

Abstract
In the present study, solid dispersion nanoparticles with a hydrophilic polymer and surfactant were developed using the supercritical antisolvent (SAS) process to improve the dissolution and oral absorption of megestrol acetate. The physicochemical properties of the megestrol acetate solid dispersion nanoparticles were characterized using scanning electron microscopy, differential scanning calorimetry, powder X-ray diffraction, and a particle-size analyzer. The dissolution and oral bioavailability of the nanoparticles were also evaluated in rats. The mean particle size of all solid dispersion nanoparticles that were prepared was <500 nm. Powder X-ray diffraction and differential scanning calorimetry measurements showed that megestrol acetate was present in an amorphous or molecular dispersion state within the solid dispersion nanoparticles. Hydroxypropylmethyl cellulose (HPMC) solid dispersion nanoparticles significantly increased the maximum dissolution when compared with polyvinylpyrrolidone K30 solid dispersion nanoparticles. The extent and rate of dissolution of megestrol acetate increased after the addition of a surfactant into the HPMC solid dispersion nanoparticles. The most effective surfactant was Ryoto sugar ester L1695, followed by D-α-tocopheryl polyethylene glycol 1000 succinate. In this study, the solid dispersion nanoparticles with a drug:HPMC:Ryoto sugar ester L1695 ratio of 1:2:1 showed >95% rapid dissolution within 30 minutes, in addition to good oral bioavailability, with approximately 4.0- and 5.5-fold higher area under the curve (0–24 hours) and maximum concentration, respectively, than raw megestrol acetate powder. These results suggest that the preparation of megestrol acetate solid dispersion nanoparticles using the supercritical antisolvent process is a promising approach to improve the dissolution and absorption properties of megestrol acetate.
Keywords: bioavailability, megestrol acetate, dissolution, supercritical technology
Introduction
Megestrol acetate, 17-(acetyloxy)-6-methyl-pregna-4,6-diene-3,20-dione, a synthetic steroidal progestin, is widely used to treat loss of appetite and weight loss in patients with AIDS.1 In addition, it is used as an antineoplastic agent to treat advanced breast cancer or endometriosis.2 As a Biopharmaceutics Classification System class II drug, it is insoluble in water (2.0 μg/mL), with high permeability.3 To improve its dissolution and bioavailability, various formulations, such as microparticles, nanoemulsions, nanosuspensions, and solid dispersions, have been studied.4–9
Amorphous solid dispersions are a well-established formulation system for poorly water-soluble active pharmaceutical ingredients (APIs). APIs exist in an amorphous form within solid dispersions that increase solubility and dissolution, thereby enhancing their oral bioavailability.10–12 Amorphous solid dispersions with a hydrophilic polymer and/or surfactant can be prepared by solvent evaporation and/or melting. In addition, supercritical fluid technology, based on the principle of solvent evaporation, can be applied to the preparation of amorphous solid dispersion nanoparticles.13,14 Supercritical carbon dioxide (SC-CO2) is widely used as an antisolvent in the supercritical antisolvent (SAS) process, owing to its relatively low critical point (critical pressure =7.38 MPa, critical temperature =31.1°C). Amorphous solid dispersion nanoparticles prepared using the SAS process have been utilized to improve the solubility, dissolution, and oral bioavailability of poorly water-soluble APIs, such as itraconazole, dutasteride, and valsartan.15–17
The present study sought to develop solid dispersion nanoparticles with a hydrophilic polymer and surfactant via the SAS process to improve the dissolution and oral absorption of megestrol acetate. The physicochemical properties of megestrol acetate solid dispersion nanoparticles were characterized using scanning electron microscopy (SEM), differential scanning calorimetry (DSC), powder X-ray diffraction (PXRD), and a particle-size analyzer. In addition, the dissolution and oral bioavailability of the nanoparticles were evaluated in rats.
Materials and methods
Megestrol acetate was kindly donated by LG Life Sciences (Seoul, South Korea). Hydroxypropylmethyl cellulose (HPMC; 2910) was purchased from Shin-Etsu Chemical Co Ltd (Tokyo, Japan). Polyvinylpyrrolidone (PVP; K30), PVP vinyl acetate (PVP VA64), and poloxamer 407 were kindly obtained from BASF Co Ltd (Ludwigshafen, Ger-many). D-α-Tocopheryl polyethylene glycol 1000 succinate (TPGS; Eastman Chemical Company, Kingsport, TN, USA), Gelucire 44/14 (Gattefossé, Saint-Priest, France), and Ryoto sugar ester L1695 (Mitsubishi-Kagaku Foods Corporation, Tokyo, Japan) were used as surfactants. Medroxypro-gesterone acetate (internal standard [IS]), polyethylene glycol 6000 (PEG 6000), and sodium lauryl sulfate were purchased from Sigma-Aldrich Co Ltd (St Louis Co, MO, USA). High-performance liquid chromatography (HPLC)-grade acetonitrile, ethanol, and methanol were used in all experiments.
Preparation of megestrol acetate solid dispersion nanoparticles
Solid dispersion nanoparticles containing megestrol acetate and hydrophilic polymers/surfactant were produced via the SAS process using Thar SAS200 equipment (Thar Process Inc, Pittsburgh, PA, USA). First, megestrol acetate was dissolved in a mixture of methylene chloride and ethanol (45:55, w/w) containing hydrophilic polymers and/or surfactant at a concentration of 5% solute (w/w). The detailed formulae of solid dispersion nanoparticles are given in Table 1. The SAS process was performed under the following conditions, based on our previous work.17–19 The drug solution was delivered through a nozzle at a flow rate of 1 mL/min using an HPLC pump into a high-pressure vessel that was filled with SC-CO2. This SC-CO2 continued to flow at a rate of 11 g/min through the vessel to maintain the steady state. The temperature and pressure in the vessel were kept constant at 40°C and 15 MPa, respectively. After the drug solution had been sprayed, pure SC-CO2 was flushed through the high-pressure vessel to remove any trace of residual organic solvent on the solid dispersion nanoparticles or within the vessel. The solid dispersion nanoparticles were recovered on the walls and bottom of the vessel following slow depressurization of the system conditions to atmospheric pressure.
Table 1.
The formulation, particle size, and specific surface area of the megestrol acetate solid dispersion nanoparticles prepared using the SAS process
| Formulation | Drug content (%) | Mean particle size (nm) | Specific surface area (m2/g) |
|---|---|---|---|
| Megestrol acetate:HPMC =1:2 | 95.1±2.6 | 135.8±18.2 | 96.16±1.69 |
| Megestrol acetate:PVP K30 =1:2 | 95.9±3.7 | 310.9±30.3 | 40.21±1.33 |
| Megestrol acetate:HPMC:Gelucire 44/14 =1:2:0.5 | 96.1±2.1 | 245.9±35.9 | 49.22±1.09 |
| Megestrol acetate:HPMC:poloxamer 407 =1:2:0.5 | 95.8±2.3 | 229.7±21.6 | 51.13±1.03 |
| Megestrol acetate:HPMC:Ryoto sugar ester L1695 =1:2:0.5 | 97.1±2.0 | 155.3±20.1 | 69.31±1.25 |
| Megestrol acetate:HPMC:TPGS =1:2:0.5 | 97.7±1.9 | 495.2±65.3 | 33.11±1.19 |
| Megestrol acetate:HPMC:Ryoto sugar ester L1695 =1:2:1 | 98.9±1.8 | 180.3±29.1 | 57.22±1.35 |
Notes:
The drug content (%) = weight of the loaded drug/weight of the feeding drug ×100. Data are expressed as mean ± standard deviation (n=3).
Abbreviations: SAS, supercritical antisolvent; HPMC, hydroxypropylmethyl cellulose; PVP, polyvinylpyrrolidone; TPGS, D-α-tocopheryl polyethylene glycol 1000 succinate.
Characterization of megestrol acetate solid dispersion nanoparticles
Megestrol acetate content within solid dispersion nanopar-ticles was assessed by dissolving approximately 20 mg solid dispersion nanoparticles in 100 mL mixture of methylene chloride and ethanol (45:55, w/w). Mixtures were filtered through a 0.45 μm syringe filter, and the concentration was analyzed using a Waters HPLC system (Milford, MA, USA). The HPLC apparatus consisted of a pump (model 600), column oven (steel column heater 600), autosampler (model 717 plus), ultraviolet detector (model 486 tunable absorbance detector), and chromatographic data processor (Empower Pro). Separations were performed on a Capcell Pak C18 MG (4.6×250 mm, 5 μm) reverse-phase column (Shiseido Fine Chemicals, Tokyo, Japan) eluted under isocratic conditions with a mobile phase consisting of acetonitrile and water (65:35 v/v). Using a 20 μL injection, chromatography was performed at 30°C with a flow rate of 1.0 mL/min. An ultraviolet detector was used at a wavelength of 280 nm. SEM images of solid dispersion nanoparticles were obtained with a JSM-7000F (JEOL, Tokyo, Japan). The specimens were sputter-coated with gold-palladium. The mean particle size and particle-size distribution of the nanoparticles were analyzed with a laser particle-size analyzer (BI-9000; Brookhaven Instruments Corporation, Holtsville, NY, USA). Prior to being sized, the solid dispersion nanoparticles were suspended in mineral oil and sonicated. Measurements were made in triplicate. The specific surface area of the nanopar-ticles was measured by nitrogen adsorption in a Quantasorb gas-adsorption apparatus (Autosorb-1; Quantachrome Instruments, Boynton Beach, FL, USA). Nitrogen surface areas were automatically calculated by a system software based on the Brunauer–Emmett–Teller method. PXRD images were obtained at 25°C with an AXS D8 Discover using a general area diffraction detector-system diffractometer (Bruker AXS Inc, Madison, WI, USA). All samples were exposed to Cu Kα radiation (40 kV, 40 mA). Data were collected over an angular range from 5° to 60° 2θ in continuous-scan mode using a step size of 0.009° 2θ. DSC measurements were carried out using a Sinco S-650 (Sinco Co Ltd, Seoul, South Korea). Approximately 4–6 mg of solid dispersion nanopar-ticles were sealed in an aluminum pan. The DSC curves were recorded by heating from 25°C to 250°C at 5°C/min under nitrogen purge (20 mL/min). The characteristics of the megestrol acetate dissolution from solid dispersion nanoparticles were investigated using the US Pharmacopoeia dissolution apparatus 2 (paddle method; Electrolab, Mumbai, India) at a stirring rate of 50 rpm and temperature of 37°C±0.5°C. The dissolution media contained 900 mL of simulated gastric fluid (pH 1.2, without pepsin) containing 0.1% sodium lauryl sulfate (w/v). Solid dispersion nanoparticles equivalent to 160 mg megestrol acetate were placed in the dissolution vessel. At appropriate intervals, 3 mL samples were withdrawn and immediately filtered through a 0.45 μm syringe filter, followed by dilution with methanol. The concentration of megestrol acetate was determined by HPLC analysis. All dissolution tests were performed in quadruplicate for each formulation.
Measurement of megestrol acetate solubility in surfactant solution
The solubility of megestrol acetate was measured in various surfactant solutions at 1% concentrations (w/v). Gelucire 44/14, poloxamer 407, Ryoto sugar ester L1695, and TPGS were tested as surfactants. Approximately 10 mg megestrol acetate was added to 10 mL surfactant solution, following by sonication for 2 hours in an ultrasonic bath. The suspensions were allowed to equilibrate by incubation in a shaking water bath at 37°C for 24 hours. After equilibrium was reached, each sample was filtered using a 0.45 μm syringe filter and diluted with methanol. The concentration of megestrol acetate was determined by HPLC analysis.
Pharmacokinetic study in rats
The animal-study protocol was complied with the institutional guidelines for the care and use of laboratory animals and approved by the ethics committee of Kyungsung University (Busan, South Korea). Fifteen male Sprague Dawley rats (250±10 g) were acquired from Orient Bio Inc (Seongnam, South Korea) and divided into three treatment groups (n=5 per group). Rats were fasted for 1 day prior to pharmacokinetic investigations, but allowed free access to water. Rats were orally administered raw megestrol acetate powder or the solid dispersion nanoparticles at a dose of 20 mg/kg megestrol acetate. Both treatments were dispersed in 1 mL water immediately prior to oral dosing. Blood (approximately 0.30 mL) was sampled from the retro-orbital plexus into heparin-treated tubes at preset time-points and centrifuged at 10,000 rpm at 4°C for 10 minutes. The resulting plasma was collected and stored at −70°C until analysis by LC with tandem mass spectrometry (MS/MS) method. The concentration of megestrol acetate and the IS (medroxyprogesterone acetate) in the plasma samples was analyzed using LC-MS/MS.20 Briefly, 100 μL plasma was mixed with the IS (100 ng/mL) and 1.9 mL of acetonitrile for protein precipitation. After the mixture was vortex-mixed briefly, the organic phase was separated from the aqueous phase by centrifugation at 13,000 rpm for 5 minutes. The supernatant (10 μL) was analyzed by LC-MS/MS. The 1100 series LC system (Agilent Technologies, Santa Clara, CA, USA) consists of a solvent degasser, binary pumps, and autosampler. Megestrol acetate was separated on Pursuit XRs Ultra C18 columns (50×2.0 mm, 2.8 μm). The mobile phase consists of acetonitrile/5 mM ammonium acetate (65:35, v/v) and was pumped at a flow rate of 0.3 mL/min. An API 4000 triple-quadrupole mass spectrometer (AB Sciex, Framingham, MA, USA) with a TurboIonSpray interface operating in positive-ionization mode was used for multiple-reaction monitoring: m/z 385.3–267.2 for megestrol acetate, and m/z 387.3–327.2 for the IS. The ion-spray voltage and nebulizer gas were set at 5,500 V. For the pharmacokinetic study, the maximum concentration (Cmax), time to Cmax, and area under the concentration–time curve for hours 0–24 (AUC0–24) were determined. Cmax and time to Cmax were directly obtained from the original data set, and AUC0–24 was calculated using the linear trapezoidal rule. Pharmacokinetic parameters were calculated by noncompartmental analysis using WinNonlin standard edition software, version 5.3 (Pharsight Corporation, St Louis, MO, USA). Statistical analysis was performed using one-way analysis of variance, followed by the Student–Newman–Keuls and least squares difference tests with SPSS 21.0 (IBM Corporation, Armonk, NY, USA).
Results and discussion
Preparation and characterization of megestrol acetate solid dispersion nanoparticles
In the present study, solid dispersion nanoparticles were manufactured using the SAS process with SC-CO2 as the antisolvent to enhance the dissolution and oral bioavailability of megestrol acetate. To select a hydrophilic polymer, megestrol acetate solid dispersion nanoparticles were fabricated using HPMC, PEG 6000, PVP K30, and PVP VA64 at a drug:polymer ratio of 1:2 (w/w) with a methylene chloride–ethanol mixture as a solvent. Both PEG 6000 and PVP VA64 solid dispersion produced irregular microparticles and had <20% production yields. PVP K30 and HPMC resulted in the formation of spherical nanoparticles with >70% yields, as shown in Figure 1 and Table 1. The megestrol acetate–HPMC formulation produced spherical nanoparticles with a mean particle size of 135.8 nm, resulting in a high specific surface area: >90m2/g. The mean particle size (310 nm) was greater for PVP K30 than the mean particle size of HPMC nanoparticles. The crystalline state of megestrol acetate within the solid dispersion nanoparticles was evaluated by PXRD and DSC measurements. As shown in Figure 1, the PXRD pattern and DSC curve of crystalline megestrol acetate exhibited characteristic diffraction peaks at 2θ =12.11°, 13.64°, 14.61°, 15.82°, 16.66°, and 22.71°, and an endothermic peak at approximately 218°C (melting point). However, characteristic diffraction peaks and endothermic peaks were not observed in either HPMC or PVP K30 solid dispersion nanoparticles prepared by the SAS process. This indicated that megestrol acetate was present in an amorphous or molecular dispersion state within the solid dispersion nanoparticles.21,22 Dissolution tests were performed for raw megestrol acetate, PVP K30, and HPMC solid dispersion nanoparticles. As shown in Figure 1, the dissolution of megestrol acetate was dramatically increased by the solid dispersion nanoparticles. Interestingly, 50% megestrol acetate from HPMC solid dispersion nanoparticles, compared with 9.3% raw megestrol acetate, dissolved in 30 minutes. Furthermore, HPMC solid dispersion nanoparticles significantly increased the maximum dissolution when compared with PVP K30 solid dispersion nanoparticles (P<0.05). Therefore, HPMC was chosen as the hydrophilic polymer for the fabrication of megestrol acetate solid dispersion nanoparticles using the SAS process.
Figure 1.

Scanning electron micrographs (A), differential scanning calorimetry thermograms (B), powder X-ray diffraction patterns (C), and dissolution profiles (D) of megestrol acetate solid dispersion nanoparticles prepared using the SAS process.
Abbreviations: SAS, supercritical antisolvent; HPMC, hydroxypropylmethyl cellulose; PVP, polyvinylpyrrolidone.
Solid dispersion nanoparticles with ternary compositions – drug:HPMC:surfactant – were fabricated using the SAS process to further enhance dissolution. The effect of the hydrophilic additives Gelucire 44/14, poloxamer 407, Ryoto sugar ester L1695, and TPGS on particle formation, physicochemical properties, and dissolution of megestrol acetate was investigated at a ratio of 1:2:0.5 (drug:HPMC:surfactant). As shown in Table 1, HPLC analysis indicated that the drug content of each solid dispersion nanoparticle was approximately equal to the theoretical value, indicating that megestrol acetate was not degraded during the SAS process. All solid dispersion nanoparticles had a spherical shape, with particle sizes ranging from 150 to 500 nm, as shown by SEM (Figure 2). The particle size increased upon the addition of surfactant, resulting in decreased specific surface area. In particular, megestrol acetate-HPMC-TPGS particles exhibited some aggregation and had a specific surface area of 33.1 m2/g owing to the low melting temperature of TPGS (approximately 37°C), as previously reported.15,18,23 Nonetheless, the mean particle size of all solid dispersion nanoparticles that were prepared was <500 nm. The major characteristic crystalline peaks and an endothermic peak at approximately 218°C of megestrol acetate were not observed for any solid dispersion nanoparticles, as evaluated by PXRD and DSC measurements and depicted in Figures 3 and 4, respectively. Moreover, megestrol acetate was present in an amorphous or molecular dispersion state within the solid dispersion nanoparticles.
Figure 2.

Scanning electron micrographs of megestrol acetate solid dispersion nanoparticles prepared using the SAS process.
Notes: (A) Megestrol acetate:HPMC (1:2); (B) megestrol acetate:HPMC:Gelucire 44/14 (1:2:0.5); (C) megestrol acetate:HPMC:poloxamer 407 (1:2:0.5); (D) megestrol acetate:HPMC:TPGS (1:2:0.5); (E) megestrol acetate:HPMC:Ryoto sugar ester L1695 (1:2:0.5); (F) megestrol acetate:HPMC:Ryoto sugar ester L1695 (1:2:1).
Abbreviations: SAS, supercritical antisolvent; SEI, secondary electron imaging; HPMC, hydroxypropylmethyl cellulose; TPGS, D-α-tocopheryl polyethylene glycol 1000 succinate.
Figure 3.
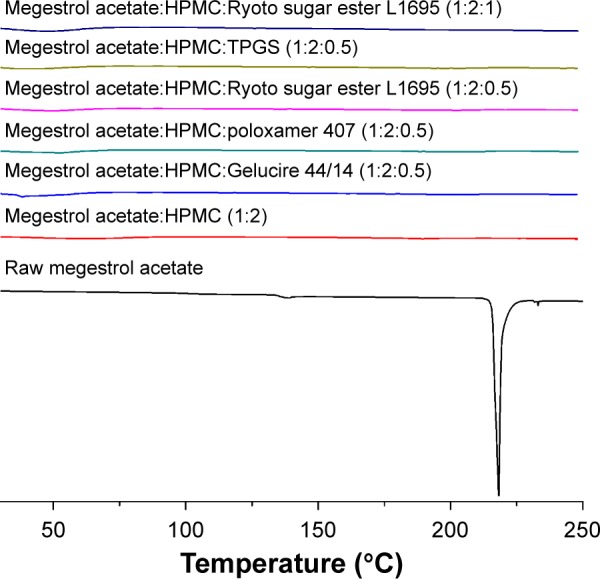
The differential scanning calorimetry thermograms of megestrol acetate solid dispersion nanoparticles prepared using the SAS process.
Abbreviations: SAS, supercritical antisolvent; HPMC, hydroxypropylmethyl cellulose; TPGS, D-α-tocopheryl polyethylene glycol 1000 succinate.
Figure 4.
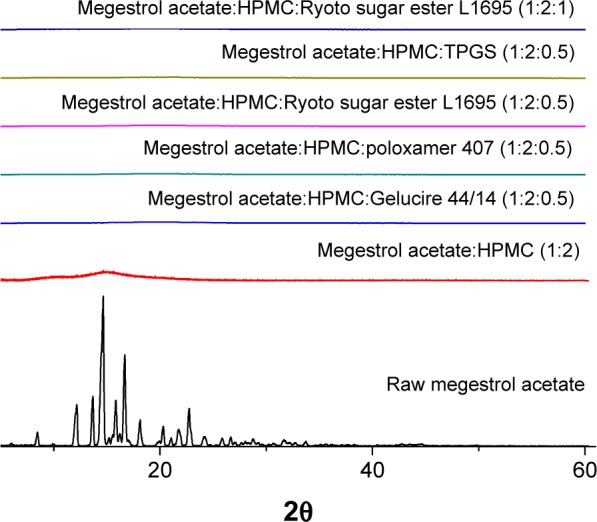
Powder X-ray diffraction patterns of megestrol acetate solid dispersion nanoparticles prepared using the SAS process.
Abbreviations: SAS, supercritical antisolvent; HPMC, hydroxypropylmethyl cellulose; TPGS, D-α-tocopheryl polyethylene glycol 1000 succinate.
In vitro dissolution
To select an optimal formulation, the dissolution profiles were obtained for raw megestrol acetate powder and solid dispersion nanoparticles (Figure 5). The dissolution data were analyzed with respect to dissolution efficiency (DE60), maximum drug dissolved (percentage), and relative dissolution rate at (RDR) 30 minutes (Table 2). The DE120 values for the raw megestrol acetate powder and solid dispersion nanopar-ticles were calculated according to the method defined by Khan and Rhodes.24 The extent and rate of dissolution of megestrol acetate increased after the addition of surfactant into the HPMC solid dispersion nanoparticles. In particular, the extent of megestrol acetate dissolution from the drug/HPMC/Ryoto ester L1695 (1:2:0.5) nanoparticles was approximately 12.6- and 1.7-fold greater, respectively, than the dissolution from raw megestrol acetate powder and drug/HPMC (1:2) nanoparticles, respectively. The DE60 ranked by the Student–Newman–Keuls test increased as follows: poloxamer 407< Gelucire 44/14< TPGS < Ryoto ester L1695 (Table 2). In this study, we found that the most effective surfactant was Ryoto ester L1695, followed by TPGS. The most effective surfactant tested was also Ryoto sugar ester L1695, followed by TPGS, as measured using a solubility test (Table 3). Megestrol acetate exhibited the greatest solubility (123.2 μg/mL) in the Ryoto ester L1695 solution, which was approximately 50-fold greater than its solubility in water without surfactant (2.5 μg/mL). Ryoto ester L1695 comprises >80% sucrose laurate, which acts as a surfactant for solubilization via micelle formation. Lerk et al reported that poorly water-soluble drugs could be incorporated into the hydrophobic 12-carbon atom chain core of micelles, formed via self-assembly, which can result in the solubilization of a poorly water-soluble drug.25 In addition, drug/HPMC/Ryoto ester L1695 (1:2:1) nanoparticles had >95% maximum dissolution within 30 minutes. Taken together, these data show that the dissolution of megestrol acetate was significantly increased by solid dispersion nanoparticles with HPMC and Ryoto ester L1695 prepared using the SAS process.
Figure 5.
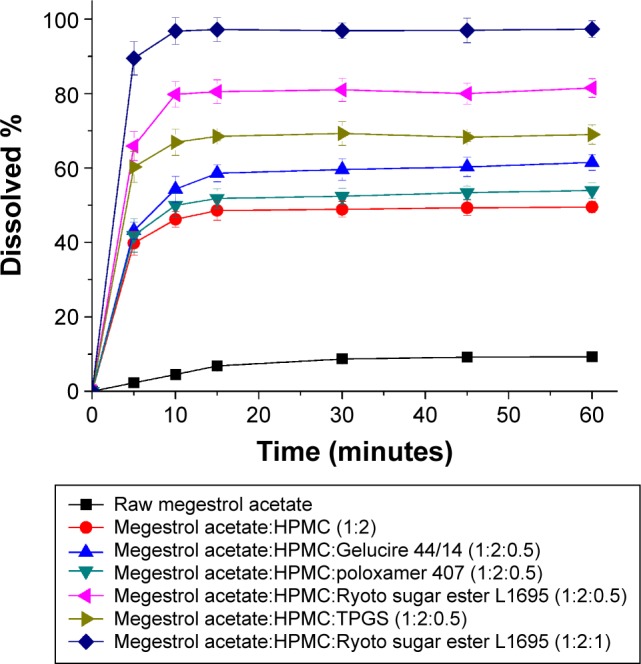
The dissolution profiles of megestrol acetate solid dispersion nanoparticles prepared using the SAS process.
Note: Data expressed as mean ± standard deviation (n=4).
Abbreviations: SAS, supercritical antisolvent; HPMC, hydroxypropylmethyl cellulose; TPGS, D-α-tocopheryl polyethylene glycol 1000 succinate.
Table 2.
The dissolution data of megestrol acetate solid dispersion nanoparticles prepared using the SAS process
| Formulation | Maximum dissolution (%) | DE60a (%) | RDRb (30 minutes) |
|---|---|---|---|
| Megestrol acetate:HPMC =1:2 | 49.5±1.9 | 46.0 | 5.6 |
| Megestrol acetate:PVP K30 =1:2 | 35.6±1.6 | 32.3 | 4.0 |
| Megestrol acetate:HPMC:Gelucire 44/14 =1:2:0.5 | 61.5±2.1 | 55.6 | 6.9 |
| Megestrol acetate:HPMC:poloxamer 407 =1:2:0.5 | 53.9±2.1 | 49.5 | 6.0 |
| Megestrol acetate:HPMC:Ryoto sugar ester L1695 =1:2:0.5 | 81.5±2.5 | 76.0 | 9.3 |
| Megestrol acetate:HPMC:TPGS =1:2:0.5 | 69.3±3.2 | 65.0 | 8.0 |
| Megestrol acetate:HPMC:Ryoto sugar ester L1695 =1:2:1 | 97.3±2.3 | 92.4 | 11.1 |
| Raw megestrol acetate | 9.3±0.6 | 7.3 | 1.0 |
Notes:
Dissolution efficiency was calculated from the area under the dissolution profile at 60 minutes, and is expressed as the percentage of the area of the rectangle described by 100% dissolution within the same period of time;
relative dissolution rate is the ratio between the amount of drug dissolved from the solid dispersion nanoparticles and that dissolved from raw megestrol acetate after 30 minutes. Data expressed as mean ± standard deviation (n=4).
Abbreviations: SAS, supercritical antisolvent; DE, dissolution efficiency; HPMC, hydroxypropylmethyl cellulose; PVP, polyvinylpyrrolidone; TPGS, D-α-tocopheryl polyethylene glycol 1000 succinate; RDR, relative dissolution rate.
Table 3.
The solubility of megestrol acetate in various surfactant solutions at 1% concentration (w/v)
| Surfactant | Solubility (μg/mL) |
|---|---|
| No additive | 2.54±0.13 |
| Gelucire 44/14 | 44.83±2.31 |
| Poloxamer 407 | 19.18±1.11 |
| Ryoto sugar ester L1695 | 123.21±1.83 |
| TPGS | 82.28±1.01 |
Notes:
Data expressed as mean ± standard deviation (n=3).
Abbreviation: TPGS, D-α-tocopheryl polyethylene glycol 1000 succinate.
Pharmacokinetic study in rats
The increased dissolution of megestrol acetate by solid dispersion nanoparticles directly increased the oral absorption of megestrol acetate. We investigated the oral absorption of megestrol acetate following oral administration of raw megestrol acetate powder, drug/HPMC (1:2) nanoparticles, and drug/HPMC/Ryoto sugar ester L1695 (1:2:1) solid dispersion nanoparticles in Sprague Dawley rats. As shown in Figure 6, solid dispersion nanoparticles exhibited a rapid drug-absorption rate and had significantly higher plasma concentration than the raw megestrol acetate powder. The Cmax ranked by the Student–Newman–Keuls test increased as follows: raw megestrol acetate powder < drug/HPMC (1:2) nanoparticles < drug/HPMC/Ryoto sugar ester L1695 (1:2:1) solid dispersion nanoparticles (Table 4). Furthermore, the oral bioavailability of the drug/HPMC/Ryoto sugar ester L1695 (1:2:1) solid dispersion nanoparticles was significantly greater than the oral bioavailability of raw megestrol acetate powder and drug/HPMC (1:2) nanoparticles, with approximately 4.0- and 1.5-fold increases in AUC0–24, respectively (P<0.05). To further evaluate the correlation between the in vitro dissolution and in vivo pharmacokinetic parameters, a linear regression analysis was performed. As shown in Figure 7, DE60 was strongly correlated with the in vivo Cmax and AUC0–24 (R2>0.98). In fact, the oral absorption of megestrol acetate increased with enhanced in vitro dissolution, as has been previously reported for poorly water-soluble drugs.26–28 The oral absorption of poorly water-soluble drugs, such as Biopharmaceutics Classification System class II, can be controlled by the solubilization and dissolution processes as a rate-limiting step.29 In this study, the enhanced dissolution and rate of megestrol acetate uptake induced by the solid dispersion nanoparticles increased the driving force on the gastrointestinal epithelial membrane, thereby enhancing the oral absorption of megestrol acetate. Furthermore, the megestrol acetate content within drug/HPMC/Ryoto sugar ester L1695 (1:2:1) solid dispersion nanoparticles was 97.6%±2.2% after 3 months at 25°C±2°C and 60%±3% relative humidity, and there were no significant changes compared to the drug content of the initial sample (98.9%±1.8%, P>0.05). We have demonstrated that the dissolution and oral absorption of megestrol acetate can be enhanced by formulating it as amorphous solid dispersion nanoparticles with HPMC and Ryoto sugar ester L1695 by using the SAS process.
Figure 6.
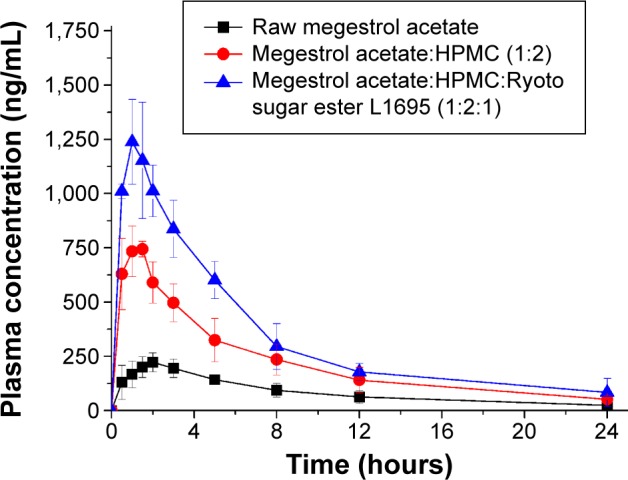
The plasma concentration–time profile of megestrol acetate in rats after oral administration of raw megestrol acetate powder or solid dispersion nanoparticles.
Note: Data expressed as mean ± standard deviation (n=5).
Abbreviation: HPMC, hydroxypropylmethyl cellulose.
Table 4.
Pharmacokinetic parameters of megestrol acetate solid dispersion nanoparticles prepared using the SAS process
| AUC0–24(ng·h/mL) | Cmax (ng/mL) | Tmax (hours) | |
|---|---|---|---|
| Raw megestrol acetate | 2,029.7±407.5 | 234.8±42.5 | 2.0±0.6 |
| Drug:HPMC =1:2 | 5,305.6±1,172.3a | 805.7±82.5a | 1.1±0.4 |
| Drug:HPMC:Ryoto sugar ester L1695 =1:2:1 | 8,175.9±1,130.1a,b | 1,288.2±198.0a,b | 1.0±0.4 |
Notes:
P<0.05 versus raw megestrol acetate;
P<0.05 versus drug:HPMC =1:2. Data expressed as mean ± standard deviation (n=5).
Abbreviations: SAS, supercritical antisolvent; AUC, area under the concentration–time curve; Cmax, peak plasma concentration; Tmax, time to Cmax; HPMC, hydroxypropylmethyl cellulose.
Figure 7.
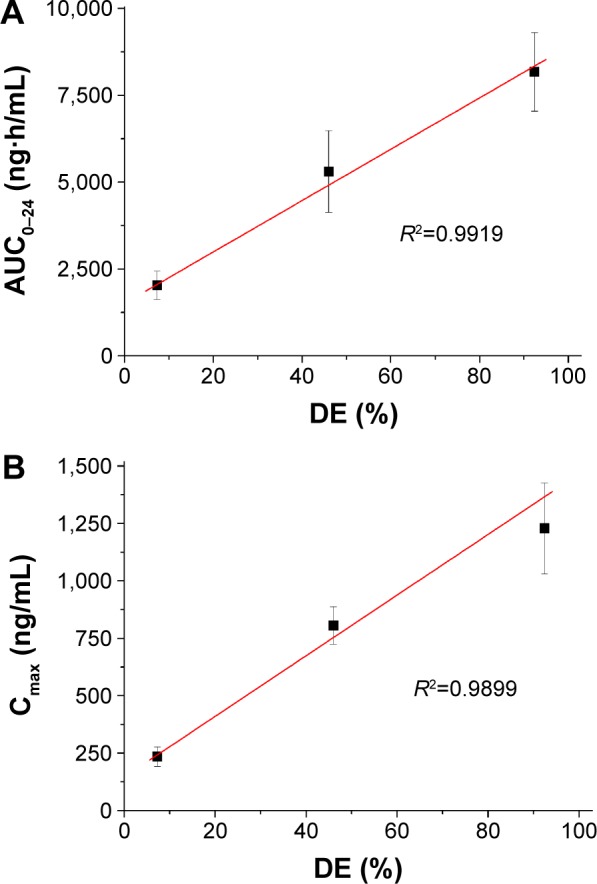
Correlation between in vitro dissolution efficiency and in vivo pharmacokinetic parameters.
Notes: (A) AUC; (B) Cmax.
Abbreviations: AUC, area under the concentration–time curve; Cmax, peak concentration; DE, dissolution efficiency.
Conclusion
In the present study, megestrol acetate solid dispersion nanoparticles were developed using the SAS process with a hydrophilic polymer and a surfactant. The dissolution and oral bioavailability of megestrol acetate significantly increased via this approach. Nanoparticles with a drug:HPMC:Ryoto sugar ester L1695 ratio of 1:2:1 showed rapid dissolution of up to 95% within 30 minutes, in addition to higher oral bioavailability than raw megestrol acetate powder. AUC0–24 and Cmax increased approximately 4.0- and 5.5-fold, respectively. Taken together, these data suggest that the preparation of megestrol acetate solid dispersion nanoparticles using the SAS process is a promising approach to improve the dissolution and absorption properties of megestrol acetate.
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the South Korean government (MSIP) (No 2009-0083538).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Yeh SS, Wu SY, Lee TP, et al. Improvement in quality-of-life measures and stimulation of weight gain after treatment with megestrol acetate oral suspension in geriatric cachexia: results of a double-blind, placebo-controlled study. J Am Geriatr Soc. 2000;48(5):485–492. doi: 10.1111/j.1532-5415.2000.tb04993.x. [DOI] [PubMed] [Google Scholar]
- 2.Tomíska M, Tomisková M, Salajka F, Adam Z, Vorlícek J. Palliative treatment of cancer anorexia with oral suspension of megestrol acetate. Neoplasma. 2003;50(3):227–233. [PubMed] [Google Scholar]
- 3.Deng Z, Zhao R, Dong LC, Wong G. Characterization of nanoparticles for drug delivery applications. Microsc Microanal. 2005;11(Suppl 2):1934–1935. [Google Scholar]
- 4.Sylvestre JP, Tang MC, Furtos A, Leclair G, Meunier M, Leroux JC. Nanonization of megestrol acetate by laser fragmentation in aqueous milieu. J Control Release. 2011;149(3):273–280. doi: 10.1016/j.jconrel.2010.10.034. [DOI] [PubMed] [Google Scholar]
- 5.Cho E, Cho W, Cha KH, et al. Enhanced dissolution of megestrol acetate microcrystals prepared by antisolvent precipitation process using hydrophilic additives. Int J Pharm. 2010;396(1–2):91–98. doi: 10.1016/j.ijpharm.2010.06.016. [DOI] [PubMed] [Google Scholar]
- 6.Jang K, Yoon S, Kim SE, et al. Novel nanocrystal formulation of megestrol acetate has improved bioavailability compared with the conventional micronized formulation in the fasting state. Drug Des Devel Ther. 2014;8:851–858. doi: 10.2147/DDDT.S62176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hong SW, Lee BS, Park SJ, et al. Solid dispersion formulations of megestrol acetate with copovidone for enhanced dissolution and oral bioavailability. Arch Pharm Res. 2011;34(1):127–135. doi: 10.1007/s12272-011-0115-2. [DOI] [PubMed] [Google Scholar]
- 8.Shekunov BY, Chattopadhyay P, Seitzinger J, Huff R. Nanoparticles of poorly water-soluble drugs prepared by supercritical fluid extraction of emulsions. Pharm Res. 2006;23(1):196–204. doi: 10.1007/s11095-005-8635-4. [DOI] [PubMed] [Google Scholar]
- 9.Zhang ZB, Shen ZG, Wang JX, Zhao H, Chen JF, Yun J. Nanonization of megestrol acetate by liquid precipitation. Ind Eng Chem Res. 2009;48(18):8493–8499. [Google Scholar]
- 10.Kim MS, Kim JS, Cho W, et al. Supersaturatable formulations for the enhanced oral absorption of sirolimus. Int J Pharm. 2013;445(1–2):108–116. doi: 10.1016/j.ijpharm.2013.01.067. [DOI] [PubMed] [Google Scholar]
- 11.Ha ES, Baek IH, Cho W, Hwang SJ, Kim MS. Preparation and evaluation of solid dispersion of atorvastatin calcium with Soluplus® by spray drying technique. Chem Pharm Bull (Tokyo) 2014;62(6):545–551. doi: 10.1248/cpb.c14-00030. [DOI] [PubMed] [Google Scholar]
- 12.de la Torre-Iglesias PM, García-Rodriguez JJ, Torrado G, Torrado S, Torrado-Santiago S, Bolás-Fernández F. Enhanced bioavailability and anthelmintic efficacy of mebendazole in redispersible microparticles with low-substituted hydroxypropylcellulose. Drug Des Devel Ther. 2014;8:1467–1479. doi: 10.2147/DDDT.S65561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cocero MJ, Martín A, Mattea F, Varona S. Encapsulation and co-precipitation processes with supercritical fluids: fundamentals and applications. J Supercrit Fluids. 2009;47(3):546–555. [Google Scholar]
- 14.Adeli E. A comparative evaluation between utilizing SAS supercriti-cal fluid technique and solvent evaporation method in preparation of azithromycin solid dispersions for dissolution rate enhancement. J Supercrit Fluids. 2014;87:9–21. [Google Scholar]
- 15.Kim MS, Baek IH. Fabrication and evaluation of valsartan-polymer-surfactant composite nanoparticles by using the supercritical antisolvent process. Int J Nanomedicine. 2014;9(1):5167–5176. doi: 10.2147/IJN.S71891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yin X, Daintree LS, Ding S, et al. Itraconazole solid dispersion prepared by a supercritical fluid technique: preparation, in vitro characterization, and bioavailability in beagle dogs. Drug Des Devel Ther. 2015;9:2801–2810. doi: 10.2147/DDDT.S81253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim MS. Influence of hydrophilic additives on the supersaturation and bioavailability of dutasteride-loaded hydroxypropyl-β-cyclodextrin nanostructures. Int J Nanomedicine. 2013;8(1):2029–2039. doi: 10.2147/IJN.S44795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ha ES, Choo GH, Baek IH, Kim MS. Formulation, characterization, and in vivo evaluation of celecoxib-PVP solid dispersion nanopar-ticles using supercritical antisolvent process. Molecules. 2014;19(12):20325–20339. doi: 10.3390/molecules191220325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ha ES, Choo GH, Baek IH, et al. Dissolution and bioavailability of lercanidipine-hydroxypropylmethyl cellulose nanoparticles with surfactant. Int J Biol Macromol. 2015;72:218–222. doi: 10.1016/j.ijbiomac.2014.08.017. [DOI] [PubMed] [Google Scholar]
- 20.Seo JH, Park JS, Jo MH, et al. Rapid and sensitive LC-MS/MS method for determination of megestrol acetate in human plasma: application to a human pharmacokinetic study. Biomed Chromatogr. 2013;27(4):409–415. doi: 10.1002/bmc.2807. [DOI] [PubMed] [Google Scholar]
- 21.Lee JY, Kang WS, Piao J, Yoon IS, Kim DD, Cho HJ. Soluplus®/TPGS-based solid dispersions prepared by hot-melt extrusion equipped with twin-screw systems for enhancing oral bioavailability of valsartan. Drug Des Devel Ther. 2015;9:2745–2756. doi: 10.2147/DDDT.S84070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palanisamy M, Khanam J. Effect of physiochemical variables on phase solubility and dissolution behavior of indomethacin solid dispersion system. J Pharm Investig. 2014;44(3):147–162. [Google Scholar]
- 23.Kim MS, Kim JS, Park HJ, Cho WK, Cha KH, Hwang SJ. Enhanced bio-availability of sirolimus via preparation of solid dispersion nanoparticles using a supercritical antisolvent process. Int J Nanomedicine. 2011;6:2997–3009. doi: 10.2147/IJN.S26546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khan KA, Rhodes CT. Effect of compaction pressure on the dissolution efficiency of some direct compression systems. Pharm Acta Helv. 1972;47(10):594–607. [PubMed] [Google Scholar]
- 25.Lerk PC, Sucker HH, Eicke HF. Micellization and solubilization behavior of sucrose laurate, a new pharmaceutical excipient. Pharm Dev Technol. 1996;1(1):27–36. doi: 10.3109/10837459609031415. [DOI] [PubMed] [Google Scholar]
- 26.Berthelsena R, Sjögrenb E, Jacobsen J. Combining in vitro and in silico methods for better prediction of surfactant effects on the absorption of poorly water soluble drugs – a fenofibrate case example. Int J Pharm. 2014;473(1–2):356–365. doi: 10.1016/j.ijpharm.2014.06.060. [DOI] [PubMed] [Google Scholar]
- 27.Baek IH, Kim JS, Ha ES, et al. Dissolution and oral absorption of pran-lukast nanosuspensions stabilized by hydroxypropylmethyl cellulose. Int J Biol Macromol. 2014;67:53–57. doi: 10.1016/j.ijbiomac.2014.03.006. [DOI] [PubMed] [Google Scholar]
- 28.Lee DH, Yeom DW, Song YS, et al. Improved oral absorption of dutasteride via Soluplus®-based supersaturable self-emulsifying drug delivery system (S-SEDDS) Int J Pharm. 2014;478(1):341–347. doi: 10.1016/j.ijpharm.2014.11.060. [DOI] [PubMed] [Google Scholar]
- 29.Junghanns JU, Müller RH. Nanocrystal technology, drug delivery and clinical applications. Int J Nanomedicine. 2008;3(3):295–310. doi: 10.2147/ijn.s595. [DOI] [PMC free article] [PubMed] [Google Scholar]