

Abstract
In patient with Alzheimer’s disease (AD), deposition of amyloid-beta Aβ, a proteolytic cleavage of amyloid precursor protein (APP) by β-secretase/BACE1, forms senile plaque in the brain. BACE1 activation is caused due to oxidative stresses and dysfunction of ubiquitin–proteasome system (UPS), which is linked to p53 inactivation. As partial suppression of BACE1 attenuates Aβ generation and AD-related pathology, it might be an ideal target for AD treatment. We have shown that both in neurons and in HEK-APP cells, BACE1 is a new substrate of E3-ligase CHIP and an inverse relation exists between CHIP and BACE1 level. CHIP inhibits ectopic BACE1 level by promoting its ubiquitination and proteasomal degradation, thus reducing APP processing; it stabilizes APP in neurons, thus reducing Aβ. CHIPUbox domain physically interacts with BACE1; however, both U-box and TPR domain are essential for ubiquitination and degradation of BACE1. Further, BACE1 is a downstream target of p53 and overexpression of p53 decreases BACE1 level. In HEK-APP cells, CHIP is shown to negatively regulate BACE1 promoter through stabilization of p53’s DNA-binding conformation and its binding upon 5′ UTR element (+127 to +150). We have thus discovered that CHIP regulates p53-mediated trans-repression of BACE1 at both transcriptional and post-translational level. We propose that a CHIP–BACE1–p53 feedback loop might control APP stabilization, which could further be utilized for new therapeutic intervention in AD.
Keywords: Alzheimer’s disease, Neurodegenerative diseases, p53, proteasome, ubiquitin pathway
Introduction
Deposition of amyloid-beta (Aβ) as a form of senile plaque in the human brain is a major pathological feature of Alzheimer’s disease (AD) (Murphy & LeVine, 2010). Aβ is produced from the amyloid precursor protein (APP) through sequential proteolytic cleavages by β- and γ-secretases (Li et al., 2006). Proteolytic cleavage of APP at the β-site is a rate-limiting step for Aβ generation; β-site APP-cleaving enzyme 1 (BACE1) is the β-secretase in vivo (Cai et al., 2001). Although the large majority of AD cases are sporadic, autosomal-dominant mutations in APP and presenilin genes might also be responsible for the rare, familial form (Gatz et al., 2006).
Oxidative stress has long been implicated in the pathogenesis of AD and is potential cause of the increased level and activity of BACE1 (Tamagno et al., 2002, 2008; Tong et al., 2005). Multiple studies have explained the molecular mechanism underlying oxidative stress-induced BACE1 activation that might be contributed by redox-sensitive transcriptional factors such as NF-kβ (Bourne et al., 2007), HIF (Sun et al., 2006; Zhang et al., 2007), STAT1 (Cho et al., 2009), PPARγ (Sastre et al., 2006), YY1 (Nowak et al., 2006), Sp1 (Christensen et al., 2004), and dysfunction of ubiquitin–proteasome system (UPS) (Upadhya & Hegde, 2007; Riederer et al., 2011). Various oxidative stress markers are found to be elevated in the AD brain, such as HNE, 3-NT, protein carbonyls (Cenini et al., 2008), and are associated with p53 tumor suppressor protein that can change its conformation, resulting in loss of DNA binding as well as transcriptional activity (Uberti et al., 2006). In response to oxidative stress, p53, a key regulator, is activated and transactivates its target genes through binding upon specific DNA sequences in their promoter region (Gambino et al., 2013). An increase in the expression of p53 and its altered conformation have been observed in brain and peripheral cells of patients with AD, leading to intense dysfunction in the p53 signaling pathway in response to various stresses, without any evidence of genetic mutations (Uberti et al., 2006; Lanni et al., 2007, 2008). While there is an increase in the total p53 level, the level of phosphorylated p53-Ser15 decreased along with a decrease in the total p21 and phosphorylated p21-Thr145 level in peripheral blood lymphocytes of patients with AD (Tan & Evin, 2012), suggestive of G1/S check point dysfunction (Zhou & Jia, 2010).
The levels and activity of BACE1 increased in the brain of patients with sporadic AD (Yang et al., 2003) as BACE1-knockout mice lack Aβ generation and are free from AD-associated pathologies (Luo et al., 2001). It is suggested that dysfunction of UPS might also be involved in AD pathogenesis (Upadhya & Hegde, 2007; Riederer et al., 2011). BACE1 proteins are degraded through the UPS (Qing et al., 2004) in which brain-specific SFC E3-ligase helps in recognition, ubiquitination, and degradation coinciding with decrease in the production of Aβ (Gong et al., 2010). Interestingly, the accumulation of Aβ is also shown to activate BACE1 expression (Piccini et al., 2012) along with a decrease in activity of UPS (Almeida et al., 2006). Thus, partial suppression of BACE1 attenuates Aβ generation and AD-related pathology (Kimura et al., 2010), suggesting that partial inhibition of BACE1 could be an ideal target for AD treatment.
We had earlier shown that CHIP, a brain-enriched E3 ligase, chaperones p53 and stabilizes its native conformation under stress (Tripathi et al., 2007). It further contributes to ubiquitination and degradation of several AD-related proteins, such as CFTR (Meacham et al., 2001), tau (Petrucelli et al., 2004), p53 (Esser et al., 2005), APP, and Aβ (Kumar et al., 2007). Its expression was shown to decrease upon Aβ accumulation in both transgenic mice and cultured cells (Oddo et al., 2008). As the increased activity of BACE1 and increased expression of p53 with altered conformation are observed in brain of patients with sporadic AD, we hypothesized whether CHIP would destabilize BACE1 via proteasomal degradation and whether it could stabilize p53 to trans-repress BACE1 gene transcription. In this report, we have shown in neurons that CHIP decreases BACE1 protein level, thus stabilizing APP in reducing Aβ production. CHIP promotes ubiquitination and proteasomal degradation of BACE1 and CHIP-mediated p53 stabilization results in negative regulation of BACE1 gene transcription. Hence, CHIP’s function in stabilizing APP at both transcription and post-translational level might open up new strategies in therapeutic control of AD.
Results
CHIP prevents β-cleavage of APP through BACE1 destabilization
CHIP, a chaperone-associated E3-ligase, regulates the stability of several diseases-associated brain proteins through the ubiquitin–proteasome pathway. Although CHIP stabilizes APP, it also helps in the ubiquitination of APP that are destined for proteasome degradation. During AD pathogenesis, the levels of CHIP decrease along with an increase in the expression of BACE1, which is degraded through UPS (Qing et al., 2004). To determine the role of CHIP in the regulation of BACE1 during AD pathogenesis, we first asked whether BACE1 is a substrate for CHIP and checked the stability of BACE1 in the presence of CHIP. We expressed Flag-BACE1 alone or in the presence of increasing amounts of myc-CHIP in human HEK 293 and H1299 cells and estimated the level of BACE1 and CHIP by Western blotting. We observed that the ectopic expression of BACE1 was significantly reduced by CHIP in a dose-dependent manner as compared to BACE1 when expressed alone (Fig.1A,B). Further, endogenous expression of BACE1 protein was also reduced by CHIP in human neuroblastoma cells (SH-SY5Y) (Fig.1C). Next, we investigated whether endogenous CHIP can directly modulate the stability of BACE1 protein. The silencing of endogenous CHIP by shRNA resulted in the stabilization of BACE1 protein as compared to control where BACE1 was transfected with either scrambled shRNA or empty plasmid (Fig.1D). This result establishes that CHIP negatively regulates BACE1 stability.
Fig 1.

CHIP prevents β-cleavage of APP through BACE1 destabilization. (A–B) Destabilization of ectopic BACE1 by CHIP (0.5, 1, 2, and 3) μg in a dose-dependent manner. (C) CHIP destabilizes endogenous BACE1 level in SH-SY5Y neuroblastoma cells. (D) Silencing of endogenous CHIP stabilizes BACE1 protein level. (E) and (F) Functional domain of CHIP responsible for BACE1 destabilization. (G–H) CHIP destabilizes BACE1 protein level and decreases APP processing at β-cleavage site. (G) HEK 293 cell stably expressing APP were co-transfected with Flag-BACE1 constructs along with increasing amounts of myc-CHIP constructs. (H) Rat primary cortical neurons were transfected with increasing amount of myc-CHIP. After 30 h of transfection, C-terminal β-site cleavage product of APP in whole-cell lysate was determined by Western blotting using anti-APP antibodies (C-terminal). All the data were expressed as mean ±SE from three independent experiments. Statistical analysis were performed by one-group t-test for the significance at the * = P < 0.05, ** = P < 0.01 and *** =P < 0.001.
CHIP contains an N-terminal TPR domain that is responsible for chaperone binding, a C-terminal U-box domain possessing E3-ubiquitin ligase activity and a central charged domain. To identify the CHIP domains that regulate BACE1 stability, a series of CHIP mutants were examined for their abilities to regulate BACE1 stability. Both TPR and U-box deletion constructs of CHIP were unable to reduce BACE1 protein level (Fig.1E). Furthermore, point mutations K30A (TPR domain) and H260Q (U-box domain), which abolish the ability of CHIP to interact with chaperones and of its ubiquitin ligase activity, respectively, also failed to reduce BACE1 protein (Fig.1F), thus suggesting that both TPR and U-box domains of CHIP are necessary for BACE1 degradation.
We further investigated the role of CHIP in the β-cleavage of APP and Aβ generation in HEK-APP stable cells and rat primary cortical neurons. HEK 293 cells stably expressing human APP695 were transfected with cDNA encoding Flag-BACE1 along with control plasmid or with myc-CHIP, and the levels of BACE1 cleavage product of APP (CTFβ 99) and other proteins were estimated by Western blotting. Further, rat primary cortical neurons were transfected with control plasmid or with increasing amount of myc-CHIP and the level of endogenous BACE1, CTFβ 99 (C-99), and other proteins were estimated. The results show that CHIP significantly reduced BACE1 cleavage product of APP (CTFβ 99) as compared to control plasmid in both HEK-APP cells (Fig.1G) and rat primary cortical neurons (Fig.1H). Thus, CHIP destabilizes BACE1 protein and decreases APP processing at the β-secretase site to attenuate Aβ generation.
CHIP promotes BACE1 ubiquitination and proteasomal degradation
To check whether the reduced stability of BACE1 by CHIP is a post-translational event, we first examined half-life of BACE1 protein using the protein synthesis inhibitor cycloheximide (CHX). HEK 293 cells were co-transfected with BACE1 cDNA along with empty vector, myc-CHIP, or myc-CHIPΔUbox. The rate of degradation of BACE1 was found to be significantly faster in the presence of CHIP as compared to BACE1 expressed alone or in the presence of CHIPΔUbox, which lacks E3-ligase activity (Fig.2A,B). Therefore, CHIP promotes destabilization of BACE1 at the post-translational level and its E3-ligase activity was essential for BACE1 degradation. CHIP-mediated destabilization of the BACE1 at post-translational level led us to investigate the mechanism through which CHIP exerts its effect upon BACE1 protein. Previous studies had established the role of CHIP in promoting proteasome-dependent degradation of its client proteins, such as hTERT (Lee et al., 2010), PTEN (Ahmed et al., 2012), and p53 (Esser et al., 2005). The turnover of BACE1 protein is also controlled through a proteasome-dependent pathway (Qing et al., 2004; Gong et al., 2010). The treatment of cells with MG132, an inhibitor of proteasome, caused stabilization of BACE1, and the level of BACE1 that was decreased by co-expression of CHIP was reverted back to normal (Fig.2C). This result clearly indicates that the treatment of cells with proteasome inhibitor rescued BACE1 from CHIP-induced destabilization, thus establishing that CHIP promoted destabilization of the BACE1 protein through the proteasome-mediated degradation. We then performed ubiquitination assay for BACE1 protein with CHIP. HEK 293 cells were transfected with Flag-BACE1 and 6X His-ubiquitin (His-Ub) in the presence of CHIP or empty plasmid. Cells were lysed and washed under denaturing conditions to ensure both inactivation of deubiquitinating enzymes and removal of contaminating proteins, except proteins that are covalently associated with Ub. Proteins that were conjugated with His-Ub were pulled down by Ni2+-NTA beads followed by Western blotting with anti-flag antibodies to detect ubiquitin-conjugated BACE1. Western blot results showed that high molecular weight ubiquitinated forms of BACE1 were considerably enhanced when BACE1 was co-expressed along with CHIP in a dose-dependent manner (Fig.2D), suggesting that CHIP was responsible for BACE1 ubiquitination. The ubiquitination of BACE1 was also examined in the presence of CHIP mutants. It was observed that only wild-type CHIP enhanced the ubiquitination of BACE1, while the deletion mutants CHIPΔTPR and CHIPΔUbox failed to ubiquitinate BACE1 (Fig.2E). The ubiquitination level of BACE1 in the presence of CHIP deletion mutants was similar as it was observed in the presence of an empty plasmid. Thus, both deletion mutants of CHIP could not enhance the ubiquitination of BACE1, suggesting that both domains of CHIP were essential for ubiquitination and degradation of BACE1. Further, in vitro ubiquitination reaction was performed to confirm that CHIP is directly involved in the ubiquitination of BACE1 (Fig.2F).
Fig 2.

CHIP promotes BACE1 ubiquitination and proteasomal degradation. (A–B) Destabilization of BACE1 at post-translational level by CHIP was determined by cycloheximide chase assay. HEK 293 cells were transfected with Flag-BACE1 along with myc-CHIP (A) or myc-CHIPΔUbox (B). After 20 h of transfection, cells were treated with 100 μg/mL of cycloheximide (CHX) at indicated time points to inhibit protein synthesis. (C) Proteasomal-dependent degradation of BACE1 by CHIP. HEK 293 cells were co-transfected with Flag-BACE1 along with myc-CHIP. After 24 h of transfection, cells were treated with 20 μm MG12 for 6 h to inhibit proteasome activity. (D–E) CHIP promotes BACE1 ubiquitination. HEK 293 cells were co-transfected with Flag-BACE1 and His-ubiquitin (His-Ub) in the presence of an increasing amount of CHIP (D) or its deleted mutants (E). Ubiquitinated BACE1 from whole-cell lysate was precipitated with Ni2+-NTA beads followed by Western blotting with anti-flag antibodies. (F) GST-BACE1 (2 μg) was incubated with E1 and E2 (UbcH5a) and ubiquitin (2 μm) in the presence or absence of His-CHIP (5 μg) for 30 min. Ubiquitination of BACE1 was analyzed by Western blotting using anti-BACE1 antibodies. All the data were expressed as mean ±SE from three independent experiments. Statistical analysis were performed by one-group t-test for the significance at the ** = P < 0.01. ns, nonsignificant.
BACE1 physically interacts with CHIP’s U-box domain
As BACE1 is shown to be a natural substrate of CHIP, we then examined the physical interaction between CHIP and BACE1. We first performed a co-immunoprecipitation assay in HEK 293 cells where Flag-BACE1 was co-transfected with myc-CHIP or its deletion mutants (Fig.3A). The deletion mutant CHIPΔTPR lacks the TPR domain (1–141 aa) and is defective in chaperone binding ability, whereas the CHIPΔUbox lacks U-box domain (198–303 aa) and is defective in E3-ligase activity. We found that CHIP physically associates with BACE1 through its U-box domain, as assessed by co-immunoprecipitation with anti-myc antibodies followed by Western blotting detection with anti-flag antibodies. Both CHIP and CHIPΔTPR were co-immunoprecipitated with BACE1, whereas CHIPΔUbox failed to co-immunoprecipitate with BACE1, revealing that U-box domain of CHIP interacts with BACE1 (Fig.3B).
Fig 3.
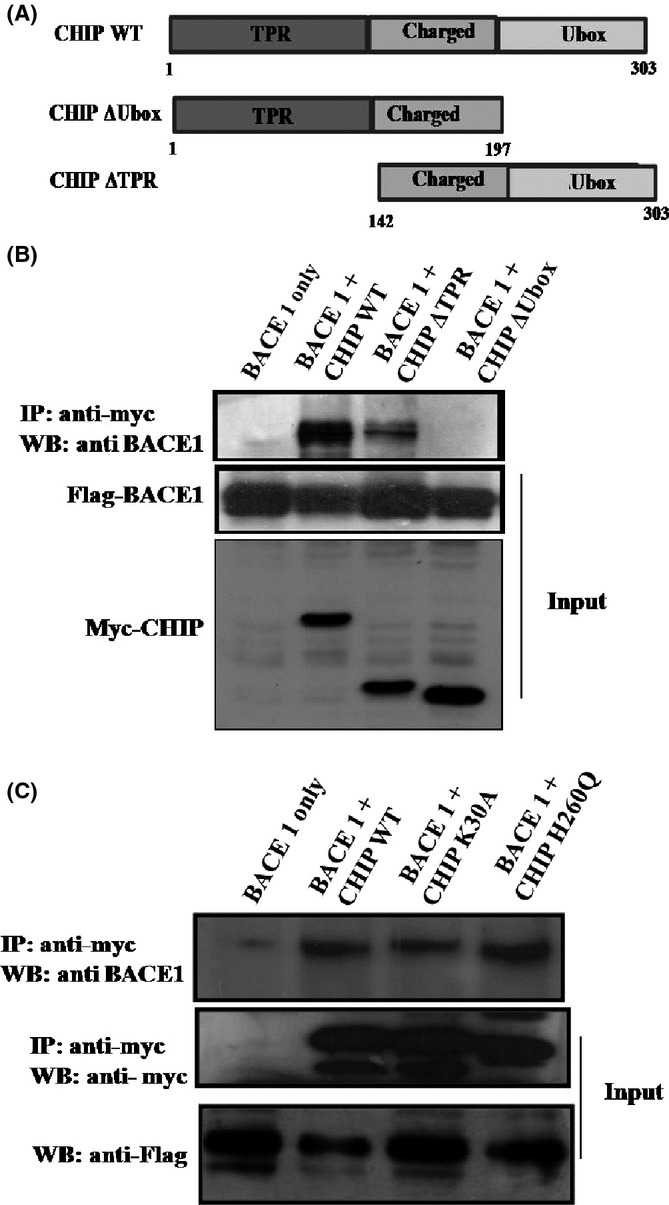
BACE1 physically interacts with CHIP. (A) Schematic representation of CHIP and its deleted mutants used in the study. (B) Interaction between BACE1 and CHIP mutants. Two micrograms of Flag-BACE1 was co-transfected with 2 μg of myc-CHIP wild-type or its mutant. Co-immunoprecipitation assay was performed using anti-myc antibodies against CHIP, followed by Western blotting with anti-flag antibodies against the BACE1. BACE1 interacts with CHIP through its U-box domain. (C) Interaction between CHIP and BACE1 was independent of point mutations.
Several studies have reported that the association of CHIP with Hsp70/90 was important for its interaction with client proteins through TPR domain (Lee et al., 2010; Ahmed et al., 2012). To check whether interaction between CHIP and BACE1 is mediated through Hsp70/90, we expressed Flag-BACE1 along with myc-CHIP, myc-CHIPK30A, or myc-CHIPH260Q mutants in HEK 293 cells. Substitution mutation K30A in the TPR domain of CHIP prevented its interaction with Hsp70/90. H260Q mutation in the U-box domain of CHIP also prevented the interaction of CHIP with E2 enzymes and results in loss of its E3 ligase activity. Co-immunoprecipitation with anti-myc antibodies followed by Western blotting with anti-flag antibodies showed that BACE1 co-immunoprecipitates with CHIP, CHIPK30A, and CHIPH260Q mutants (Fig.3C). These results implied that the interaction between BACE1 and CHIP is independent of Hsp70/90.
Transcriptional repression of BACE1 by p53
The expression of BACE1 is tightly regulated at the transcriptional level and its dysregulation leads to its overexpression during AD pathogenesis (Sun et al., 2012). Enhanced transcription of BACE1 in patients with AD was shown to be synchronized with functional inactivation of p53 (Uberti et al., 2006; Lanni et al., 2007, 2008) and that led us to ask whether p53 negatively regulates BACE1 gene transcription. SH-SY5Y cells were transfected with increasing dose of p53 followed by quantitative real-time PCR (qPCR) and Western blotting analysis. The cDNA synthesis was carried out from total RNA, and qPCR was performed in triplicate using cDNA. Relative BACE1 mRNA expression level was normalized to an endogenous housekeeping gene (GAPDH). The result showed that exogenous expression of p53 decreases the expression of BACE1 at mRNA level in a dose-dependent manner (Fig.4A). This result confirmed that p53 suppresses BACE1 expression at the transcriptional level.
Fig 4.
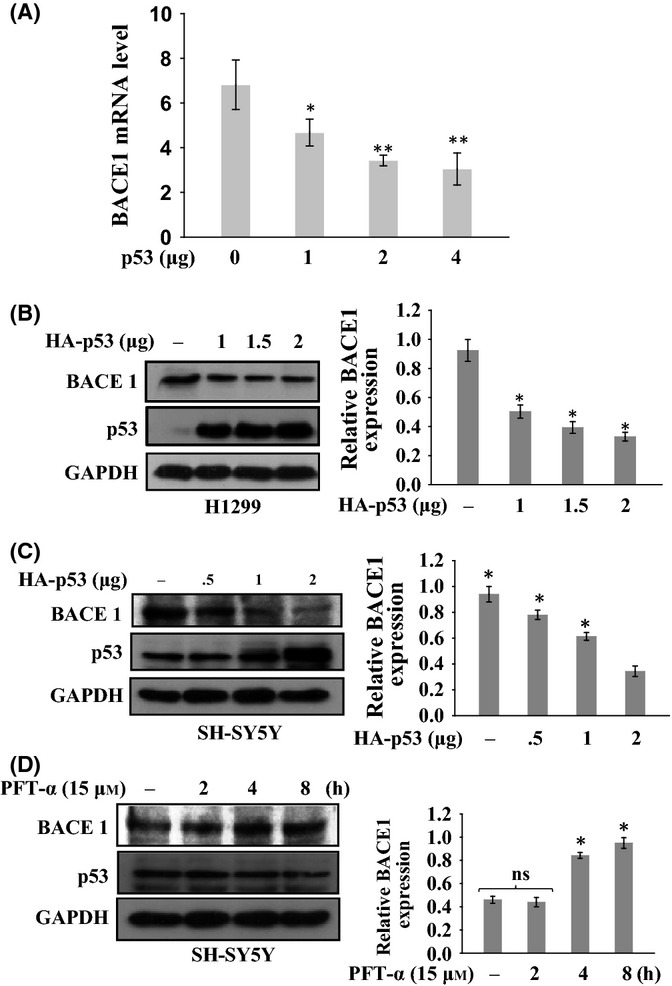
Transcriptional repression of BACE1 by p53. (A) Effect of p53 on BACE1 mRNA level. SH-SY5Y cells were transfected with increasing amount (1, 2, and 3 μg) of p53. Quantitative real-time PCR analysis was performed. The endogenous BACE1 mRNA level decreases with increasing p53 expression. (B–C) Effect of p53 on BACE1 protein level. (A) H1299 and (C) SH-SY5Y cells were transfected with increasing amount (1, 1.5, and 2 μg) of p53 expression constructs. (D) Effect of inhibited endogenous p53 on BACE1 level. SH-SY5Y cells were treated with PFT-α (15 μm) for indicated time period to inhibit endogenous p53. Endogenous BACE1 protein levels were determined by Western blotting using anti-BACE1 antibodies. Data represent the mean ± SE from three independent experiments. Statistical analysis were performed by one-group t-test for the significance at the *=P < .005 and **=P < 0.001. ns, nonsignificant.
As BACE1 transcription is downregulated by p53, as revealed by mRNA level of BACE1, we then investigated the effects of p53 on the BACE1 protein level. We transfected H1299 cells (p53−/−) and SH-SY5Y cells (p53+/+) with increasing amount of p53 to examine the effect of overexpression of p53 on the endogenous BACE1 level. As the expression of p53 increased, the level of endogenous BACE1 protein was decreased in a dose-dependent manner in both cell lines (Fig.4B,C). Inhibition of p53 expression by pifithrin-α in SH-SY5Y cells showed that BACE1 protein level was increased with increasing duration of treatment (Fig.4D). This clearly indicated that the p53 downregulates the expression of BACE1 protein.
p53 downregulates BACE1 promoter through selective binding at +127 to +150 position
As p53 negatively regulates BACE1 expression, we searched for p53 binding sites upon BACE1 promoter through in silico analysis using MatInspector tool (Cartharius et al., 2005). Two sites were found: the first one was located downstream of the start site (TSS) at (+150 to +127, score: 0.942), whereas the second one was at (−2670 to −2693, score: 0.847) (Fig5A). Chromatin immunoprecipitation (ChIP) assay was performed in H1299 cells that were transfected with p53 cDNA. Anti-p53 antibodies were used for immunoprecipitation followed by PCR with flanking primers for both p53DBS. Amplification of a 182-bp region of putative p53 binding site I (+150 to +127) (Fig.5B) was observed; no band was observed with primers for the site II at (−2570 to −2693). Similar results were observed with controls that contain no antibodies or nonspecific antibodies; PCR with input sample showed amplification against both the regions (Fig.5B). This result clearly shows that p53 exclusively binds to the BACE1 promoter in the region of 5′ UTR (+150 to +127 bp). In some genes, 5′ UTR is a part of core promoter that contains transcriptional factor binding sites and regulates its transcription (Yu et al., 2001). This result indicates that p53 transcriptional factor directly binds to the BACE1 promoter at the site of +150 to +127 (5′ UTR) and not upon the site of −2670 to −2693. The effect of p53 binding upon DBS of the 5′ UTR in the BACE1 promoter was then analyzed by luciferase assay. H1299 (p53−/−) cells were transfected with either the pGL2 BACE1 P1-Luc (P1) or pGL2 BACE1 P2-Luc (P2) (Fig5A) along with the p53 expression plasmid. The co-transfection of BACE1 promoter constructs along with p53 resulted in significant repression (5-fold) of BACE1 promoter activity (Fig.5C). By contrast, co-transfection of pGL2 BACE1 promoter constructs along with p53 R175H, a p53 mutant that does not bind to DNA, resulted in partial inhibition of the BACE1 promoter activity as compared with wild-type p53 (Fig.5C). Transcriptional repression ability of mutant p53 might not depend exclusively on functional sequence-specific DNA-binding domain of p53. An earlier study shows that p53 R175H mutant exerted repression activity on MAD1 promoter with weaker DNA-binding activity as compared to wild-type p53 (Chun & Jin, 2003). An inactivation of endogenous p53 by pifithrin-α (PFT-α) in MCF-7 (p53+/+) cells caused activation of BACE1 promoter activity as compared to untreated cells (Fig.5D). These findings clearly demonstrate that p53 interacts with BACE1 promoter and downregulates its transcriptional activity.
Fig 5.
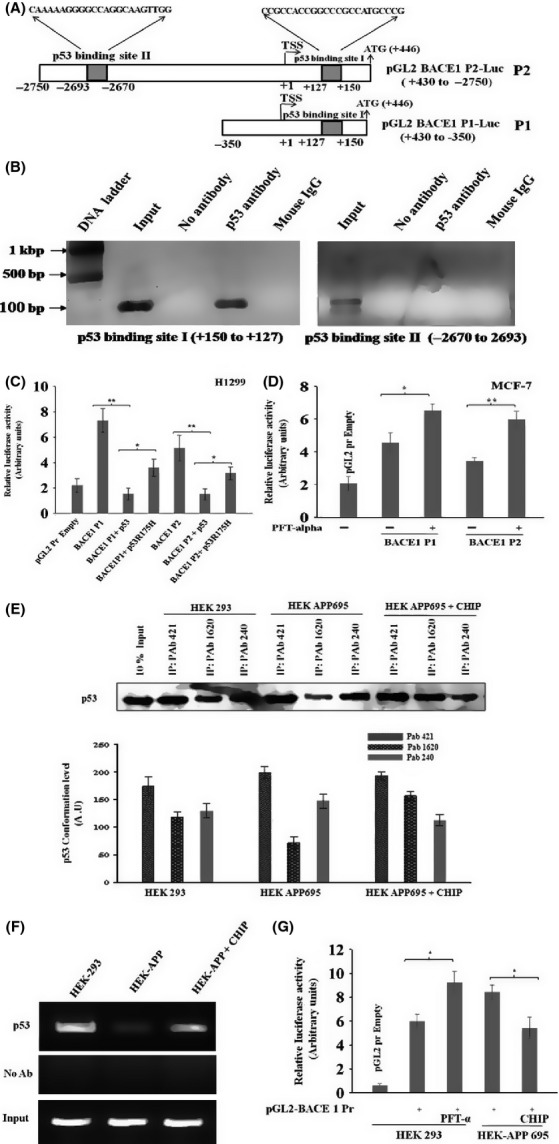
p53 selectively binds to BACE1 promoter. (A). Schematic representation of BACE1 promoter. Boxes shown are the two potential p53 binding sites in the BACE1 promoter, identified by MatInspector software. (B) ChIP assay shows that p53 binding site on BACE1 promoter. H1299 cells were transfected with p53 expression plasmid. Cells were fixed and DNA was precipitated with anti-p53 antibodies followed by PCR amplification. (C) Luciferase reporter plasmids carrying the BACE1 promoter regions were co-transfected with wild-type p53 and mutant p53 into H1299 cells. (D) The p53 inhibitor PFT-α prevents the repression of BACE1 transcriptional activity in MCF-7 (p53+/+) cells. Cells were transfected with plasmid carrying the BACE1 promoter region and treated with PFT-α (15 μm, 6 h) to inhibit endogenous p53. (E–F) Downregulation of BACE1 is due to CHIP-mediated p53 stabilization. (E) HEK-APP stable cells were transfected with CHIP expression plasmids. p53 was immunoprecipitated with conformation-specific antibodies PAb 1620 (wild-type) and PAb 240 (mutant) to analyze the conformational state of p53 followed by Western blotting with anti-p53 (FL-393) antibodies. (F) ChIP assay were performed with anti-p53 antibodies (FL-293) on HEK 293 and HEK-APP cells. PCRs were performed on the immunoprecipitated DNA samples using specific primers for the BACE1 promoter. A sample representing linear amplification of the total input chromatin (Input) was included as control. Additional controls included immunoprecipitation performed with nonspecific immunoglobulins (no Ab). (G) Luciferase reporter plasmids carrying the BACE1 promoter regions were transfected alone or with CHIP in to HEK 293 and HEK-APP cells. Cells were treated with PFT-α (15 μm) for next 6 h. Relative luciferase activity was determined in triplicate, and β-galactosidase is used for normalization of transfection efficiency. Data represent the mean ± SE from three independent experiments. Statistical analysis were performed by one-group t-test for the significance at the *=P < .005 and **=P < 0.001.
CHIP-mediated p53 stabilization downregulates BACE1 gene promoter
Previous studies have reported that HEK-APP stable cells have high levels of oxidative stress markers and unfolded p53 conformation, which may be due to its nitration at tyrosine residues. It is assumed that unfolded p53 is responsible for the loss of its transcriptional activity and reduced sensitivity to various cytotoxic insults (Uberti et al., 2007; Buizza et al., 2013). The expression level of CHIP increases significantly in cells upon exposure to various stresses and this could be an adaptive response of the cell to deal with the excess burden of misfolded protein (Dikshit & Jana, 2007). CHIP also protects folded p53 conformation and restores its DNA-binding and transcription activity (Tripathi et al., 2007). To investigate whether CHIP can stabilize the folded p53 conformation and restores its transcriptional activity in HEK-APP stable cells, the effect of CHIP on p53 conformation was analyzed. For this, HEK-APP cells were transfected with CHIP expression plasmid and cell lysates were immunoprecipitated using two p53 conformation-specific antibodies, PAb1620 (folded p53) and PAb240 (unfolded p53). The level of unfolded p53 conformation is high (PAb 240) in HEK-APP cells (Fig.5E) as compared to mock HEK 293 cells. However, when HEK-APP cells were transfected with CHIP, the folded conformation (PAb 1620) of p53 was restored and at the same time, a decrease in the expression of unfolded p53 was observed. This result clearly demonstrates that CHIP protects folded p53 conformation, which was getting unfolded due to APP metabolism in HEK-APP cell. Further, we investigated whether p53 binding upon target promoters is somehow compromised in HEK-APP cells. As shown in Fig.5F, ChIP assay shows that the p53 recruitment onto BACE1 promoter was present in HEK 293 cell, whereas it was reduced in HEK-APP cells. Of note, upon transfection of CHIP to HEK-APP restored p53 binding activity to DNA, likely counteracting the misfolding of p53 confirmation and thus suggesting that CHIP was able to restored the DNA-binding activity of p53. Next, we assessed the transcription activity of BACE1 in HEK-APP cells. We transfected the plasmid carrying BACE1 promoter into HEK-APP and HEK 293 cells, and transcriptional activity of p53 was analyzed. As it was expected, in mock HEK 293 cells, inactivation of p53 by treating with PFT-α increased BACE1 promoter activity as compared to untreated cells (Fig.5G). But, in HEK-APP stable cells, BACE1 promoter activity is increased corresponding to mock HEK 293 cells that were treated with PFT-α. However, exogenous expression of CHIP in HEK-APP cells decreased BACE1 promoter activity (Fig.5G). This result shows that CHIP protects p53 transcriptional activity in HEK-APP cells and represses BACE1 promoter activity.
Discussion
BACE1 plays a central role in the AD pathogenesis by processing APP to Aβ (Cai et al., 2001). Therefore, understanding the mechanism of regulation of APP and BACE1 is critical for designing therapeutic strategies for AD. In this study, we have shown that BACE1 is a new substrate of CHIP which binds to CHIP’s U-box domain and CHIP promotes BACE1 destabilization through UPS by promoting its ubiquitination (Fig.6). CHIP and HSPs were earlier shown to interact with β-APP in a proteasome-dependent manner and influence Aβ metabolism (Kumar et al., 2007). We have also demonstrated the functional consequence of BACE1 regulation by CHIP on APP processing in both neurons and HEK-APP stable cells in which overexpression of CHIP reduced BACE1 activity through decreased β-cleavage product (CTFβ-99) and Aβ generation. Recently, it was reported that Aβ induces BACE1 expression in primary astrocytes as well as in human astrocytoma cell line (Piccini et al., 2012; Tan & Evin, 2012). One might thus presume that an increased level of BACE1 during AD pathogenesis could be due to decreased expression of CHIP, which results in CHIP-mediated BACE1 degradation.
Fig 6.
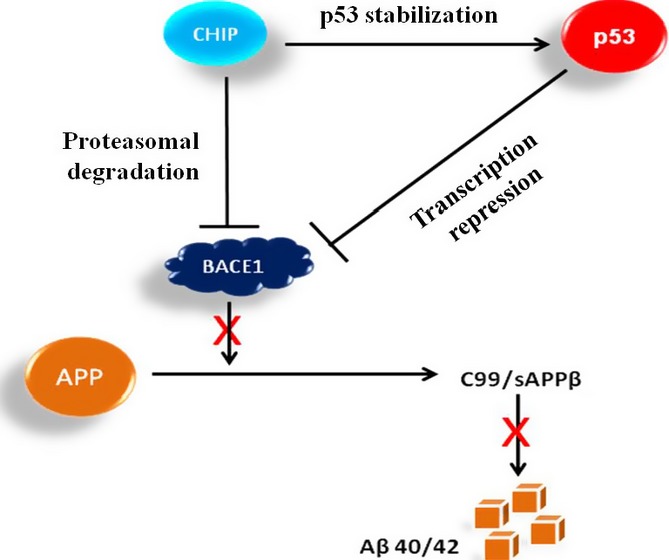
CHIP-mediated p53 stabilization prevents APP degradation. Model proposing that a CHIP–p53–BACE1 loop might exist in AD.
An impairment of proteasome activity was earlier reported in patients with AD which could be a possible reason for the elevation of BACE1 during AD pathogenesis (Upadhya & Hegde, 2007). Moreover, another E3-ligase complex SFCFbx2 that degrades BACE1 through proteasome was found to be reduced in patients with AD and AD mouse model (Gong et al., 2010). Thus, there might exist an inverse relation between CHIP and BACE1. Recently, it was also reported that mutant ubiquitin B (UBB+1) was found in the brain of patients with AD (Fischer et al., 2003), thus causing attenuation of proteasome activity along with reduced BACE1 degradation (Zhang et al., 2010). Although interaction of CHIP with Hsp70/90 helps in recognizing new substrates and plays an important role in the client protein regulation, CHIP physically interacts with substrates through either its TPR or U-box domain. Our results show that BACE1 interact with CHIP’s U-box domain and this interaction is independent of molecular chaperones Hsp70/90. In a similar manner, highly charged region and U-box domain of CHIP interacts with interferon regulatory factor-1 (IRF-1), indicating no role of chaperones in this interaction (Narayan et al., 2011). Ubiquitination and degradation of Tal1/SCL are induced by notch signaling and depend on Skp2 and U-box domain (Nie et al., 2008). By contrast, CHIP interacts with RunX1, a member of the Runt transcription factor, through its TPR and charged domain and with interferon regulatory factor-1 (IRF-1) (Shang et al., 2009).
During progression of AD, in the brain of patients with AD, oxidative stress markers associate with p53, leading it into unfolded conformation and impaired transcriptional activity (Lanni et al., 2007; Cenini et al., 2008; Zhou & Jia, 2010). These unfolded p53 increases APP metabolism and Aβ load and vice versa (Uberti et al., 2007; Cai & Ratka, 2011; Buizza et al., 2013). Level of CHIP mRNA was shown to be significantly increased in cells exposed to oxidative stress, and it could be an adaptive response to deal with the burden of misfolded proteins (Dikshit & Jana, 2007). By contrast, selective accumulation of Aβ42 with a reduction in CHIP expression markedly accelerates the progression of tau pathology, which is rescued by restoring CHIP level (Oddo et al., 2008). In HEK-APP stable cells, Aβ changes conformation of p53 to unfolded state through degradation of homeodomain interacting protein kinase 2 (HIPK2) (Lanni et al., 2010). We had earlier shown that CHIP preferentially binds to the unfolded p53 conformation and restores its DNA-binding activity, which is independent of Hsp70 (Tripathi et al., 2007). But, when CHIP was expressed in HEK-APP cell, the DNA-binding activity of p53 was restored due to chaperone activity of CHIP. This suggests that CHIP might be responsible for partially restoring p53 wild-type conformation, thus enhancing DBS binding upon BACE1 promoter and promoting transcriptional repression.
Furthermore, oxidative stress elevates transcription of BACE1 gene through redox-sensitive transcription factors (Christensen et al., 2004; Sastre et al., 2006; Sun et al., 2006; Bourne et al., 2007; Zhang et al., 2007). Our results show that an overexpression of p53 suppresses BACE1 expression via its binding upon BACE1 promoter; the suppression of BACE1 expression by p53 is one of pathways by which p53 might inhibit APP processing and Aβ generation during AD pathogenesis. Interestingly, BACE1 promoter activity is increased in HEK-APP stable cells as compared to HEK 293 cells. Our finding could be correlated by an earlier study which showed that an impairment of p53 signaling pathway and G1/S check point dysfunction was found in patients with AD (Uberti et al., 2002; Zhou & Jia, 2010).
In summary, our study shows that BACE1 is a p53 downstream gene and can be downregulated at both transcriptional and post-translational level by CHIP-mediated p53 activation and CHIP-mediated degradation. We purpose that an inverse relation between CHIP and BACE1 through a possible p53–CHIP–BACE1 feedback loop (Fig. 6) might be an indicator during AD pathogenesis; the stabilization of p53 as well as of APP seems to be linked to AD pathogenesis. This study might bring new direction in developing new therapeutic protocols during AD pathogenesis through novel intervention through CHIP–p53–BACE1 loop.
Experimental procedures
Cell culture, antibodies, and transfection assay
For luciferase assay, we generated reporter constructs of BACE1 promoter. About 3.2-kb fragment of the 5′-flanking region (+430 to −2750) of the BACE1 gene was amplified from the genomic DNA of H1299 cells by PCR using Phusion polymerase. This amplified product was further used as a template for amplification of deletion constructs and cloned into pGL2 promoter vector at NheI/BglII site. The 3XFLAG-BACE1 plasmid was generated using specific primers to amplify BACE1 insert and ligated into 3XFLAG-CMV-10 vector at HindIII/BamHI restriction site. HA-p53, His-Ub, myc-CHIP, myc-CHIPΔTPR, myc-CHIPΔUbox, myc-CHIPK30A, and myc-CHIPH260Q were available in our laboratory. H1299, MCF-7, HEK 293, and SH-SY5Y cell lines were obtained from National Centre for Cell Sciences Pune (India). The HEK 293 cells were stably transfected with APP (Uberti et al., 2007). The medium used for these cell lines was Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum. Anti-p53 PAb DO1, FL-393 antibodies (nonconformational), anti-p53 PAb1620 antibodies (wild-type), anti-p53 PAb240 antibodies (mutant conformation), anti-myc antibodies, anti-BACE1 antibodies (Z-183 and M-83), and anti-APP antibodies (c-terminus) were purchased from Santa Cruz, and anti-flag antibodies were from Sigma. All transfections were carried out using Lepofectamine LTX (Invitrogen, USA), except in primary cortical neurons using Lipofectamine Messenger MAX (Invitrogen) according to manufacturer’s instructions. Luciferase assay was performed using luciferase reporter gene assay kit (Promega, USA) according to manufacturer’s instructions. One day before transfection, 2 × 105 cells were seeded in 12-well culture plates, and after 30 h of transfection, the assay was performed. Cells were washed with cold PBS and lysed. Equal volume of luciferin solution and lysate was mixed and luminescence was measured immediately in a luminometer. The experiment was performed in triplicate and data were expressed as mean ± SD.
Primary cortical neuron culture
Primary cultures of embryonic rat cortical neurons were prepared as described (Kao et al., 2004). In brief, dissociated embryonic neurons from pregnant rat were plated onto poly-D-lysine/laminin-coated 35-mm plates and cultured at a density of 4 × 105 cells/plates in a neurobasal medium (Invitrogen) supplemented with B27, L-glutamine, and 1% penicillin–streptomycin sulfate. The neurons were transfected with myc-CHIP using Lipofectamine Messenger MAX. The medium was changed after transfection, and after 48 hrs of transfection, cell lysates were prepared for detecting C99/C89 generation using Western blot analysis.
Western blot
After protein samples were resolved on SDS-PAGE, proteins were transferred on to the nitrocellulose membrane by wet blot system (Bio-Rad, USA). Post-transfer, membrane was transferred to blocking buffer (PBS, 5% skimmed milk and 0.1% Tween-20) for 1 h at room temperature. After incubation, blot was washed three times (5 min each) with washing buffer (PBS containing 0.1% Tween-20). Subsequently, the membrane was incubated with primary antibodies diluted in PBS with 1% BSA and 0.05% Tween-20 for 1–2 h followed by washing thrice (5 min each) with washing buffer. The membrane was then incubated with secondary antibodies conjugated to poly-horse radish peroxidase (HRP) for another 1 h. After subsequent washing, blot was developed with ECL™ (Millipore, Germany) Western blotting detection reagents. We used GeneSnap/GeneTools software (Syngene, USA) to quantify band density of the blot.
Immunoprecipitation (IP)
Cells were harvested and lysed in NP-40 buffer (50 mm Tris–HCl, pH 7.4, 150 mm NaCl, 1% NP-40, supplemented with cocktail protease inhibitor) at 4 °C for 20 min. After centrifugation, supernatant was transferred to fresh tubes. Approx 10% of whole-cell lysate was used as input. About 0.5–1 mg of whole-cell lysate was incubated with 1.0 μg of anti-myc antibodies and incubated for 2–3 h at 4 °C. Twenty-five microliters of protein A agarose (50%) was added to the lysate and further incubated at 4 °C for 2 h. Washing was carried out 5 times with NP-40 buffer. Immunocomplex was released by the addition of SDS loading dye, boiled, and analyzed by Western blotting.
In vivo ubiquitination assay
To precipitate ubiquitinated proteins, cells were lysed in 1 mL of buffer I (8 m Urea, 0.1 m Na2HPO4/NaH2PO4 pH 8.0, 0.01 m Tris–HCl, pH 8.0, 150 mm NaCl, 0.2% Triton X-100, 20 mm imidazole, and 10 mm β-mercaptoethanol) for 15 m followed by sonication. Fifty microliters of Ni2+-NTA-agarose beads (50%) (Qiagen, Germany) was added to cell lysates and incubated at room temperature (RT) for 2–3 h with continuous rotation. The beads were pelleted and supernatant was discarded. Beads were then washed two times with buffer I for 5 m in each step at RT followed by washing two times with buffer II (8 m Urea, 0.1 m Na2HPO4/NaH2PO4 pH 6.3, 150 mm NaCl, 0.2% Triton X-100, 20 mm imidazole, and 10 mm β-mercaptoethanol). His-tagged ubiquitinated proteins were eluted by incubating the beads in 50 μl of elution buffer (200 mm imidazole, 0.15 m Tris–HCl, pH 6.7, 30% glycerol, 0.72 m β-mercaptoethanol, 0 5% SDS) for 20 m at RT and was analyzed by Western blotting.
In vitro ubiquitination assay
The ubiquitination assay of BACE1 was carried out using an ubiquitination kit from Enzo Life Science. For this, we have purified recombinant GST-BACE1 and His-CHIP from E. coli and ubiquitination reaction was carried out with 1.5 μL E1, 3 μL E2 (UbcH5a), 1.5 μL Mg-ATP buffer, 3 μL 10X ubiquitination buffer, 1.5 μL Ub, 2 μg GST-BACE1, 5 μg of His-CHIP, and H2O in a 30-μL volume at 30 °C for 1 h. The ubiquitinated BACE1 proteins were detected by Western blotting using the anti-BACE1 antibody.
Chromatin immunoprecipitation (ChIP) assay
To fix the DNA–protein complex, cells were cross-linked with 1% formaldehyde at 37 °C for 10 m and then stopped by the adding 125 mm glycine. Cells was resuspended in 0.6 mL of lysis buffer (1% SDS, 10 mm EDTA, 50 mm Tris–HCl, pH 8.0, and cocktail protease inhibitor) and incubated for 15 m on ice. Sonication was carried out for 2 m (pulse on 5 s and pulse off 5 s) followed by centrifugation. The supernatant was diluted in dilution buffer (1% Triton X-100, 2 mm EDTA, 150 mm NaCl, 20 mm Tris–HCl, pH 8.0), and immunoprecipitation was carried out over night with p53 antibodies at 4 °C. Twenty-five microliters of protein A–Sepharose beads (saturated with salmon sperm DNA) was added and incubated on a rotary shaker for 2 h at 4 °C. Agarose beads were pelleted and washed sequentially for 10 m each in TSE I (0.1% SDS, 1% Triton X-100, 2.0 mm EDTA, 20 mm Tris–HCl, pH 8.0, and 150 mm NaCl), TSE II (0.1% SDS, 1% Triton X-100, 2.0 mm EDTA, 20 mm Tris–HCl, pH 8.0, and 500 mm NaCl) and buffer III (0.25 m LiCl, 1% NP-40, 1% Na deoxycholate, 1 mm EDTA, 10 mm Tris–HCl, pH 8.0). Beads were then washed once with TE buffer. The immunocomplex was eluted twice with 150 μl of elution buffer (1% SDS and 0.1 m NaHCO3) after 15 m incubation at RT. De-cross-linking was performed at 65 °C in a water bath. The DNA was extracted and PCR was carried out with appropriate primers. Equal amount of chromatin solution was precipitated with no antibody as a negative control.
Real-time PCR
Quantitative PCRs were performed on an ABI 7000, using SYBR-Green Master Mix (Applied Biosystems), ABI PRISM® 96-well optical reaction plates and ABI PRISM™ optical adhesive plate sealers. All reactions were completed in triplicate. Each 20 μL PCR contained 0.02–0.1 μg cDNA, 2X SYBR-Green Master Mix, and primers diluted to a final concentration of 0.5 μm. The following cycling parameters were used: 50 °C for 10 min, then 95 °C for 10 min. This was followed by 40 cycles of 95 °C for 10 s and a combined annealing/extension temperature of 60 °C for 2 m. During each cycle of the PCR, the fluorescence emitted by the binding of SYBR-Green dye to the double-stranded DNA produced in the reaction was measured. To confirm the specificity of the reactions, dissociation curves were constructed for each primer pair at 0.1 °C intervals between the temperatures of 60 °C and 95 °C.
Statistical analysis
Comparisons of the difference in mean of two groups (±SEM) were carried out using the Student’s t-test. Comparisons were two-tailed. All statistical tests were carried out using Sigma Plot, version 11 statistics software. P < 0.05 was accepted as significant. Mean values (±SEM) for each experiment with more than two groups were calculated using one-way ANOVA.
Acknowledgments
We thank to N. Khan, DIPAS, New Delhi for rat primary cortical neurons, BD. Strooper for BACE1-Flag vector, and N. Hooper for HA-APP plasmid. AK Singh is a recipient of research fellowship from Department of Biotechnology (DBT) and Indian council of medical research (ICMR), New Delhi.
Author contributions
AKS and UP conceived the project; AKS performed the experiment; AKS and UP analyzed the data and wrote the manuscript.
Funding
This study was supported by University Grant Commission (UPOE), Govt of India.
Conflict of interest
None declared.
References
- Ahmed SF, Deb S, Paul I, Chatterjee A, Mandal T, Chatterjee U, Ghosh MK. The chaperone-assisted E3 ligase C terminus of Hsc70-interacting protein (CHIP) targets PTEN for proteasomal degradation. J. Biol. Chem. 2012;287:15996–16006. doi: 10.1074/jbc.M111.321083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida CG, Takahashi RH, Gouras GK. Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J. Nerosci. 2006;26:4277–4288. doi: 10.1523/JNEUROSCI.5078-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne KZ, Ferrari DC, Lange-dohna C, Roßner S, Wood TG, Perez-polo JR. Differential regulation of BACE1 promoter activity by nuclear factor-kappaB in neurons and Glia upon exposure to beta-amyloid peptides. J. Neurosci. Res. 2007;85:1194–1204. doi: 10.1002/jnr.21252. [DOI] [PubMed] [Google Scholar]
- Buizza L, Prandelli C, Bonini SA, Delbarba A, Cenini G, Lanni C, Buoso E, Racchi M, Govoni S, Memo M, Uberti D. Conformational altered p53 affects neuronal function: relevance for the response to toxic insult and growth-associated protein 43 expression. Cell Death Dis. 2013;4:1–10. doi: 10.1038/cddis.2013.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Ratka BZA. Oxidative stress and beta-amyloid protein in Alzheimer’s disease. NeuroMol. Med. 2011;13:223–250. doi: 10.1007/s12017-011-8155-9. [DOI] [PubMed] [Google Scholar]
- Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC. BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat. Neurosci. 2001;4:233–234. doi: 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics. 2005;21:2933–2942. doi: 10.1093/bioinformatics/bti473. [DOI] [PubMed] [Google Scholar]
- Cenini G, Sultana R, Memo M, Butterfield DA. Elevated level of pro-apoptotic p53 and its oxidative modification by the lipid peroxidation product, HNE, in brain from subjects with amnestic mild cognitive impairment and Alzheimer’s. J. Cell Mol. Med. 2008;12:987–994. doi: 10.1111/j.1582-4934.2008.00163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HJ, Jin SM, Son SM, Kim YW, Hwang JY, Hong HS, Mook-Jung I. Constitutive JAK2/STAT1 activation regulates endogenous BACE1 expression in neurons. Biochem. Biophys. Res. Commun. 2009;386:175–180. doi: 10.1016/j.bbrc.2009.06.006. [DOI] [PubMed] [Google Scholar]
- Christensen MA, Zhou W, Qing H, Lehman A, Philipsen S, Song W. Transcriptional regulation of BACE1, the beta-amyloid precursor protein β – secretase, by Sp1. Mol. Cell. Biol. 2004;24:865–874. doi: 10.1128/MCB.24.2.865-874.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun AC, Jin D. Transcriptional regulation of mitotic checkpoint gene MAD1 by p53 transcriptional regulation of mitotic checkpoint gene MAD1 by p53. J. Biol. Chem. 2003;278:37439–37450. doi: 10.1074/jbc.M307185200. [DOI] [PubMed] [Google Scholar]
- Dikshit P, Jana NR. The co-chaperone CHIP is induced in various stresses and confers protection to cells. Biochem. Biophys. Res. Commun. 2007;357:761–765. doi: 10.1016/j.bbrc.2007.04.018. [DOI] [PubMed] [Google Scholar]
- Esser C, Scheffner M, Höhfeld J. The chaperone-associated ubiquitin ligase CHIP is able to target p53 for proteasomal degradation. J. Biol. Chem. 2005;280:27443–27448. doi: 10.1074/jbc.M501574200. [DOI] [PubMed] [Google Scholar]
- Fischer DF, De Vos RI, Van Dijk R, De Vrij FMS. Disease-specific accumulation of mutant ubiquitin as a marker for proteasomal dysfunction in the brain. FASEB J. 2003;17:2014–2024. doi: 10.1096/fj.03-0205com. [DOI] [PubMed] [Google Scholar]
- Gambino V, De Michele G, Venezia O, Migliaccio P, Dall’Olio V, Bernard L, Minardi SP, Della Fazia MA, Bartoli D, Servillo G, Alcalay M, Luzi L, Giorgio M, Scrable H, Pelicci PG, Migliaccio E. Oxidative stress activates a specific p53 transcriptional response that regulates cellular senescence and aging. Aging Cell. 2013;12:435–445. doi: 10.1111/acel.12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatz M, Reynolds C, Fratiglioni L, Johansson B, Mortimer J, Berg S, Pedersen NL. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry. 2006;63:168–174. doi: 10.1001/archpsyc.63.2.168. [DOI] [PubMed] [Google Scholar]
- Gong B, Chen F, Pan Y, Arrieta-Cruz I, Yoshida Y, Haroutunian V, Pasinetti GM. SCFFbx2-E3-ligase-mediated degradation of BACE1 attenuates Alzheimer’s disease amyloidosis and improves synaptic function. Aging Cell. 2010;9:1018–1031. doi: 10.1111/j.1474-9726.2010.00632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao S, Krichevsky AM, Kosik S, Tsai L. Mechanisms of signal transduction: BACE1 suppression by RNA interference in primary cortical neurons BACE1 suppression by RNA interference in primary cortical neurons. J. Biol. Chem. 2004;279:1942–1949. doi: 10.1074/jbc.M309219200. [DOI] [PubMed] [Google Scholar]
- Kimura R, Devi L, Ohno M. Partial reduction of BACE1 improves synaptic plasticity, recent and remote memories in Alzheimer’s disease transgenic mice. J. Neurochem. 2010;113:248–261. doi: 10.1111/j.1471-4159.2010.06608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Ambasta RK, Veereshwarayya V, Rosen KM, Kosik KS, Band H, Mestril R, Patterson C, Querfurth HW. CHIP and HSPs interact with β-APP in a proteasome-dependent manner and influence Aβ metabolism. Hum. Mol. Genet. 2007;16:848–864. doi: 10.1093/hmg/ddm030. [DOI] [PubMed] [Google Scholar]
- Lanni C, Uberti D, Racchi M, Govoni S, Memo M. Unfolded p53: a potential biomarker for Alzheimer’s disease. J. Alzheimers Dis. 2007;12:93–99. doi: 10.3233/jad-2007-12109. [DOI] [PubMed] [Google Scholar]
- Lanni C, Racchi M, Mazzini G, Ranzenigo A, Polotti R, Sinforiani E, Olivari L, Barcikowska M, Styczynska M, Kuznicki J, Szybinska A, Govoni S, Memo M, Uberti D. Conformationally altered p53: a novel Alzheimer’s disease marker? Mol. Psychiatry. 2008;13:641–647. doi: 10.1038/sj.mp.4002060. [DOI] [PubMed] [Google Scholar]
- Lanni C, Nardinocchi L, Puca R, Stanga S, Uberti D, Memo M, Govoni S, D’Orazi G, Racchi M. Homeodomain interacting protein kinase 2: a target for Alzheimer’s beta-amyloid leading to misfolded p53 and inappropriate cell survival. PLoS ONE. 2010;5:e0171. doi: 10.1371/journal.pone.0010171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Khadka P, Baek SH, Chung IK. CHIP promotes human telomerase reverse transcriptase degradation and negatively regulates telomerase activity. J. Biol. Chem. 2010;285:42033–42045. doi: 10.1074/jbc.M110.149831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhou W, Tong Y, He G, Song W. Control of APP processing and Aβ generation level by BACE1 enzymatic activity and transcription. FASEB J. 2006;20:285–292. doi: 10.1096/fj.05-4986com. [DOI] [PubMed] [Google Scholar]
- Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, Denis P, Fan W, Kha H, Zhang J, Gong Y, Martin L, Louis JC, Yan Q, Richards WG, Citron M, Vassar R. Mice deficient in BACE1, the Alzheimer’s beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat. Neurosci. 2001;4:231–232. doi: 10.1038/85059. [DOI] [PubMed] [Google Scholar]
- Meacham GC, Patterson C, Zhang W, Younger JM, Cyr DM. The Hsc70 co- chaperone CHIP targets immature CFTR for proteasomal degradation. Nat. Cell Biol. 2001;3:100–105. doi: 10.1038/35050509. [DOI] [PubMed] [Google Scholar]
- Murphy MP, LeVine H. Alzheimer’s disease and the β-Amyloid Peptide. J. Alzheimers Dis. 2010;19:311–323. doi: 10.3233/JAD-2010-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan V, Pion E, Landré V, Müller P, Ball KL. Docking-dependent ubiquitination of the interferon regulatory factor-1 tumor suppressor protein by the ubiquitin ligase CHIP. J. Biol. Chem. 2011;286:607–619. doi: 10.1074/jbc.M110.153122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie L, Wu H, Sun X-H. Ubiquitination and degradation of Tal1/SCL are induced by notch signaling and depend on Skp2 and CHIP. J. Biol. Chem. 2008;283:684–692. doi: 10.1074/jbc.M704981200. [DOI] [PubMed] [Google Scholar]
- Nowak K, Lange-Dohna C, Zeitschel U, Günther A, Lüscher B, Robitzki A, Perez-Polo R, Rossner S. The transcription factor Yin Yang 1 is an activator of BACE1 expression. J. Neurochem. 2006;96:1696–1707. doi: 10.1111/j.1471-4159.2006.03692.x. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Tseng B, Cheng D, Vasilevko V, David H, Cribbs DH, LaFerla FM. Blocking Aβ42 accumulation delays the onset and progression of tau pathology via the CHIP: a mechanistic link between Aβ and tau pathology. J. Neurosci. 2008;28:12163–12175. doi: 10.1523/JNEUROSCI.2464-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrucelli L, Dickson D, Kehoe K, Taylor J, Snyder H, Grover A, De Lucia M, McGowan E, Lewis J, Prihar G, Kim J, Dillmann WH, Browne SE, Hall A, Voellmy R, Tsuboi Y, Dawson TM, Wolozin B, Hardy J, Hutton M. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum. Mol. Genet. 2004;13:703–714. doi: 10.1093/hmg/ddh083. [DOI] [PubMed] [Google Scholar]
- Piccini A, Borghi R, Guglielmotto M, Tamagno E, Cirmena G, Garuti A, Pollero V, Cammarata S, Fornaro M, Messa M, Colombo L, Salmona M, Perry G, Tabaton M. Beta-amyloid 1-42 induces physiological transcriptional regulation of BACE1. J. Neurochem. 2012;122:1023–1031. doi: 10.1111/j.1471-4159.2012.07834.x. [DOI] [PubMed] [Google Scholar]
- Qing H, Zhou W, Christensen MA, Sun X, Tong Y, Song W. Degradation of BACE1 by the ubiquitin-proteasome pathway. FASEB J. 2004;18:1571–1583. doi: 10.1096/fj.04-1994fje. [DOI] [PubMed] [Google Scholar]
- Riederer BM, Leuba G, Vernay A, Riederer IM. The role of the ubiquitin proteasome system in Alzheimer’s disease. Exp. Biol. Med. (Maywood) 2011;236:268–276. doi: 10.1258/ebm.2010.010327. [DOI] [PubMed] [Google Scholar]
- Sastre M, Dewachter I, Rossner S, Bogdanovic N, Rosen E, Borghgraef P, Evert BO, Dumitrescu-Ozimek L, Thal DR, Landreth G, Walter J, Klockgether T, van Leuven F, Heneka MT. Nonsteroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPARgamma. Proc. Natl Acad. Sci. USA. 2006;103:443–448. doi: 10.1073/pnas.0503839103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Y, Zhao X, Xu X, Xin H, Li X, Zhai Y, He D, Jia B, Chen W, Chang Z. CHIP functions an E3 ubiquitin ligase of Runx1. Biochem. Biophys. Res. Commun. 2009;386:242–246. doi: 10.1016/j.bbrc.2009.06.043. [DOI] [PubMed] [Google Scholar]
- Sun X, He G, Qing H, Zhou W, Dobie F, Cai F, Staufenbiel M, Huang LE, Song W. Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc. Natl Acad. Sci. USA. 2006;103:18727–18732. doi: 10.1073/pnas.0606298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Bromley-Brits K, Song W. Regulation of β-site APP-cleaving enzyme 1 gene expression and its role in Alzheimer’s disease. J. Neurochem. 2012;120S:62–70. doi: 10.1111/j.1471-4159.2011.07515.x. [DOI] [PubMed] [Google Scholar]
- Tamagno E, Bardini P, Obbili A, Vitali A, Borghi R, Zaccheo D, Pronzato MA, Danni O, Smith MA, Perry G, Tabaton M. Oxidative stress increases expression and activity of BACE1 in NT2 neurons. Neurobiol. Dis. 2002;10:279–288. doi: 10.1006/nbdi.2002.0515. [DOI] [PubMed] [Google Scholar]
- Tamagno E, Guglielmotto M, Aragno M, Borghi R, Autelli R, Giliberto L, Muraca G, Danni O, Zhu X, Smith MA, Perry G, Jo DG, Mattson MP, Tabaton M. Oxidative stress activates a positive feedback between the gamma- and beta-secretase cleavages of the beta-amyloid precursor protein. J. Neurochem. 2008;104:683–695. doi: 10.1111/j.1471-4159.2007.05072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Evin G. Beta-site APP-cleaving enzyme-1 trafficking and Alzheimer’s disease pathogenesis. J. Neurochem. 2012;120:869–880. doi: 10.1111/j.1471-4159.2011.07623.x. [DOI] [PubMed] [Google Scholar]
- Tong Y, Zhou W, Fung V, Christensen MA, Qing H, Sun X, Song W. Oxidative stress potentiates BACE1 gene expression and Aβ generation. J. Neural. Transm. 2005;112:455–469. doi: 10.1007/s00702-004-0255-3. [DOI] [PubMed] [Google Scholar]
- Tripathi V, Ali A, Bhat R, Pati U. CHIP chaperones wild type p53 tumor suppressor protein. J. Biol. Chem. 2007;282:28441–28454. doi: 10.1074/jbc.M703698200. [DOI] [PubMed] [Google Scholar]
- Uberti D, Carsana T, Bernardi E, Rodella L, Grigolato P, Lanni C, Racchi M, Govoni S, Memo M. Selective impairment of p53-mediated cell death in fibroblasts from sporadic Alzheimer’s disease patients. J. Cell Sci. 2002;115:3131–3138. doi: 10.1242/jcs.115.15.3131. [DOI] [PubMed] [Google Scholar]
- Uberti D, Lanni C, Carsana T, Francisconi S, Missale C, Racchi M, Govoni S, Memo M. Identification of a mutant-like conformation of p53 in fibroblasts from sporadic Alzheimer’s disease patients. Neurobiol. Aging. 2006;27:1193–1201. doi: 10.1016/j.neurobiolaging.2005.06.013. [DOI] [PubMed] [Google Scholar]
- Uberti D, Cenini G, Olivari L, Ferrari-Toninelli G, Porrello E, Cecchi C, Pensalfini A, Liguri G, Govoni S, Racchi M, Maurizio M. Over-expression of amyloid precursor protein in HEK cells alters p53 conformational state and protects against doxorubicin. J. Neurochem. 2007;103:322–333. doi: 10.1111/j.1471-4159.2007.04757.x. [DOI] [PubMed] [Google Scholar]
- Upadhya SC, Hegde AN. Role of the ubiquitin proteasome system in Alzheimer’s disease. BMC Biochem. 2007;8:S1–S12. doi: 10.1186/1471-2091-8-S1-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL, Beach T, Sue L, Wong P, Price D, Li R, Shen Y. Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat. Med. 2003;9:3–4. doi: 10.1038/nm0103-3. [DOI] [PubMed] [Google Scholar]
- Yu JJ, Thornton K, Guo Y, Kotz H, Reed E. An ERCC1 splicing variant involving the 5′-UTR of the mRNA may have a transcriptional modulatory function. Oncogene. 2001;20:7694–7698. doi: 10.1038/sj.onc.1204977. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhou K, Wang R, Cui J, Lipton SA, Liao FF, Xu H, Zhang YW. Hypoxia-inducible factor 1α (HIF-1 alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation. J. Biol. Chem. 2007;282:10873–10880. doi: 10.1074/jbc.M608856200. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xiong M, Yan RQ, Sun FY. Mutant ubiquitin-mediated beta-secretase stability via activation of caspase-3 is related to beta-amyloid accumulation in ischemic striatum in rats. J. Cereb. Blood Flow Metab. 2010;30:566–675. doi: 10.1038/jcbfm.2009.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Jia J. p53-mediated G(1)/S checkpoint dysfunction in lymphocytes from Alzheimer’s disease patients. Neurosci. Lett. 2010;468:320–325. doi: 10.1016/j.neulet.2009.11.024. [DOI] [PubMed] [Google Scholar]