

Summary
Background and Objectives
Although leukocytes and platelets adhere to fibrin with alacrity in vitro, these cells do not readily accumulate on the surfaces of fibrin clots in vivo. The difference in the capacity of blood cell integrins to adhere to fibrin in vivo and in vitro is striking and implies the existence of a physiologic antiadhesive mechanism. The surfaces of fibrin clots in the circulation are continually exposed to plasma proteins, several of which can bind fibrin and influence cell adhesion. Recently, we have demonstrated that adsorption of soluble fibrinogen on the surface of a fibrin clot results in its deposition as a soft multilayer matrix, which prevents attachment of blood cells. In the present study, we demonstrate that another plasma protein, plasminogen, which is known to accumulate in the superficial layer of fibrin, exerts an antiadhesive effect.
Results
After being coated with plasminogen, the surfaces of fibrin clots became essentially non-adhesive for U937 monocytic cells, blood monocytes, and platelets. The data revealed that activation of fibrin-bound plasminogen by the plasminogen-activating system assembled on adherent cells resulted in the generation of plasmin, which decomposed the superficial fibrin layer, resulting in cell detachment under flow. The surfaces generated after the initial cell adhesion remained non-adhesive for subsequent attachment of leukocytes and platelets.
Conclusion
We propose that the limited degradation of fibrin by plasmin generated by adherent cells loosens the fibers on the clot surface, producing a mechanically unstable substrate that is unable to support firm integrin-mediated cell adhesion.
Keywords: adhesion, fibrin, fibrinolysis, integrins, plasminogen, thrombus
Introduction
Fibrin, a major component of normal hemostatic thrombus, performs an important mechanical task by sealing a breach after vascular damage. It is generally believed that fibrin serves as a highly adhesive substrate for leukocyte and platelet integrins. Indeed, in the purified system in vitro, fibrin and its mimic, immobilized fibrinogen, can support strong adhesion of leukocytes and platelets, under either static or flow conditions [1–3]. However, rather different observations have been reported regarding the relationship between the dynamics of fibrin accumulation and adhesion of blood cells following vascular injury in vivo. It was first reported by Arfors et al. [4] and Baumgartner [5], and later confirmed and extended by others [6,7], that the kinetics of platelet and fibrin deposition are different. Platelet accumulation peaks within 20–30 min of experimental arterial injury, after which time it ceases. In contrast, fibrin accumulation continues for several hours; remarkably, the surface of the clot is no longer thrombogenic. The intracoronary thrombi isolated from patients with acute myocardial infarction also exhibit dramatic differences in their cellular composition, depending on the time after the onset of the episode; whereas an early thrombus is platelet-rich and contains little fibrin, the surface of a late clot is covered with extensive fibrin and contains almost no adherent platelets and leukocytes [8]. Furthermore, it is well known in biomaterial applications that vascular prostheses prepared by a ‘preclotting method’, with a flow surface consisting of a layer of fibrin, are paradoxically hypothrombogenic [9,10]. Fibrin clots are relatively poor adhesive substrates not only for platelets but also for leukocytes, when examined in vivo or in non-fractionated blood in vitro. Studies using experimental models of thrombosis have shown that few leukocytes adhere to and invade the thrombus within the first hours following its formation [11]. Moreover, although numerous monocytes migrate into and accumulate within the fibrin matrix of venous thrombi over several days after an experimental injury, these cells appear to arrive there through the vessel wall surrounding the thrombus, but not through the lumenal surface of the clot [12]. These data suggest that the processes occurring in the superficial layer of the fibrin clot may play important roles in the control of thrombus formation under both normal hemostatic and pathologic conditions. However, the reasons for the poor adhesive properties of fibrin in vivo and antiadhesive mechanisms operating at the surface of blood clots are poorly understood.
We and others have recently shown that soluble plasma fibrinogen is a potent inhibitor of integrin-mediated leukocyte and platelet adhesion to fibrin clots and immobilized fibrinogen [2,13,14]. We have also provided evidence that fibrinogen reduces cell adhesion by binding to the fibrin(ogen) substrates [13]. Further studies revealed that an alteration of the physical properties of fibrin(ogen) surfaces was the mechanism underlying this antiadhesive effect [15]. Accordingly, the deposition of fibrinogen creates a soft multilayer substrate consisting of loosely bound fibrinogen molecules that are unable to sustain firm cell adhesion. Consequently, the cells that engage this friable matrix detach when subjected to shear stress.
Another plasma protein, plasminogen, interacts with fibrin [16,17], is present in plasma at a relatively high concentration (approximately 0.2 mg mL−1), and could potentially modify the surfaces of fibrin clots. Furthermore, the affinity of plasminogen for fibrin increases when the surface of a clot is ‘nicked’ with plasmin, resulting in the generation of new binding sites [18,19] and strong superficial accumulation of plasminogen [20,21]. As a consequence, fibrin lysis is restricted to a very narrow zone from the clot surface [21,22]. In addition to fibrin, various cells, including leukocytes and platelets, can assemble plasminogen and its activators on their surfaces [23]. Therefore, it is reasonable to assume that when these cells contact a clot, they activate fibrin-bound plasminogen to plasmin, resulting in lysis of the superficial fibrin layer and the alteration of its architecture. Hence, the plasminogen-accumulating shell could potentially influence cell adhesion. In this study, we show that binding of plasminogen to fibrin inhibits cell adhesion and that the conversion by leukocytes and platelets of fibrin-bound plasminogen into plasmin is the process preventing stable cell adhesion to a clot. Thus, the study reveals that plasminogen is not only involved in fibrin lysis but may also control cell adhesion to the clot surface.
Materials and methods
Materials
Human plasminogen (depleted of plasmin activity), plasmin, thrombin and fibrinogen, depleted of fibronectin and plasminogen, were obtained from Enzyme Research Laboratories (South Bend, IN, USA). Tissue-type plasminogen activator (t-PA) was from Calbiochem (Darmstadt, Germany). Plasminogen and fibrinogen were labeled with 125I, using IODO-GEN (Thermo Scientific Pierce Protein Research Products, Rockford, IL, USA), dialyzed against phosphate-buffered saline (PBS), and stored at −20 °C. The plasminogen concentration was determined on the basis of its extinction coefficient, which is 1.7 at 1 mg mL−1. Phenylalanyl-L-prolyl-L-arginine chlormethyl ketone (PPACK), tranexamic acid and poly(vinylpyrrolidone) were from Sigma (St Louis, MO, USA). The chromogenic plasmin substrate S-2251 was from Chromogenix Diapharma Group (Franklin, OH, USA). Calcein-AM was purchased from Molecular Probes (Eugene, OR, USA).
Cell culture
U937 monocytoid cells were obtained from the ATCC and cultured in RPMI-1640 supplemented with 10% fetal bovine serum. Human platelets were isolated as described previously [24]. Briefly, platelets were collected from fresh aspirin-free human blood in the presence of 2.8 μM prostaglandin E1, and isolated by differential centrifugation followed by gel filtration on Sepharose 2B in divalent cation-free Tyrode’s buffer (pH 7.2) containing 0.1% bovine serum albumin (BSA). Peripheral blood monocytes were isolated using a modification of the previously described method [25]. Briefly, after removal of the platelet-rich plasma, the buffy coat and red blood cells were diluted two-fold with PBS, and mononuclear cells were separated by centrifugation through Ficoll-Paque. Monocytes were isolated from the mononuclear cells by adherence to the wells of 12-well plates for 30 min at 37 °C. The non-adherent cells were removed and adherent cells were collected using the cell dissociation buffer (Cellgro Mediatech, Inc., Manassas, VA, USA). The cells were resuspended in Hank’s balanced salt solution (HBSS), washed, and counted. The protocol for blood donations was approved by the Institutional Review Board of Arizona State University.
Static cell adhesion assays
The fibrin gels were formed in siliconized wells of 96-well microtiter plates by mixing 100 μL of 2 mg mL−1 fibrinogen in HBSS with 0.25 U mL−1 thrombin for 2 h at 37 °C. The cells were labeled with 5 μM calcein for 30 min, and washed twice with HBSS + 0.1% BSA; assays were then performed as described previously [13]. Briefly, aliquots (100 μL) of labeled cells (5 × 105 mL−1) were added to the wells. After 30 min of incubation at 37 °C, the non-adherent cells were removed by two washes with PBS. Fluorescence was measured in a CytoFluorII fluorescence plate reader (Applied Biosystems, Foster City, CA, USA). For inhibition experiments, the wells containing fibrin gels were incubated with different concentrations of soluble plasminogen (treated with PPACK to block residual plasmin) or PPACK-inhibited plasmin for 15 min at 22 °C, after which protein solutions were aspirated, the gels were washed with HBSS, and cells were added. Alternatively, cells were incubated for 15 min at 22 °C with soluble plasminogen before they were added to the wells. Data are expressed as fluorescence of adherent cells.
Flow-based adhesion assays
Perfusions with U937 cells were performed as previously described [13]. Briefly, a flow chamber consisted of a transparent glass tube (50 × 2.5 mm) lined with the fibrin gel containing a fabricated 0.9-mm capillary (flow tube) inside the gel. U937 cells were resuspended at 5 × 105 or 107 mL−1 in HBSS + 0.1% BSA supplemented with 50 μg mL−1 plasminogen, and perfused through fibrin gels for 5 and 10 min at flow rates of 0.1 and 1.5 mL min−1, respectively. After these times, the flow tubes were flushed with HBSS + 0.1% BSA for an additional 9 min at the respective flow rates, and then inserted into a holder for mounting of 96-well microtiter well strips. Fluorescence was measured using a fluorescence plate reader. The wall shear rate was calculated as previously described [26]. Adherent cells within the flow tubes were visualized using a fluorescence microscope (Leica DM4000B; Leica, Bannockburn, IL, USA) or with an inverted microscope (Leica DMIL) and digital camera (Leica DFC 340 FX).
Results
Pretreatment of fibrin clots with plasminogen inhibits adhesion of U937 monocytic cells, platelets, and blood monocytes
To assess how plasminogen might influence cell adhesion to fibrin, two approaches were used. In the initial experiments, U937 monocytic cells in suspension were incubated with the physiologic concentration of plasminogen (200 μg mL−1), and cell adhesion to fibrin clots was then examined. As shown in Fig. 1A, pretreatment of cells with plasminogen resulted in blocking of adhesion (approximately 90%). As U937 cells assemble the components of the plasminogen system on their surface and convert plasminogen into plasmin [23], the antiadhesive effect arising from plasmin activity was considered. The addition of 0.5 mM PPACK to cell suspensions restored adhesion to a level near that observed with cells incubated in the absence of plasminogen (Fig. 1A). In addition, tranexamic acid, which is known to interact with lysine-binding sites of plasmin(ogen) and prevents its binding to U937 cells [27], almost completely abolished the inhibitory effect of plasminogen (Fig. 1A). In the next set of analyses, fibrin gels were first overlaid with plasminogen for 15 min at 22 °C, solutions above the gels were aspirated, the gels were washed to remove non-bound plasminogen, and adhesion of U937 cells was examined. Pretreatment of fibrin with plasminogen (200 μg mL−1) produced approximately 85% inhibition of cell adhesion as compared with non-treated gels (Fig. 1B). PPACK added to cell suspensions at 0.5 mM restored adhesion to the levels obtained with non-treated fibrin gels, and tranexamic acid added during the incubation of plasminogen with fibrin gels abolished the inhibitory effect of plasminogen. Thus, by two approaches, the fluid phase (Fig. 1A) and the solid phase (Fig. 1B), plasminogen was capable of blocking cell adhesion. These results suggested that a potent antiadhesive effect of plasminogen may depend on the generation of plasmin. Indeed, pretreatment of fibrin clots with active plasmin for 15 min resulted in the loss of cell adhesion (Fig. 1C), and its inactivation with PPACK eliminated this effect. Thus, the conversion of fibrin-bound plasminogen into plasmin by adherent cells probably contributes to the loss of cell adhesion.
Fig. 1.
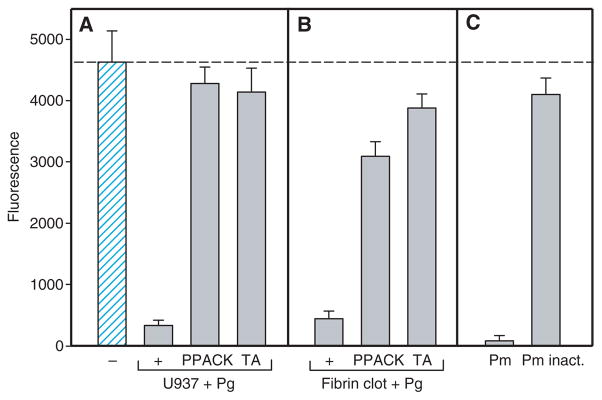
Effect of plasmin(ogen) on adhesion of U937 cells to fibrin clots. (A) U937 cells (5 × 105 mL−1) were first preincubated with 200 μg mL−1 plasminogen (+, Pg) for 15 min at 22 °C without or with tranexamic acid (TA, 5.0 mM) or phenylalanyl-L-prolyl-L-arginine chlormethyl ketone (PPACK) (0.5 mM), and aliquots of cells were added to intact fibrin gels. Cell adhesion was determined as described in Materials and methods. A hatched bar shows adhesion of control untreated U937 cells to intact fibrin gels. (B) Plasminogen (+, 200 μg mL−1) without or with tranexamic acid (5.0 mM) was preincubated with fibrin gels for 15 min at 22 °C. The liquid above the gels was aspirated, and the gels were washed with Hank’s balanced salt solution (HBSS); this was followed by the addition of a suspension of calcein-labeled U937 cells in the absence or presence of PPACK (0.5 mM). (C) Five micrograms per milliliter of active plasmin (Pm) or PPACK-inactivated plasmin (Pm inact.) was preincubated with fibrin gels for 15 min, the liquid above the gels was aspirated, the gels were washed, and adhesion of calcein-labeled U937 cells was examined. The lack of proteolytic activity in PPACK-inactivated plasmin was confirmed by using the plasmin-specific substrate S-2251. A dotted line, which shows control adhesion of U937 cells in (A), also applies to control cell adhesion in (B) and (C). The data shown are the average of six measurements at each experimental data point and are representative of four experiments.
Figure 2A shows that the antiadhesive effect of plasminogen added to the fibrin clot was concentration-dependent, with an IC50 of approximately 45 μg mL−1 (0.5 μM). This concentration corresponds to the range of Kd values (0.3–1.2 μM) previously reported for the interaction of plasminogen with fibrin clots [28] or with fibrin immobilized on plastic [29]. The amount of plasminogen bound to the surface of fibrin, assessed with 125I-labeled protein, correlated with its antiadhesive effect (Fig. 2B). Furthermore, the concentration of tranexamic acid required to obtain a 50% reversal of the effect of plasminogen was approximately 0.2 mM (Fig. 2A, inset), which is close to the Kd (0.22 mM) for the interaction of tranexamic acid with plasminogen lysine-binding sites [30].
Fig. 2.

Effect of different concentrations of plasminogen (Pg) on adhesion of U937 cells to fibrin gel. (A) Fibrin gels were preincubated with different concentrations of plasminogen for 15 min, washed with Hank’s balanced salt solution (HBSS) to remove non-bound protein, and used as substrates for adhesion of calcein-labeled U937 cells. Inset: plasminogen (200 μg mL−1) was added to fibrin gels in the presence of different concentrations of tranexamic acid, gels were washed, and cell adhesion was determined. (B) Correlation between the amount of plasminogen bound to the fibrin gel and cell adhesion. Solutions of 125I-labeled plasminogen (100 μL, 50 μg mL−1, 800 cpm μg−1) were incubated with fibrin gels formed in the wells of 96-well microtiter plates for the indicated times. The wells were washed with HBSS, and calcein-labeled U937 cells were added. Bound plasminogen (closed circles) and cell adhesion (open circles) were determined as described in Materials and methods.
In addition to leukocytes, platelets adhere to fibrin via an integrin-dependent mechanism and assemble the components of the plasminogen system, including plasminogen and t-PA [31]. Therefore, we have sought to examine whether activation of fibrin-bound plasminogen by platelets can alter their adhesion. Platelets isolated by gel filtration from freshly drawn blood adhered firmly to untreated fibrin gels (Fig. 3A). However, coating of the fibrin gel with plasminogen did not decrease platelet adhesion (Fig. 3A). This is in marked contrast to U937 cells, whose adhesion was abolished by this procedure (Fig. 3B; see also Fig. 1). To understand the difference between adhesion of platelets and that of U937 cells, we measured plasmin activity generated during adhesion of these cells to plasminogen-treated fibrin. As assessed with S-2251, whereas plasmin was generated during the incubation of U937 cells with plasminogen, no activity was detected with platelets isolated by gel filtration (Fig. 3A,B) as well as with platelets centrifuged directly from the platelet-rich plasma (not shown). However, the addition of t-PA (0.1 μg mL−1) to platelet suspensions resulted in the generation of plasmin and inhibition of platelet adhesion to plasminogen-coated fibrin (Fig. 3A). As the generation of plasmin activity by isolated platelets required the addition of t-PA, we considered whether isolated peripheral blood monocytes depend on the exogenously added t-PA to activate fibrin-bound plasminogen. In initial experiments, we verified that isolated monocytes did not activate plasminogen. Furthermore, in the presence of plasminogen, these cells adhered to fibrin gels to the same extent as control cells (in the absence of plasminogen) (Fig. 4A). Next, suspensions of monocytes were preincubated with t-PA (0.1 μg mL−1), diluted with HBSS, and centrifuged. Monocytes were resuspended in the buffer containing plasminogen (100 μg mL−1), and their adhesion to fibrin gels was determined. As shown in Fig. 4A, coinciding with the production of plasmin was the loss of monocyte adhesion. To corroborate the lack of adhesion with conversion of fibrin-bound plasminogen into plasmin, monocytes adherent for 30 min were removed by washing, and the plasmin substrate S-2251 was added to fibrin gels. Exposure of fibrin-bound plasminogen to the t-PA-bearing monocytes resulted in the generation of plasmin, which remained bound to fibrin (Fig. 4B). These results indicate that monocytes and platelets are capable of activating fibrin-bound plasminogen to plasmin, which reduces adhesion of these blood cells to the surfaces of clots.
Fig. 3.
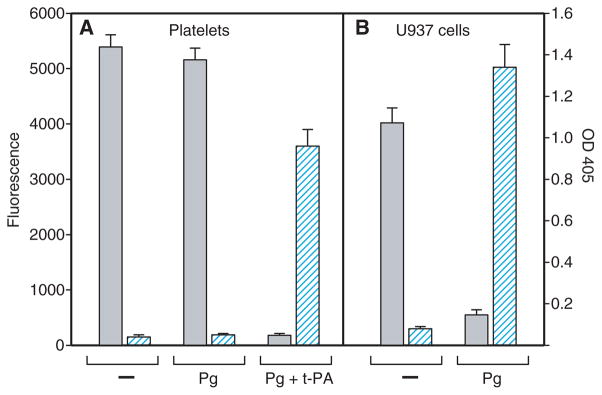
Relationship between adhesion of platelets and U937 cells to plasminogen-coated fibrin gels and their ability to activate plasminogen. Suspensions of platelets (5 × 105 per 0.1 mL) (A) or U937 cells (5 × 104 per 0.1 mL) (B) were added to untreated (−) or plasminogen (Pg)-coated fibrin gels, and cell adhesion was determined as described in Materials and methods. Gray bars show the fluorescence of adherent cells (left ordinate). Hatched bars (right ordinate) show the plasmin activity of platelets and U937 cells incubated with or without plasminogen. Aliquots of cells (106 per 0.2 mL) were incubated for 60 min without or with Pg (10 μg mL−1), the samples were centrifuged, and the generated plasmin was determined by measuring the optical density (OD) at 405 nm with the plasmin substrate S-2251. In additional experiments, suspended platelets were preincubated with tissue-type plasminogen activator (t-PA) (0.1 μg mL−1) and then added to plasminogen-coated fibrin gels (Pg + t-PA). Data are means of triplicate determinations, and error bars represent standard errors. The result shown is representative of three independent experiments.
Fig. 4.

Inhibition of adhesion of peripheral blood monocytes to fibrin clots as a consequence of plasmin generation. (A) Suspensions of calcein-labeled monocytes were preincubated for 15 min with tissue-type plasminogen activator (t-PA) (0.1 μg mL−1), diluted five-fold with Hank’s balanced salt solution, and centrifuged. Cells were resuspended in the same buffer containing plasminogen (Pg) (100 μg mL−1), and aliquots of cells (5 × 104 per 0.1 mL) were added to fibrin gels (Pg + t-PA). Control cells without (−) or in the presence of plasminogen are shown. Cell adhesion was determined as described in Materials and methods. Gray bars show the fluorescence of adherent cells (left ordinate). Hatched bars (right ordinate) show the plasmin activity of monocytes incubated with or without plasminogen. Plasmin activity was determined as in Fig. 3. (B) Plasmin activity associated with fibrin gels after cell adhesion is shown. Monocytes (treated with t-PA or not) were mixed with plasminogen as described in (A), allowed to adhere to fibrin clots for 30 min, and then removed by washing. The activity of fibrin-bound plasmin was determined by measuring the optical density (OD) at 405 nm with the plasmin substrate S-2251.
Effect of plasminogen on cell adhesion under flow
To demonstrate the effect of plasminogen under permanent shear forces, a situation more relevant to physiologic conditions, we used a flow adhesion assay. In this assay, cells are perfused through a flow chamber consisting of a capillary preformed within the fibrin gel. The fibrin tubes were attached to the perfusion system, and calcein-labeled U937 cells resuspended in HBSS + 0.1% BSA containing 50 μg mL−1 plasminogen were perfused at a flow rate of 0.1 or 1.5 mL min−1 (corresponds to wall shear rates of 23.1 and 350 s−1) at 37 °C. After 5 min, the tubes were flushed with HBSS + 0.1% BSA for 9 min at the respective flow rate to remove non-adherent cells. Cell adhesion was observed in a microscope (Fig. 5A, shown for 23.1 s−1) and quantified by measuring fluorescence (Fig. 5B, shown for 350 s−1). With both flow rates, the plasminogen coat markedly reduced cell adhesion (> 95%). In fact, after washing, fibrin gels perfused with plasminogen retained few cells. The effect of plasminogen on adhesion of U937 cells is also shown in a video that, for demonstrative purposes, was produced at a flow rate of 0.1 mL min−1 (Video S1).
Fig. 5.

Effect of plasminogen on adhesion of U937 cells to fibrin gel under flow conditions. The fibrin gels containing the inner capillary were prepared in glass tubes as described in Materials and methods. U937 cells were resuspended at 5 × 105 mL−1 (A) or 107 mL−1 (B) in Hank’s balanced salt solution (HBSS)–bovine serum albumin without (control) or with 50 μg mL−1 plasminogen and perfused through fibrin tubes for 5 min at a flow rate of 0.1 mL min−1 (A) and for 10 min at a flow rate of 1.5 mL min−1 (B). After these times, the tubes were flushed for an additional 9 min at the respective flow rates to remove non-adherent cells (open arrow). Adherent cells were viewed with a Leica DMIL microscope with a × 10 objective and a Leica DFC 340 FX digital camera (A). To quantify cell adhesion (B), calcein-labeled U937 cells were perfused through fibrin tubes at 1.5 mL min−1 in the absence (control) or presence of plasminogen (Pg). The fluorescence of adherent cells was determined using a CytoFluor plate reader. The results shown are from two separate experiments performed with quadruplicate measurements for each flow tube.
Lysis of the superficial fibrin layer renders the clot non-adhesive
The above experiments suggested that cell attachment to fibrin-bound plasminogen causes rapid generation of plasmin, which may degrade the surface of a fibrin clot and render it non-adhesive. Figure 6A shows that the surface of the fibrin clot that was made non-sticky by plasmin generated by adherent U937 cells remained non-adhesive for the second round of adhesion of U937 cells and platelets, even in the presence of PPACK or tranexamic acid. This is in contrast to the experiments illustrated in Fig. 1, in which PPACK and tranexamic acid eliminated the inhibitory effect of plasminogen during the first round of adhesion. Also, washed platelets, which do not generate plasmin in the absence of t-PA, also did not adhere to the ‘postadhesion’ gels. These results suggested that during the second round of adhesion, plasmin per se was not responsible for the antiadhesive effect. Furthermore, the data indicated that plasmin-mediated cleavage of cell surface integrins did not contribute to the defective cell adhesion. Therefore, we considered that the structural alterations of the surface produced by cells during the first round of adhesion might result in the inability of clots to support subsequent cell adhesion. To assess the extent of proteolysis, we determined the amount of fibrin cleaved by plasmin. Fibrin gels were prepared from 125I-labeled fibrinogen in the wells of 24-well plates, and non-polymerized fibrinogen was removed by extensive washing with PBS. The fibrin gels were then coated with plasminogen, and U937 cells were allowed to adhere for 30 min at 37 °C. The amount of digested fibrin was estimated by measuring radioactivity released in the solution above the gels during adhesion (0.14% of total [125I]fibrin). Furthermore, additional radioactivity was extracted from the gels using 0.06% sodium dodecylsulfate (SDS) (0.07% of total [125I]fibrin). The results demonstrated that only approximately 0.2% of total [125I]labeled fibrin could be extracted from the gel as a result of proteolysis by adherent cells. Thus, plasmin generated by adherent cells appears to produce only minor decomposition of the surface.
Fig. 6.

Initial adhesion of U937 cells to plasminogen-coated gels renders them non-adhesive for subsequent cell adhesion. (A) To produce non-adhesive fibrin gels, non-labeled U937 cells were allowed to adhere to plasminogen-treated fibrin for 30 min at 37 °C (first adhesion). The cells were completely removed by washing gels with phosphate-buffered saline (PBS), and the gels were used for the second round of adhesion. Calcein-labeled U937 cells (left panel) and platelets (right panel) in the absence (−) or in the presence of phenylalanyl-L-prolyl-L-arginine chlormethyl ketone (PPACK, 0.2 mM) and tranexamic acid (TA, 5.0 mM) were added to the postadhesion fibrin gels. After 30 min at 37 °C, cell adhesion was measured (gray bars). Adhesion of control U937 cells and platelets to intact fibrin gels untreated with plasminogen was assigned a value of 100% (closed bars). (B) Restoration of the adhesive properties of fibrin gels. The surfaces of fibrin gels were first rendered non-adhesive as in (A). Different concentrations of sodium dodecylsulfate (SDS) (0–0.08%) were then added to the gels for 20 min at 22 °C. Gels were washed extensively with PBS, and the second round of adhesion of calcein-labeled U937 cells in the presence of PPACK was performed. When non-adhesive surfaces were cleaned with increasing concentrations of SDS, adhesion was gradually restored to the level observed with untreated fibrin gels (shown as a dashed line). As a control, rinsing the intact fibrin gels with 0.08% SDS did not alter cell adhesion.
To examine the possibility that after the first round of adhesion the surface of fibrin becomes non-adhesive as a result of the generation of degradation products loosely connected with the bulk of the fibrin matrix, the ‘postadhesion’ gels were rinsed with SDS and washed with PBS, and U937 cells were added again. As shown in Fig. 6B, the adhesiveness of fibrin clots was restored. Adhesion of cells (added in the presence of PPACK) progressively increased after the ‘postadhesion’ fibrin gels were cleaned with increasing concentrations of SDS (0–0.08%). To confirm that SDS removes degradation products, the liquid above the ‘postadhesion’ gels was collected and analyzed by SDS polyacrylamide gel electrophoresis. The analyses revealed high molecular weight fibrin degradation products in the material eluted from the gels (not shown). As a control, the incubation of intact gels with 0.08% SDS did not modify cell adhesion (Fig. 6B). Therefore, SDS is unlikely to modify the surface of fibrin, resulting in exposure of additional binding sites for cells. Thus, the data suggest that the surface of plasminogen-treated fibrin becomes non-adhesive as a result of its alteration by plasmin generated by cells that were in contact with this surface.
Discussion
In previous studies, we demonstrated that binding of soluble fibrinogen to and its deposition on the surfaces of fibrin clots or immobilized fibrinogen produces a strong antiadhesive effect and prevents accumulation of leukocytes and platelets under both static and flow conditions [13,15]. The major finding of this study is that another plasma protein, plasminogen, produces a phenotypically similar effect. However, in contrast to fibrinogen, plasminogen exerts its antiadhesive effect by a proteolytic mechanism. We show that when plasminogen binds its biological substrate, fibrin, the antiadhesive effect results from the conversion of plasminogen into plasmin by adherent cells. Moreover, plasminogen protects fibrin clots from adhesion of both leukocytes and platelets, blood cells known to assemble the components of the fibrinolytic system on their surfaces [23]. The inhibitory effect of fibrin-bound plasminogen arises from the decomposition of a superficial layer of the clot surface, which loses its ability to sustain firm adhesion of cells under flow. These studies identify plasminogen as a second protein that can exert an antiadhesive effect and further define the mechanisms whereby plasma proteins function as antiadhesive molecules.
The loss of cell adhesion produced by fibrin-bound plasmin(ogen) resulted from degradation of a very small fraction of fibrin (approximately 0.2%) that occurred at the clot surface apparently in direct contact with adherent cells. Remarkably, the surfaces produced by initial cell adhesion remain non-adhesive for subsequent attachment of leukocytes and platelets (Fig. 6A). These surfaces evidently lose their adhesive properties because they contain partially degraded fibers loosely attached to the bulk of the fibrin network, and thus present a mechanically unstable substrate for cellular integrins. When the surfaces of fibrin clots were washed with low concentrations of SDS, cell adhesion was completely restored to the level observed with non-treated fibrin (Fig. 6B). The regeneration of the surface apparently resulted from the removal of degradation products and restoration of the substrate in which all fibers are interconnected within the stable network. Previous studies, performed in a cell-free system in which lysis was induced by t-PA added from outside a clot, demonstrated that degradation was spatially restricted to a very narrow zone (5–8 μm) from the clot surface [21]. Examination of the dynamics of fibrin clot lysis by confocal fluorescence microscopy showed that this superficial zone of lysis consisted of mobile fibers that formed friable agglomerates on the clot surface [21]. Fiber bundle aggregates generated by plasmin have also been observed by scanning electron microscopy [32]. Conceivably, a similar agglomerated material was formed on the surfaces of fibrin clots by plasmin generated by adherent cells in our experiments. Sakharov et al. [20,21] have found that plasminogen accumulated in the superficial layers of lysing clots at a concentration that was about 30-fold higher than its concentration in the surrounding plasma. Furthermore, substantial accumulation of plasminogen at the flow surface of mural fibrin in vivo has also been reported [22]. On the basis of our data, it is reasonable to propose that the mobile plasminogen shell formed on the surfaces of fibrin clots may control cell adhesion and prevent excessive accumulation of leukocytes and platelets.
The activation of fibrin-bound plasminogen has been observed with U937 human monocytoid cells, as well as with isolated peripheral blood monocytes. However, in contrast to U937 cells, isolated blood monocytes generated plasmin only in the presence of added t-PA. Likewise, platelets activated fibrin-bound plasminogen only after they had been supplemented with t-PA. U937 cells, which are widely used to study the regulation of the fibrinolytic system of monocytes, produce plasminogen activators and express urokinase plasminogen activator receptor; thus, they may be more efficient at generating plasmin than platelets and blood monocytes. Furthermore, plasma levels of t-PA are low (1–9 ng mL−1) under normal physiologic conditions [33,34] and become high only after venous occlusion or after the administration of agents that induce its acute secretion from endothelial cells [34]. As isolated blood monocytes and platelets bind t-PA with low affinity (Kd values in the range 0.3–0.9 μM) [35,36]) these cells are unlikely to bind t-PA under basal conditions and/or retain it after cell isolation. It has been proposed that acute release of t-PA from endothelial cells near the forming thrombus may result in a local concentration that is much higher than the basal level of t-PA and also than that of its inhibitor plasminogen activator inhibitor-1 (PAI-1), and will therefore accelerate thrombolysis [34]. The locally secreted t-PA may also bind to monocytes and platelets, leading to activation of plasminogen deposited on the surface of fibrin by adherent cells. It is noteworthy that previous studies evaluated separately either the mechanisms that regulate the assembly of the plasminogen system on blood cells or the activation of fibrin-bound plasminogen by t-PA. However, the interplay between adherent cells, the fibrin surface and the plasminogen system remains unknown.
Even though the antiadhesive effects of two fibrin-binding proteins, plasminogen and fibrinogen [13], involve proteolytic and non-proteolytic mechanisms, respectively, which would seem to make them different, they are thought to be based on a common underlying mechanism: the alteration of the physical properties of the fibrin clot surface. It is now well established that the physical properties of substrates, that is, soft vs. rigid, are sensed by cells [37]. This mechanosensing is transduced into intracellular signaling that results in differential cell adhesion. We have recently demonstrated that binding of fibrinogen to various surfaces results in its deposition as a multilayer material that alters the physical properties of the substrate, making it mechanically soft [15]. This ‘softening’ of the substrate reduces forces developed by integrins and leads, in turn, to insufficient signaling and the inability of cells to spread and adhere firmly. The generation by plasmin of the cleaved fibrils loosely bound to the surface of the fibrin network might result in the same outcome. Accordingly, when integrins pull on the severed fibers, these structures may yield, resulting in a weak rigidity response and weak signaling. Consequently, cells adherent to this soft matrix are not able to attach firmly and are washed away by flow. Thus, the surface of fibrin clots modified by fibrinogen and/or plasmin may potentially regulate integrin-mediated mechanosensing of blood cells and their responses.
Numerous studies in animals have demonstrated that the dynamics of platelet and fibrin accumulation after experimental arterial injury are very different. Platelet accumulation at a lesion of a blood vessel wall peaks within 20–30 min of injury, and following this initial phase, the injury site is no longer thrombogenic for platelets [4–7]. By contrast, fibrin deposition continues over several hours, resulting in the complete coverage of the initial platelet aggregate with no accumulation of platelets on the surface of the clot. Very similar morphology and apparently similar dynamics of platelet and fibrin deposition have been reported in patients with acute myocardial infarction [8]. In addition, studies using experimental models of thrombosis have documented that leukocytes, especially monocytes, do not readily accumulate on the surfaces of fibrin clots, at least within the first hours following their formation [11,12]. The reasons for the loss of reactivity of the vessel surface for platelets and leukocytes following the initial injury have not been defined. We propose that the gradual accumulation of fibrin may be responsible for this effect. The antiadhesive mechanisms operating at the surface of the fibrin clot may be beneficial for maintaining the stability of clots, which is critical in early hemostasis. Indeed, since phagocytic leukocytes contain potent fibrinolytic proteases, their rapid adhesion to the highly adhesive fibrin matrix could be detrimental to the stability of initial clots. Therefore, under physiologic conditions, fibrin-bound plasminogen may limit the accumulation of leukocytes at the flow surfaces of blood clots. Likewise, uncontrolled adhesion of platelets to fibrin would result in the unlimited expansion of the thrombus. The findings of the present study that activation of fibrin-bound plasminogen by adherent blood cells prevents cell adhesion, in conjunction with our previous data showing that binding of soluble fibrinogen limits cell adhesion, raise the intriguing idea that these processes may represent the elements of a physiologic antiadhesive mechanism guarding clots from excessive cell accumulation.
On another front, there are a wide variety of pathophysiologic circumstances that could counteract this protective mechanism. Among them, the changes in the level and activity of various molecules constituting the plasminogen system can influence fibrinolysis and, consequently, alter the qualities of the fibrin surface, shifting the balance towards cell accumulation. An illustrative example may be PAI-1, whose elevated levels are often found in patients with venous and arterial thrombosis (Reviewed in [38]). Studies in PAI-1-deficient mice have indicated that PAI-1 has important roles in the control of thrombosis [39]. Another example is thrombin-activatable fibrinolysis inhibitor (TAFI), which removes C-terminal lysines from partially degraded fibrin and cells, thereby preventing plasminogen and t-PA binding. High levels of TAFI are often associated with venous and arterial thrombosis [38]. Moreover, as well as its activators and inhibitors, plasminogen itself may play roles. It is interesting that deficiency of plasminogen resulted in a several-fold greater retention of neutrophils within both arterial and venous thrombi generated by experimental vascular injury [40]. It is also noteworthy that the inhibitory effect of Apo(a), a component of Lp(a) and a risk factor for atherothrombosis, was suggested to result from its competition with plasminogen for binding to fibrin [41]. In recent years, numerous studies have linked the impaired fibrinolytic function with venous and arterial thrombosis [38]. The antiadhesive plasmin(ogen)-dependent mechanism identified in this study may play important roles in normal hemostasis. Furthermore, its disturbance may contribute to thrombotic disease. It is obvious that more studies are necessary to clarify the antiadhesive role of fibrin-bound plasminogen identified in this study and its relationship with other processes that may contribute to the antiadhesive properties of fibrin clots in the circulation.
Supplementary Material
Acknowledgments
We are grateful to V. Novokhatny for valuable discussions during the course of these studies.
Footnotes
Disclosure of Conflict of Interests
This work was supported by the National Institutes of Health.
Additional Supporting Information may be found in the online version of this article:
Video S1. Effect of plasminogen on adhesion of U937 cells to fibrin gel under flow (0.1 ml min−1).
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Kuijper PHM, Torres HIG, van der Linden JAM, Lammers JWJ, Sixma JJ, Zwaginga JJ, Koenderman L. Neutrophil adhesion to fibrinogen and fibrin under flow conditions is diminished by activation and L-selectin shedding. Blood. 1997;89:2131–8. [PubMed] [Google Scholar]
- 2.Kuijper PHM, Torres G, Lammers J-WJ, Sixma JJ, Koenderman L, Zwaginga JJ. Platelet and fibrin deposition at the damaged vessel wall: cooperative substrates for neutrophil adhesion under flow conditions. Blood. 1997;89:166–75. [PubMed] [Google Scholar]
- 3.Hantgan RR, Endenburg SC, Cavero I, Marguerie G, Uzan A, Sixma JJ, de Groot PG. Inhibition of platelet adhesion to fibrin(ogen) in flowing whole blood by arg–gly–asp and fibrinogen gamma-chain carboxy terminal peptides. Thromb Haemost. 1992;68:694–700. [PubMed] [Google Scholar]
- 4.Arfors K-E, Dhall DP, Engeset J, Hint H, Matherson NA, Tangen O. Biolaser endothelial trauma as a means of quantifying platelet activity in vivo. Nature. 1968;218:887–8. [Google Scholar]
- 5.Baumgartner HR. The role of blood flow in platelet adhesion, fibrin deposition, and formation of mural thrombi. Microvasc Res. 1973;5:167–79. doi: 10.1016/0026-2862(73)90069-1. [DOI] [PubMed] [Google Scholar]
- 6.Groves HM, Kinlough-Rathbone RL, Richardson M, Jorgensen L, Moore S. Thrombin generation and fibrin formation following injury to rabbit neointima. Lab Invest. 1982;46:605–12. [PubMed] [Google Scholar]
- 7.van Ryn J, Lorenz M, Merk H, Buchanan M, Eisert WG. Accumulation of radiolabeled platelets and fibrin on the carotid artery of rabbits after angioplasty: effects of heparin and dipyridamole. Thromb Haemost. 2003;90:1179–86. doi: 10.1160/TH03-05-0305. [DOI] [PubMed] [Google Scholar]
- 8.Beygui F, Collet JP, Nagaswami C, Weisel JW, Montalescot G. Architecture of intracoronary thrombi in ST-elevation acute myocardial infarction. Time makes difference. Circulation. 2006;113:e21–3. doi: 10.1161/CIRCULATIONAHA.105.551705. [DOI] [PubMed] [Google Scholar]
- 9.Yates SG, D’Sa A, Berger K, Fernandes LG, Wood SJ, Rittenhouse EA, Davis CC, Mansfield PB, Sauvage LR. The preclotting of porous arterial prostheses. Ann Surg. 1978;188:611–22. doi: 10.1097/00000658-197811000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patel M, Arnell RE, Sauvage LR, Wu HD, Shi Q, Wechezak AR, Mungin D, Walker M. Experimental evaluation of ten clinically used arterial prostheses. Ann Vasc Surg. 1992;6:244–51. doi: 10.1007/BF02000270. [DOI] [PubMed] [Google Scholar]
- 11.van Aken PJ, Emeis JJ. Organization of experimentally induced arterial thrombosis in rats: the first six days. Artery. 1982;11:156–73. [PubMed] [Google Scholar]
- 12.McGuiness CL, Humphries J, Waltham M, Burnand KG, Collins M, Smith A. Recruitment of labelled monocytes by experimental venous thrombi. Thromb Haemost. 2001;85:1018–24. [PubMed] [Google Scholar]
- 13.Lishko VK, Burke T, Ugarova TP. Anti-adhesive effect of fibrinogen: a safeguard for thrombus stability. Blood. 2007;109:1541–9. doi: 10.1182/blood-2006-05-022764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Endenburg SC, Lindeboom-Blokzijl L, Zwaginga JJ, Sixma JJ, de Groot PG. Plasma fibrinogen inhibits platelet adhesion in flowing blood to immobilized fibrinogen. Arterioscler Thromb Vasc Biol. 1996;16:633–8. doi: 10.1161/01.atv.16.5.633. [DOI] [PubMed] [Google Scholar]
- 15.Podolnikova NP, Yermolenko IS, Fuhrmann A, Lishko VK, Magonov S, Bowen B, Enderlein J, Podolnikov A, Ros R, Ugarova TP. Control of integrin αIIbβ3 outside-in signaling and platelet adhesion by sensing the physical properties of fibrin(ogen) substrates. Biochemistry. 2009;49:68–77. doi: 10.1021/bi9016022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thorsen S. Differences in the binding to fibrin of native plasminogen and plasminogen modified by proteolytic degradation influence of omega-aminocarboxylic acids. Biochim Biophys Acta. 1975;393:55–65. doi: 10.1016/0005-2795(75)90216-0. [DOI] [PubMed] [Google Scholar]
- 17.Lucas MA, Fretto LJ, McKee PA. The binding of human plasminogen to fibrin and fibrinogen. J Biol Chem. 1983;258:4249–56. [PubMed] [Google Scholar]
- 18.Suenson E, Lutzen O, Thorsen S. Initial plasmin-degradation of fibrin as a basis of a positive feed-back mechanism in fibrinolysis. Eur J Biochem. 1984;140:513–22. doi: 10.1111/j.1432-1033.1984.tb08132.x. [DOI] [PubMed] [Google Scholar]
- 19.Tran-Thang C, Kruithof EK, Atkinson J, Bachmann F. High-affinity binding sites for human Glu-plasminogen unveiled by limited plasmic degradation of human fibrin. Eur J Biochem. 1986;160:599–604. doi: 10.1111/j.1432-1033.1986.tb10080.x. [DOI] [PubMed] [Google Scholar]
- 20.Sakharov DV, Rijken DC. Superficial accumulation of plasminogen during plasma clot lysis. Circulation. 1995;92:1883–90. doi: 10.1161/01.cir.92.7.1883. [DOI] [PubMed] [Google Scholar]
- 21.Sakharov DV, Nagelkerke JF, Rijken DC. Rearrangements of the fibrin network and spacial distribution of fibrinolytic components during plasma clot lysis. J Biol Chem. 1996;271:2133–8. doi: 10.1074/jbc.271.4.2133. [DOI] [PubMed] [Google Scholar]
- 22.Sasajima T, Takano Y, Hiraishi Y, Goh K, Inaba M, Azuma N, Sasajima Y, Yamazaki K, Yamamoto H. High accumulation of plasminogen and tissue plasminogen activator at the flow surface of mural fibrin in the human arterial system. J Vasc Surg. 2000;32:374–82. doi: 10.1067/mva.2000.105677. [DOI] [PubMed] [Google Scholar]
- 23.Plow EF, Redlitz A, Hawley SB, Xue S, Herren T, Hoover-Plow JL, Miles LA. Assembly of the plasminogen system on cell surfaces. In: Bachmann F, editor. Fibrinolytics and Antifibrinolytic. Berlin: Springer-Verlag; 2000. pp. 141–70. [Google Scholar]
- 24.Ugarova TP, Budzynski AZ, Shattil SJ, Ruggeri ZM, Ginsberg MH, Plow EF. Conformational changes in fibrinogen elicited by its interaction with platelet membrane glycoprotein GPIIb–IIIa. J Biol Chem. 1993;268:21080–7. [PubMed] [Google Scholar]
- 25.Schober JM, Chen N, Grzeszkiewicz T, Emeson EE, Ugarova TP, Ye RD, Lau LF, Lam SCT. Identification of integrin αMβ2 as an adhesion receptor on peripheral blood monocytes for Cyr61 and connective tissue growth factor, immediate-early gene products expressed in atherosclerotic lesions. Blood. 2002;99:4457–65. doi: 10.1182/blood.v99.12.4457. [DOI] [PubMed] [Google Scholar]
- 26.Olson JD, Zaleski A, Herrimann D, Flood PA. Adhesion of platelets to purified solid-phase von Willebrand factor: effects of wall shear rate, ADP, thrombin, and risrocetin. J Lab Clin Med. 1989;14:6–18. [PubMed] [Google Scholar]
- 27.Ellis V, Behrendt N, Dano K. Cellular receptor for urokinase-type plasminogen activator: function in cell-surface proteolysis. Methods Enzymol. 1993;223:223–33. doi: 10.1016/0076-6879(93)23048-r. [DOI] [PubMed] [Google Scholar]
- 28.TranThang C, Kruithof EKO, Bachman F. Tissue-type plasminogen activator increases the binidng of glu-plasminogen to clots. J Clin Invest. 1984;74:2009–16. doi: 10.1172/JCI111623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fleury V, Angles-Cano E. Characterization of the binding of plasminogen to fibrin surfaces: the role of carboxy-terminal lysines. Biochemistry. 1991;30:7630–8. doi: 10.1021/bi00244a035. [DOI] [PubMed] [Google Scholar]
- 30.Takada A, Sugawara Y, Takada Y. Degradation of Glu- and Lys-plasminogen by elastase in the presence of absence of tranexamic acid. Thromb Res. 1988;50:285–94. doi: 10.1016/0049-3848(88)90229-0. [DOI] [PubMed] [Google Scholar]
- 31.Miles LA, Plow EF. Binding and activation of plasminogen on the platelet surface. J Biol Chem. 1985;260:4303–11. [PubMed] [Google Scholar]
- 32.Veklich Y, Francis CW, White J, Weisel JW. Structural studies of fibrinolysis by electron microscopy. Blood. 1998;12:4721–9. [PubMed] [Google Scholar]
- 33.Hashimoto Y, Kobayashi A, Yamazaki N, Sugawara Y, Takada Y, Takada A. Relationship between age and plasma t-PA, PA-inhibitor, and PA activity. Thromb Res. 1987;46:625–33. doi: 10.1016/0049-3848(87)90264-7. [DOI] [PubMed] [Google Scholar]
- 34.van den Eijnden-Schrauwen Y, Kooistra T, de Vries REM, Emeis JJ. Studies on the acute release of tissue-type plasminogen activator from human endothelial cells in vitro and in rats in vivo: evidence for a dynamic storage pool. Blood. 1995;85:3510–17. [PubMed] [Google Scholar]
- 35.Felez J, Chanquia CJ, Levin EG, Miles LA, Plow EF. Binding of tissue plasminogen activator to human monocytes and monocytoid cells. Blood. 1991;78:2318–27. [PubMed] [Google Scholar]
- 36.Vaughan DE, Mendelsohn ME, Declerck PJ, van Houtte E, Collen D, Loscalzo J. Characterization of the binding of human tissue-type plasminogen activator to platelets. J Biol Chem. 1989;264:15869–74. [PubMed] [Google Scholar]
- 37.Vogel V, Sheetz M. Local force and geometry sensing regulate cell functions. Nat Rev. 2006;7:265–75. doi: 10.1038/nrm1890. [DOI] [PubMed] [Google Scholar]
- 38.Meltzer ME, Doggen CJM, de Groot PG, Rosendaal FR, Lisman T. Fibrinolysis and the risk of venous and arterial thrombosis. Curr Opin Hematol. 2007;14:242–8. doi: 10.1097/MOH.0b013e3280dce557. [DOI] [PubMed] [Google Scholar]
- 39.Eitzman DT, Westrick RJ, Nabel EG, Ginsburg D. Plasminogen activator inhibitor-1 and vitronectin promote vascular thrombosis in mice. Blood. 2000;95:577–80. [PubMed] [Google Scholar]
- 40.Zeng B, Bruce D, Kril J, Ploplis V, Frredman B, Brieger D. Influence of plasminogen deficiency on the contribution of polymorphonuclear leukocytes to fibrin/ogenolysis. Thromb Haemost. 2002;88:805–10. [PubMed] [Google Scholar]
- 41.Angles-Cano E, de la Pena Diaz A, Loyau S. Inhibition of fibrinolysis by lipoproteins(a) Ann NY Acad Sci. 2001;936:261–75. doi: 10.1111/j.1749-6632.2001.tb03514.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.