

Abstract
Objectives
Misfolding of key disease proteins to an insoluble state is associated with most neurodegenerative conditions, such as prion, Parkinson, and Alzheimer’s diseases. In this work, and by studying animal models of multiple sclerosis, we asked whether this is also the case for myelin basic protein (MBP) in the late and neurodegenerative phases of demyelinating diseases.
Methods
To this effect, we tested whether MBP, an essential myelin component, present prion-like properties in animal models of MS, as is the case for Cuprizone-induced chronic demyelination or chronic phases of Experimental Autoimmune Encephalomyelitis (EAE).
Results
We show here that while total levels of MBP were not reduced following extensive demyelination, part of these molecules accumulated thereafter as aggregates inside oligodendrocytes or around neuronal cells. In chronic EAE, MBP precipitated concomitantly with Tau, a marker of diverse neurodegenerative conditions, including MS. Most important, analysis of fractions from Triton X-100 floatation gradients suggest that the lipid composition of brain membranes in chronic EAE differs significantly from that of naïve mice, an effect which may relate to oxidative insults and subsequently prevent the appropriate insertion and compaction of new MBP in the myelin sheath, thereby causing its misfolding and aggregation.
Interpretation
Prion-like aggregation of MBP following chronic demyelination may result from an aberrant lipid composition accompanying this pathological status. Such aggregation of MBP may contribute to neuronal damage that occurs in the progressive phase of MS.
Introduction
Perturbation of the myelin sheath, or demyelination, leads to several severe brain diseases, including multiple sclerosis (MS).1 While different mechanisms, including autoimmune pathways may lead to demyelination and its clinical consequences,2 recent evidence also suggests that a significant part of the neurological damage in MS may be of neurodegenerative nature.3–5 In recent years, a variety of neurodegenerative diseases has been associated with the accumulation of misfolded key disease proteins.6–8 This effect, first established for PrPSc in prion diseases,9 was next expanded to proteins associated with other brain conditions such as A-beta for Alzheimer’s disease (AD), α-synuclein for Parkinson’s disease, phosphorylated Tau for tauopathies and other diseases,10–13 and several others. Most of these proteins are natively unstructured when prepared as monomers in vitro, but form β-sheath-rich aggregates when incubated at high concentrations.13,14
Myelin basic protein (MBP) is an indispensable protein of myelinated axons.1 The sequence of MBP predicts the formation of an intrinsically disordered protein carrying a large positive charge, which allows for countless protein surface interactions.15 This property is specific for MBP and is not recognized in other abundant myelin proteins, such as myelin oligodendrocyte glycoprotein (MOG) or Myelin proteolipid protein (PLP).1 To preclude MBP from nonspecific binding to molecules, MBP mRNA assembles into ribonucleoprotein complexes termed “granules” which are targeted into oligodendrocyte extensions,1 and translated in-situ. Consequent MBP-membrane interactions rely on an accurate stoichiometrical balance between the basic residues of MBP and the acidic headgroups of the lipid bilayer components, as well as on copper ions inducing the compaction of the MBP structure.16 This suggests that reduced myelin stability may relate to changes in levels of diverse myelin components, including designated lipids and their oxidized products,17 copper ions or expression levels of MBP. Upon demyelination, MBP may either rapidly degrade or otherwise aggregate with other myelin components.
In this work, we tested in mouse models of demyelination whether MBP can present in aggregated forms, and whether such effect may relate to changes in myelin components.18 In one model, chronic demyelination was induced by Cuprizone, an established copper chelator.19 Cuprizone administration to naïve mice may promote the removal of copper ions from MBP and thereby generate loss of myelin compaction.16 In the Experimental Autoimmune Encephalomyelitis model (EAE), induced by immunization of naïve C57BL/6 mice with a peptide form MOG,20 mice suffer from acute paralytic neurological disease ∼10 days after the immunization, which remits partially while leaving the mice with a disability associated with chronic demyelination and axonal injury.21 Our results show that MBP can precipitate and aggregate in brains affected by chronic forms of demyelination and that aggregation of MBP occurred concomitant with accumulation of insoluble and phosphorylated Tau. Of special note, flotation experiments demonstrated that the membrane microenvironment of PrPC, an established raft protein,22,23 is less resistant to solubilization by Triton X-100 during chronic EAE, suggesting a reduction in cholesterol and glycosphingolipids levels, as shown for MS.24,25 Indeed, depletion of cholesterol from myelin membranes may induce the formation of abnormal myelin.26 These aberrant alterations in membrane components may explain the exit of MBP form the demyelinating sheath and its subsequent aggregation.27
Materials and Methods
Animal experiments: ethical statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Committee on the Ethics of Animal Experiments of the Hebrew University Medical School, and all efforts were made to minimize suffering.
Induction of demyelination by cuprizone
Induction of chronic demyelination disease by Cuprizone was done as previously described.28 Shortly, 5–6-month-old female C57BL/6 mice were feed with 0.3% w/w Cuprizone (biscyclohexane oxaldihydrazone, Sigma-Aldrich, St. Louis, MO) in a diet of ground mouse chow for a period of 10 weeks. Subsequently, mice were sacrificed for western blot analysis (n = 3) or for histological analysis (n = 6). An age-matched control group of normal diet to serve as naïve, nontreated controls (n = 9).
Induction of EAE
Induction of MOG EAE was done as previously described.29,30 Shortly, 8 to 9-week-old female C57BL/6 mice were immunized with an emulsion containing 300 μg of MOG35-55 in phosphate-buffered saline and an equal volume of complete Freund’s adjuvant containing 5 mg/mL H37RA (Difco Laboratories, Detroit, MI). The inoculum (0.2 mL) was injected subcutaneously into the left flank. One hundred nanograms of pertussis toxin (List Biological Labs, Campbell, CA) in 0.1 mL phosphate-buffered saline was also injected i.p. on day 0 and 48 h later. Mice were observed daily for the appearance of neurological symptoms, which were scored as follows: 0, asymptomatic; 1, partial loss of tail tonicity; 2, limp tail; 3, impaired righting reflex; 4, hind limb weakness (ataxia); 5, complete hind limb paralysis; 6, moribund or dead.
Floatation experiments
Flotation of detergent insoluble complexes was performed as described previously.31 In brief, mouse brains were homogenized 1:10 wt/vol in an ice-cold buffer containing 150 mmol/L sodium chloride, 25 mmol/L TRIS hydrochloride (pH 7.5), 5 mmol/L EDTA (Ethylenediaminetetraacetic acid), and 1% Triton X-100 (Sigma-Aldrich, St. Louis, MO). Insoluble particles were spun down and the lysate was loaded at the bottom of ultracentrifuge tubes (TLS-55; Beckman Instruments, Inc, Fullerton, CA). An equal volume of ice-cold Nycodenz (Biological Industries, Beit Haemek, Israel), 70%, in TNE (25 mmol/L TRIS hydrochloride [pH 7.5], 150 mmol/L sodium chloride, and 5 mmol/L EDTA) was added and mixed with the lysate. An 8% to 35% Nycodenz linear step gradient in TNE was then overlaid above the lysate (200 mL each of 35%, 25%, 22.5%, 20%, 18%, 15%, 12%, and 8% Nycodenz). The tubes were spun at 55,000 rpm for 4 h at 4°C in a TLS-55 rotor (200,000 g). Fractions were collected from the top to the bottom of the tube. Each fraction was applied to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotted with α-PrP polyclonal antibody RTC, as well as with a α-MBP antibody.
Preparation of pellets and supernatants for immunoblots
Brains were homogenized at 10% (W/V) in 10 mmol/L Tris-HCl, pH 7.4 and 0.3 mol/L sucrose. For ultracentrifugation experiments (100,000 g for 1 h), homogenates were first extracted with 2% sarkosyl. For Proteinase K (PK) digestions, samples were incubated with 30 mg/mL PK for 30 min at 37°C. Samples were subsequently subjected to SDS-PAGE and immunoblotted with diverse antibodies, as described in the text.
Histopathological analysis
Histological evaluations were performed on paraffin-embedded sections of brains and spinal cords sampled at two time points after MOG immunization (acute phase and chronic phase, 15 or 100 days post EAE induction, respectively) and 10-weeks of Cuprizone treatment. In some experiments, sections were stained with Luxol fast blue (LFB)/periodic acid-Schiff stain,32 to assess demyelination. In consecutive sections, immunohistochemistry was performed with antibodies against MBP.
Mass spectrometry analysis
Identification of proteins in bands excised from SDS polyacrylamide gels was performed as previously described.33,34 Shortly, mass spectrometry of digested samples was carried out with Qtof2 (Micromass, Manchester, England) using nanospray attachment.35 Data analysis was done using the biolynx package (Micromass, Manchester, England) and database searches were performed with Mascot package (Matrix Science, London, England) (Figure S1).
Results
Cuprizone administration to mice induced intracellular accumulation of MBP in oligodendrocytes
Six-month-old mice (C57BL/6, 9 mice per group) were treated with Cuprizone for 10 weeks (see Methods) before they were sacrificed and their brains compared to control untreated mice for demyelination parameters. Myelination status of treated and untreated brains was first examined in Paraffin-embedded brain sections, stained with LFB, a copper phthalocyanine dye that is soluble in alcohol and is attracted to bases found in the lipoproteins of the myelin sheath.32 Cuprizone-treated mice exhibited extensive demyelination, particularly in the corpus callosum, as depicted by the reduced levels of LFB staining in the treated versus the control samples (Fig.1A and B). While immunofluorescent staining with an α-MBP body indicated that the levels of MBP in brains of Cuprizone-treated mice (Fig.1E and F) were mostly preserved, the staining pattern was different from that of untreated brains (Fig.1C and D). In control mice, MBP was, as expected, localized in the oligodendrocyte extension, while in Cuprizone-treated mice MBP was present not only in cell extensions, but also in a peri-nuclear distribution (Fig.1E and F). This abnormal localization is contrary to the notion that MBP mRNA is usually translated only in oligodendrocyte extension forming axonal myelin.1 The pathologic concentration of MBP in Cuprizone-demyelinated brains suggested that the protein may be aggregated in a form that prevents its degradation following demyelination. To test this possibility, we extracted brain homogenates of treated and untreated brains with Sarkosyl, and subsequently separated supernatants and pellets after ultracentrifugation. As positive controls for brain samples comprising a key disease aggregated protein, we also extracted by the same method homogenates of prion-infected brains, both from a transmissible36 and for a genetic TgMHu2ME199K model.37 The normal PrP protein is present in all brains as a detergent soluble membrane protein (PrPC). However, in prion affected brains an aberrantly aggregated form, PrPSc, constituting the key component and marker of prions and their diseases, is detected in pellets. All samples (pellets and supernatants) of the different experimental groups were immunoblotted with an α-MBP and an α-PrP antibody.
Figure 1.

Cuprizone treatment results in demyelination and aggregation of MBP in oligodendrocyte’s cell bodies. (A) (naive) and (B) (treated) brain sections are stained with LFB. The (C–F) sections were immunostained with α-MBP antibody: (A, C, and D): naïve brains; (B, E, and F): Cuprizone-treated brains. Scale bars: (C and F) (in F) = 100 μm, (D and E) (in E) = 10 μm. MBP, myelin basic protein; LFB, Luxol fast blue.
Figure2 shows the results of these experiments. As expected, while PrPC was detected in all sarkosyl soluble brain samples, its aggregated isoforms were present only in the pellets of the prion affected mice, both in the transmissible (Rocky mountains laboratory strain) and in the genetic TgMHu2ME199K model (Fig.2A). Respectively, only the pellet of the Cuprizone mice brains presented pelletable MBP (Fig.2B). In combination with the results described in Figure1, we may conclude that intracellular MBP in oligodendrocytes of Cuprizone-treated mice is accumulated in an aggregated–insoluble form.
Figure 2.
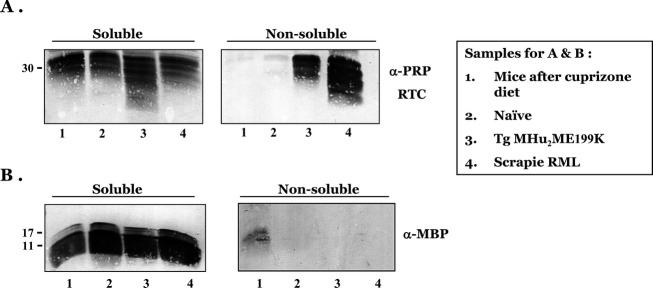
Myelin basic protein (MBP) precipitates in brains of Cuprizone-treated mice. Brain samples from diverse mice were extracted with sarkosyl as described in Methods. Subsequently pellets and supernatants were immunoblotted with α-PrP (A) and α-MBP (B) antibodies. Samples were from: 1, Cuprizone-treated mice; 2, Naïve mice; 3, TgMHu2ME199K mice and 4, scrapie RML-infected mice. As expected PrP forms precipitate in the prion models and MBP in the demyelinating model.
MBP aggregation in chronic EAE
We next tested the biochemical properties of MBP in mice affected with the most popular model of demyelination, EAE.20 To this effect, naïve mice were immunized with a MOG35–55 peptide (see Methods) resulting in an acute neurological paralytic disease, followed by partial remission and chronic phase (Fig.3A).38 In the present experiments, mice were sacrificed either at the end of the acute phase (15 days) or 100 days thereafter in the chronic state (see arrows in Fig.3A). Brain samples from naïve, acute, and chronic EAE mice were homogenized in the presence of Sarkosyl, as described above for the Cuprizone mice, and following ultracentrifugation, tested for the levels of pelletable MBP accumulation as well as its resistance to PK digestion. Resistance to protease digestion of aggregated key proteins is a common but not an obligatory property for prion and alike disease-related proteins in neurodegenerative conditions.39 Panels I and II of Figure3B depicts immunoblots of pellets and supernatants of naïve, acute, and chronic EAE brains, challenged with α-MBP antibodies, and demonstrates that while only traces of MBP were present in the nonsoluble fractions of acute EAE brains, significant levels of MBP were pelleted in the chronic phase. In Panel III of Figure3B, pellets and supernatants from naïve, acute and chronic EAE brains were compared to that of prion-infected brains. Samples from soluble and nonsoluble brain fractions were digested in the presence or absence of PK, and immunoblotted first with an α-MBP and then with an α-PrP antibody. The figure shows that only PrPSc in the brains of scrapie-infected mice was resistant to PK digestion in these experiments, whereas aggregated MBP in chronic EAE brains was PK sensitive, as is the case for some of the prion pathological isoforms.40
Figure 3.

(A) Clinical scores of EAE-induced mice. EAE was induced in C57B/6 mice as described in Methods and subsequently the affected mice were followed up to 120 days for their clinical scores as described in Methods. Mice were sacrificed at designated points and their brains processed for biochemistry of MBP. (B) Aggregated MBP is not resistant to protease digestion. Brain samples from naïve, acute, and chronic EAE were extracted with sarkosyl as described in Methods and subsequently pellets and supernatants were immunoblotted with α-MBP antibodies. I: soluble samples; II: nonsoluble samples. III: samples as described in the inserted table were digested in the presence or absence of Proteinase K (PK) as described in Methods and immunoblotted for both MBP and PrP. No PK-resistant MBP was observed in any of the fractions, while PK-resistant PrP was present in scrapie-infected brains. EAE, Experimental Autoimmune Encephalomyelitis; MBP, myelin basic protein.
Aggregated MBP binds to neuronal cells
Next, brain sections from naïve mice, as well as from those suffering from acute and chronic EAE were subjected to immunohistochemistry with an α-MBP antibody. Figure4 shows extensive MBP immunostaining in cerebellar white matter of all mice. However, in chronic EAE sections MBP accumulated also around neuronal cells, such as Purkinje cells, as can be seen in sections Figure4E and F. Indeed, it was recently shown that MBP binding may induce neuron-specific toxicity by direct damaging the neuronal plasma membrane integrity.41 Consistent with this, our results indicate that following demyelination, new MBP aggregates may damage neuronal cells by a prion-like mechanism.42
Figure 4.

MBP immunostaining of naïve and EAE brains. Paraffin-embedded brain sections of Naïve mice and of mice in acute and chronic phases of EAE were immunostained for α-MBP. Sections (A and B): naive mice; (C and D): acute phase of EAE; (E and F): chronic phase of EAE. MBP, myelin basic protein; EAE, Experimental Autoimmune Encephalomyelitis.
Aggregated tau accumulates in chronic EAE concomitant with MBP
To look for additional proteins that may aggregate in the brains of chronic EAE mice, we subjected sarkosyl pellets (as above) of brain homogenates from naïve, acute, and chronic EAE mice to SDS-PAGE and subsequently stained the gels with coomassie blue. Figure5A shows the profile of bands appearing in acute and chronic EAE brains. Bands specific for EAE samples were excised from the gels and subjected to novel mass spectrometry examination (MS –MS) (see Methods). Some of the identified proteins are listed in Figure5B. Of special note is the accumulation of the Tau protein in the nonsoluble fraction of chronic EAE. Tau is a glial and neuronal43 cytoskeletal protein involved in microtubule assembly and stabilization, which accumulates in several neurodegenerative disorders, including Alzheimer’s and human prion diseases in an hyperphospho-rylated and aggregated form.44–46 Accumulation of insoluble Tau is associated with disease progression also in MS and in EAE.47,48 To assess the mass spectrometry results, we immunoblotted total brain homogenates, as well as soluble and nonsoluble fractions with an antibody against phosphorylated Tau. Figure5C shows that while total and soluble Tau levels were present at similar levels in all samples, insoluble Tau was mostly present in chronic EAE, as is the case for the insoluble MBP. These results are consistent with the notion that insoluble MBP may be a marker of the neurodegenerative stage in demyelinating diseases, as is the case for Tau.
Figure 5.

Aggregated Tau precipitates in EAE chronic brains concomitant with MBP. (A) Nonsoluble fractions from naïve, acute, and chronic EAE brains were subjected to SDS Page and stained with coomassie blue. (B): Bands 1 and 2 from (A) were excised from the gel and subjected to MS–MS analysis, as described in Methods. (C) Total, soluble, and nonsoluble fractions from naïve, acute, and chronic EAE brains were immunoblotted with an antibody recognizing phosphorylated Tau. EAE, Experimental Autoimmune Encephalomyelitis; MBP, myelin basic protein; SDS, sodium dodecyl sulfate.
Chronic EAE brains may differ in their lipid composition from naïve brains
Lipid–protein interactions are pivotal for myelin maintenance, since they regulate the molecular organization within the myelin sheath.18 Therefore, alteration in correlative lipid levels may well affect myelin stability and function, as well as the capability of inserting myelin proteins such as MBP to the normally compacting myelin. Indeed, reduction in cholesterol and glycosphingolipids levels interferes with oligodendrocyte differentiation and myelination.25,26 To test the stability of lipid composition in the EAE affected brains, we subjected Triton X-100 extracted brain homogenates of naïve, acute, and chronic EAE brains to Nicodentz floatation gradients.31 Under these conditions, Triton X-100 detergent-resistant membranes (DRMs) float to upper fractions together with their associated proteins. For example, PrPC, a well-established DRM or raft protein may be found in the floating fractions.23 MBP does not normally float in Triton X-100 gradients,49 but does show affinity to Raft lipids in other detergent systems.49,50 Following ultracentrifugation, the gradient fractions were immunoblotted first for PrP and then for MBP. Figure6 shows that while PrP floats to the lighter gradient fractions in the naïve brains, and somewhat less so in acute EAE brains, in the chronic state the location of PrP changed dramatically to denser fractions of the gradient, indicating altered levels of cholesterol and glycosphingolipids, the main DRM components. Indeed, similar results for PrP were obtained when naïve membranes were extracted with methyl-β-cyclodextrin (not shown), a reagent that depletes cholesterol from membranes.51,52 Also MBP was pushed to denser fractions of the gradient in chronic EAE brain extracts, indicating weaker association of the myelin protein with DRM fractions. The changes in lipid composition of chronic EAE brain membranes, which origin is not totally understood, may well disrupt MBP insertion and compaction in myelin sheaths and subsequently promote its aggregation.
Figure 6.
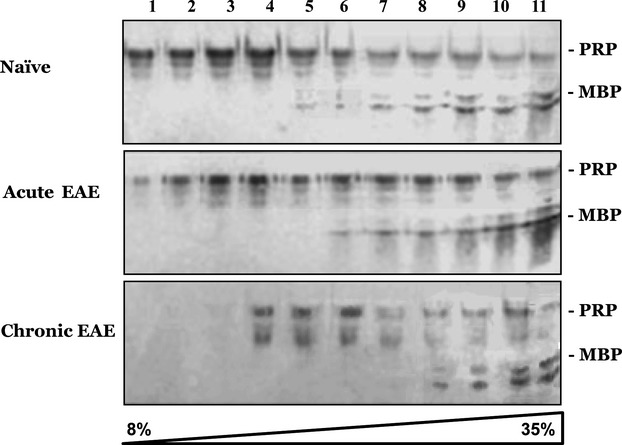
PrP and MBP change their membrane location in chronic EAE brains. Brain homogenates from naïve, acute, and chronic EAE mice were subjected to floatation gradients (as described in Methods). Samples collected from the gradients were subsequently immunoblotted with an α-PrP and an α-MBP antibody. MBP, myelin basic protein; EAE, Experimental Autoimmune Encephalomyelitis.
Discussion
We have shown in this work that MBP, a main component of the myelin membrane, may aggregate and precipitate during chronic demyelinating conditions, similarly to key proteins in well-established neurodegenerative diseases.13 While in these diseases, and particularly in prion diseases featuring the conversion of PrPC to PrPSc, such aggregation occurs concomitantly with protein oxidation and aberrant conformational changes,53 MBP misfolding to an insoluble condition may be secondary to alterations in levels of myelin components responsible for the compaction of MBP between the myelin sheaths.16,26 Indeed, in the Cuprizone model of demyelination, this copper chelator may reduce the levels of copper ions required for MBP compaction and thereby promote its aggregation outside the membrane.19 In the brains of mice suffering from chronic EAE, it can be the reduced levels of cholesterol and other lipids which may preclude the entrance of MBP to its appropriate location in the myelin sheath and induce its aggregation outside the membrane. Outside the membrane, aggregated MBP may damage neighboring neurons,41 promoting further neurodegeneration. It remains to be established whether changes in lipid composition result or encourage the demyelinating process.
In addition to key protein misfolding and aggregation, oxidative stress is also a common factor in both neurodegenerative conditions and chronic demyelination.54,55 Indeed, most of the aggregated key proteins in neurodegenerative conditions are oxidized in some of their residues, as is the case for Met residues in PrPSc.53,56 While we have no knowledge at this point about direct oxidation of MBP, oxidation of other brain proteins was shown to occur both in MS and in EAE.57 Importantly, it was shown convincingly that lipids in MS plaques are oxidized, as demonstrated by immunostaining with an antibody against oxidized phospholipids.54 In addition, several studies have demonstrated a significant increase in lipid peroxidation products in brain, plasma, and cerebrospinal fluid of MS patients. Since oxidized lipoproteins are neurotoxic and have pro-inflammatory properties, lipid peroxidation products could be involved in demyelination and axonal injury in MS.17,58 The EO6 antibody (for oxidized lipids)54 also reacts extensively with brain slices from a mouse model of genetic prion disease,59 suggesting that lipid oxidation may be a general feature of neurodegeneration. Oxidation of lipids in the myelin sheath and other membranes may also contribute to the change in lipid composition that generates less stable rafts, thereby disturbing the location of their associated proteins and even their function. This may promote the instability of MBP within the membrane and its aggregation.
We conclude that due to the complex interactions of MBP with other myelin components, small alterations in lipid composition/oxidation or copper concentration may affect the affinity of MBP to the myelin membrane.60,61 Next, decompacted forms of MBP released from the myelin sheath may misfold into new entities, which may subsequently cause additional damage to neuronal cells. Misfolded and basically charged MBP forms may aggregate with lipid structures or complex with negative GAGs, (Glycosaminoglycans) for example, in autophagic lysosomes.18,62 In summary, we suggest a mechanistic model for progressive neurodegeneration in MS, in which prion-like aggregated MBP formed during demyelination may cause neuronal toxicity and induce further demyelination, eventually resulting in more misfolded MBP and more neuronal damage. Indeed, it would be nice to test this hypothesis in sample form human patients.
Conflict of Interest
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. MS–MS data for tau on chronic EAE. The figure 5 shows the original MS–MS data for band 1 in Figure comprising sequences of the Tau protein.
References
- Baron W, Hoekstra D. On the biogenesis of myelin membranes: sorting, trafficking and cell polarity. FEBS Lett. 2010;584:1760–1770. doi: 10.1016/j.febslet.2009.10.085. [DOI] [PubMed] [Google Scholar]
- Lassmann H. Mechanisms of inflammation induced tissue injury in multiple sclerosis. J Neurol Sci. 2008;274:45–47. doi: 10.1016/j.jns.2008.04.003. [DOI] [PubMed] [Google Scholar]
- Lassmann H, van Horssen J. The molecular basis of neurodegeneration in multiple sclerosis. FEBS Lett. 2011;585:3715–3723. doi: 10.1016/j.febslet.2011.08.004. [DOI] [PubMed] [Google Scholar]
- Ellwardt E, Zipp F. Molecular mechanisms linking neuroinflammation and neurodegeneration in MS. Exp Neurol. 2014;262:8–17. doi: 10.1016/j.expneurol.2014.02.006. [DOI] [PubMed] [Google Scholar]
- Herz J, Zipp F, Siffrin V. Neurodegeneration in autoimmune CNS inflammation. Exp Neurol. 2010;225:9–17. doi: 10.1016/j.expneurol.2009.11.019. [DOI] [PubMed] [Google Scholar]
- Hardy J. A hundred years of Alzheimer’s disease research. Neuron. 2006;52:3–13. doi: 10.1016/j.neuron.2006.09.016. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Cell biology of protein misfolding: the examples of Alzheimer’s and Parkinson’s diseases. Nat Cell Biol. 2004;6:1054–1061. doi: 10.1038/ncb1104-1054. [DOI] [PubMed] [Google Scholar]
- Goedert M. Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci. 2001;2:492–501. doi: 10.1038/35081564. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- Piccardo P, Cervenak J, Bu M, et al. Complex proteinopathy with accumulations of prion protein, hyperphosphorylated tau, alpha-synuclein and ubiquitin in experimental bovine spongiform encephalopathy of monkeys. J Gen Virol. 2014;95:1612–1618. doi: 10.1099/vir.0.062083-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10(suppl):S10–S17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Shattuck lecture–neurodegenerative diseases and prions. N Engl J Med. 2001;344:1516–1526. doi: 10.1056/NEJM200105173442006. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Biology and genetics of prions causing neurodegeneration. Annu Rev Genet. 2013;47:601–623. doi: 10.1146/annurev-genet-110711-155524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost B, Diamond MI. The expanding realm of prion phenomena in neurodegenerative disease. Prion. 2009;3:74–77. doi: 10.4161/pri.3.2.8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harauz G, Ladizhansky V, Boggs JM. Structural polymorphism and multifunctionality of myelin basic protein. Biochemistry. 2009;48:8094–8104. doi: 10.1021/bi901005f. [DOI] [PubMed] [Google Scholar]
- Baran C, Smith GS, Bamm VV, et al. Divalent cations induce a compaction of intrinsically disordered myelin basic protein. Biochem Biophys Res Commun. 2010;391:224–229. doi: 10.1016/j.bbrc.2009.11.036. [DOI] [PubMed] [Google Scholar]
- Ferretti G, Bacchetti T. Peroxidation of lipoproteins in multiple sclerosis. J Neurol Sci. 2011;311:92–97. doi: 10.1016/j.jns.2011.09.004. [DOI] [PubMed] [Google Scholar]
- D’Antonio M, Feltri ML, Wrabetz L. Myelin under stress. J Neurosci Res. 2009;87:3241–3249. doi: 10.1002/jnr.22066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakemore WF. Demyelination of the superior cerebellar peduncle in the mouse induced by cuprizone. J Neurol Sci. 1973;20:63–72. doi: 10.1016/0022-510x(73)90118-4. [DOI] [PubMed] [Google Scholar]
- Gold R, Linington C, Lassmann H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain. 2006;129:1953–1971. doi: 10.1093/brain/awl075. [DOI] [PubMed] [Google Scholar]
- Siffrin V, Brandt AU, Herz J, Zipp F. New insights into adaptive immunity in chronic neuroinflammation. Adv Immunol. 2007;96:1–40. doi: 10.1016/S0065-2776(07)96001-0. [DOI] [PubMed] [Google Scholar]
- Sarnataro D, Campana V, Paladino S, et al. PrP(C) association with lipid rafts in the early secretory pathway stabilizes its cellular conformation. Mol Biol Cell. 2004;15:4031–4042. doi: 10.1091/mbc.E03-05-0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naslavsky N, Stein R, Yanai A, et al. Characterization of detergent-insoluble complexes containing the cellular prion protein and its scrapie isoform. J Biol Chem. 1997;272:6324–6331. doi: 10.1074/jbc.272.10.6324. [DOI] [PubMed] [Google Scholar]
- van de Kraats C, Killestein J, Popescu V, et al. Oxysterols and cholesterol precursors correlate to magnetic resonance imaging measures of neurodegeneration in multiple sclerosis. Mult Scler. 2014;20:412–417. doi: 10.1177/1352458513499421. [DOI] [PubMed] [Google Scholar]
- Wheeler D, Bandaru VV, Calabresi PA, et al. A defect of sphingolipid metabolism modifies the properties of normal appearing white matter in multiple sclerosis. Brain. 2008;131:3092–3102. doi: 10.1093/brain/awn190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier O, De Jonge J, Nomden A, et al. Lovastatin induces the formation of abnormal myelin-like membrane sheets in primary oligodendrocytes. Glia. 2009;57:402–413. doi: 10.1002/glia.20769. [DOI] [PubMed] [Google Scholar]
- Lee DW, Banquy X, Kristiansen K, et al. Lipid domains control myelin basic protein adsorption and membrane interactions between model myelin lipid bilayers. Proc Natl Acad Sci USA. 2014;111:E768–E775. doi: 10.1073/pnas.1401165111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einstein O, Friedman-Levi Y, Grigoriadis N, Ben-Hur T. Transplanted neural precursors enhance host brain-derived myelin regeneration. J Neurosci. 2009;29:15694–15702. doi: 10.1523/JNEUROSCI.3364-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Nun A, Mendel I, Bakimer R, et al. The autoimmune reactivity to myelin oligodendrocyte glycoprotein (MOG) in multiple sclerosis is potentially pathogenic: effect of copolymer 1 on MOG-induced disease. J Neurol. 1996;243:S14–S22. doi: 10.1007/BF00873697. [DOI] [PubMed] [Google Scholar]
- Friedman-Levi Y, Ovadia H, Hoftberger R, et al. Fatal neurological disease in scrapie-infected mice induced for experimental autoimmune encephalomyelitis. J Virol. 2007;81:9942–9949. doi: 10.1128/JVI.00780-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naslavsky N, Shmeeda H, Friedlander G, et al. Sphingolipid depletion increases formation of the scrapie prion protein in neuroblastoma cells infected with prions. J Biol Chem. 1999;274:20763–20771. doi: 10.1074/jbc.274.30.20763. [DOI] [PubMed] [Google Scholar]
- Kluver H, Barrera E. A method for the combined staining of cells and fibers in the nervous system. J Neuropathol Exp Neurol. 1953;12:400–403. doi: 10.1097/00005072-195312040-00008. [DOI] [PubMed] [Google Scholar]
- Rosenfeld J, Capdevielle J, Guillemot JC, Ferrara P. In-gel digestion of proteins for internal sequence analysis after one- or two-dimensional gel electrophoresis. Anal Biochem. 1992;203:173–179. doi: 10.1016/0003-2697(92)90061-b. [DOI] [PubMed] [Google Scholar]
- Kariv-Inbal Z, Halimi M, Dayan Y, et al. Characterization of light chain immunoglobulin in urine from animals and humans infected with prion diseases. J Neuroimmunol. 2005;162:12–18. doi: 10.1016/j.jneuroim.2004.12.013. [DOI] [PubMed] [Google Scholar]
- Wilm M, Mann M. Analytical properties of the nanoelectrospray ion source. Anal Chem. 1996;68:1–8. doi: 10.1021/ac9509519. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Prions. Proc Natl Acad Sci USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman-Levi Y, Meiner Z, Canello T, et al. Fatal prion disease in a mouse model of genetic E200K Creutzfeldt-Jakob disease. PLoS Pathog. 2011;7:e1002350. doi: 10.1371/journal.ppat.1002350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman L, Zamvil SS. Virtues and pitfalls of EAE for the development of therapies for multiple sclerosis. Trends Immunol. 2005;26:565–571. doi: 10.1016/j.it.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Sajnani G, Silva CJ, Ramos A, et al. PK-sensitive PrP is infectious and shares basic structural features with PK-resistant PrP. PLoS Pathog. 2012;8:e1002547. doi: 10.1371/journal.ppat.1002547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saverioni D, Notari S, Capellari S, et al. Analyses of protease resistance and aggregation state of abnormal prion protein across the spectrum of human prions. J Biol Chem. 2013;288:27972–27985. doi: 10.1074/jbc.M113.477547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Sun X, Zheng S, et al. Myelin basic protein induces neuron-specific toxicity by directly damaging the neuronal plasma membrane. PLoS One. 2014;9:e108646. doi: 10.1371/journal.pone.0108646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom GS. Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014;71:505–508. doi: 10.1001/jamaneurol.2013.5847. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Lopez-Gonzalez I, Carmona M, et al. Glial and neuronal tau pathology in tauopathies: characterization of disease-specific phenotypes and tau pathology progression. J Neuropathol Exp Neurol. 2013;73:81–97. doi: 10.1097/NEN.0000000000000030. [DOI] [PubMed] [Google Scholar]
- Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- Gendron TF, Petrucelli L. The role of tau in neurodegeneration. Mol Neurodegener. 2009;4:13. doi: 10.1186/1750-1326-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs GG, Seguin J, Quadrio I, et al. Genetic Creutzfeldt-Jakob disease associated with the E200K mutation: characterization of a complex proteinopathy. Acta Neuropathol. 2010;121:39–57. doi: 10.1007/s00401-010-0713-y. [DOI] [PubMed] [Google Scholar]
- Anderson JM, Patani R, Reynolds R, et al. Evidence for abnormal tau phosphorylation in early aggressive multiple sclerosis. Acta Neuropathol. 2009;117:583–589. doi: 10.1007/s00401-009-0515-2. [DOI] [PubMed] [Google Scholar]
- Anderson JM, Hampton DW, Patani R, et al. Abnormally phosphorylated tau is associated with neuronal and axonal loss in experimental autoimmune encephalomyelitis and multiple sclerosis. Brain. 2008;131:1736–1748. doi: 10.1093/brain/awn119. [DOI] [PubMed] [Google Scholar]
- DeBruin LS, Haines JD, Wellhauser LA, et al. Developmental partitioning of myelin basic protein into membrane microdomains. J Neurosci Res. 2005;80:211–225. doi: 10.1002/jnr.20452. [DOI] [PubMed] [Google Scholar]
- Uruse M, Yamamoto M, Sugawa M, et al. Phase separation of myelin sheath in Triton X-114 solution: predominant localization of the 21.5-kDa isoform of myelin basic protein in the lipid raft-associated domain. J Biochem. 2014;155:265–271. doi: 10.1093/jb/mvu005. [DOI] [PubMed] [Google Scholar]
- Asakura K, Ueda A, Shima S, et al. Targeting of aquaporin 4 into lipid rafts and its biological significance. Brain Res. 2014;1583:237–244. doi: 10.1016/j.brainres.2014.08.014. [DOI] [PubMed] [Google Scholar]
- Mahammad S, Parmryd I. Cholesterol depletion using methyl-beta-cyclodextrin. Methods Mol Biol. 2015;1232:91–102. doi: 10.1007/978-1-4939-1752-5_8. [DOI] [PubMed] [Google Scholar]
- Canello T, Frid K, Gabizon R, et al. Oxidation of Helix-3 methionines precedes the formation of PK resistant PrP. PLoS Pathog. 2010;6:e1000977. doi: 10.1371/journal.ppat.1000977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider L, Fischer MT, Frischer JM, et al. Oxidative damage in multiple sclerosis lesions. Brain. 2011;134:1914–1924. doi: 10.1093/brain/awr128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte ME, Bol JG, Gerritsen WH, et al. Parkinson’s disease-associated parkin colocalizes with Alzheimer’s disease and multiple sclerosis brain lesions. Neurobiol Dis. 2009;36:445–452. doi: 10.1016/j.nbd.2009.08.009. [DOI] [PubMed] [Google Scholar]
- Hajieva P, Bayatti N, Granold M, et al. Membrane protein oxidation determines neuronal degeneration. J Neurochem. 2014;133:352–367. doi: 10.1111/jnc.12987. [DOI] [PubMed] [Google Scholar]
- Castegna A, Palmieri L, Spera I, et al. Oxidative stress and reduced glutamine synthetase activity in the absence of inflammation in the cortex of mice with experimental allergic encephalomyelitis. Neuroscience. 2011;185:97–105. doi: 10.1016/j.neuroscience.2011.04.041. [DOI] [PubMed] [Google Scholar]
- Leitinger N. The role of phospholipid oxidation products in inflammatory and autoimmune diseases: evidence from animal models and in humans. Subcell Biochem. 2008;49:325–350. doi: 10.1007/978-1-4020-8830-8_12. [DOI] [PubMed] [Google Scholar]
- Mizrahi M, Friedman-Levi Y, Larush L, et al. Pomegranate seed oil nanoemulsions for the prevention and treatment of neurodegenerative diseases: the case of genetic CJD. Nanomedicine. 2014;10:1353–1363. doi: 10.1016/j.nano.2014.03.015. [DOI] [PubMed] [Google Scholar]
- Saher G, Brugger B, Lappe-Siefke C, et al. High cholesterol level is essential for myelin membrane growth. Nat Neurosci. 2005;8:468–475. doi: 10.1038/nn1426. [DOI] [PubMed] [Google Scholar]
- Viquez OM, Lai B, Ahn JH, et al. N, N-diethyldithiocarbamate promotes oxidative stress prior to myelin structural changes and increases myelin copper content. Toxicol Appl Pharmacol. 2009;239:71–79. doi: 10.1016/j.taap.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee R, Beal MF, Thomas B. Autophagy in neurodegenerative disorders: pathogenic roles and therapeutic implications. Trends Neurosci. 2010;33:541–549. doi: 10.1016/j.tins.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. MS–MS data for tau on chronic EAE. The figure 5 shows the original MS–MS data for band 1 in Figure comprising sequences of the Tau protein.