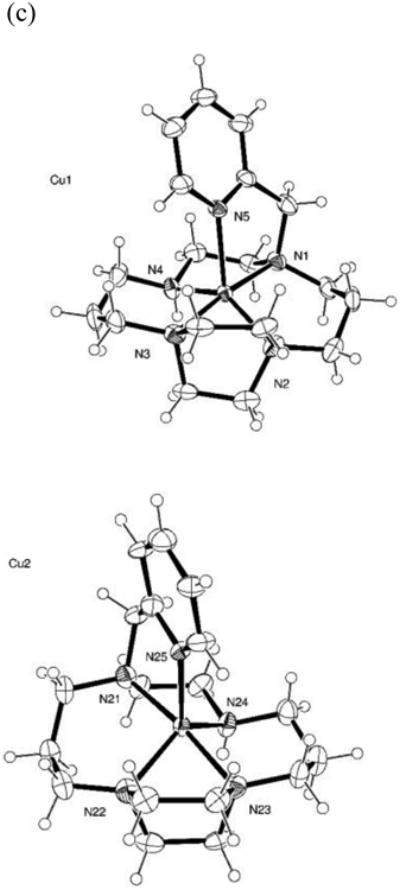
(a) Structure of H23Cl2 • 2 H2O (b) Structure of Cu(3)2+ depicted as an approximate trigonal bipyramidal geometry (c) Structure of Cu(3)2+ depicted as an approximate square-based pyramidal geometry and showing that the two slightly different ligand orientations are approximately enantiomers.
Upon coordination of Cu2+, the pyridine nitrogen is oriented into the center of the cyclam ring and coordinates the Cu2+ ion located there. This compound contains Cu2+ in two different coordination geometries. The ligand coordinates through each of the nitrogen atoms but in a slightly different orientation. The two copper complexes are approximately enantiomers, as shown in Figure 1(c). The coordination geometry around the copper(II) ion is somewhat between trigonal bipyramidal (as shown in Figure 1(b)) and square pyramidal (as shown in Figure 1(c)). To illustrate the slight differences between the two different metal ion coordination geometries of this structure, square pyramidal type bond angles for the two different copper(II) ions are given: Cu(1) N–Cu–N angles / °: (in plane) 100.9(2), 74.0(2), 94.75(19), 87.79(19); (to N5) 82.91(17), 118.02(18), 101.94(17), 97.03(18). Cu(2) N-Cu-N angles / °: (in plane) 102.10(19), 73.4(2), 91.7(2), 86.2(2); (to N25) 80.5(2), 107.85(18), 107.37(19), 120.5(2). Complete metrical parameters for both structures are given in the Supplementary Information.
Unfortunately, crystal structures of the Mn2+ and Fe2+ complexes were not obtained. We sought other hints at their structures and turned to solid state magnetic moments to help determine them. First, the magnetic moment of [Cu(3)][PF6]2 was determined to be 2.32 μB, which is consistent with n = 1 for a d9 Cu2+ ion. [43] [Mn(3)Cl]PF6 was determined to have μ = 5.52 μB, which is consistent with n = 5 for a high spin octahedral d5 Mn2+ metal ion. [43] High spin n = 5 is not diagnostic of a particular coordination geometry for Mn2+, but the consistent presence of one chloride in the elemental analysis of this complex led us to conclude that the most likely structure is a 6-coordinate pseudo-octahedral one. If the chloride is not coordinated and the complex is only 5-coordinate, precipitation by PF6- would have led to the dihexafluorophosphate salt.
Perhaps the most interesting contribution to structural information came from the magnetic moment of [Fe(3)]Cl2. μ = 3.10 μB, which is most consistent with n = 2 for a low spin 5-coordinate d6 Fe2+ metal ion. [44] In our previous experience with Mn2+ and Fe2+ complexes of bridged tetraazamacrocycles, [1] [3] [45] both ions agree in that they were high spin and 6-coordinate. However, in this case, Mn(3)Cl+ appears 6-coordinate and high spin, while Fe(3)2+ appears 5-coordinate and low spin. Two supporting pieces of evidence for this perhaps unexpected geometry for the Fe2+ complex are (1) the 5-coordinate Cu(3)2+ crystal structure presented above for this ligand system; and (2) an unpublished crystal structure in which Fe2+ is coordinated to the four nitrogens of a similar ethylene side-bridged ligand having a methyl group in place of the pyridylmethyl group of 3, and a chloro ligand. [39] Interestingly, a six-coordinate 1,8-bispyridylmethylcyclam Fe2+ complex with spin-crossover properties and a transition temperature to low spin of 150 K is known. [46] Of course, low spin d6 Fe2+ complexes are ubiquitous in organometallic chemistry. A number of 5-coordinate low-spin Fe2+ examples have been published. [47] [48] [49] [50]
In anticipation of carrying out oxidation studies, we obtained cyclic voltammograms (Figure 2) on these complexes in acetonitrile looking for multiple stabilized oxidations states if catalytic processes were to be likely. Unfortunately, only one reversible redox wave was observed for each complex. Relative to SHE, Cu(3)2+ gave a reversible Cu1+/2+ redox wave at E1/2 = -0.586 V (ΔE = 77 mV). No oxidation to Cu3+ was observed, which is perhaps not surprising, as there are no negatively charged ligands to help stabilize the Cu3+ cation. Cu(Me2EBC)Cl+ has an irreversible Cu2+ to Cu+ reduction wave at Ered = -0.544 V and an irreversible Cu2+ oxidation to Cu3+ at Eox = +1.530 V. [51] In comparison, Cu(3)2+ is more difficult to oxidize (due to lack of negatively charged ligands) and more reversibly reduced.
Figure 2.

Cyclic voltammograms in acetonitrile vs. SHE for (a) Cu(3)2+, (b) Fe(3)2+, and (c) Mn(3)Cl+.
Mn(3)Cl+ gave a reversible Mn2+/3+ redox wave at E1/2 = +0.856 V (ΔE = 93 mV) and an irreversible reduction at Ered = -0.685 V. This can be compared to the well-known Mn(Me2EBC)Cl2 catalyst [1] which has reversible Mn2+/3+ and Mn3+/4+ waves at E1/2 = +0.585 V (ΔE = 61 mV) and E1/2 = +1.343 (ΔE = 65 mV), respectively. The single chloro ligand allows only Mn3+ to be accessed for Mn(3)Cl+, and at a higher potential, since only one negatively charged chloro ligand is present to stabilize the growing positive charge. Reduction is possible here, when not observed for Mn(Me2EBC)Cl2, but not reversible, likely due to the loss of the chloro ligand upon reduction to Mn+.
5-coordinate Fe(3)2+ exhibits a complex cyclic voltammogram with a reversible Fe2+/3+ wave at E1/2 = +0.456 V (ΔE = 78 mV), two irreversible reductions at Ered = -0.802 V and -1.671 V, respectively, and a large return oxidation at Eox = -0.252 V. A much simpler behavior is observed for Fe(Me2EBC)Cl2, with only E1/2 = 0.110 (ΔE = 63 mV) observed for Fe2+/3+. [1] Oxidation is obviously much easier for the latter complex, where two negatively charged chloro ligands stabilize the positive charge. Reduction is observed only for Fe(3)2+, where no negatively charge ligands inhibit it. The complex behavior of Fe(3)2+ after the initial reduction is the subject of ongoing study.
Finally, we present initial oxidation screening data on these complexes. As shown in Table 1, compared with its analog, Mn(Me2EBC)Cl2 complex, the redox activities of Cu(3)2+, and Mn(3)Cl+ are quite poor. In sulfide oxidation using H2O2 as oxidant, while the Mn(Me2EBC)Cl2 catalyst provided nearly complete conversion of thioanisole (99.8%) with 44.3% yield of sulfoxide and 46.5% of sulfone, the Cu(3)2+ and Mn(3)Cl+ complexes are almost inactive for sulfide oxidation. However, Fe(3)2+ demonstrated some activity, providing 9.7% yield of sulfoxide, and 2.9% of sulfone with 15.6% conversion. Similarly, in hydrogen abstraction from 1,4-cyclohexadiene, Mn(Me2EBC)Cl2 gave 71.4% yield of benzene with 86.2% conversion, and Cu(3)2+ and Mn(3)Cl+ complexes were still inactive for benzene formation (∼3% yields represent natural benzene content in commercial 1,4-cyclohexadiene). Again, Fe(3)2+ provides more encouraging results, generating 23.6% yield of benzene with 44.5% conversion.
Table 1.
Oxidation catalysis screening results for manganese(II), iron(II), and copper(II) complexes of 3, in comparison to Mn(Me2EBC)Cl2.
| |||
|---|---|---|---|
| Complex | Conversion % | Yield % Sulfoxide | Yield % Sulfone |
| Mn(Me2EBC)Cl2 | 99.8 | 44.3 | 46.5 |
| [Mn(3)Cl]+ | 8.3 | 0.2 | 0.02 |
| [Fe(3)]2+ | 15.6 | 9.7 | 2.9 |
| [Cu(3)]2+ | 6.1 | 3.4 | 1.9 |
| |||
| Complex | Conversion % | Yield % Benzene | |
| Mn(Me2EBC)Cl2 | 86.2 | 71.4 | |
| [Mn(3)Cl]+ | 23.1 | 3.5 | |
| [Fe(3)]2+ | 44.5 | 23.6 | |
| [Cu(3)]2+ | 29.6 | 4 | |
These results are perhaps not surprising, when reconciled with the electrochemical studies (Figure 2). Mn(3)Cl+ has only a redox couple of Mn2+/3+ at E1/2 = +0.856 V, which is much higher than that of the corresponding Mn2+/3+ couple of Mn(Me2EBC)Cl2 (E1/2 = +0.585 V), and no access to Mn(IV). Therefore, the catalytic cycle of the Mn(3)Cl+ complex is very sluggish in oxidations. Similar sluggish redox behavior is observed in Cu(3)2+ since its oxidation to Cu(3)3+ is not observed. For the Fe(3)2+ complex, although its catalytic activity in sulfide oxidation, an oxygen transfer process, is poor, it still demonstrates a relatively good activity in hydrogen abstraction because its redox couple for Fe2+/3+ is modest (E1/2 = +0.456 V) and even lower than that of Mn(Me2EBC)Cl2. Consistent with the hydrogen abstraction activity of the Fe(3)2+ complex, Stack has reported[55] that Fe(PY5)(OH)3+, which has a potential of Epc = + 0.555 V, is capable of stoichiometric hydrogen abstraction from 9,10-dihydroanthracene (PY5 = 2,6-bis-(bis(2-pyridyl)methoxymethane)pyridine). This result encourages us to continue the exploration of the Fe(3)2+ complex and its biological relevance to lipoxygenase[56] in the future.
In conclusion, a new pyridylmethyl N-pendant arm, side-bridged cyclam ligand, 3, has been synthesized, and its diprotonated salt structurally characterized, with a key synthetic step the use of non-polar chloroform to decrease the self-reactivity of picolyl chloride in the presence of bisaminal 4. Divalent Mn, Fe, and Cu complexes were synthesized and the copper(II) complex structurally characterized by X-ray crystallography as 5-coordinate Cu(3)2+ showing the cyclam ring of 3 in a trans-II configuration and the chelated pyridine nitrogen bound to the Cu2+ ion. Solid state magnetic moment determination and elemental analysis revealed a high spin, octahedral Mn(3)Cl+ cation, but a 5-coordinate, low-spin Fe(3)2+ cation. Electrochemical studies in acetonitrile revealed reversible access to only two oxidation states for each metal ion, whereas optimal behavior for successful oxidation catalysis is likely a greater redox range. Preliminary screens for oxidation catalysis using H2O2 as the oxidant carried out on all three complexes showed promising results only in the hydrogen atom abstraction of 1,4-cyclohexadiene by Fe(3)2+, which is consistent with another iron complex from the literature having a similar redox potential. [55] Future work will include expanding the range of oxidation reactions possible with this catalyst and determination of its oxidation catalysis mechanisms.
Supplementary Material
Scheme 2.

Synthesis of 3. a) i. CHCl3, 2 eq. picolyl chloride hydrochloride, 4 eq. NaHCO3, 1 h, filter to remove solids; ii. 1 eq. of (1), 2 eq. KI, reflux 6 d; b) i. 95% EtOH,5 eq. NaBH4, N2, reflux, 1.5 h; ii. 12 M HCl(aq), 30% KOH(aq), benzene extraction.
Research Highlights.
-The first 2-pyridylmethyl side-bridge cyclam ligand and its X-ray crystal structure
-The first transition metal complex of a 2-pyridylmethyl side-bridge cyclam ligand and its X-ray crystal structure
-Cyclic voltammetry and oxidation screening of Mn2+, Fe2+, and Cu2+ complexes of this new ligand showing promising hydrogen atom abstraction for the Fe2+ complex
Acknowledgments
TJH acknowledges the Donors of the American Chemical Society Petroleum Research Fund; Health Research award for project number HR13-157, from the Oklahoma Center for the Advancement of Science and Technology; and Grant Number P20RR016478 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH) for partial support of this research. TJH also acknowledges the Henry Dreyfus Teacher-Scholar Awards Program for support of this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hubin TJ, McCormick JM, Collinson SR, Buchalova M, Perkins CM, Alcock NW, Kahol PK, Raghunathan A, Busch DH. J Am Chem Soc. 2000;122:2512–2522. [Google Scholar]
- 2.Hubin TJ, McCormick JM, Alcock NW, Busch DH. Inorg Chem. 2001;40:435–444. doi: 10.1021/ic9912225. [DOI] [PubMed] [Google Scholar]
- 3.Hubin TJ, McCormick JM, Collinson SR, Alcock NW, Clase HJ, Busch DH. Inorg Chim Acta. 2003;346:76–86. [Google Scholar]
- 4.Hubin TJ. Coord Chem Rev. 2003;241:27–46. [Google Scholar]
- 5.Yin G, Buchalova M, Danby AM, Perkins CM, Kitko D, Carter JD, Scheper WM, Busch DH. J Am Chem Soc. 2005;127:17170–17171. doi: 10.1021/ja055413k. [DOI] [PubMed] [Google Scholar]
- 6.Yin G, Danby AM, Kitko D, Carter JD, Scheper WM, Busch DH. Inorg Chem. 2007;46:2173–2180. doi: 10.1021/ic061957r. [DOI] [PubMed] [Google Scholar]
- 7.Yin G, Danby AM, Kitko D, Carter JD, Scheper WM, Busch DH. J Am Chem Soc. 2008;130:16245–16253. doi: 10.1021/ja804305x. [DOI] [PubMed] [Google Scholar]
- 8.Chattopadhyay S, Geiger RA, Yin G, Busch DH, Jackson TA. Inorg Chem. 2010;49:7530–7535. doi: 10.1021/ic101014g. [DOI] [PubMed] [Google Scholar]
- 9.Shi S, Wang Y, Xu A, Wang H, Zhu D, Roy SB, Jackson TA, Busch DH, Yin G. Angew Chem Int Ed. 2011;50:7321–7324. doi: 10.1002/anie.201100588. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, Shi S, Wang H, Zhu D, Yin G. Chem Commun. 2012;48:7832–7834. doi: 10.1039/c2cc33615d. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, Sheng J, Shi S, Zhu D, Yin G. J Phys Chem C. 2012;116:13231–13239. [Google Scholar]
- 12.Wang Y, Shi S, Zhu D, Yin G. Dalton Trans. 2012;41:2612–2619. doi: 10.1039/c2dt11814a. [DOI] [PubMed] [Google Scholar]
- 13.Dong L, Wang Y, Lu Y, Chen Z, Mei F, Xiong H, Yin G. Inorg Chem. 2013;52:5418–5427. doi: 10.1021/ic400361s. [DOI] [PubMed] [Google Scholar]
- 14.Yin G. Acc Chem Res. 2013;46:483–492. doi: 10.1021/ar300208z. [DOI] [PubMed] [Google Scholar]
- 15.Weisman GR, Rogers ME, Wong EH, Jasinski JP, Paight ES. J Am Chem Soc. 1990;112:8604–8605. [Google Scholar]
- 16.Feng Y, England J, Que LJ. ACS Catal. 2011;1:1035–1042. [Google Scholar]
- 17.Wainwright KP. Inorg Chem. 1980;19:1396–1398. [Google Scholar]
- 18.Helps IM, Parker D, Murphy JR, Chapman J. Tetrahedron. 1989;45:219–226. [Google Scholar]
- 19.Kolinski RA. Polish J Chem. 1995;69:1039–1045. [Google Scholar]
- 20.Kowallick R, Neuburger M, Zehnder M, Kaden TA. Helv Chem Acta. 1997;80:948–959. [Google Scholar]
- 21.Boiocchi M, Bonizzoni M, Fabbrizzi L, Foti F, Licchelli M, Poggi A, Taglietti A, Zema M. Chem Eur J. 2004;10:3209–3216. doi: 10.1002/chem.200305717. [DOI] [PubMed] [Google Scholar]
- 22.Khan A, Silversides JD, Madden L, Greenman J, Archibald SJ. Chem Commun. 2007:416–418. doi: 10.1039/b614557d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McRobbie G, Valks GC, Empson CJ, Khan A, Silversides JD, Pannecouque C, De Clercq E, Fiddy SG, Bridgeman AJ, Young NA, Archibald SJ. Dalton Trans. 2007:5008–5018. doi: 10.1039/b705800d. [DOI] [PubMed] [Google Scholar]
- 24.Silversides JD, Allan CC, Archibald SJ. Dalton Trans. 2007;9:971–978. doi: 10.1039/b615329a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Plutnar J, Havlickova J, Kotek J, Hermann P, Lukes I. New J Chem. 2008;32:496–504. [Google Scholar]
- 26.Boswell CA, Regino CAS, Baidoo KE, Wong KJ, Milenic DE, Kelley JA, Lai CC, Brechbiel MW. Bioorg & Medicinal Chem. 2009;17:548–552. doi: 10.1016/j.bmc.2008.11.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thibon A, England J, Martinho M, Young VGJ, Frisch JR, Guillot R, Girerd JJ, Munck E, Que LJ, Banse F. Angew Chem Int Ed. 2008;47:7064–7067. doi: 10.1002/anie.200801832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alcock NW, Balakrishnan KP, Moore P. J Chem Soc. Dalton Trans; 1986. pp. 1743–1745. [Google Scholar]
- 29.Asato E, Hashimoto S, Matsumoto N, Kida S. J Chem Soc. Dalton Trans; 1990. pp. 1741–1746. [Google Scholar]
- 30.Vuckovic G, Asato E, Matsumoto N, Kida S. Inorg Chim Acta. 1990;171:45–52. [Google Scholar]
- 31.Royal G, Dahaoui-Gindrey V, Dahaoui S, Tabard A, Guilard R, Pullumbi P, Lecomte C. Eur J Org Chem. 1998:1971–1975. [Google Scholar]
- 32.Bucher C, Royal G, Barbe JM, Guilard R. Tetrahedron Lett. 1999;40:2315–2318. [Google Scholar]
- 33.Goeta AE, Howard JAK, Maffeo D, Puschmann H, Williams JAG, Yufit DS. J Chem Soc. Dalton Trans; 2000. pp. 1873–1880. [Google Scholar]
- 34.Batsanov AS, Goeta AE, Howard JAK, Maffeo D, Puschmann H, Williams JAG. Polyhedron. 2001;20:981–986. [Google Scholar]
- 35.El Ghachtouli S, Cadiou C, Dechamps-Olivier I, Chuburu F, Aplincourt M, Roisnel T. Eur J Inorg Chem. 2006:3472–3481. [Google Scholar]
- 36.El Ghachtouli S, Cadiou C, Dechamps-Olivier I, Chuburu F, Aplincourt M, Patinec V, Le Baccon M, Handel H, Roisnel T. New J Chem. 2006;30:392–398. [Google Scholar]
- 37.Narayanan J, Solano-Peralta A, Ugalde-Salvidar VM, Escudero R, Hopfl H, Sosa-Torres ME. Inorg Chim Acta. 2008;361:2747–2758. [Google Scholar]
- 38.Morfin JF, Tripier R, Le Baccon M, Handel H. Polyhedron. 2009;28:3691–3698. [Google Scholar]
- 39.Jones DG, Wilson KR, Cannon-Smith DJ, Shircliff AD, Zhan Z, Chen Z, Prior TJ, Yin G, Hubin TJ. Inorg Chem. 2015;54:2221–2234. doi: 10.1021/ic502699m. [DOI] [PubMed] [Google Scholar]
- 40.Almassio MF, Sarimbalis MN, Garay RO. Designed Monomers and Polymers. 2005;8:287–296. [Google Scholar]
- 41.Synthetic Details: 3a-(pyridin-2-ylmethyl)-decahydro-5a,8a,10a-triaaza-3a-azoniapyrenium iodide (5). 13.28 g (0.08096 mol, 2 eq.) of picolyl chloride hydrochloride and 13.60 g (0.1619 mol, 4 eq.) of anhydrous NaHCO3 were stirred in 700 ml chloroform for 1 h. Solids were removed by filtration and the filtrate was added to 9.00 g (0.04048 mol, 1 eq.) of (4) [52] and 13.44 g (0.08096 mol, 2 eq.) of KI. The reaction was stirred and heated to reflux for 6 d under nitrogen, during which it became an orange color. After cooling, minimal solids were removed by filtration and discarded. The filtrate was evaporated to 100 ml volume and excess diethyl ether was added to precipitate the yellow solid product, which was filtered on a glass frit, washed with diethyl ether, and dried under vacuum. Yield = 11.841 g (66%). Electrospray mass spectrometry gave a single peak at m/z = 314 corresponding to (M-I)+. Anal. Calc. for C18H28N5I • H2O: C 47.06, H 6.58, N 15.25; found: C 46.87, H 6.54, N 14.88. 1H NMR (300 MHz, CDCl3) δ 1.36 (d, 1H), 1.84 (d, 1H), 2.25 (m, 2H), 2.41 (d, 1H), 2.56 (d, 1H), 2.66 (m, 2H), 3.03 (m, 6H), 3.23 (t, 1H), 3.65 (m, 2H), 3.89 (d, 1H), 4.21 (m, 2H), 4.40 (td, 1H), 4.58 (s, 1H), 5.45 (m, 2H), 7.41 (m, 1H), 7.84 (m, 1H), 8.31 (d, 1H), 8.66 (d, 1H). 13C{1H} NMR (75.6 MHz, D2O) δ 18.0, 18.4, 41.9, 46.6, 49.4, 51.3, 51.9, 53.2, 54.0, 60.5, 62.9, 69.5, 82.2, 125.8, 129.0, 138.6, 146.6, 150.3.5-(pyridin-2-ylmethyl)-1,5,8,12-tetraazabicyclo[10.2.2]hexadecane (3). 14.138 g (0.0320 mol, 1 eq.) of 5 was stirred in 1 L of 95% EtOH in a 2 L roundbottom flask. 6.059 g (0.160 mol, 5 eq.) of NaBH4 was added and the reaction was stirred at reflux for 1.5 h under N2. Upon cooling, 12 M HCl was added to a pH of 2. Solvent was removed under vacuum and the residue was dissolved in 100 ml of water to which 200 ml of 30% KOH was added. The cloudy white suspension was extracted into benzene, dried over Na2SO4, and evaporated to a tan oil. Yield = 7.947 g (78%). Electrospray mass spectrometry gave a single peak at m/z = 318 corresponding to (MH)+. Anal. Calc. for C18H31N5 • 1.3H2O • 0.1C6H6: C 64.07, H 9.89, N 20.08; found: C 63.80, H 10.00, N 19.87. 1H NMR (300 MHz, CDCl3) δ 1.65 (m, 4H), 2.16 (m, 2H), 2.45-2.65 (m, 11H), 2.86 (m, 4H), 3.14 (m, 2H), 3.73 (s, 2H), 3.82 (m, 2H), 7.09 (m, 1H), 7.30 (t, 1H), 7.56 (t, 1H), 8.45 (d, 1H). 13C{1H} NMR (75.6 MHz, D2O) δ 23.5, 26.1, 48.0, 48.1, 50.4, 51.0, 55.0, 55.1, 55.7, 56.9, 58.9, 121.8, 123.6, 135.9, 148.9, 159.1. X-ray quality crystals of the diprotonated salt H232+ were grown from evaporation of the acetonitrile solvent from a complexation reaction of 3 with FeCl2 which yielded, in addition to the complex product, a small amount of colorless crystals of H23Cl2 • 2 H2O.[Fe(3)]Cl2: 0.317 g (0.001 mol) of (3) and 0.127 g anhydrous FeCl2 were added to 10 ml of anhydrous acetonitrile in an inert atmosphere glovebox. The reaction was stirred at room temperature for 1 day during which the brown FeCl2 beads dissolved and formed a yellow precipitate. This product was filtered on a glass frit, washed with ether, and allowed to dry open to the atmosphere of the glovebox. A second crop was obtained from partial evaporation of the filtrate followed by filtration of the additional yellow powder that precipitated. Combined Yield = 0.275 g (62%). Electrospray mass spectrometry (in MeOH/H2O) gave a peak at m/z = 421 corresponding to (Fe(3)(OH)(OCH3))2+. Anal. Calc. for [Fe(C18H31N5)]Cl2 • 0.5 H2O: C 47.70, H 7.12, N 15.45; found C 47.58, H 7.46, N 15.31.[Mn(3)Cl]PF6 and [Cu(3)][PF6]2: The general procedure for [Fe(3)]Cl2 above was followed, however, these reactions gave little or no precipitation. The solutions were filtered to remove trace solids, which were discarded. The filtrates were then evaporated under vacuum to give crude [M(3)] chloride salts that was dissolved in a minimum MeOH in the glovebox. To these solutions were added 0.815 g (0.005 mol, 5 eq.) of NH4PF6 likewise dissolved in a minimum of MeOH. Precipitation of the PF6 salt products was immediate, but the suspensions were allowed to stir approximately 1 h to complete precipitation. The solid products were filtered off, washed with diethyl ether, and allowed to dry overnight open to the glovebox atmosphere.[Mn(3)Cl]PF6: Yield = 0.393 g (71%) of white powder. Electrospray mass spectrometry gave peaks at m/z = 393 corresponding to Mn(3)(H2O)+ and m/z = 421 corresponding to (Mn(3)(CH3OH)(H2O)+. Anal. Calc. for [Mn(C18H31N5)Cl]PF6 • 3 H2O: C 35.62, H 6.15, N 11.54; found C 35.62, H 6.10, N 11.67.[Cu(3)][PF6]2 Yield = 0.457 g (68%) of blue powder. Electrospray mass spectrometry gave peaks at m/z = 379 corresponding to (Cu(3))+ and m/z = 190 corresponding to (Cu(3))2+. Anal. Calc. for [Cu(C18H31N5)][PF6]2 • 0.2 H2O: C 32.05, H 4.69, N 10.38; found C 31.69, H 4.66, N 10.32. X-ray quality crystals were obtained from the diffusion of ether into an acetone solution.
- 42.Le Baccon M, Chuburu F, Toupet L, Handel H, Soibinet M, Deschamps-Olivier I, Barbier JP, Aplincourt M. New J Chem. 2001;25:1168–1174. [Google Scholar]
- 43.X-ray Crystallographic Details: Single crystal X-ray diffraction data were collected in series of ω-scans using a Stoe IPSD2 image plate diffractometer utilising monochromated Mo radiation (λ = 0.71073 Å). Standard procedures were employed for the integration and processing of the data using X-RED. [53] Samples were coated in a thin film of perfluoropolyether oil and mounted at the tip of a glass fibre located on a goniometer. Data were collected from crystals held at 150 K in an Oxford Instruments nitrogen gas cryostream.Crystal structures were solved using routine automatic direct methods implemented within SHELXS-97. [54] Completion of structures was achieved by performing least squares refinement against all unique F2 values using SHELXL-97. [54] All non-H atoms were refined with anisotropic displacement parameters. Hydrogen atoms were placed using a riding model. Where the location of hydrogen atoms was obvious from difference Fourier maps, C H bond lengths were refined subject to chemically sensible restraints.Crystal Data for H23Cl2 • 2 H2O: C18H37N5Cl2O2, Mr = 426.42, Z = 4, T = 150(2) K, Monoclinic, P 21/c, a = 17.1551(16) Å, b = 10.2884(11) Å, c = 13.0825(11) Å, α = 90°, β = 100.328(7)°, γ = 90°, V = 2271.6(4) Å3, F(000) = 920, GOF = 0.822. A total of 12083 reflections were collected, of which 4630 were unique (R(int)) = 0.0828). R1 (I>2σ(I)) = 0.0421, wR2 = 0.1297. Crystal Data for [Cu(3)][PF6]2 • C3H6O: Cu2C39H68N10OP4F24, Mr = 1399.99, Z = 4, T = 150(2) K, Orthorhombic, P c a 21, a = 17.6639(8) Å, b = 9.8148(5) Å, c = 31.3088(18) Å, α = 90°, β = 90°, γ = 90°, V = 5427.6(4) Å3, F(000) = 2856, GOF = 0.798. A total of 33224 reflections were collected, of which 9229 were unique (R(int)) = 0.0554). R1 (I>2σ(I)) = 0.0318, wR2 = 0.0569.
- 44.X-AREA v 1.64. Darmstadt: STOE & Cie GmbH; 2012. [Google Scholar]
- 45.Sheldrick G. Acta Crystallogr Sect A: Found Crystallogr. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 46.Burger K. Coordination Chemistry: Experimental Methods. London: Butterworth; 1973. [Google Scholar]
- 47.Barefield KE, Busch DH, Nelson SM. Quart Rev Chem Soc. 1968;22:457–498. [Google Scholar]
- 48.Collinson SR, Alcock NW, Hubin TJ, Busch DH. Journal of Coordination Chemistry. 2001:317–331. [Google Scholar]
- 49.El Hajj F, Sebki G, Patinec V, Marchivie M, Triki S, Handel H, Yefsah S, Tripier R, Gomez-Garcia C, Coronado E. Inorg Chem. 2009;48:10416–10423. doi: 10.1021/ic9012476. [DOI] [PubMed] [Google Scholar]
- 50.Di Vaira M, Midollini S, Sacconi L. Inorg Chem. 1977;1518-1524:16. [Google Scholar]
- 51.Sacconi L, Di Vaira M. Inorg Chem. 1978;17:810–815. [Google Scholar]
- 52.Bianchini C, Laschi F, Masi D, Ottavianci FM, Pastor A, Peruzzini M, Zanello M, Zanello P, Zanobini F. J Am Chem Soc. 1993;115:2723–2730. [Google Scholar]
- 53.Divaira M, Stoppioni PMJA. Gazzetta Chimica Italiana. 1995;125:277–281. [Google Scholar]
- 54.Hubin TJ, Alcock NW, Morton MD, Busch DH. Inorg Chim Acta. 2003;348:33–40. [Google Scholar]
- 55.Goldsmith CR, Stack TDP. Inorg Chem. 2006;45:6048–6055. doi: 10.1021/ic060621e. [DOI] [PubMed] [Google Scholar]
- 56.McGinley CM, van der Donk WA. Chem Commun. 2003:2843–2846. doi: 10.1039/b311008g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.