

Abstract
Aicardi–Goutières syndrome is a leukoencephalopathy with calcifications and increased cerebrospinal fluid interferon-α. The relation between interferon-α and brain pathology is poorly understood. We report a patient with mutations in the disease-associated gene SAMHD1. Neuropathology showed an extensive microangiopathy with calcifications consistently associate with blood vessels. In an in vitro model of the microangiopathy, interferon-α enhanced vascular smooth muscle cell-derived calcifications. The noninfarcted white matter harbored apoptotic oligodendrocytes and increased numbers of oligodendrocyte progenitors. These findings better define the white matter pathology and provide evidence that interferon-α plays a direct pathogenetic role in the calcifying angiopathy typical of this disease.
Introduction
Aicardi–Goutières syndrome (AGS; OMIM 225750) is an inherited leukoencephalopathy characterized by calcifications in the white matter, basal ganglia and thalami, and increased cerebrospinal fluid (CSF) interferon (IFN)-α with lymphocytosis.1,2 AGS is clinically heterogeneous. The classic severe form begins in the first months with psychomotor delay, feeding difficulties, irritability, episodes of aseptic febrile illness, and chilblain skin lesions. Symptoms progress over months to years with secondary microcephaly, brain atrophy and spastic tetraparesis, and then stabilize. Death usually occurs before the age of 10 years. Milder forms have onset after the first year and show slower progression and longer survival.2
AGS is genetically heterogeneous. About 90% of patients have mutations in any of seven genes (TREX1, RNASEH2B, RNASEH2C and RNASEH2A, SAMHD1, ADAR1, and IFIH1) leading to a perturbation of the innate immune response with increased intrathecal production of the IFN-type I cytokine IFN-α.3,4> Typical neuropathological findings include brain atrophy, lack of myelin, vascular changes, infarctions, and calcifications.5–7
We report the neuropathology of an AGS patient. The observation of calcifications consistently associated with blood vessels prompted us to investigate in vitro if IFN-α promotes a calcifying phenotype in vascular smooth muscle cells (VSMCs).
Patient and Methods
The patient was a girl with early-infantile severe encephalopathy and later also chill blains. CSF analysis at age 1 year revealed pleocytosis (61 cells/mm3, normal <3 cells/mm3) and increased IFN-α (37 IU/mL, normal <2 IU/mL). Imaging studies at 3 years showed bilateral signal change and atrophy of the cerebral white matter, and calcifications in the basal ganglia and periventricular regions (Fig.1A and B). Repeated imaging at 5 and 11 years showed attenuation of the white matter signal abnormalities and a new lesion in the left frontal cortex (Fig.1C and D). The patient died at 17 years due to pneumonia. The clinical picture, imaging and laboratory data were typical of AGS. Genetic analysis revealed compound-heterozygous mutations in SAMHD1 (p.Met216fs and p.Arg408Pro). The first mutation is a null-mutation; the second mutation results in an amino acid change affecting a residue poorly conserved amongst species. The mutation is not listed in the 1000 Genomes database (www.1000genomes.org), making it unlikely that it represents a common polymorphism. Conventional prediction programs are heavily influenced by interspecies amino acid conservation and do not confirm, but also do not exclude pathogenicity. However, proline has an exceptional conformational rigidity and in silico analysis suggests an effect on the tertiary structure of the protein and possibly on tetramer formation. The patient had one healthy and two affected siblings; the mutations segregated with the disease in the family.
Figure 1.
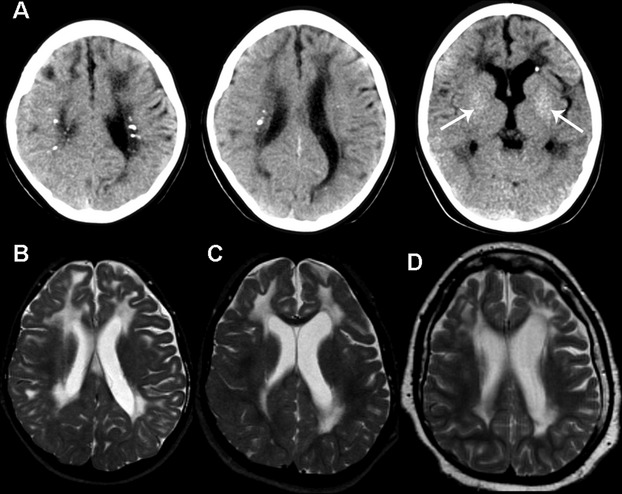
Imaging findings. (A) Axial CT scans at age 3 years demonstrate focal calcifications in the periventricular white matter and subtle finely dispersed calcifications in the basal nuclei (arrows). (B–D) MRI axial T2-weighted images at age 3 years (B) shows diffuse high signal in the cerebral white matter extending frontally into the subcortical regions as well as white matter atrophy. Serial scans at 5 (C) and 11 years (D) reveal attenuation of the white matter signal changes and focal involvement of the cortex in the left more than in the right frontal lobe (D).
Neuropathology
Brain tissue of the patient and three age-matched controls without confounding neuropathology was obtained with informed consent within 5 h postmortem. Formalin-fixed, paraffin-embedded tissue sections were routinely stained with Hematoxylin & Eosin (H&E) and immunostained as described8 for proteolipid protein (PLP; AbD Serotec, 1:3000), glial fibrillary acidic protein (GFAP; Millipore, Pucheim, Germany, 1:1000), smooth muscle actin (SMA; Dako, Amsterdam, The Netherlands, 1:200), IFN-α (PBL Biochemical Labs, 1:100), CD31 (Dako, Heverlee, Belgium, 1:50), CD3 (Dako, Heverlee, Belgium, 1:100), CD68 (Dako, Heverlee, Belgium, 1:3000), human leukocyte antigen-DR (Abcam, Cambridge, United Kingdom, 1:50) and caspase3 (Dako, Heverlee, Belgium, 1:500). Fluorescence immunohistochemistry was performed on cryosections as described9 against oligodendrocyte transcription factor 2 (olig2; Abcam, Cambridge, United Kingdom, 1:400) and platelet-derived growth factor receptor α (PDGFRα; BD Pharmingen, Breda, The Netherlands, 1:100). Known positive controls and negative controls by omitting the primary antibody were included to verify specificity of immunohistochemical labeling. Images were acquired on a Leica DM6000B microscope (Leica Microsystems, Eindhoven, The Netherlands). Control tissue was employed for selected stains and to compare oligodendrocyte lineage cell numbers and apoptotic cells in the white matter. Caspase3-, PDGFRα-, and Olig2-immunopositive cells were counted individually in 10 fields using a 20× objective, expressed as percentage of the total number of oligodendrocytes or nuclei and compared using an independent-samples t-test.
Mineralization assay
Human VSMCs (hVSMCs) isolated from umbilical artery explants were grown on 0.1% gelatin-coated (Sigma, Zwijndrecht, The Netherlands) six-well plates in culture medium supplemented with 20% 1:1 heat-inactivated fetal calf serum (FCS, Hyclone, Bodinco, Alkmaar, The Netherlands)/human serum (Millipore, Amsterdam, The Netherlands), as described.10 SMA-immunoreactivity confirmed hVSMC identity (not shown). For the mineralization assay, fresh (one experiment, two donors, 5000 cells/well) or frozen (two experiments, one donor each, passage two or three, 20,000 cells/well) hVSMCs were grown on gelatin-coated 96-well plates until 80–90% confluence. Following standard methods with minor adjustments,11 mineralization was induced in culture medium containing 1% FCS/human serum (1:1) and a concentration gradient of calcium (0.0, 0.9, 1.3, 2.0, 3.0, 4.4, 6.7 and 10 mmol/L CaCl2; mammalian extracellular calcium range 1.2–1.6 mmol/L12) with or without IFN-α2b (100 ng/mL or 200 pg/mL, Miltenyi Biotec, Leiden, The Netherlands) for 3 weeks. Mineralization was detected with von Kossa or Alizarin red staining as described13,14 and quantified by determining average pixel intensities per well using ImageJ (v1.44, imagej.nih.gov/ij/). For each condition, a third well was used to stain nuclei with Hoechst (H1399, Life Technologies, Bleiswijk, The Netherlands 0.2 μg/mL) to visualize cell density.
Results
Neuropathology
Brain weight was 750 g (mean normal for age: 1300 g). Inspection on sectioning showed mild white matter atrophy, numerous cortico-subcortical infarcts, and small basal nuclei bleedings. No infarcts were detected in the brainstem and cerebellum.
Microscopically, infarcts coincided with thrombotic occlusion of meningeal and intraparenchymal vessels. Microangiopathy was widespread in the striatum, cerebral white matter (Fig.2A), and cerebellum (Fig.2B). Small arterioles and precapillary vessels showed variably thickened tunica media and adventitia with focal or circular calcific deposits. Presence of calcifications coincided with loss of SMA-immunoreactivity in smooth muscle cells (Fig.2C). Notably, calcifications were always associated with blood vessels. Stain for the endothelial marker CD31 revealed that many microcalcifications apparently not adjacent to blood vessels were also centred on endothelial cells (Fig.2D).
Figure 2.

Neuropathology. (A and B) H&E stain show perivascular calcifications (purple) with a concentric pattern around blood vessels in the parietal white matter (A) and in the cerebellum (B). (C) Stain against SMA (brown) of the cerebellar white matter shows that calcifications (blue) are located in the tunica media and lined by SMA-positive smooth muscle cells. Note that the normal SMA-immunoreactivity (upper right) is progressively lost coincident with increasing vessel calcification (lower left). (D) Stain against CD31 of the frontal white matter reveals that apparently perivascular small calcifications are often centered on a CD31-immunoreactive core (arrowheads). (E) At high magnification, GFAP stain of the frontal white matter shows reactive astrocytes, one of which is multinucleated (arrowhead). (F) In the same region, a multinucleated astrocyte (arrowhead) shows a similarly intense IFN-α-immunoreactivity compared to mononucleated astrocytes. (G) Control frontal white matter shows no IFN-α-positive astrocytes. (H) Whole mount of a noninfarcted area in the frontal lobe stained against PLP reveals mild lack of myelin in the deep white matter (arrow) compared to the U-fibers. (I) Stain against CD3 of the frontal white matter shows few infiltrating T-cells mostly restricted to the perivascular space. (J) Caspase3 stain of the same region reveals numerous immunoreactive cells with small, round nuclei, and a perinuclear halo, compatible with apoptotic oligodendrocytes. (K) Caspase3-positive oligodendrocytes are virtually absent in control fronto-parietal white matter (P < 0.0001). (L) Cell counts of cells expressing the oligodendrocyte lineage-specific marker olig2 in the fronto-parietal white matter shows mild loss of oligodendrocytes in AGS compared to controls (P = 0.036). (M) There are higher numbers of OPCs in the noninfarcted white matter of the AGS patient compared to the controls as revealed by PDGFRα immunoreactivity (P < 0.0001). Bars: (A, B, I and J) 50 μm, (C and D) 40 μm, (E–G) 30 μm. Bars in the graphs indicate the standard error of the mean. H&E, Hematoxylin & Eosin; SMA, smooth muscle actin; GFAP, glial fibrillary acidic protein; IFN, interferon; PLP, proteolipid protein; AGS, Aicardi–Goutières syndrome; OPC, oligodendrocyte progenitor cell; PDGFRα, platelet-derived growth factor α receptor. *, P < 0.05; *** P < 0.001.
In the noninfarcted cerebral white matter, GFAP stain revealed numerous reactive astrocytes, some of which multinucleated, that also expressed IFN-α (Fig.2E and F). No IFN-α-positive astrocytes were found in the control white matter (Fig.2G). Stain against PLP showed mild lack of myelin with relative sparing of U-fibers (Fig.2H). Inflammatory signs were inconspicuous, with scarce HLA-DR- and CD68-positive activated microglia/macrophages (not shown) and CD3-positive T-lymphocytes (Fig.2I). To better characterize myelin pathology, we assessed oligodendrocyte lineage cell numbers in frontal and parietal white matter. Compared to controls, there was mild loss of olig2-positive oligodendrocytes (P = 0.036) with numerous caspase3-positive apoptotic cells (P < 0.0001) (Fig.2J–L); however, a count of cells expressing the oligodendrocyte progenitor cell (OPC) marker PDGFRα revealed that OPC numbers were markedly higher in AGS than in control white matter (P < 0.0001, Fig.2M).
Mineralization assay
Cultured hVSMCs underwent cumulative calcification induced by increasing concentrations of calcium in the medium (Fig.3A–C). Addition of IFN-α2b-enhanced hVSMC calcification at lower calcium concentrations in a dose-dependent manner. Notably, this effect was already evident at IFN-α2b concentration of 200 pg/mL, comparable to that in AGS patients’ CSF.6,15–17 Cultures of fresh or frozen hVSMCs gave comparable results.
Figure 3.
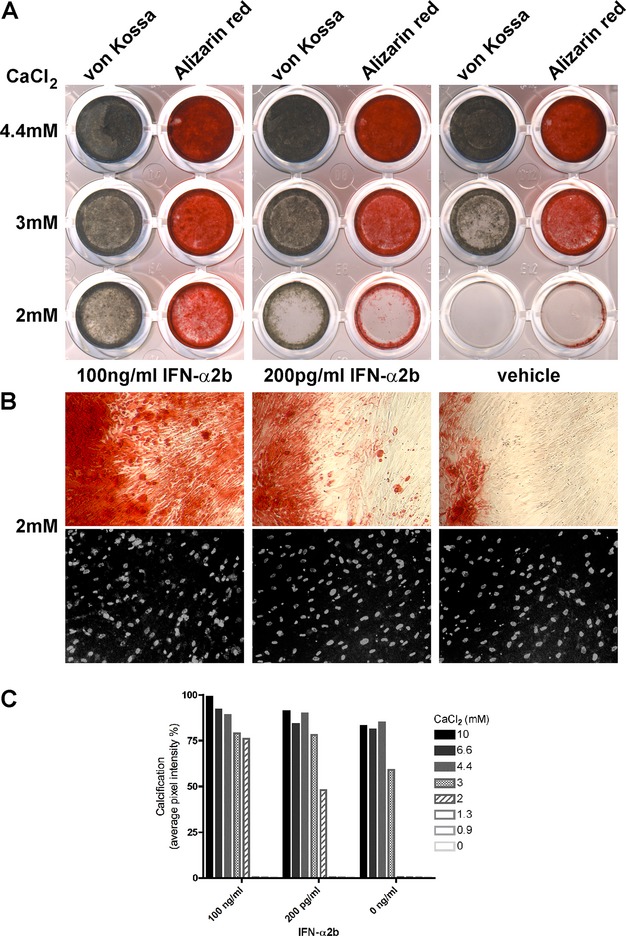
IFN-α enhances calcification in cultured hVSMCs. (A) hVSMCs were exposed to increasing concentrations of calcium (0, 0.9, 1.3, 2 [shown], 3 [shown], 4.4 [shown], 6.7, and 10 mmol/L CaCl2), with or without IFN-α2b (200 pg/mL or 100 ng/mL). Calcification was detected by von Kossa (gray-brown) or Alizarin red stain. IFN-α2b promoted hVSMC calcification particularly at 2 and 3 mmol/L calcium, with 100 ng/mL IFN-α2b having a stronger effect than 200 pg/mL IFN-α2b. Calcification was similar at calcium concentrations of 4.4 mmol/L or higher, with IFN-α2b having a small inducing effect. No calcification was detected at 1.3 mmol/L calcium concentration or lower, with or without IFN-α. Representative results are shown for three comparable experiments. (B) Hundred-times magnification pictures of the hVSMCs cultured with 2 mmol/L calcium, for the wells stained with Alizarin red (top) or for cell density with Hoechst (grayscale, bottom). (C) Calcification quantification of wells stained with von Kossa measured in average pixel intensity per well. An effect of IFN-α2b was particularly detected at 3 mmol/L CaCl2, with an intensity increase of 20% with 200 pg/mL or 100 ng/mL IFN-α2b compared to 0 ng/mL IFN-α2b, or at 2 mmol/L CaCl2, with an intensity increase of 48% with 200 pg/mL IFN-α2b or 76% with 100 ng/mL IFN-α2b, respectively. Representative results are shown for three comparable experiments. IFN, interferon; hVSMCs, human vascular smooth muscle cells.
Discussion
The findings of wedge-shaped infarcts, inhomogeneous loss of myelin and dystrophic calcifications in and around small vessels led to the suggestion that AGS represents a primary microangiopathy.5 Neuropathology of our case confirms extended involvement of small vessels and widespread calcifications. Most calcifications were within vessel walls, surrounding CD31-expressing intimal endothelial cells and adjacent to SMA-positive smooth muscle cells. Calcifications apparently not associated with blood vessels also surrounded a CD31-immunoreactive core, implying that they represent completely occluded calcified small vessels. This suggests that the calcific deposits in AGS arise in vascular walls, in particular in smooth muscle cells of the tunica media.
Vascular calcification is a cell-mediated process associated with dedifferentiation of smooth muscle cells to an osteoblast/chondrocytic phenotype.10,18 Dedifferentiation coincides with loss of contractile properties and SMA-immunoreactivity, as observed in our patient. Inflammation is a known risk factor for vascular calcification; innate immune response mediators, including cytokines and tumor necrosis factor α, induce calcification of vascular cells in vitro.18 Little is known about the mechanisms underlying the calcifying microangiopathy in AGS. To experimentally investigate a role for IFN-α, we established an in vitro model of AGS-microangiopathy, using hVSMCs chronically exposed to IFN-α. At a concentration comparable to that in patient’s CSF, IFN-α2b enhanced calcification of cultured hVSMCs. This indicates that IFN-α promotes the generation of calcium deposits by smooth muscle cells and supports the proposition that IFN-α acts directly on VSMCs in AGS, thus contributing to the calcifying microangiopathy. A similar mechanism may operate in other brain disorders with excessive IFN-α, including congenital viral infections and autoimmune disorders like systemic lupus erythematosus, that feature neuropathological changes and calcifications evocative of AGS.3
In AGS patients’ brains, astrocytes produce the excessive IFN-α.17 Mice overexpressing IFN-α in astrocytes develop a leukoencephalopathy similar to AGS.19 The myelin pathology has been related to overexpression of cathepsin-D in CSF lymphocytes, a pro-apoptotic factor with myelin-directed protease activity induced by IFN-α.20 CSF of our patient was not available to verify IFN-α and cathepsin-D levels, but astrocytic IFN-α expression and apoptotic loss of oligodendrocytes were conspicuous in the white matter, whereas influx of lymphocytes was negligible. This points to mechanisms other than lymphocytic neurotoxicity in maintaining oligodendrocytic pathology in AGS, at least in older patients in whom CSF lymphocyte numbers have fallen to normal.2 In our patient’s noninfarcted white matter, lack of myelin was only mild and associated with increased OPC numbers. No myelin-laden macrophages were found, excluding ongoing demyelination. These findings suggest that the OPCs actively proliferate in response to oligodendrocyte loss and can mature into myelin-forming cells. Such remyelination effort highlights a previously unrecognized repair potential in AGS, which is in line with the progressive attenuation of the MRI white matter signal abnormalities.
In conclusion, present evidence indicates that astrocyte-derived IFN-α directly contributes to the calcifying microangiopathy typical of AGS. Further in vivo and in vitro studies are warranted to understand if IFN-α exerts a direct effect also on oligodendrocytic pathology and OPC differentiation throughout the disease course.
Acknowledgments
The authors are grateful to the mortuary assistants of the VU University Medical Centre for their skilful help during the autopsies.
Author Contribution
M. D. K., M. S. v. d. K. and M. B. designed the study. M. D. K., N. L. P. and R. M. L. v. S. performed the genetic analysis and experimental work. M. B. performed the autopsy and autopsy diagnostics. M. D. K., H. S. B., M. S. v. d. K. and M. B. analyzed the data. M. D. K., H. S. B., M. S. v. d. K. and M. B. wrote the manuscript. N. L. P. and R. M. L. v. S. edited the manuscript.
Conflict of Interest
None declared.
References
- Aicardi J, Goutières F. A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol. 1984;15:49–54. doi: 10.1002/ana.410150109. [DOI] [PubMed] [Google Scholar]
- Crow YJ. Aicardi-Goutières syndrome. Handb Clin Neurol. 2013;113:1629–1635. doi: 10.1016/B978-0-444-59565-2.00031-9. [DOI] [PubMed] [Google Scholar]
- Hofer MJ, Campbell IL. Type I interferon in neurological disease-the devil from within. Cytokine Growth Factor Rev. 2013;24:257–267. doi: 10.1016/j.cytogfr.2013.03.006. [DOI] [PubMed] [Google Scholar]
- Diamond J. Autosomal dominant IFIH1 gain-of-function mutations cause Aicardi-Goutières syndrome. Clin Genet. 2014;86:473–474. doi: 10.1111/cge.12471. [DOI] [PubMed] [Google Scholar]
- Barth PG, Walter A, van Gelderen I. Aicardi-Goutières syndrome: a genetic microangiopathy? Acta Neuropathol. 1999;98:212–216. doi: 10.1007/s004010051071. [DOI] [PubMed] [Google Scholar]
- Goutières F, Aicardi J, Barth PG, et al. Aicardi-Goutières syndrome: an update and results of interferon-alpha studies. Ann Neurol. 1998;44:900–907. doi: 10.1002/ana.410440608. [DOI] [PubMed] [Google Scholar]
- Kumar D, Rittey C, Cameron AH, et al. Recognizable inherited syndrome of progressive central nervous system degeneration and generalized intracranial calcification with overlapping phenotype of the syndrome of Aicardi and Goutières. Am J Med Genet. 1998;75:508–515. [PubMed] [Google Scholar]
- Kevelam SH, Bugiani M, Salomons GS, et al. Exome sequencing reveals mutated SLC19A3 in patients with an early-infantile, lethal encephalopathy. Brain. 2013;136:1534–1543. doi: 10.1093/brain/awt054. [DOI] [PubMed] [Google Scholar]
- Bugiani M, Postma N, Polder E, et al. Hyaluronan accumulation and arrested oligodendrocyte progenitor maturation in vanishing white matter disease. Brain. 2013;136:209–222. doi: 10.1093/brain/aws320. [DOI] [PubMed] [Google Scholar]
- Proudfoot D, Shanahan C. Human vascular smooth muscle cell culture. Methods Mol Biol. 2012;806:251–263. doi: 10.1007/978-1-61779-367-7_17. [DOI] [PubMed] [Google Scholar]
- Reynolds JL, Joannides AJ, Skepper JN, et al. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J Am Soc Nephrol. 2004;15:2857–2867. doi: 10.1097/01.ASN.0000141960.01035.28. [DOI] [PubMed] [Google Scholar]
- Jones HC, Keep RF. Brain interstitial fluid calcium concentration during development in the rat: control levels and changes in acute plasma hypercalcaemia. Physiol Bohemoslov. 1988;37:213–216. [PubMed] [Google Scholar]
- Bonewald LF, Harris SE, Rosser J, et al. von Kossa staining alone is not sufficient to confirm that mineralization in vitro represents bone formation. Calcif Tissue Int. 2003;72:537–547. doi: 10.1007/s00223-002-1057-y. [DOI] [PubMed] [Google Scholar]
- Proudfoot D, Skepper JN, Hegyi L, et al. Apoptosis regulates human vascular calcification in vitro: evidence for initiation of vascular calcification by apoptotic bodies. Circ Res. 2000;87:1055–1062. doi: 10.1161/01.res.87.11.1055. [DOI] [PubMed] [Google Scholar]
- Rubinstein M, Levy WP, Moschera JA, et al. Human leukocyte interferon: isolation and characterization of several molecular forms. Arch Biochem Biophys. 1981;210:307–318. doi: 10.1016/0003-9861(81)90194-6. [DOI] [PubMed] [Google Scholar]
- Hobbs DS, Pestka S. Purification and characterization of interferons from a continuous myeloblastic cell line. J Biol Chem. 1982;257:4071–4076. [PubMed] [Google Scholar]
- van Heteren JT, Rozenberg F, Aronica E, et al. Astrocytes produce interferon-alpha and CXCL10, but not IL-6 or CXCL8, in Aicardi-Goutières syndrome. Glia. 2008;56:568–578. doi: 10.1002/glia.20639. [DOI] [PubMed] [Google Scholar]
- Chen NX, Moe SM. Vascular calcification: pathophysiology and risk factors. Curr Hypertens Rep. 2012;14:228–237. doi: 10.1007/s11906-012-0265-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akwa Y, Hassett DE, Eloranta ML, et al. Transgenic expression of IFN-alpha in the central nervous system of mice protects against lethal neurotropic viral infection but induces inflammation and neurodegeneration. J Immunol. 1998;161:5016–5026. [PubMed] [Google Scholar]
- Pulliero A, Marengo B, Longobardi M, et al. Inhibition of the de-myelinating properties of Aicardi-Goutières syndrome lymphocytes by cathepsin D silencing. Biochem Biophys Res Commun. 2013;430:957–962. doi: 10.1016/j.bbrc.2012.11.131. [DOI] [PubMed] [Google Scholar]