

Summary
Discovering the structure and dynamics of transcriptional regulatory events in the genome with cellular and temporal resolution is crucial to understanding the regulatory underpinnings of development and disease. We determined the genomic distribution of binding sites for 92 transcription factors (TFs) and regulatory proteins across multiple stages of C. elegans development by performing 241 ChIP-seq experiments. Integrating regulatory binding and cellular-resolution expression data yielded a spatiotemporally-resolved metazoan TF binding map. Using this map, we explore developmental regulatory circuits that encode combinatorial logic at the levels of co-binding and co-expression of TFs, characterizing (1) the genomic coverage and clustering of regulatory binding, (2) the binding preferences of and biological processes regulated by TFs, (3) the global TF co-associations and genomic subdomains that suggest shared patterns of regulation, and (4) key TFs and TF co-associations for fate specification of individual lineages and cell-types.
Keywords: Transcription Factor, Gene Regulation, ChIP-seq, Cellular Expression, Development
In multi-cellular organisms, transcription factors (TFs) bind at cis-regulatory elements in the genome to mediate diverse gene expression programs with exquisite spatiotemporal control1–3. However, owing to the paucity of in vivo developmental stage TF binding data and cellular TF expression data, the integrated maps required to study transcriptional control of development with spatiotemporal resolution are lacking.
In this work, we analyzed regulatory activity of a broad set of C. elegans TFs in one or more developmental stages. Exploiting recently developed methods4–6, we integrate TF binding data with an initial cellular-resolution map of TF expression in the embryo. Our integrated analyses support the discovery of many key TFs and candidate TF co-associations for fate specification, providing insights into the temporal and spatial dynamics of regulatory interactions in development.
Large-scale survey of regulatory binding
As part of the modENCODE consortium, we performed 241 chromatin immunoprecipitation-sequencing (ChIP-seq) experiments to identify in vivo binding sites for 92 (10%) C. elegans TFs and regulatory proteins (collectively termed factors) in one or more stages of development or treatments (Fig. 1a, Supplementary Table 1). To identify factor binding from the ~5.1 billion raw reads, we developed a uniform processing pipeline (Extended Data Fig. 1a–e, see Methods) that allows comparison of orthologous TF properties7, such as sequence preferences (Extended Data Fig. 1f–h). Eight previously reported8 experiments failed to pass our quality controls and were thus removed from consideration.
Figure 1.

Large-scale regulatory analysis of the C. elegans genome. (a) Factors assayed per developmental stage (or treatment) in 241 quality-filtered ChIP-seq experiments. Stages and treatments are abbreviated as Early Embryo (EE), Late Embryo (LE), Embryo Mixed (EM; EE and LE), larval L1 (L1), larval L2 (L2), larval L3 (L3), larval L4 (L4), Young Adult (YA), mixed Larval and Young Adults (LY), Day 4 Adult (D4), and Starved L1 (S1). Embryonic datasets were combined into a compiled embryonic stage (EX). Analyses in this report focus on embryonic (yellow) and larval (blue) experiments (NA=187). (b) Genomic coverage (percent of genomic bases) of regulatory binding (excluding RNA polymerases) in 181 C. elegans (outer circle) and 339 H. sapiens (inner circle) ChIP-seq experiments. Genomic coverage of cHOT, HOT, and other regulatory binding (RGB) regions are highlighted in red, yellow, and blue, respectively. cXOT and XOT percentages are shown in parenthesis. cHOT, HOT and RGB region coverage in the human genome are 0.17%, 1.4%, and 6.1%, respectively7. (c) Cutoff-normalized, occupancy levels in 126 embryo-specific (yellow) and 91 larval L4-specific (blue) HOT regions. Error bars indicate the 25th and 75th percentiles. (d) Chromatin state14 distribution of L3 larvae binding regions by occupancy. RGB, HOT, and XOT region occupancy levels are indicated along the x-axis as blue, yellow, and red bars, respectively. (e, f) Signal densities near enzymatically-derived TSSs30 for BLMP-1 and ALY-2, and RNA Pol II. (g) Functional (GO term) enrichment for gene targets of binding31. A subset of biological process (BP) terms (levels ≥ 4) are shown for factors enriched (BH-corrected, P < 0.01) in synaptic transmission; Early MEP-1 and DPL-1 data sets are included for comparison. (h) Example signal tracks near the UNC-104 locus.
We focused our analyses on embryonic and larval (L1-L4) stages, examining a total of 397,539 reproducible binding sites distributed across 33,833 binding regions in the genome. Collectively, factor binding (excluding RNA polymerases) is spread throughout 21.7% of the C. elegans genome (Fig. 1b), an upper-bound defined by ChIP-seq resolution9. We estimate that –within our ChIP-seq resolution and sensitivity– we have identified ~90% of the regulatory binding regions (albeit not the majority of binding events; Extended Data Fig. 1i). Consistent with this estimate, we observe binding within 2 kb upstream of a transcription start site (TSS) for 91.3% of genes (Extended Data Fig. 1j) 10.
HOT regions are dynamic in development
Previous studies8,11–13 have revealed regions in metazoan genomes with heavily clustered factor binding, termed high-occupancy target (HOT) regions. The availability of multiple data sets across stages allowed us to examine the dynamics of HOT regions through development. We identified high- (HOT) and extreme- (XOT) occupancy target regions for each developmental stage, where significant (5% and 1%, respectively) enrichments in TF binding sites are observed (Extended Data Fig. 2a–c). We found a total of 9,142 HOT regions in at least one developmental stage, and 858 constitutive HOT (cHOT) regions occurring across all stages assayed (Supplementary Table 2). cHOT regions are enriched in promoters of genes with housekeeping functions (Extended Data Fig. 2d, Supplementary Table 3). However, most HOT regions are dynamic across development: 31–56% of HOT regions change between sequential stages and occupancy at larval L4-specific HOT regions increases as development progresses (Fig. 1c).
Across developmental stages, 77–85% of HOT regions occur within 2 kb upstream of an annotated TSS (Extended Data Fig. 2e). Furthermore, 88.7% and 88.8% of cHOT regions occur in promoter or enhancer states in L3 larvae (Fig. 1d) and embryos, respectively (Extended Data Fig. 2f,g). These results indicate that HOT regions reside at important regulatory locations in the genome and are dynamic during development (Extended Data Fig. 3a–c).
Preferences and roles of regulators
Factors displayed a range of chromatin state14 preferences, with a general bias towards promoter and enhancer states (Extended Data Fig. 3d,e). Though generally clustered near TSSs, many factors display enrichments for upstream or downstream binding (Extended Data Fig. 3f). Proximal and downstream binders include RNA Pol II (AMA-1) and other regulators of transcription initiation and elongation, respectively. Upstream binders may be enriched for chromatin remodelers and factors that recruit the transcriptional machinery. For example, binding of BLMP-1 – the ortholog of the human repressor PRDM115,16– is tightly concentrated upstream of TSSs (Fig. 1e). Likewise, ALY-2, a human THOC4 mRNA export factor ortholog17, exhibits an enrichment in binding downstream of TSSs during development (Fig. 1e) and is increased at elongation chromatin states relative to other factors. Generally, TFs assayed in multiple stages retain their upstream and downstream binding preferences. Remarkably, RNA Pol II positioning shifts (Fig. 1f) from a strong elongating distribution in the early embryo to weaker elongation distributions in later stages, consistent with its previously observed continued presence at promoters that are down-regulated during development8,18.
Gene ontology (GO) analysis of the candidate protein-coding gene targets revealed 6,347 functional associations (BH-corrected, P < 0.05) for 75 factors (Extended Data Fig. 4a, Supplementary Table 4), suggesting biological roles for TFs of previously unknown function. The unstudied factors FKH-10, KLU-1, and C34F6.9 group with the established neuronal fate regulators SEM-4 (ZNF236), MAB-5 (HOXA2/B2), CES-1 and ZAG-1 in targeting neurotransmission genes, with C34F6.9 additionally regulating muscle development and sex differentiation (Fig. 1g). Most of these factors, including SEM-4 and C34F6.9 in L2 larvae, appear to regulate the neuronal kinesin UNC-104 (human KIF1A/C, Fig. 1h). Although expression of FKH-10 is restricted to six neuronal cells near the terminal bulb of the pharynx19, its specific molecular role in neuronal regulation and its regulatory targets were heretofore unknown.
Functional associations also demonstrate malleability of regulation. For example, UNC-62 transitions from regulating diverse muscle and neuronal development genes in embryos to regulating lipid metabolism processes in L4 larvae (Extended Data Fig. 4b). These changes are consistent with known diverse UNC-62 roles in motor neuron and vulval development, as well as locomotion, and aging20,21. Similarly, the C2H2 zinc-finger protein KLU-1 transitions from targeting neuronal genes in L2 larvae to targeting carbohydrate and lipid metabolism genes in L4 larvae. The change in UNC-62 regulatory targets coincides with increased expression of the UNC-62 (7a) isoform in late larval and adult intestine20, which has been shown to affect lifespan 22. Such early development regulators may often target metabolic regulation in later developmental stages23.
Global and sub-domain TF co-associations
Global analyses of pairwise TF co-associations24 revealed a multitude of established and novel co-associations (Fig. 2a), many stage-specific clusters of co-association (Extended Data Fig. 5a), as well as differences in co-associations between expressed and repressed promoters (Extended Data Fig. 5b). FOS-1:JUN-1 as well as GEI-11:LIN-15B co-associations are readily apparent in L1 and L3 larvae, but not in L4 larvae. Likewise, ELT-3 and BLMP-1, which preferentially reside at molting and cuticle development gene promoters, co-localize in L1 larvae but not in embryos. The neuronal regulators CES-1 and FKH-10 co-associate across larval stages (L1, L3-L4) but their co-association is not apparent in late embryogenesis (Fig. 2a). Changes in co-association are often correlated with the presence of additional factors, e.g., in the embryo to larval L1 transition, the increased ELT-3:BLMP-1 co-association is also accompanied by increased GEI-11 co-associations with these factors (Extended Data Fig. 5c–f). Other factors remain largely invariant through multiple stages, e.g., ZTF-11, a human MTF1 ortholog.
Figure 2.
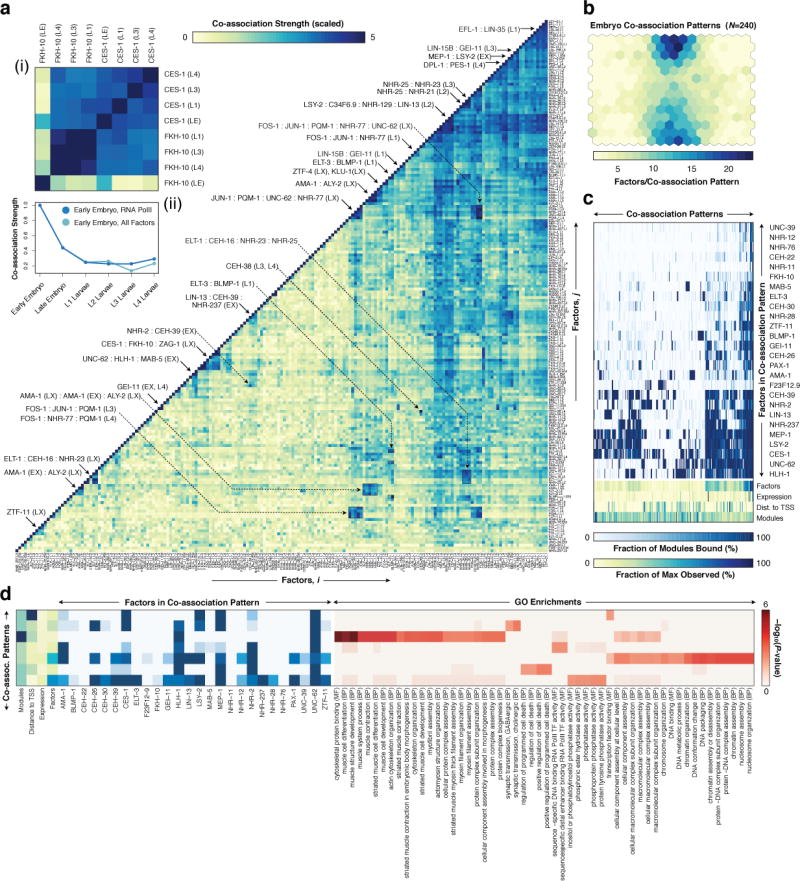
Global and domain-specific patterns of TF co-association. (a) Global pairwise TF co-association matrix (NT=17,391) as defined by promoter interval statistics24. Co-association scores are scaled by the standard deviation (uncentered) for visualization purposes. Co-associations of interest and discussed in the text are highlighted. LX indicates larval stages L1-L4. A higher-resolution version is available in Extended Data Fig. 9. CES-1:FKH-10 co-associations are highlighted in inset (i). Co-association strengths (unscaled) between early embryo and later stages are shown in inset (ii) for RNA Pol II-specific binding (blue), and for all factor-specific binding (light-blue). (b) Embryonic (EX) binding regions (NR=6,555) were clustered into a SOM describing 240 co-association patterns among 26 factors. (c) Binding signatures (fraction of modules bound by each factor) of the learned co-association patterns are shown. The relative number of factors per co-association pattern, expression from overlapping promoters, distance to TSSs, and number of modules with each co-association pattern are indicated as a fraction of the maximum observed across co-association patterns. (d) Functional enrichment for regions with UNC-62-bound co-association patterns of the embryo SOM.
Functionally-related factors were often co-associated. For example, FOS-1, NHR-77 and PQM-1 target promoters of genes in cellular lipid and ketone metabolic processes. Similarly, EFL-1 and LIN-35, the known interacting orthologs of human E2F and RB, show a strong co-association in L1 larvae, where they target membrane organization and endocytosis genes.
We observed strong similarities in RNA Pol II binding within embryonic (early and late embryo) and within larval L1-L4 stages, but larval RNA Pol II binding is only marginally and weakly co-associated with embryonic binding, reflecting the dynamic establishment of the transcriptional machinery through development (Fig. 2a).
To uncover higher-order co-associations (involving ≥ 2 factors), and their genomic subdomains, we applied self-organizing maps (SOMs), an unsupervised machine learning technique25. For each developmental stage, we trained SOMs to cluster genomic regions with shared TF co-association patterns (Fig. 2b, Extended Data Fig. 6a–d), thereby concomitantly identifying TF co-association patterns (Fig. 2c) and their target regulatory regions.
We performed GO analysis of the target regulatory regions for 240, 390, 439, 390, and 409 clusters in the embryo, larval L1, L2, L3, and L4 SOMs, respectively, revealing enrichments across 1,209 GO terms (BH-corrected, P < 0.05, Extended Data Fig. 6e, Supplementary Table 5). As illustrated in the embryo, higher-order co-association patterns show a richness of functional associations, with 137 clusters spanning 273 GO terms. A close examination of UNC-62 co-association patterns reveals how diverse patterns for individual factors can result in specialized functional targeting (Fig. 2d). Regions bound exclusively by UNC-62 and HLH-1 are highly-enriched at muscle development promoters. In contrast, genes targeted by more complex UNC-62 co-associations are enriched in synaptic transmission, regulation of cell death, and chromatin assembly functions. Higher-order co-associations are largely stage-specific (Fig. 3), a feature modulated by changes in the observed number of binding sites for individual factors between stages (Extended Data Fig. 7).
Figure 3.

Stage-specificity in higher-order TF co-associations. (a) Embryonic (EX) and larval L1 binding SOM with raw binding sites. Binding data for factors (NF=15) assayed in embryos and L1 larvae was assigned to stage-specific binding modules (NM=25,261) as diagramed in the inset. Stage-specific binding modules were clustered into an SOM describing 192 co-association patterns. The SOM is colored by the embryonic (versus L1) stage-specificity of the learned co-association patterns, measured as the fraction of binding modules that are embryonic. (b) Histogram of preceding (T1) versus subsequent (T2) stage-specificities.
Spatiotemporal TF expression analysis
Although studies in C. elegans and D. melanogaster have led analyses of organismal-level regulatory binding circuits, such studies have generally lacked cell-type and tissue resolution. We sought to remedy this deficiency by tracking5,6 the expression of 180 diverse genes (mostly TFs) through early embryogenesis with cellular resolution(Extended Data Fig. 8a–d, Supplementary Table 6). Our expression data, from previously published5,6 and newly acquired series, includes 36 factors with genome-wide binding measurements (13 embryo, 23 larval).
We observed common and distinctive cellular expression patterns amongst a wide distribution of broadly- and narrowly-expressed genes (Extended Data Fig. 8e,f). For example, expression of DMD-4, an ortholog of the vertebrate spinal circuit configuration regulator DMRT326, is tightly limited to posterior regions of the pharynx. Similarly, F49E8.2 expression is exclusive to the Z2/Z3 germ cells (Extended Data Fig. 8a). 95.7% of pairs of tracked cells show distinct gene expression signatures (R < 0.75).
Cellular expression mapped the regulatory activity of 16 assayed factors to specific tissues (Fig. 4a). As expected, the known regulators of pharynx and muscle development, PHA-4 and HLH-1, were respectively enriched in these tissues. The co-associated factors, MEP-1 and DPL-1 (human TFDP1/2), although broadly-expressed, are enriched in neuronal lineages. This is consistent with the observed MEP-1 targeting of neuronal function genes in the larvae, and provides further support for the coordinate activities of MEP-1 and DPL-1 in targeting membrane organization, receptor-mediated endocytosis, and cell-cycle genes (Fig. 1f, Supplementary Table 4).
Figure 4.

Cell-type and lineage resolution of regulator activity and TF co-associations. (a) Tissue enrichment (−log10, P-value) and depletion (log10, P-value) scores for the expressing population of each gene are shown (Fisher’s exact, Bonferroni-corrected). Only genes with significant enrichments (or depletions) are shown. (b) Co-association strength (Fig. 2a) versus cellular overlap coefficient for 13 focus factors. The Jaccard index for the cellular overlap is indicated for each gene pair by ring size and color. (c) Cellular-resolution regulatory binding SOM. Cellular-resolution binding modules were generated by annotating in each cell, the binding of focus factors expressed in the cell. Cellular-resolution binding modules (inset) were clustered into a SOM with 268 learned co-association patterns, 161 (68%) of which were discovered in the data. The SOM is colored by the number of factors in the learned co-association patterns. (d) Tissue classes and co-association signatures are shown for 43 co-association patterns with significant enrichments. Tissue enrichments of interest are highlighted red.
More complex patterns of co-expression and co-association were observed in epidermal tissues, where CEH-16 (human Engrailed), and particularly ELT-1 and NHR-25 expression is concentrated. In both L2 and L3 larvae, ELT-1 and NHR-25 are modestly co-associated. ELT-1 targets transcriptional regulators, including NHR-25, and tail morphogenesis genes, whereas NHR-25 targets nuclear organization and genitalia development genes (Supplementary Table 4). However, L2 larval binding of ELT-1 and NHR-25 is co-associated with that of CEH-16, whose early embryonic expression is primarily concentrated in a subset of pharynx and epidermal cells.
In early embryo (Fig. 4a) and L1 larvae27, HLH-1, is primarily and broadly expressed in muscle tissues whereas posterior-specific HOX factors MAB-5, and EGL-5 are expressed in a small subset of posteriorly placed muscle, epidermal and neuronal precursors. We observed modest co-association signals between embryonic HLH-1 and MAB-5 binding, and larval L3 EGL-5 binding, perhaps reflecting the intersection of tissue-specific and positional regulatory programs. As expected, HLH-1 targets muscle differentiation genes (together with UNC-62); however in GO analysis, we only detect MAB-5 targeting of diverse neuronal functions (in mixed embryos and L2 larvae), consistent with its later role in neuron specification28. CO5D10.1, whose early embryonic expression is also restricted to muscle tissues is not co-associated with the above factors, and neither C05D10.1 or EGL-5 showed specific functional associations. Thus, although co-associated factors were often expressed in the same tissue, this is not pervasive and factors with diverse binding targets are frequently co-expressed. Moreover, these co-expression patterns are dynamically established during embryogenesis (data not shown).
Refinement of embryonic co-associations
Despite extensive studies in metazoan regulatory networks, the relationship between regulator binding in overlapping genomic regions and co-expression in cell-types is not well-studied. We examined this relationship among 13 ‘focus’ factors, for which both embryonic binding and cellular expression were assayed. This analysis is limited to the first half of embryogenesis, where expression was directly measured in 696 ‘focus’ cells. Later events may occur that would not be identified in our analysis. We found a poor correlation between TF co-expression and co-association (R=0.07, Fig. 4b), consistent with precise coordination of these separate processes underlying the differential establishment of cell- and lineage-specific regulatory circuits (Fig. 4b).
Integrated analysis show that MEP-1 is co-associated and co-expressed with similarly broadly-expressed factors (LIN-13, CEH-39) and narrowly-expressed factors (CES-1, CEH-26), suggesting MEP-1 often works in cis with these additional factors. MEP-1 binding is co-associated with CES-1 and CEH-26 in embryos, and expression of these factors is narrowly restricted within the MEP-1 expressing population. These MEP-1:CES-1 and MEP-1:CEH-26 co-associations are reminiscent of MEC-3:UNC-86 interactions in which the classic ‘terminal selector’ MEC-3 heterodimerizes with the broadly-expressed UNC-86 exclusively in touch sensory neurons29. Thus, the co-association and co-expression of MEP-1:CEH-26 suggests CEH-26 may function as a terminal selector in head and tail neurons, and the excretory cell. The spatiotemporally-resolved co-association analyses demonstrate how broadly-expressed factors, such as MEP-1 and LIN-13 –which targets both neurotransmission functions and genitalia development– can play diversified functional roles during development through co-associations with narrowly-expressed factors.
To determine how co-binding and co-expression coordinately define regulatory patterning in distinct cell-types and genomic regions, we intersected cellular expression and binding data by mapping focus factor binding to in silico genomes for cells where the factors are expressed. This procedure resulted in 4,779,810 binding sites distributed across 2,858,477 cell-resolved binding modules. We applied an SOM approach to cluster the cell-resolved binding modules by co-association patterns, uncovering 161 TF co-association patterns and the genomic subdomains and specific cellular subsets of the embryo in which they may occur (Fig. 4c). The cellular distribution of TF co-association patterns revealed co-associations shared among and specific to particular cell-fates (Fig. 4d). For example, we found that distinct MEP-1, CEH-26, and NHR-2 co-associations were specific to neuronal tissues. Similarly, muscle cells were enriched in various HLH-1 co-associations (Fig. 4d).
We identified 39 co-association patterns whose cellular distribution coincides with the cellular expression of at least one of 124 target genes (non-focus factors; Bonferroni-corrected, P < 0.01). Focus factor binding allowed us to analyze co-association patterns at the promoters of 44 of these genes (where binding is observed). For 28 (63.6%) of these genes, co-association patterns were detected at the promoter and the gene’s cellular expression matched the cellular distribution. Moreover, the overlap between the expression cells for a gene and the co-association cells is higher in cases where the co-association occurs in the promoter of the gene (Wilcoxon, P=5.1×10−6, Extended Data Fig. 8g). This result suggests that co-associations at promoters are correlated with cellular expression patterns for genes, and indicates a functional regulatory role of the discovered co-associations.
Discussion
We have generated a high-coverage TF binding map of the C. elegans genome, revealing regulatory targets, co-associations, and dynamics across five developmental windows for 92 diverse factors. Gene targets suggest a multitude of functional associations for 75 factors, many previously unannotated and with clear mammalian homologs. Our work reveals extensive regulatory rewiring through development, with temporal differentiation of HOT regions in the genome, factor positioning preferences, regulatory targets, and co-associations.
A systematic analysis of TF co-associations through developmental reveals sets of factors that assemble at genomic regions associated with >1,200 biological functions (GO terms), with likely spatiotemporal specificity. As illustrated with UNC-62, these higher-order co-associations reveal how individual TFs can participate in distinct TF co-associations patterns at promoters of functionally diverse genes.
Lastly, cellular-resolution expression tracking allowed us to map the activity of 35 factors to precise cell- and tissue-types, demonstrating lineage-specific activities for 16 factors in the early embryo. Importantly, co-associations that are observed in whole-organism binding data are not always evident at the cellular level, highlighting the need to incorporate such information in our understanding of regulatory circuits. As additional expression patterns and TF binding sites are determined, and methods to track TF binding with cell-type specificity are developed, the broader and more precise regulatory logic of development should emerge.
Methods
Strain construction
C. elegans strains were constructed essentially as described by Sarov et al. 33. Briefly, each transgene fosmid constructed contains the entire transcription factor tagged at its C-terminus with an in-frame GFP:3xFLAG tag. Transgenic strains were generated by microparticle bombardment of transgene fosmids. 20–50 μg of fosmid DNA was use per transformation. The fosmid contained the unc-119 marker for selection of transgenic animals.
Strain growth and staging
Worms were grown on NGM media using standard growth protocols. Worms were synchronized by bleaching and L1 starvation, and grown to the desired developmental stage as determined by visual inspection34. Briefly, animal populations consisting mostly of embryo-bearing adults were bleached and eggs were harvested. Embryos were hatched in the absence of food to synchronize larval development, and then placed on food and grown for specified times to reach the appropriate larval or adult stage for collection and ChIP. To collect early stage embryos, young adult animals bearing relatively few embryos were collected and bleached. The subsequent embryos were mesh-purified and immediately fixed. To collect predominantly late stage embryos, the same procedure was used, except the embryos were incubated for six hours before fixing. All procedures result in a population synchronized within a 2-hour developmental window. The vast majority of animals (>80%) are within this window.
Chromatin-immunoprecipitation
C. elegans ChIP-Seq assays were performed essentially as described by Zhong et al. 23. Briefly, wild-type (N2) and transgenic worms expressing GFP tagged factors were grown to the desired developmental stage under controlled conditions (Extended Data Fig. 10) and cross-linked with 2% formaldehyde. Cell extracts were sonicated to yield predominantly DNA fragments in the range of 200–500 bp. For most experiments (~93%), factor expression was driven by the endogenous promoters. With the exception of RNA Pol II (AMA-1), RNA Pol III (RPC-1), TBP-1, EOR-1, and EFL-1, where native antibodies were used, the sonicated lysates were immunoprecipitated using α-GFP antibody. Most immunoprecipitations were performed in 5% Triton, although a few were performed in 1% Triton. Direct comparison indicated that different concentrations of Triton had minimal effect on IP efficiency (data not shown). At least two biological replicates were performed for each ChIP, with parallel genomic DNA controls prepared from the same strain.
Library construction and sequencing
Sequencing libraries were prepared from independent biological replicates of IP enriched and input DNA fragments. Libraries were multiplexed using four 4bp barcodes35 and sequenced on an Illumina Genome Analyzer II.
Sequencing data pre-processing
FASTQ files were aligned to the C. elegans ws220 genome with BWA36 and quality-filtered to retain only high-quality alignments (Q ≥ 30). Because numerous ChIP and input DNA libraries were sequenced multiple times, we merge the sequencing files of re-sequenced libraries using the heuristics that follow. For each library with multiple re-sequencing files (instances), the following parameters are determined for each instance:
aligned.reads: number of aligned reads qc.reads: number of quality-filtered reads qc.percent: percent of reads that pass quality-filtering qc.duplicates: fraction of quality-filtered reads that are duplicates (non-distinct)
For these libraries, these same metrics are calculated for all possible combinations of instances. Two additional metrics are calculated. Status is defined as “pass” unless any of the constituent instances has <106 aligned reads or <20% quality-filtered reads (in which case the combination status is set to “fail”). In addition, we calculate the percent of effective alignments (qc.score) as a quality control score for each combination.
qc.score: qc.percent x (1 - qc.duplicate)
To select the best combination of instances, we choose the “passing” combination that has ≥ 106 uniquely aligned reads. If no combination has status equal to “pass”, we choose the combination that has ≥ 106 uniquely aligned reads with the highest percent of effective alignments (qc.score). If no combination yields ≥ 106 uniquely aligned reads, all instances are used (i.e., the combination with the highest number of reads is chosen). To perform uniform binding site identification on each data set (see below), we merge input DNA files from replicates into a single merged input DNA control.
Uniform binding site identification
All ChIP-seq experiments were scored against an appropriate input DNA control. For worm datasets, we used the SPP binding site caller to identify and score (rank) potential binding sites37. As described in7, we used the Irreproducible Discovery Rate (IDR) framework for obtaining optimal thresholds and determine high confidence binding events by leveraging the reproducibility and rank consistency of binding site identifications across replicate experiments of each dataset38. Code and detailed step-by-step instructions to call binding sites using the IDR framework are available at https://sites.google.com/site/anshulkundaje/projects/idr.
The SPP caller37 was used with a relaxed threshold (FDR=0.9) to obtain a large number of binding sites (maximum of 30K for worm) that span true signal as well as noise (false identifications). Binding sites were ranked using the signal score output from SPP (which is a combination of enrichment over control with a penalty for binding site shape). The IDR method analyzes a pair of replicates, and considers binding sites that are present in both replicates to belong to one of two populations: a reproducible signal group or an irreproducible noise group. Binding sites from the reproducible group are expected to show relatively higher ranks (ranked based on signal scores) and stronger rank-consistency across the replicates, relative to binding sites in the irreproducible groups. Based on these assumptions, a two-component probabilistic copula-mixture model is used to fit the bivariate binding site rank distributions from the pairs of replicates38.
The method adaptively learns the degree of binding site rank consistency in the signal component and the proportion of binding sites belonging to each component. The model can then be used to infer an IDR score for every binding site that is found in both replicates. The IDR score of a binding site represents the expected probability that the binding site belongs to the noise component, and is based on its ranks in the two replicates. Hence, low IDR scores represent high-confidence binding sites. An IDR score threshold of 5% for worm datasets was used to obtain an optimal binding site rank threshold on the replicate binding site sets (cross-replicate threshold). If a dataset had more than two replicates, all pairs of replicates were analyzed using the IDR method. The maximum binding site rank threshold across all pairwise analyses was used as the final cross-replicate binding site rank threshold.
Any thresholds based on reproducibility of binding site calling between biological replicates are bounded by the quality and enrichment of the worst replicate. Valuable signal is lost in cases for which a dataset has one replicate that is significantly worse in data quality than another replicate. Hence, we used a rescue strategy to overcome this issue. In order to balance data quality between a set of replicates, mapped reads were pooled across all replicates of a dataset, and then randomly sampled (without replacement) to generate two pseudo-replicates with equal numbers of reads. This sampling strategy tends to transfer signal from stronger replicates to the weaker replicates, thereby balancing cross-replicate data quality and sequencing depth. These pseudo-replicates were then processed using the same IDR pipeline as was used for the true biological replicates to learn a rescue threshold. For datasets with comparable replicates (based on independent measures of data quality), the rescue threshold and cross-replicate thresholds were found to be very similar. However, for datasets with replicates of differing data quality, the rescue thresholds were often higher than the cross-replicate thresholds, and were able to capture more binding sites that showed statistically significant and visually compelling ChIP-seq signal in one replicate but not in the other. Ultimately, for each dataset, the best of the cross-replicate and rescue thresholds were used to obtain a final rank threshold. Reads from replicate datasets were then pooled and SPP was once again used to call binding sites on the pooled data with a relaxed FDR of 0.9. Pooled-data binding sites were once again ranked by signal-score. The final rank threshold (best of cross-replicate and rescue threshold) was then used to threshold the ranked set of pooled-data binding sites.
All binding site sets were then screened against specially curated empirical blacklists for the worm genome. Briefly, these blacklist regions typically show the following characteristics:
Unstructured and extreme high signal in sequenced input DNA and control datasets as well as open chromatin datasets irrespective of developmental stage/treatment.
An extreme ratio of multi-mapping to unique mapping reads from sequencing experiments.
The worm blacklist can be downloaded from: http://www.broadinstitute.org/~anshul/projects/worm/blacklist/ce10-blacklist.bed.gz
ChIP-seq quality control
A number of quality metrics for all replicate experiments of each dataset were computed39. In brief, these metrics measure ChIP enrichment and signal-to-noise ratios, sequencing depth and library complexity and reproducibility of binding site identification. These metrics will be available through the ENCODE portal at http://encodeproject.org/ENCODE/qualityMetrics.html. We examined multiple quality control thresholds, flagging datasets with low signal-to-noise ratios as determined by normalized strand cross-correlation scores (NSC < 1.03), low rank correlations between binding site scores across replicates (BSR < 0.3), or poor IDR models as indicated by a low correlation between binding site ranks and IDR ranks (BIR < 0.3). A poor IDR model fit is a result of a pair of replicates having inseparable signal and noise components and abnormally low binding site rank consistency. Experiments that passed all quality control thresholds were automatically scored as high-quality experiments. Experiments that passed most but not all quality control thresholds where scored as medium-quality experiments. Experiments that did not pass multiple quality control thresholds were discarded, excluded from further analyses with a few exceptions. Because factors with genuinely few binding sites inherently have lower genome-wide signal-to-noise ratios, datasets with low NSC scores were rescued if the number of binding sites was low (<1,000). Analogously, high reproducibility scores (i.e. low NP/NT ratios) were occasionally allowed to rescue experiments where the IDR models appeared to have poor BIR values (<0.3) due to low numbers of binding sites.
A summary of relevant quality metrics computed is provided below:
NP/NT : Ratio of number of binding sites passing 5% IDR thresholds based on comparison of pairs of pooled pseudo-replicates to pairs of biological replicates. The NP/NT ratio is a measure of reproducibility, computed as max(NP) / max(NT), where:
NP = Number of binding sites passing the 5% IDR threshold by comparing binding sites from a pair of pooled pseudo-replicates. The pair of pseudo-replicates are created by pooling reads from all replicates of a sample and randomly subsampling two equally sized sets of reads.
NT = Number of binding sites passing the 5% IDR threshold by comparing binding sites from the best pair of biological replicates.
A high NP/NT ratio indicates that pooling replicates and subsampling substantially increased reproducibility in comparison to true replicates. This usually implies that at least one of the replicates has significantly higher enrichment as compared to others. The correlation between NP and NT across all experiments analyzed is shown in Extended Data Fig. 1b.
Normalized strand cross-correlation (NSC)
A genome-wide measure of ChIP enrichment or signal-to-noise ration measure. A strand cross-correlation profile is computed as the Pearson correlation (y-axis) between per-base read-start count vectors on the + and – strand over a wide range of strand shifts (x-axis). The cross-correlation profile peaks at the predominant ChIP fragment length. The NSC is computed as the ratio of this maximal strand cross-correlation at the estimated fragment length (signal) to the minimum background cross-correlation over all shifts (noise). Samples for which both replicates had NSC < 1.03 are flagged as potential low signal-to-noise datasets. However, these can be rescued if the sample passes peak reproducibility criteria especially in cases where the number of binding sites is low (<1,000).
Binding Site Rank Correlation (BSR)
Using the pre-IDR relaxed set of binding sites from the best pair of replicates, we find all binding sites that are present in both replicates. This set includes binding sites from the signal and noise components learned by the IDR model. We then compute the rank correlation of the binding site scores across the pair of replicates. Datasets with BSR < 0.3 are flagged as potentially low in binding site reproducibility.
Binding Site vs. IDR Rank Correlation (BIR)
Using the pre-IDR relaxed set of binding sites from the best pair of replicates, we find all binding sites that are present in both replicates. These binding sites have scores from each of the replicates as well as an IDR score indicating the likelihood that the binding sites are not from the signal component. We rank the binding sites using the IDR scores and original binding site scores. For valid IDR models with good fits, the IDR scores and original binding site scores have a strong monotonic relationship and hence high rank correlation. Hence, we compute BIR as the rank correlation between the IDR scores and the original binding site scores as a measure of stability of the IDR models. Poor IDR model fits are usually a sign of abnormal rank consistency of binding sites and poor reproducibility. BIR is estimated as the primary data quality metric in that if a sample shows a poor IDR model fit no statements can be made about reproducibility. Datasets with BIR < 0.3 are considered to have poor IDR models. We make one exception for samples involving factors that bind few sites (<1000) in the genome. In such cases, stable IDR models can obtain artificially low BIR scores. We perform additional tests of model stability for such samples, and allow for rescue if the models are deemed stable and if the NP/NT ratio is low.
ChIP-seq experiment selection
We uniformly processed ~5.1 billion raw reads from 323 worm ChIP-seq experiments, removing 82 (25%) low quality experiments that failed to meet our quality control standards (described above, Extended Data Fig. 1c). Examining approved experiments (NR=241), ~89% of the binding sites are shared between a pair of duplicate (redundant) experiments where binding was assayed for the same TF and development stage (NU=22, Extended Data Fig. 1d). True biological duplicates –in which binding was assayed for the same developmental stage and factor, as driven by same promoter, and assayed with the same ChIP protocol– share 77–92% of the binding sites. Thus, the identified binding sites have demonstrably reliable reproducibility rates.
We focused our analysis on a refined set of approved experiments (for 86 factors), selecting the highest-quality ChIP-seq data to produce a non-redundant set of embryo and larval experiments (NA=187) with unique factor and developmental stage combinations, prepared with the same ChIP protocol, and in which TF expression is driven by the native promoter (Extended Data Fig. 1e). As such, the released collection corresponds to the top ~75% highest-quality worm ChIP-seq experiments performed by the modENCODE consortium. Furthermore, the biological observations presented in this work stem from analysis of a top, non-redundant selection of embryo and larval experiments that collectively encompass ~58% of the worm ChIP-seq experiments performed.
Binding sites and reports for the released (NR=241) and analyzed (NA=187) sets of non-redundant ChIP-seq experiments are available online through the modENCODE DCC (www.modencode.org) and at tapanti.stanford.edu/cetrn.
Signal profiles
We generated signal track files for each ChIP-seq experiment using MACS2 (available at https://github.com/taoliu/MACS/) on pooled data (for ChIP and control), as follows:
macs2 callpeak -t ChIP.bam -c CONTROL.bam -B --nomodel --shiftsize round(FRAGLEN/2) --SPMR -g ce
Where, (1) --nomodel and --shiftsize round(FRAGLEN/2) tell MACS2 to use the estimated fragment length as fragment size (FRAGLEN, estimated in the uniform binding site identification pipeline) to pileup sequencing reads, (2) -g ce lets MACS2 consider the C. elegans genome as background, and (3) -B --SPMR indicate MACS2 to generate pileup signal files of ‘fragment pileup per million reads’ in bedGraph format.
To examine factor positioning preferences at high-resolution in each ChIP-seq experiment, we collected signal values per position (bp) within 1000 bp of enzymatically-enriched TSSs30 for protein coding genes. For visualization purposes(Fig. 1e, Extended Data Fig. 3f–h), we graph the scaled, mean signal density at each position, P(signal.density), calculated as:
P(signal.density) = (P(signal.mean) - min(signal.mean))/(max(signal.mean)-min(signal.mean))
Where the average signal at any given position, P(signal.mean), is normalized to represent the fraction of the signal distance between the maximal average signal, max(signal.mean), and the minimal average signal, min(signal.mean). This normalization serves to correct signal:noise differences between ChIP-seq experiments.
For each factor and each ChIP-seq experiment, we calculated the log2-ratio of upstream to downstream binding in the windows >50 bp upstream and downstream from TSSs, respectively (Fig. 1e).
Sequence preferences (motifs)
We examined C. elegans and H. sapiens binding sequence preferences7 among 21 TF families, available from http://www.broadinstitute.org/~pouyak/motif-disc/integrate-cold/. Briefly, these sequence preferences (motifs) were obtained by analyzing sequence enrichment in the top 200 TF binding sites from uniformly processed C. elegans (analyzed here) and H. sapiens ChIP-seq experiments7. Sequence preferences were determined7 from TF binding sites outside of HOT regions, un-mappable and blacklist regions, 3′ UTRs, and exons, and motif discovery was conducted using five discovery tools: AlignACE48 (v4.0 with default parameters), MDscan49 (v2004 with default parameters), MEME50 (v4.7.0 with -maxw 26 and -nmotifs 6), Weeder51 (v1.4.2 with option large), and Trawler52 (v1.2 with 200 random intergenic blocks for background). The top three motifs for each factor (and species) are selected after ranking by the enrichment in the datasets for the species and excluding motifs for which a similar motif was already selected (R > 0.7). The discovered motifs were augmented with known literature motifs in each gene family.
Among the 21 TF families evaluated, C. elegans motifs were discovered for 15 TF families (Extended Data Fig. 1f). We evaluated the prevalence of the discovered sequence preferences among binding sites from corresponding factors, scoring the fraction of binding sites with matches to the discovered motif for the top 200, 400, 600, 800 and 1,000 binding sites (Extended Data Fig. 1g). Motif matches in sequences were scored using the MAST module40 from MEME (v4.4), and applying an E-value cutoff equivalent to 10% of the input binding sites (FDR=10%). For TF families with multiple ChIP-seq experiments, we report the prevalence for the motif/ChIP-seq experiment combination with the highest correspondence. Across all binding site numbers evaluated, ~85% of the learned motifs have a prevalence exceeding 30% of the binding sites.
The C. elegans and H. sapiens motifs discovered for 12 TF families in the Boyle et al. study allow direct analysis of sequence preference conservation between these distant species (Extended Data Fig. 1f,h). We scored the similarity between the sequence preferences (motifs) of C. elegans and H. sapiens orthologous TFs within each family using the TOMTOM module41 from MEME (v4.4), qualifying significantly similar (P < 0.05) orthologous TF sequence preferences as conserved (Extended Data Fig. 1f,h).
Chromatin states
Chromatin state and enhancer calls from C. elegans early embryos (EE) and stage 3 larvae (L3) were obtained from Ho et al., 201314. As recommended by the authors, we make use of the hierarchically-linked infinite hidden Markov model (iHMM) segmentations reported14, examining 16 chromatin states derived from 8 histone marks.
Transcript expression analysis (RNA-seq)
The RNA-seq predicted transcripts per developmental stage, DCPM (depth of coverage per million reads) expression measurements for each gene/exon, transcription start site (TSS), transcription end site (TES), splice junctions, polyAs, and splice leader sites for C. elegans N2 early embryos (EE), late embryos (LE), and L1-L3 larvae were obtained as integrated transcript files from: ftp://encodeftp.cse.ucsc.edu/users/akundaje/worm/transcription/.
HOT and XOT region determination
To identify regions with higher than expected binding occupancies, we first determined for each developmental stage the number and size distribution of observed binding sites for each factor assayed, as well as the total number and size distribution of binding regions in which these binding sites from all factors are clustered. For each developmental stage, we first analyzed the number and size distribution of target binding regions (in which factor binding sites are concentrated). For each developmental stage simulation, we randomly select an equivalent number of random binding regions with a matched size distribution. Next, for each factor assayed (in the target developmental stage), we evaluated the number and size of observed binding sites, and simulated an equivalent number and size distribution of target binding sites, restricting their placement to the simulated binding regions. We collapsed simulated binding sites from all factors into binding regions, verifying that these cluster into a similar number of simulated binding regions as the target binding regions. For each developmental stage simulation, the occupancy (number of binding sites), density (binding sites per kb), and complexity (diversity of factors) in the simulated binding regions are annotated. This procedure was repeated 1000x for each developmental stage. For each developmental stage, we constructed expected binding region occupancy (and density) distributions from the corresponding simulations (NS=1000). We determined the cutoffs at which fewer than 5% and 1% of the simulated binding regions have higher occupancies (Extended Data Fig. 2a). We classified observed binding regions with occupancies higher than the 5% and 1% cutoffs as high-occupancy target (HOT) and extreme-occupancy target (XOT) regions, respectively (Extended Data Fig. 2b,c). As such, HOT regions include XOT regions.
Recently, Teytelman et al.42 have suggested regions with artefactual enrichment of ChIP-seq signals calling into question the validity of regions of high-occupancy where multiple ChIP-seq experiments produce enrichments. Using uniformly processed from human cell-lines, we have dutifully established that our HOT regions are not an artifact of “hyper-ChIPable” regions as described by Teytelman et al. In Boyle et al. 7, we have demonstrated that (a) there is no correlation between our non-specific binding controls (IgG) and our measured transcription factor occupancy, (b, c) our HOT regions are not enriched in non-specific binding at any cutoff, and (d) that non-specific binding can account at most for 0.5% of the binding signal as observed in RNA PolII experiments. We note that the Teytelman et al. procedures are very different from ours and many others in the field. A brief discussion of these differences, and their potential relationship to the Teytelman et al. results ensues:
The regions determined by Teytelman et al. 42 represent regions with a very low enrichment (2x or less) of non-specific immuno-precipation (IP) in anti-GFP antibody controls over input DNA evaluated using a non-standard sliding-window approach. Importantly, IP/Input ratios at this level are typically not considered enriched for binding at all in modern peak-calling procedures. For example, the median IP/Input ratio for our human RNA PolII experiments is 20x, and only 0.033% of human RNA PolII peaks contain an IP/Input ratio ≤ 2x. Thus, it is essential to note that the term ‘hyper-ChIPable’ coined by Teytelman et al. is quite misleading, as a correctly performed ChIP experiment will evaluate statistically enriched regions, with higher IP/Input ratios. Put bluntly, the so-called hyper-ChIPable regions in the Teytelman et al. study are not even binding regions as determined under ChIP-seq best practices. Tellingly, when Teytelman et al., performed statistical peak-calling (using the established MACS peak-caller) to evaluate signals only at significantly-enriched regions (Table S1) only 17 (<7.5%) of the 238 claimed “hyper-ChIPable” regions were called significant by all three Sir proteins. In fact, 68% of their 238 regions do not contain a binding site for any Sir protein as determined with MACS, despite even very liberal settings used (P-value <10−5, no fold enrichment cutoff). Thus, their own data contradict their major claim that all three Sir proteins showed enrichment at the 238 sites. Furthermore, as indicated in Teytelman et al. Table S3, the Sir2, Sir3, and Sir4 ChIP-seq experiments were performed only once each, which raises the question as to whether enrichment of Sir proteins at the 238 sites is reproducible. More rigorously, even for the remaining 17 genomic loci, their status as hyper-ChIPable is questionable as each region would first have to be established as a reproducible binding site in replicate experiments for each individual Sir protein. If you consider Sir2-4 ChIP-seq are three replicates of Sir proteins, their data show that most of their claimed sites were not reproducibly enriched.
In addition to the analytical differences outlined above, other potential sources for the marked differences between our data and the Teytelman et al. Sir-enriched regions are deviations from a typical ChIP protocol. In particular, Teytelman et al. employed a significantly longer cross-link time (1 hour as opposed to the typical 10–20 min). This might contribute to formation of large non-specific protein-DNA complexes, which can in turn increase non-specific immunoprecipitation.
We believe HOT regions (as other binding regions) likely reflect something other than a simple static model of TF binding to DNA. Naturally, in the light of steric hindrances for large numbers of TFs in and the dynamic nature of molecular interactions, these high-occupancy regions may represent regions with diverse transient, or population-level diverse, binding. Such model is consistent with a known affinity for accessible DNA (as would be present in enhancer and promoter regions) and scanning mechanisms of TF binding43. An alternative argument proposes HOT regions arise from multimeric TF complexes that coordinately enrich genomic DNA from distinct loci. Thus, it is not clear that these regions are a meaningless artifact. In particular, these regions seem to segregate to enhancer and promoter regions with different chromatin architectures and different sets of TFs. Understanding how association and dissociation rates coordinately define residence time of TF binding at individual sites, genome-wide and how chromosomal interactions relate to ChIP-seq signals will prove paramount to regulation but such analyses are outside the scope of this study.
Nonetheless, we have excluded HOT and XOT regions from sequence preference, functional, and global pairwise co-association analyses of factor binding. However, HOT/XOT regions were retained in self-organizing map (SOM) analyses since these analyses separate regions of high and lower occupancy.
Functional (GO term) enrichment analyses
To evaluate the functional role of regulators we performed GO enrichment analysis on the targets of binding of each ChIP-seq experiment. Briefly, we applied ChIPpeakAnno31 to (1) assign factor binding to genic targets as defined by binding within 1 kb of TSSs, and (2) evaluate the enrichment of genic targets for GO ontologies using standard procedures. We required a minimum of 20 binding sites per ChIP-seq experiment to evaluate enrichment and report Benjamini-Hochberg (BH) corrected P-values of enrichment (hypergeometric testing). We report GO terms in which at least one ChIP-seq experiment was significantly enriched (BH-corrected, P < 0.05). The specific enrichments per HOT regions, per ChIP-seq experiment, and per stage-specific SOMs (see below) are provided in Supplementary Tables 3, 4, and 5, respectively. Because high-occupancy can mask the biological significance of co-binding, sequence and target-gene specificity12, we focused our GO analysis on the 292,466 binding sites outside of XOT regions. Although we highlight GO terms in levels ≥4 in our figures, we report GO term enrichments in Supplementary Tables 3–5 without correcting for redundancy. As such, the raw GO term counts represent a serious overestimate, several-fold, of the number of distinct biological processes, molecular activities, or cellular components targeted by TF binding but facilitate queries and analyses.
Global pairwise TF co-associations
We determined the similarity in binding sites between ChIP-seq experiments applying recently-developed interval statistics methods that allow calculation of exact P-values for proximity between binding sites24. Using this method, we performed all pairwise, directional comparisons of ChIP-seq experiments evaluating binding similarity in 34,782 comparisons. To exclude the possibility of promiscuous binding regions and generate more conservative co-association estimates, we excluded binding sites from XOT regions in each developmental stage from these analyses. (as above, see ‘Functional (GO) enrichment analyses’). We restrained interval analyses to the promoter domains by excluding binding intervals outside promoter regions, defined as 2000 bp to 200 bp downstream of annotated TSSs. Focusing co-association analyses on the promoter domains serves to (1) focus co-association evaluations on transcriptional regulatory interactions and to (2) account for the known biases in binding at TSSs, producing more conservative estimates of co-association significance. For each ChIP-seq experiment comparison (NC=34,782), the intervals of the query ChIP-seq experiment are compared individually against all reference intervals of the reference ChIP-seq experiment, calculating the probability that a randomly located query interval of the same length would be at least as close to the reference set. For each ChIP-seq experiment comparison, we compute the fraction of proximal binding events in promoter domains that are significant (P < 0.05). Because these comparisons are asymmetric –depending on the assignment of experiments as query or reference sets– we report the mean values of the complementary (inverted query and reference) comparisons and report this value as the ‘co-association strength’ (NT=17,391) between ChIP-seq experiments. We refer to binding sites from pairs of ChIP-seq experiments as ‘co-associated’ if the co-association strength (unscaled) exceeds the 95th percentile of co-association strengths (CS95%=0.4266, Extended Data Fig. 9) among comparisons of ChIP-seq experiments from distinct factors.
We examined co-association dynamics further by quantifying changes in co-associations (ΔCo-association) between factors assayed in sequential developmental stages. We were able to track 21 pairwise co-associations across all developmental stages and 78 across larval stages (Extended Data Fig. 5b–e). On average, 10% of the examined co-associations changed by more than 23.3% between sequential stages of development. Global co-association analysis was performed with an updated LIN-35 (L1) dataset.
Stage-specific SOM analyses
Although global co-associations are useful surveys of factor co-binding, co-associations can have higher-order complexities involving three or more factors and vary between genomic subdomains. To uncover higher-order co-associations and the specific genomic subdomains in which they occur we applied self-organizing maps (SOMs), an unsupervised machine learning technique, in R using the kohonen package. Specifically, for each stage of development, we collapsed factor binding into developmental stage binding regions. For each binding region, we generate a binding module (e.g. EX:I:10001174-10001734) with a binary signature indicating the presence or absence of binding (in the region) for each factor assayed in the developmental stage. For each stage, we generated a matrix of binding modules, and randomly seeded and trained 100 large, fine-grained SOMs to cluster binding modules by their binary signatures into coherent units (clusters) within a toroidal map. SOMs concomitantly discover common combinations of co-associated factors from the binary signatures (which we refer to as TF co-association patterns) and assign binding modules (i.e., the target regulatory regions) in which these combinations occur. Therefore, each cluster has a TF co-association pattern (i.e., a common set of co-associated factors) and a collection of putative target regulatory regions.
For each stage, we select the SOM with the lowest quantization error from the 100 trials for downstream analysis. Because we are interested in identifying TF co-associations, we exclude binding modules from regions in which only one factor is bound from the matrix prior to SOM analyses. This approach generated maps with regulatory clusters that reveal how diverse TF co-association patterns relate to target regulatory regions in the C. elegans genome at each stage (Fig. 2b,c, Extended Data Fig. 6a–d). For visualization and analysis of SOMs, we used a modified kohonen package25 and custom scripts.
Stage-comparison SOM analyses
To compare higher-order co-associations between sequential stages of development (T1 versus T2), we evaluated the relative representation of co-association patterns involving factors assayed in both stages of development. First, we collapsed binding across developmental stages into stage-independent binding regions. For each pair of stages to be compared (T1, T2), we generated a matrix combining stage-specific binding modules. Specifically, for each binding region we generated a T1 and a T2 binding modules (e.g., EX:I:10001174-10001734 and L1:I:10001174-10001734) with the respective T1 and T2 binary signatures indicating the presence or absence of binding for each factor assayed in the two stages. We exclude binding modules from regions in which ≤1 factor is bound. We applied this approach to perform two types of comparative SOMs. In the first, we constructed such binding modules using all binding sites for each factor (i.e., raw binding site model). In the second, we corrected for differences in binding site numbers for individual factors by sub-sampling binding sites from the stage with the higher binding site count (to those of the stage with lower binding site count). For this second approach (matched binding site model), we generated 100 such sub-sampled binding matrixes, and select the most representative matrix as that in which frequency of the individual binary signatures is best correlated with the frequency of binary signatures across the 100 sub-sampled matrixes (R > 0.9997). For both analyses, we then randomly seed and execute 100 SOMs to cluster binary signatures and select the SOM with the lowest quantization error for downstream analysis. To examine the stage-specificity of co-association patterns, we examined the relative abundance of T1 versus T2 binding modules per SOM cluster for each approach. Such stage-comparison SOMs were performed for sequential stages of development only (Fig. 3, Extended Data Fig. 7a–c).
Cellular-resolution expression imaging and tracing
Embryonic lineage tracing and gene expression tracking were performed from both promoter reporter and protein fusion strains as previously described4–6,33,44. Briefly, for target genes in promoter reporter strains, we cloned 2250–5750 bp upstream intergenic sequences (UIS) into pJIM20 (containing a cloning site followed by histone-mCherry and a permissive let-858 3′ UTR) 44 using standard cloning methods. For each target promoter, we fixed the gene-proximal primer to the translation start site (including up to 6 aa of the endogenous protein). The resulting plasmids were used to generate transgenic C. elegans by microparticle bombardment of the strain CB4845[unc-119(ed3)] and histone::mCherry expression was tracked for at least three generations to verify stable inheritance. Promoter reporter strains were crossed with RW10029 to generate strains homozygous for the H3.3-GFP lineage tracing marker as well as for the histone-mCherry reporter. For protein fusion strains generated as part of the modENCODE project, we used strain RW10226 for the lineage tracing marker, and the colors were reversed for downstream analyses.
Strain imaging and lineage tracing was performed as previously described5,6,44, with lineages curated to at least the 350-cell stage. Expression values per cell were corrected for z-bias using a calculated attenuation level of 3.3% per plane5,44. Lineage data from each embryo was aligned to a reference lineage with standard cell cycle lengths45. We combined these data with previously published lineage data. The number of genes and image series from which expression data was derived is indicated below. The corresponding numbers of genes and image series previously published5,6 and recently acquired is shown:
Compiled tracked genes: 180 (512 image series)
Previously published genes: 130 (324 image series)
Original report genes: 50 (188 image series)
The cellular-resolution gene expression data are freely available for download through the Expression Patterns in C. elegans (EPIC) database (http://epic.gs.washington.edu) and via WormBase.
Cellular-resolution expression post-processing
For each gene, we obtained cellular-resolution expression measurements by assigning to each cell the average fluorescence signal from corresponding reporter experiments, and normalizing the signal in each cell by the maximum signal observed among imaged cells (Extended Data Fig. 8a,b, Supplementary Table 6).
We combed our imaging data to identify the set of cells tracked across all genes assayed (‘tracked’ cells), as well as the developmental time-point with the highest number of tracked cells. We directly measured expression of all 180 genes in a common set of 596 tracked cells, with maximal coverage of the embryo at 244 minutes of development, when 344 (98.3%) of the existing cells in the embryo have fluorescence measurements for all genes (Extended Data Fig. 8c,d). We refer to the set of factors (FF=13) whose binding by ChIP-seq and expression by GFP reporters was measured in the embryo as the ‘focus’ factors. We identified the set of 696 cells for which expression of all 13 focus factors was directly measured and refer to this set of cells as the ‘focus’ cells.
As a heuristic to determine the population of cells in which a gene is expressed, referred to as the expressing population for the gene, we explored a range of expression cutoffs. We required a fluorescence signal ≥2000 and chose 10% of maximal expression as the cellular expression cutoff on the basis of previous analysis5, as well as the strong and broad correlation in expression overlap with higher expression cutoffs, and its robust correlation with the quantitative expression of genes (Extended Data Fig. 8f). These expression calls revealed both distinctive and shared expressing populations for individual genes, and clusters of genes (such as a MEP-1, CEH-39, NHR-2, NHR-28, and F23F12.9-containing cluster) with similar expressing populations (Extended Data Fig. 8e).
We derived gene expression values for the 671 terminal cells born during embryogenesis by ascribing to each cell its measured expression signal or that of its last measured ancestor. To examine lineage-specificity of regulatory factors, we evaluated the enrichment of broad tissue classes in the expressing population of terminal cells of each gene.
Cellular-resolution expression data quality
For the vast majority of genes (~80%), cellular expression signals were derived from multiple time-series (Extended Data Fig. 8b). Genes with multiple time-series have, on average, 5 time-series recorded. Replicate time-series (for 145 genes), allowed us to examine the correlation in cellular-resolution expression-signals between NPR=762 pairs of replicates (Extended Data Fig. 8b), revealing a median replicate signal correlation of R=0.83. For all genes with replicate time-series (NGR=145), replicate time-series correlated at a significance level of P < 10−11 were found. The cellular overlap coefficient and Jaccard index between expressing populations of cells (A, B) as shown in Fig. 4b are calculated as:
Coefficient(A, B) = (A ∩ B)/min(A, B) Jaccard(A, B) = (A ∩ B)/(A ∪ B)
As with the binding data, our embryonic, cellular expression data is unique in both its resolution and scale. As such, homologous –quantitative, cellular-resolution, embryonic expression– measurements are not available (do not exist) for direct comparison. Nonetheless, we observe a high-degree of correspondence between the cellular expression patterns of factors and previously published lineage involvements. Owing to our focus on integrating binding and expression data, only examples of correspondence for factors with both data types are highlighted in the main text. These include the previously known regulator of pharynx and muscle, PHA-4 and HLH-1, respectively. Our expression data show also shows consistencies between known, wide-spread roles of factors and cellular-expression breadth, as illustrated for MEP-1, an oocyte development zinc-finger protein required for maintenance of somatic versus germline differentiation46 that is broadly-expressed (Ncells=379, 52% of examined cells). Undiscussed (but correlated) controls include the known regulators of intestine fate initiation and maintenance, ELT-2 and ELT-747, the cell-body muscle-expressed helix-loop-helix factor, HND-148, the pharyngeal-cell expression factor, CEH-3449, the human NeuroD homolog, CND-150, and the hypodermally-expressed molting factor, NHR-2551, among others.
Cellular-resolution SOM analyses
To integrate cellular-resolution expression and binding data, we simulated in silico genomes for each focus cell (FC=696) and mapped (embryonic stage) focus factor (FF=13) binding to the genome of cells in which factors are expressed in the early embryo. To examine physically plausible TF co-associations and the cellular contexts wherein these may occur, we compiled the cellular-resolution binding data annotating binding modules per binding region, per cell. For each focus cell, we generate binding modules spanning each of the observed binding regions from the embryonic, organism-wide data, and annotate it with a binary signature describing which of the factors bound in the region (in the embryo) are expressed in the cell (in the early embryo). This approach resulted in 2,858,477 cellular-resolution binding modules (binding regions with cell identity; e.g. ABalaa:I:10001174-10001734). We clustered cellular-resolution binding modules by their binding signatures in 100 separate SOMs, and selected the SOM with the lowest quantization error for downstream analysis (Fig. 4c,d). As before, we exclude binding modules from regions in which ≤1 factor is bound.
Lineage enrichment analyses
We constructed 3,915,749 cellular lineages in silico from the C. elegans embryogenesis cell-division tree. For each of the 696 focus cells, we generated up to 100,000 descendant lineages. We mined the cellular-resolution co-association map (Fig. 4c) for lineage-specific TF co-association patterns by examining the enrichment (hyper-geometric) of cells in the co-association patterns discovered among the cells of each cellular lineage. We discovered significant overlaps involving 8 TF co-association patterns and 5 lineage nodes (Bonferroni-corrected, P < 0.01).
Extended Data
Extended Data Figure 1.

(a) ChIP-seq raw read data were processed using a uniform processing pipeline with identical alignment, filtering criteria, and standardized IDR binding site identification using SPP. (b) Comparison of conservative (replicate) and pooled (pseudo-replicate) binding site calls –from the cross-replicate and rescue thresholds, respectively. (c) Distribution of NSC scores across 323 ChIP-seq experiments. Experiments are classified as high- (blue, NHI=181), medium- (green, NMD=60), and low-quality (yellow, NLO=82), and the relative fractions of each is indicated in the inset. High and medium quality experiments were approved for downstream analysis. (d) The fraction of binding sites shared between duplicate, approved ChIP-seq experiments with (NU=22) unique factor and stage combinations is shown. The fraction shared between the best-overlapping pairs of experiments with matched factor, stage combinations is shown in the light blue distribution. The fraction shared among all duplicates experiments (NP=24) with matched factor, stage, and promoter-driven TF expression is shown in dark blue. The range of fractions shared between true biological duplicates (ND=2) with matched factor, stage, promoter, and ChIP protocol is indicated in dashed lines. For comparison, the fraction shared between randomly sampled pairs (NS=500) of approved experiments from distinct factors is shown in gray. The median fractions for each distribution are shown. (e) Binding site histogram for 187 embryo and larval ChIP-seq experiments with unique factor-stage combinations, and a common ChIP protocol, selected for analysis in this work. The fraction of high- (blue, NHI=138) and medium-quality (green, NMD=49) ChIP-seq experiments selected is indicated (inset). (f) Analysis of sequence preferences for 21 C. elegans factors (NO) with human ortholog binding data7. The fraction of C. elegans factors (NM,=71.4%) for which sequence preferences could be determined is shown (left). The fraction of factors with conserved sequence preferences (66.7%, P < 0.05) from 12 human/worm orthologs with determined sequence preferences is shown (right). (g) The distribution in the fraction of binding sites with matches to the discovered preferred sequence (motif) is shown for 15 factors. The prevalence of the preferred sequence is evaluated among the top 200, 400, 600, 800, and 1000 binding sites for each factor (see Methods). (h) Discovered sequence preferences for 12 human/worm orthologs. Factors with similar (P < 0.05) and distinct sequence preferences are indicated in dark blue and light blue, respectively. †The consensus sequence preference for the ONECUT3 homeobox factor was obtained from Jolma et al. 32 (i) Saturation analysis of regulatory binding data. Using either binding data from embryonic and larval (L1-L4) stages or L2 larvae only (inset), k ChIP-seq experiments were randomly sampled (50 times each), collapsing overlapping binding sites into binding regions. For each k ChIP-seq experiments, the number of binding regions from 50 iterations is plotted (red points, ±1 S.D.). For each series, a exponential curve (blue, dashed line) was fit to the data and used to estimate the total number of binding regions. The percent of the binding regions (CBP) observed in the acquired data is reported for each series. (j) Amongst genes with annotated TSSs, the fraction of genes with binding observed within the specified window upstream of a TSS is shown. Promoter regions examined correspond to the windows (1) 1000/100 bp, (1) 2000/200 bp, (3) 3000/300 bp, (4) 4000/400 bp, and (5) 5000/500 bp upstream/downstream of the TSS, respectively.
Extended Data Figure 2.

Stage-dependent determination and analysis of HOT and XOT regions. (a) Correlations in occupancy (number of binding sites, x-axis) and density (number of binding sites per kb, y-axis) in embryo and larval L1-L4 binding regions. Quantiles for occupancy and density derived from binding site simulations are indicated on each axis. The fraction of binding regions (b) and the fraction of binding sites in regions (c) exceeding the significance cutoffs (quantiles from simulations) is indicated for both occupancy (yellow) and density (blue). Fractions exceeding cutoffs for both metrics are shown in red. Specific occupancy and density cutoffs for each significance level are indicated above each point. HOT (5% significance) and XOT (1% significance) regions exceed the specific occupancy thresholds indicated with arrows. (d) GO enrichment analysis of constitutive HOT (cHOT), embryo, and larval L1-L4 HOT regions. For each stage, the non-cHOT stage-derived HOT regions were analyzed. GO enrichments in stage-specific HOT regions are available in Supplementary Table 3. (e) The distribution of HOT region distances from annotated TSS in the C. elegans genome (ws220) is indicated for cHOT regions, non-constitutive HOT regions (non-cHOT), and stage-specific HOT regions. With the exception of larval L1-specific HOT regions, stage-specific HOT regions tend to be more distal. The overlap of HOT regions with embryonic (f) and larval L3 (g) chromatin states14 is indicated for cHOT, stage-derived HOT regions, and stage-specific HOT regions. With the exception of larval L1-specific HOT regions, cHOT regions show stronger promoter-associated chromatin states than non-constitutive HOT regions.
Extended Data Figure 3.

(a) Chromatin state distribution (y-axis) of embryonic binding regions as a function of binding region occupancy (x-axis). Embryonic binding regions with occupancies spanning 1–20 were mapped to 16 hiHMM chromatin states14 discovered in embryos. Regulator binding regions (RGB), HOT region, and XOT region occupancy levels are indicated along the x-axis as blue, yellow, and red bars, respectively. Chromatin state identities are indicated underneath. (b, c) Fold change in frequency of chromatin states as a function of occupancy in embryos (b) and in L3 larvae (c). HOT and XOT cutoffs for each stage are indicated in dashed lines. (d, e) Chromatin state distribution of factor binding in embryonic and larval L3 stages. Embryonic (d) and larval L3 (e) binding sites from individual ChIP-seq experiments were mapped to chromatin states derived from embryos and L3 larvae, respectively14. (f) Signal densities near enzymatically-derived TSSs30. The log2-ratio of upstream (red) versus downstream (blue) binding is color-coded below. Factors discussed in the text are highlighted.
Extended Data Figure 4.

(a) Gene ontology (GO) enrichment matrix for 150 binding experiments (75 factors) spanning 6,347 significant GO enrichments (BH-corrected, P < 0.05) across 713 GO terms (level ≥4). For each experiment, GO-term enrichment was performed on gene targets as defined by binding within 1 kb of TSSs (ChipPeakAnno) 31. Enrichments for biological process (BP) and molecular function (MF) ontologies are shown, with distinct sets of enrichments highlighted (i–viii). (b) GO term enrichments among targets of UNC-62 binding show dramatical changes in the functional role of UNC-62 regulatory activity through development. Biological process (BP) terms (levels ≥4) enriched in UNC-62 libraries are shown. The number of UNC-62 binding sites identified per stage is indicated in parenthesis. Although changes in targets between mid-larval and adult stages have been suggested previously22, our analyses (performed with uniformly called binding sites) and expanded data indicate that the most dramatic changes occur between embryo and L4 larval stages. (+) MEP-1 indicates experiments performed in strain OP102.
Extended Data Figure 5.

(a) Clustering patterns in pairwise TF co-associations. Clustered libraries from shared factors are colored blue. Clustered embryonic libraries are colored yellow. ChIP-seq libraries that cluster in embryonic groups and with distinct stages for the same factor are colored green. BLMP-1 and ELT-3 libraries are colored purple. FOS-1 and JUN-1 libraries are colored red. All other libraries are colored gray in the dendrogram. The clustering dendrogram is derived from Fig. 2a. (b) Difference in pairwise TF co-associations at expressed and repressed promoter domains. For embryonic and larval L1 stages, we computed co-association strength 2kb upstream and 200bp downstream domains of TSSs associated with expressed and repressed genes, from stage-specific binding experiments with IntervalStats24. For each comparison (and each domain), the difference in the strength of co-associations between the expressed and repressed domains is shown for embryo (bottom left) and larval L1 stages (top right). Positive values indicate stronger co-associations in the expressed domain whereas negative values indicate stronger co-associations in the domain of repressed promoters. (c–f) Change in pairwise TF co-associations across sequential developmental stages. For factors assayed in sequential developmental stages, the difference in the co-association strengths for pairs of factors is shown. The change in co-association strengths are shown for the embryo to larval L1 (c), larval L1 to L2 (d), larval L2 to L3 (e), and larval L3 to L4 transitions (f). Co-association strengths for pairs of factors at each stage are derived from Fig. 2a.
Extended Data Figure 6.

Stage-specific analysis of higher-order co-associations in the larvae. For each larval stage of development, binding regions were annotated with binary signatures indicating the presence or absence of factor binding and clustered into SOMs describing the co-association patterns amongst factors assayed in each stage. SOMs (a–d) are colored by number of factors per co-association pattern with respective patterns in each cluster are indicated underneath. (e) For each co-association pattern discovered in stage-specific SOMs, GO enrichment analysis was performed on genes associated by binding within 1 kb of TSSs (ChipPeakAnno) 31. GO terms are arranged along the circumference of the graph, and their enrichment is indicated in each stage. The inner-most layer contains the gene ontology color key as indicated and subsequent layers (from the center) indicate embryonic (EX), L1, L2, L3, and L4 enrichment of each GO term. For visualization purposes, only GO terms with 5 ≤ annotated genes ≤25 (NGO= 419) are shown.
Extended Data Figure 7.

Stage-comparison SOMs highlight patterns in the specificity of higher-order TF co-associations. (a) Abundance of co-association patterns is graphed as function of the number of factors in each co-association in stage-comparison SOMs for the embryo versus larval L1 stage comparison. Similar patterns are observed in all stage-comparisons SOMs. (b) Difference in binding sites between embryos and L1 larvae for each factor (gray dots). The fractional difference, calculated as fraction of the larger set of binding sites represented by the difference in binding sites, is shown. Factors are rank-ordered by their difference in binding sites. The fraction of co-association patterns that are stage-specific (≥90% embryonic or larval L1) in SOMs is indicated for the raw binding sites with all factors (Fig. 3a, dashed line), in SOMs with individual factors removed (blue), and in SOMs with factors sequentially removed (red). (c) Embryonic and larval L1 binding SOM with matched numbers of binding sites. Briefly, binding data for the 15 factors assayed in the embryo and L1 larvae was sub-sampled to generate stage-specific binding modules with equal numbers of binding sites for each factor (see Methods). Stage-specific binding modules with matched binding sites were clustered in an SOM describing 140 co-association patterns. SOM is colored as in (Fig. 3a). (d) Binding signatures (fraction of modules bound by each factor) are shown for each co-association pattern from (c). Sidebar indicates the embryonic (versus L1) stage-specificity of each co-association pattern as in (c). Stage-comparison SOMs with raw and matched binding sites are presented for the (e) larval L1 versus L2 comparison, (f) larval L2 versus L3 comparison, and (g) larval L3 versus L4 comparison. Binding region comparisons are performed as in Fig. 3. Briefly, binding data for factors assayed in sequential stages are assigned to stage-resolved binding modules (i.e. L1:I:10001174-10001734). Stage-resolved binding modules are clustered into SOMs describing shared and stage-specific co-association patterns. SOMs are colored by the T1 versus T2 (for example, L1 versus L2) stage-specificity of the learned co-association patterns, measured as the fraction of binding modules that are T1. T1- and T2-specific co-association patterns are shown in red and blue, respectively. Sidebars indicate the T1 (versus T2) stage-specificity of each co-association pattern. As in Fig. 3, SOMs with matched binding sites were generated by sub-sampling binding sites to generate stage-resolved binding modules with equal numbers of binding sites for each factor. For each comparison, the most representative sampling (from 100 iterations) was selected to seed SOM analyses. For each of the stage-comparison SOMs with matched binding sites (e–g), the matrix of learned co-association patterns (fraction of modules bound by each factor) are shown below each SOM. (h–j) The fraction of co-association patterns that are stage-specific (≥90% either stage) in SOMs is indicated for the raw binding sites with all factors assayed in both stages (dashed line), in SOMs with individual factors removed (blue), and in SOMs with factors sequentially removed (red) are shown for the larval L1 and L2 stage (h), larval L2 and L3 stage (i), and larval L3 and L4 stage (j) comparisons.
Extended Data Figure 8.

(a) Cellular-resolution, protein expression levels for 180 genes (x-axis) in terminal embryo cells (N=671, y-axis). For each gene, the normalized expression signal in each cell is shown (see Methods). For each gene, expression signals in cells not measured directly corresponds to the expression signal of the last measured ancestor. Focus factors (FF=13) whose binding was assayed in embryonic stages are labeled red. Factors whose binding was assayed only in larval stages are labeled blue (FL=23). The broad tissue class of each cell is indicated in the sidebar. (b) Embryonic, cellular-resolution expression data quality controls. The number of time-series recorded per gene (x-axis) is shown. For genes with multiple time-series (NGR=145), the Pearson correlation coefficient (R) in the fluorescence signals of cells recorded was calculated between NPR=762 pairs of time-series (replicates). The distribution of correlation coefficients is shown. The median correlation co-efficient among replicate experiments is shown (R = 0.8310). The number (c) and percentage (d) of embryonic cells with expression measurements across any of the assayed genes (assayed cells, gray), all of the assayed genes (tracked cells), and all of the 13 genes (focus factors) for which both embryonic binding data and cellular-resolution expression data was acquired (focused cells) are plotted as a function of developmental time (Sulston minutes). The specific developmental times with the maximum coverage of the cells in the embryo are indicated for the tracked (TT) and focused cells (TF). (e) Previous reports5 have suggested that a robust heuristic to identify cells in which individual genes are expressed can be obtained by requiring a fluorescence signal ≥ 2000 and a fluorescence signal that is ≥ 10% of the maximum signal observed for each reporter (gene). To confirm these recommendations, we calculated the overlap in the expressing cell populations for pairs of genes at 10% (f=0.1) and 20% (f =0.2) of the maximal signal for each gene, and computed the correlation between calculated overlaps per gene-pair between the two thresholds (R=0.94). This analysis was extended to compare a wide range of expression cutoffs (f) in (e), where we observed robust correlations for the 10% cutoff (f =0.1). (f) Cellular expression overlap matrix for 180 genes in the early embryo. For each pairwise gene comparison, we calculated the significance of the overlap between the population of cells expressing each gene. The overlap enrichment and depletion P-values between gene pairs were determined using directional Fisher’s exact tests and were Benjamini-Hochberg corrected. To generate a final overlap score, we select the most significant of the enrichment and depletion scores, reporting either the -log10(P-value of enrichment) or the log10(P-value of depletion) to obtain positive and negative values for enrichment and depletion, respectively. (g) Overlap between co-association cells and the gene-expressing cells (the expressing population) for non-focus factors (NNF=168). For each cellular-resolution co-association pattern discovered (Fig. 4c), the set of co-association cells is defined as the population of cells in which the co-association is observed in the SOM. For 39 co-association patterns, co-association cells significantly overlap (hypergeometric test, Bonferroni-corrected, P < 0.01) the gene-expression cells of at least one of 124 non-focus factor target genes. Co-association patterns and target gene pairs with significant overlaps between the co-association cells and gene-expression cells were classified as ‘Co-association in promoter’ if the co-association pattern with the significant enrichment was observed at the promoter at the target gene, and as ‘Co-association not in promoter’ if this was not the case. The distribution of overlap significance values for the two classes and the respective Wilcoxon test P-value for similarity between the two distributions is shown. MEP-1 (+) indicates experiments performed with strain OP102.
Extended Data Figure 9.

Full-resolution view of global pairwise TF co-association matrix. As outlined in Fig. 2a, the significance of co-binding (co-association strength) 2kb upstream and 200bp downstream of TSSs was measured reciprocally between all binding experiments (IntervalStats24, see Methods). For each comparison (NC=34,782), the fraction of significant (P < 0.05) co-binding events was computed and the mean fraction of reciprocal tests is reported (NT=17,391). Co-association scores are scaled by the standard deviation (uncentered) for visualization purposes. Co-associations were examined among 292,466 binding sites outside of XOT regions. Inset (i) shows the distribution of global TF co-association strengths from pairwise comparisons of 187 ChIP-seq experiments. The distribution of co-association strengths is shown from comparisons of all (distinct) ChIP-seq experiments (NDE=17,391, light blue) and from comparisons of ChIP-seq experiments from distinct factors (NDF=17,197, dark blue). The 75th, 90th, and 95th percentiles from comparisons between distinct factors (CS75%=0.2437, CS90%=0.3589, and CS95%=0.4266) are indicated as light red, red, and dark red dashed-lines, respectively. Co-association strengths between FOS-1:JUN-1 in L1, L3 and L4 larvae are indicated with arrows. Inset (ii) highlights the similarity (Wilcoxon test, P=0.4913) between distributions from distinct factors and distinct experiments.
Extended Data Figure 10.

Representative samples of staged, transgenic C. elegans embryos and larvae expressing GFP-tagged fusion proteins. GFP fluorescence images, DIC images, and merged (GFP/DIC) images are labeled with green, white, and blue dots, respectively. The 10 μm scale bar is shown in GFP fluorescence images. Images were selected independent of binding experiment results. Approved binding experiments include: MEP-1 (mixed embryo, L2 larvae), DPL-1 (L1 larvae), C27D6.4 (L2 larvae), NHR-23 (L3 larvae), and CEH-16 (L4 larvae) experiments.
Supplementary Material
Acknowledgments
We thank Ghia Euskirchen at the Stanford Center for Genomics and Personalized Medicine for sequencing ChIP libraries and members of the Reinke lab for contributing ChIP-seq data. We thank members of the Waterston lab, Sarov lab, and Kim lab for tagged constructs and generating C. elegans strains. We thank J. Reuter, D. Phanstiel, H. Tilgner, and D. Fowler for critical comments on the manuscript. This work is supported by the NHGRI as part of the modENCODE project (U01 HG004267).
Footnotes
Author Contributions
C.L.A., T.K., J.I.M., V.R., R.H.W., and M.P.S. designed experiments. T.K., L.J. L.M., W.N., and B.W. constructed strains and prepared ChIP libraries for sequencing. J.I.M., D.V., R.T., P.W., L.G., and D.M. constructed strains and performed reporter imaging experiments. A.K., C.L.A., and A.P.B. performed alignments, uniform binding-site identification and quality-filtering. C.L.A. organized data, pre-processed binding site and expression data, and executed all analyses. A.P.B., D.X., and C.L.A developed SOM analysis techniques. C.L.A., V.R., J.I.M., R.H.W., and M.P.S. wrote the manuscript.
Author Information
ChIP-seq data reported here is available through the modENCODE DCC (www.modencode.org) and at tapanti.stanford.edu/cetrn.
The authors declare no competing financial interests.
Readers are welcome to comment on the online version of the paper.
References
- 1.Davidson EH. Emerging properties of animal gene regulatory networks. Nature. 2010;468:911–920. doi: 10.1038/nature09645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spitz F, Furlong EEM. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet. 2012;13:613–626. doi: 10.1038/nrg3207. [DOI] [PubMed] [Google Scholar]
- 3.Lee TI, Young RA. Transcriptional regulation and its misregulation in disease. Cell. 2013;152:1237–1251. doi: 10.1016/j.cell.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bao Z, et al. Automated cell lineage tracing in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2006;103:2707–2712. doi: 10.1073/pnas.0511111103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murray JI, et al. Multidimensional regulation of gene expression in the C. elegans embryo. Genome Res. 2012;22:1282–1294. doi: 10.1101/gr.131920.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mace DL, Weisdepp P, Gevirtzman L, Boyle T, Waterston RH. A High-Fidelity Cell Lineage Tracing Method for Obtaining Systematic Spatiotemporal Gene Expression Patterns in Caenorhabditis elegans. G3 (Bethesda) 2013;3:851–863. doi: 10.1534/g3.113.005918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boyle AP, et al. Comparative analysis of regulatory information and circuits across diverse species. doi: 10.1038/nature13668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerstein MB, et al. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science. 2010;330:1775–1787. doi: 10.1126/science.1196914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rhee HS, Pugh BF. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell. 2011;147:1408–1419. doi: 10.1016/j.cell.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Allen MA, Hillier LW, Waterston RH, Blumenthal T. A global analysis of C. elegans trans-splicing. Genome Res. 2011;21:255–264. doi: 10.1101/gr.113811.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moorman C, et al. Hotspots of transcription factor colocalization in the genome of Drosophila melanogaster. Proc Natl Acad Sci USA. 2006;103:12027–12032. doi: 10.1073/pnas.0605003103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nègre N, et al. A cis-regulatory map of the Drosophila genome. Nature. 2011;471:527–531. doi: 10.1038/nature09990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yip KY, et al. Classification of human genomic regions based on experimentally determined binding sites of more than 100 transcription-related factors. Genome Biol. 2012;13:R48. doi: 10.1186/gb-2012-13-9-r48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ho JWK, et al. Comparative analysis of metazoan chromatin architecture [Google Scholar]
- 15.Ohinata Y, et al. Blimp1 is a critical determinant of the germ cell lineage in mice. Nature. 2005;436:207–213. doi: 10.1038/nature03813. [DOI] [PubMed] [Google Scholar]
- 16.Smith MA, et al. PRDM1/Blimp-1 controls effector cytokine production in human NK cells. J Immunol. 2010;185:6058–6067. doi: 10.4049/jimmunol.1001682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuersten S, Segal SP, Verheyden J, LaMartina SM, Goodwin EB. NXF-2, REF-1, and REF-2 affect the choice of nuclear export pathway for tra-2 mRNA in C. elegans. Mol Cell. 2004;14:599–610. doi: 10.1016/j.molcel.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 18.Baugh LR, Demodena J, Sternberg PW. RNA Pol II accumulates at promoters of growth genes during developmental arrest. Science. 2009;324:92–94. doi: 10.1126/science.1169628. [DOI] [PubMed] [Google Scholar]
- 19.Hope IA, Mounsey A, Bauer P, Aslam S. The forkhead gene family of Caenorhabditis elegans. Gene. 2003;304:43–55. doi: 10.1016/s0378-1119(02)01175-7. [DOI] [PubMed] [Google Scholar]
- 20.Van Auken K, et al. Roles of the Homothorax/Meis/Prep homolog UNC-62 and the Exd/Pbx homologs CEH-20 and CEH-40 in C. elegans embryogenesis. Development. 2002;129:5255–5268. doi: 10.1242/dev.129.22.5255. [DOI] [PubMed] [Google Scholar]
- 21.Curran SP, Ruvkun G. Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet. 2007;3:e56. doi: 10.1371/journal.pgen.0030056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Nostrand EL, Sánchez-Blanco A, Wu B, Nguyen A, Kim SK. Roles of the developmental regulator unc-62/Homothorax in limiting longevity in Caenorhabditis elegans. PLoS Genet. 2013;9:e1003325. doi: 10.1371/journal.pgen.1003325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhong M, et al. Genome-wide identification of binding sites defines distinct functions for Caenorhabditis elegans PHA-4/FOXA in development and environmental response. PLoS Genet. 2010;6:e1000848. doi: 10.1371/journal.pgen.1000848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chikina MD, Troyanskaya OG. An effective statistical evaluation of ChIPseq dataset similarity. Bioinformatics. 2012;28:607–613. doi: 10.1093/bioinformatics/bts009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie D, et al. Dynamic trans-acting factor colocalization in human cells. Cell. 2013;155:713–724. doi: 10.1016/j.cell.2013.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andersson LS, et al. Mutations in DMRT3 affect locomotion in horses and spinal circuit function in mice. Nature. 2012;488:642–646. doi: 10.1038/nature11399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu X, et al. Analysis of cell fate from single-cell gene expression profiles in C. elegans. Cell. 2009;139:623–633. doi: 10.1016/j.cell.2009.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salser SJ, Kenyon C. A C. elegans Hox gene switches on, off, on and off again to regulate proliferation, differentiation and morphogenesis. Development. 1996;122:1651–1661. doi: 10.1242/dev.122.5.1651. [DOI] [PubMed] [Google Scholar]
- 29.Hobert O. Regulatory logic of neuronal diversity: terminal selector genes and selector motifs. Proceedings of the National Academy of Sciences. 2008;105:20067–20071. doi: 10.1073/pnas.0806070105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gu W, et al. CapSeq and CIP-TAP identify Pol II start sites and reveal capped small RNAs as C. elegans piRNA precursors. Cell. 2012;151:1488–1500. doi: 10.1016/j.cell.2012.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu LJ, Gazin C, Green MR. 7. ChIPpeakAnno: a Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinformatics. 2010;11:237–237. doi: 10.1186/1471-2105-11-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jolma A, et al. DNA-binding specificities of human transcription factors. Cell. 2013;152:327–339. doi: 10.1016/j.cell.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 33.Sarov M, et al. A Genome-Scale Resource for In Vivo Tag-Based Protein Function Exploration in C. elegans. Cell. 2012;150:855–866. doi: 10.1016/j.cell.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lefrançois P, et al. Efficient yeast ChIP-Seq using multiplex short-read DNA sequencing. BMC Genomics. 2009;10:37. doi: 10.1186/1471-2164-10-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kharchenko PV, Tolstorukov MY, Park PJ. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat Biotechnol. 2008;26:1351–1359. doi: 10.1038/nbt.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Q, Brown JB, Huang H, Bickel PJ. Measuring reproducibility of high-throughput experiments. The Annals of Applied Statistics. 2011;5:1752–1779. [Google Scholar]
- 39.Landt SG, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bailey TL, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–8. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS. Quantifying similarity between motifs. Genome Biol. 2007;8:R24. doi: 10.1186/gb-2007-8-2-r24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Teytelman L, Thurtle DM, Rine J, van Oudenaarden A. Highly expressed loci are vulnerable to misleading ChIP localization of multiple unrelated proteins. Proceedings of the National Academy of Sciences. 2013;110:18602–18607. doi: 10.1073/pnas.1316064110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hammar P, et al. Direct measurement of transcription factor dissociation excludes a simple operator occupancy model for gene regulation. Nat Genet. 2014 doi: 10.1038/ng.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murray JI, et al. Automated analysis of embryonic gene expression with cellular resolution in C. elegans. Nat Methods. 2008;5:703–709. doi: 10.1038/nmeth.1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sulston JE, Schierenberg E, White JG, Thomson JN. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol. 1983;100:64–119. doi: 10.1016/0012-1606(83)90201-4. [DOI] [PubMed] [Google Scholar]
- 46.Unhavaithaya Y, et al. MEP-1 and a homolog of the NURD complex component Mi-2 act together to maintain germline-soma distinctions in C. elegans. Cell. 2002;111:991–1002. doi: 10.1016/s0092-8674(02)01202-3. [DOI] [PubMed] [Google Scholar]
- 47.Sommermann EM, Strohmaier KR, Maduro MF, Rothman JH. Endoderm development in Caenorhabditis elegans: the synergistic action of ELT-2 and -7 mediates the specification→differentiation transition. Dev Biol. 2010;347:154–166. doi: 10.1016/j.ydbio.2010.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mathies LD, Henderson ST, Kimble J. The C. elegans Hand gene controls embryogenesis and early gonadogenesis. Development. 2003;130:2881–2892. doi: 10.1242/dev.00483. [DOI] [PubMed] [Google Scholar]
- 49.Hirose T, Galvin BD, Horvitz HR. Six and Eya promote apoptosis through direct transcriptional activation of the proapoptotic BH3-only gene egl-1 in Caenorhabditis elegans. Proceedings of the National Academy of Sciences. 2010;107:15479–15484. doi: 10.1073/pnas.1010023107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hallam S, Singer E, Waring D, Jin Y. The C. elegans NeuroD homolog cnd-1 functions in multiple aspects of motor neuron fate specification. Development. 2000;127:4239–4252. doi: 10.1242/dev.127.19.4239. [DOI] [PubMed] [Google Scholar]
- 51.Brooks DR, Appleford PJ, Murray L, Isaac RE. An essential role in molting and morphogenesis of Caenorhabditis elegans for ACN-1, a novel member of the angiotensin-converting enzyme family that lacks a metallopeptidase active site. J Biol Chem. 2003;278:52340–52346. doi: 10.1074/jbc.M308858200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.