

Abstract
Since its discovery, diverse functions have been attributed to the G0/G1 switch gene 2 (G0S2), from lipid metabolism to control of cell proliferation. Our group showed for the first time that G0S2 promotes quiescence in hematopoietic stem cells by interacting with and retaining nucleolin around the nucleus. Herein, we report the role of G0S2 in the differentiation and function of CD8+ T cells examined in mice with an embryonic deletion of the G0s2 gene. G0S2 expression in naïve CD8+ T cells decreased immediately after T-cell receptor activation downstream of the MAPK, calcium/calmodulin, PI3K, and mTOR pathways. Surprisingly, G0S2-null naïve CD8+ T cells displayed increased basal and spare respiratory capacity that was not associated with increased mitochondrial biogenesis but with increased phosphorylation of AMPKα. Naïve CD8+ T cells showed increased proliferation in response to in vitro activation and in vivo lymphopenia; however, naïve CD8+ T cells expressing the OT-1 transgene exhibited normal differentiation of naïve cells to effector and memory CD8+ T cells upon infection with Listeria monocytogenes in a wild type or a G0s2-null environment, with increased circulating levels of free fatty acids. Collectively, our results suggest that G0S2 inhibits energy production by oxidative phosphorylation to fine-tune proliferation in homeostatic conditions.
Keywords: G0S2, metabolism, proliferation, CD8+ T cells
INTRODUCTION
Quiescence and proliferation are actively regulated processes that are able to control bursts of naïve CD8+ T cell expansion in response to homeostatic and pathogen-driven stimuli without leading to lymphoproliferative disorders 1. The emerging paradigm is that the modulation of energy metabolism is critical for lymphocyte proliferation, as well as for the transcriptional regulation of cell cycle and lineage determination genes 2, 3. The identification of inhibitors of T cell proliferation is critical for better understanding immune responses, lymphoproliferative disorders, and leukemia.
Naïve CD8+ T cells produced in the thymus circulate in peripheral tissues in a non-proliferative state of quiescence and enter the cell cycle to restore the numbers of T cells during homeostasis or upon encountering activated dendritic cells. A transient lymphopenia induces homeostatic polyclonal proliferation of T cells by increasing the availability of IL-7 and interactions with specific self-pMHC (reviewed in 4). However, pathogens induce the clonal expansion of antigen-specific CD8+ T cells, which is followed by a contraction that aims to reduce the numbers of activated T cells after pathogen clearance, and finally, the establishment of immunological memory. Memory T cells reacquire the properties of naïve T cells, such as quiescence and self-renewal, and remain in a ‘primed’ state pre-charged with cyclins involved in the G1 to S transition, which allows for faster responses during secondary exposures to the same antigen 5. There is emerging evidence that T cell proliferation is modulated by the choice of energy source in addition to the transcriptional activation of genes involved in the exit from quiescence and entry into cell cycle 6, 7. Naïve T cells oxidize pyruvate via oxidative phosphorylation (OXPHOS) to generate ATP, whereas activated T cells switch to glycolysis because this catabolic pathway allows cell growth by producing intermediate metabolites required for the synthesis of macromolecules 3. A balanced metabolism between OXPHOS and oxidation of free fatty acids and glycolysis most likely plays a critical role in the maintenance and release of naïve T cells from a quiescent state.
The G0/G1 switch gene 2 (G0S2) is a small protein first identified in activated lymphocytes with ascribed functions in the lipid metabolism and proliferation of hematopoietic and leukemic cells 8-12. Studies of subcellular localization indicate that G0S2 can be found in the endoplasmic reticulum and mitochondria of hematopoietic cells 10. It is believed that G0S2 exerts these seemingly disparate functions, at least in part, via protein-protein interactions of its hydrophobic domain with different partners: adipose triglyceride lipase 12, nucleolin 10, bcl-2 13, and more recently, F0F1-ATP synthase 14. In addition to its physiological functions, G0S2 has also been associated with tumor suppression in head and neck cancers, squamous lung cancer, and acute myeloid leukemia 15-17. The function of G0S2 in immune cells is largely unknown, even though G0S2 expression was found to be elevated in various inflammatory disorders, such as endometriosis 18, rheumatoid arthritis 19, vasculitis 20, and psoriasis 21.
We recently identified a previously unknown function of G0S2 as an inducer of quiescence in hematopoietic stem cells and chronic myeloid leukemia cells, mediated at least in part by a protein-to-protein interaction with nucleolin in the cytosol 10, 22. In this study, we report the generation of G0s2-null mice to study T cell proliferation. We found that G0s2−/− CD8+ T cells are endowed with higher basal and spare respiratory capacity (SRC) than wild type controls, which was not due to increased mitochondrial mass or membrane potential but rather due to a deregulated activity of AMPK and mTOR pathways. Therefore, G0S2 fine-tunes the exit from quiescence during homeostatic T cell proliferation and in vitro TCR activation. However, loss of G0S2 seems to have a redundant role in antigen-driven proliferation in primary and secondary infection with Listeria monocytogenes. Our study reveals a novel function for G0S2 in the energy metabolism and proliferation of CD8+ T cells.
RESULTS
Expression of G0S2 in CD8+ T cells
In a previous study, we found that resting naïve CD8+ T cells express high levels of G0S2 23. Herein, we show that G0S2 transcripts rapidly decreased in CD8+ T cells activated in vitro with plate-bound anti-CD3 and anti-CD28 (Figure 1a). Furthermore, G0S2 levels inversely correlated with the expression of cyclin E2, used as surrogate marker of cell proliferation, suggesting a potential role in the regulation of cell division. To elucidate which pathway downstream of the TCR leads to suppression of G0S2 transcription, we then activated naïve CD8+ T cells in the absence or presence of the inhibitors PD98059 (MAPK), cyclosporin A (calcium/calcineurin), LY294002 (PI3K), and rapamycin (mTOR). Inhibition of G0S2 expression triggered by TCR activation was prevented by all inhibitors, suggesting that the MAPK, calcium/calcineurin, PI3K, and mTOR pathways are all involved in G0S2 suppression during activation of CD8+ T cells (Figure 1b,c). This observation was consistent with a previous report that cyclosporin A inhibits the expression of G0S2 in human mononuclear cells 24. Based on our findings, we hypothesized that G0S2 may have an inhibitory role in T cell proliferation, and thus its expression needs to be repressed following TCR-mediated activation. Consistent with this model, we found increased reconstitution of T cells in mice transplanted with G0S2-silenced bone marrow cells 10.
Figure 1. G0S2 expression is regulated downstream of TCR signaling pathways.

(a) Kinetics of G0S2 and cyclin E2 transcripts were measured by qPCR in CD8+ T cells activated with plate-bound anti-CD3 and anti-CD28. (b) G0S2 expression in the presence of PD98059 (PD), cyclosporin A (CyA), LY294002 (LY), and rapamycin (Rapa). (c) Diagram depicting regulation of G0S2 expression downstream of TCR signals. Data are representative of three independent experiments.
Generation of G0S2-null mice
For this study, we generated G0s2−/− mice using embryonic stem cells with a targeted deletion of the entire G0s2 gene generated by the insertion of a LacZ cassette (Velocigene) (Figure 2a,b). At the time of manuscript preparation, a publication reported the generation of G0s2−/− mice using a similar approach 25. Mice with homozygous G0s2 deletion are born healthy, although heterozygous females were used for breeding because of the perinatal mortality of G0s2−/− pups born from a homozygous null mother. Consistent with G0S2 inhibition of adipocyte lipolysis 11, 26, the levels of free fatty acids, but not triglycerides, were significantly elevated in G0s2−/− mice kept on a normal chow diet (Figure 2c). We evaluated potential alterations of blood cells because ectopic G0S2 expression lowers the multi-lineage reconstitution of HSCs 10. However, flow cytometric analysis showed no alterations in the distribution of granulocytes, B cells, and T cells in the peripheral blood of G0s2−/− mice monitored up to 8 months of age (Figure 2d,e).
Figure 2. Targeted deletion of the G0s2 gene.

(a) G0s2−/− mice were generated using Velocigene ES cells with deletion of the entire G0s2 gene. (b) Mice were genotyped by PCR using genomic DNA from tail biopsies and primers for the G0S2 hydrophobic domain and LacZ-gene insertion as indicated in (a). (c) Serum was collected from 8-10 week old mice for measurements of triglyceride, glucose, and free fatty acid (FFA) levels (n = 5). (d,e) Flow cytometric analysis of Gr1 and CD11b (granulocytes, monocytes), CD19 (B cells), and CD3 (T cells) content in the peripheral blood of 10-12 week old mice. *, P < 0.05 (two-tailed Student’s t-test).
Naïve CD8+ T cells have increased mitochondrial respiratory capacity
In the last few years, many reports have indicated that the modulation of fatty acid metabolism is critical for CD8+ T cell function 27, 28. Based on the localization of G0S2 in the mitochondria 10, we investigated whether G0S2 modulates energy metabolism in lymphocytes. The oxygen consumption rate (OCR) of activated CD8+ T cells was measured in real time in the presence of mitochondrial inhibitors to block ATP synthesis (oligomycin), uncouple ATP synthesis from the electron transport chain (FCCP) and to block complexes I and III of the electron transport chain (rotenone and antimycin A, respectively). The basal OCR and spare respiratory capacity (SRC) were significantly higher in G0s2−/− CD8+ T cells (Figure 3a,c), suggesting that G0S2 inhibits OXPHOS. In addition, the extracellular acidification rate (ECAR) after the addition of glucose was higher in G0s2−/− CD8+ T cells compared to wild type CD8+ T cells (Figure 3b,c). Because ECAR is predominantly driven by lactic acid production during glycolysis, this finding suggests that G0S2 modulates glycolytic activity during T cell activation to a lower extent than OXPHOS, and therefore, that the increase in SRC is primarily due to OXPHOS (Figure 3c). To further investigate whether G0s2 modulates glycolysis, we examined the expression levels of enzymes involved in glucose metabolism (glycolysis, gluconeogenesis, tricarboxylic acid cycle, pentose phosphate pathway, and glycogen metabolism) both in resting and activated CD8+ T cells. In non-activated G0s2−/− CD8+ T cells, we observed increased levels, greater than 2-fold, in the glycolytic enzymes HK3 (hexokinase 3) and Pklr (pyruvate kinase), which feed the TCA cycle and OXPHOS. On the other hand, we found that the enzyme Fbp1 involved gluconeogenesis was downregulated in activated G0s2−/− CD8+ T cells in addition to Phkg-1 (glycogen metabolism), with no other significant changes of glycolytic enzymes (Figure 3d). Collectively, our findings support a greater role of G0s2 in OXPHOS. Contrary to our finding, a recent report proposed that G0S2 positively regulates OXPHOS in cardiomyocytes via direct interaction with the F0F1-ATP synthase 14. These data indicate that G0S2 likely interacts with proteins involved in OXPHOS in the mitochondria in a cell type-dependent manner.
Figure 3. Increased mitochondrial respiratory capacity in G0s2−/− CD8+ T cells.

(a) Oxygen consumption rates (OCR) were measured in real time in wild type and G0s2−/− CD8+ T cells activated with plate-bound anti-CD3 under basal conditions and after addition of oligomycin, FCCP, and rotenone (n = 4). (b) Extracellular acidification rates (ECAR) were measured as described in (a). (c) Calculated spare respiratory capacity (SRC) and glycolysis. (d) Scatter plot of glucose metabolism qPCR array comparing resting and activated (18 hours) wild type and G0s2−/− CD8+ T cells. (e) Cell growth of activated CD8+ T cells was determined using the colorimetric MTT assay (n = 4). (f) ATP levels of activated CD8+ T cells (n = 4). (g) Content of NAD and NADH in activated CD8+ T cells (n = 3). (h) Relative Cpt1a mRNA expression was determined by qPCR (n = 4). *, P < 0.05; **, P < 0.01 (two-tailed Student’s t-test).
To evaluate whether the improved metabolic fitness endowed G0s2−/− CD8+ T cells with a proliferative advantage, we measured the proliferation of CD8+ T cells cultured in similar conditions to those listed above for the determination of oxygen consumption. The MTT colorimetric assay revealed increased proliferation of G0s2−/− CD8+ T cells (Figure 3e). In addition, we found increased ATP production in G0s2−/− CD8+ T cells compared to wild type controls, correlating with the increased OXPHOS (Figure 3f). Because the oxidation of pyruvate in the tricarboxylic acid cycle (TCA) generates NADH, the quantification of NAD+ and NADH demonstrated an increase in the NAD/NADH ratio consistent with augmented OXPHOS (Figure 3g). Finally, no significant differences were found in the transcript levels of the carnitine palmitoyltransferase 1 (Cpt1a) in wild type and G0s2−/− CD8+ T cells (Figure 3h). Since Cpt1a transports fatty acids from the cytosol into the mitochondria for fatty acid oxidation, this data suggests no changes in the usage of fatty acids for energy generation in the absence of G0s2.
To correlate G0S2 function with mitochondrial metabolism, we examined the co-localization of COXIV and G0S2 in CD8+ T cells. For this experiment, we overexpressed V5-tagged G0S2 by retroviral transduction and transplantation of bone marrow cells; then, CD8+ T cells were isolated from the spleen after hematological reconstitution of the ablated mice. Immunofluorescence analysis of COX IV and G0S2 in resting CD8+ T cells showed mitochondrial co-localization similar to that observed in hematopoietic stem cells (Figure 4a) 10. Next, we determined the mitochondrial content because an increased respiratory capacity could be caused by increased mitochondrial biogenesis. Flow cytometric detection of mitochondria was carried out in resting and activated CD8+ T cells (plate-bound anti-CD3 and anti-CD28) using MitoTracker. As expected, T cell activation led to a clear increase in mitochondrial mass; however, the loss of G0S2 did not affect the mitochondrial mass in CD8+ T cells (Figure 4b). To further demonstrate that there was no change in mitochondrial biogenesis, we quantified mitochondrial and nuclear DNA by qPCR. The mDNA/nDNA ratio in G0s2−/− CD8+ T cells was similar to that in control cells (Figure 4c). Next, we determined the mitochondrial membrane potential (Δψ) by flow cytometry using the lipophilic fluorochrome JC-1 to rule out functional differences. Resting and activated G0s2−/− CD8+ T cells showed similar patterns of mitochondrial membrane potential than wild type CD8+ T cells (Figure 4d). Since apoptosis can be associated with depolarization of mitochondrial membrane potential, we also examined cell death by detection of Annexin V positive cells by flow cytometry and found no significant differences between naïve and activated CD8+ T cells (Figure 4e). Furthermore, intracellular levels of the anti-apoptotic Bcl2 protein in G0s2−/− CD8+ T cells showed normal levels compared to controls (Supplementary Figure 1).
Figure 4. Normal mitochondrial biogenesis in G0s2−/− CD8+ T cells.

(a) Subcellular localization of G0S2 (red), COX IV (green), and DAPI (blue) in CD8+ T cells with retroviral expression of V5-tagged G0S2. (b) Flow cytometric detection of mitochondrial content in resting and anti-CD3/anti-CD28 activated CD8+ T cells using MitoTracker. (c) Mitochondrial DNA (mDNA) and nuclear DNA (nDNA) were determined by qPCR in CD8+ T cells (n = 3). n.s., no statistical significance. (d) Flow cytometric detection of mitochondrial membrane potential using JC-1 dye in resting and activated CD8+ T cells. (e) Detection of apoptotic cells by Annexin V staining in resting and activated CD8+ T cells.
Naïve CD8+ T cells oxidize pyruvate, converted from glucose during glycolysis, via OXPHOS and alternatively via fatty acid oxidation. CD8+ T cells switch their source of energy upon activation from OXPHOS to an anabolic metabolism to allow for the generation of glycolytic byproducts required for the synthesis of biomolecules; in this way, activated T cells can increase cell mass despite the inefficient production of ATP compared to OXPHOS 27-30. This balance between OXPHOS and glycolysis is regulated by the AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) signals activated by the deprivation of intracellular ATP or TCR activation, respectively 31. AMPK is a heterotrimeric complex (α, β, γ subunits) that becomes activated by phosphorylation at threonine 172 in the catalytic subunit AMPKα by LKB1 or CAMKK2 32, which results in the stimulation of fatty acid oxidation to replenish cellular ATP levels 33. Immunoblot analysis showed increased levels of phosphorylated AMPKα in G0s2−/− CD8+ T cells activated in plate-bound anti-CD3 and anti-CD28 for 12, 24, and 30 hours (Figure 5). Interestingly, freshly isolated G0s2−/− CD8+ T cells also exhibited increased levels of phosphorylated AMPKα as early as 12 hours of activation, suggesting a state primed for proliferation (Figure 5). In addition, this activation of AMPKα correlated with increased phosphorylation of its target, acetyl-CoA carboxylase (ACC), which is an enzyme inhibited by phosphorylation that promotes oxidation of fatty acid in mitochondria (Figure 5). G0s2−/− CD8+ T cells also showed increased phosphorylation of S6K, a downstream target of the mTOR complex 1 (TORC1), after 24 and 30 hours of activation (Figure 5). These data indicate that activation of AMPK may precede mTOR signals in G0s2−/− CD8+ T cells, supporting our finding of increased basal OXPHOS and spare respiratory capacity in G0s2−/− CD8+ T cells. It seems as if activated CD8+ T cells retain OXPHOS as source of energy instead of switching to aerobic glycolysis, implying an impaired Warburg effect. Finally, the activation of AMPKα in G0s2−/− CD8+ T cells correlated with accelerated release from quiescence as evidenced by increased levels of cyclin E and phosphorylated retinoblastoma (Figure 5).
Figure 5. Loss of G0S2 deregulates activation of AMPK and mTOR pathways in CD8+ T cells.

CD8+ T cells from wild type and G0s2−/− mice were activated by plate-bound anti-CD23 and anti-CD28 for 12, 24, and 30 hours. Immunoblot analysis of AMPKα (AMP-activated protein kinase α), ACC (Acetyl-CoA carboxylase), S6K (ribosomal protein S6 kinase), Cyclin E, cdk2, phospho-Rb (Ser 780), and tubulin is shown. Data represent three independent experiments.
G0S2 controls in vitro proliferation of CD8+ T cells
The suppression of G0S2 expression upon activation and increased mitochondrial respiratory capacity and levels of cyclin E in G0s2−/− cells suggested that G0S2 might control proliferation in CD8+ T cells. To investigate the role of G0S2 in T cell proliferation, naïve CD44low CD8+ T cells harvested from the spleens of G0s2−/− mice were analyzed for DNA content and nuclear expression of the proliferation marker Ki67 by flow cytometry. Purification of naïve CD8+ T cells yielded greater than 95% purity with an immunophenotype corresponding to naïve cells: CD44lo CD69− CD25lo CD62L+ (Supplementary Figures 2 and 3). Freshly isolated CD44low CD8+ T cells showed a significant increase in the number of proliferating CD8+ T cells, Ki67 positive, in G0s2−/− mice (Figure 6a). In addition, G0s2−/− CD8+ T cells activated for 24 and 30 hours also showed increased percentages of Ki67 positive cells (Figure 6b). Of note, the levels of cyclin E were higher in “resting” G0s2−/− CD8+ T cells (Figure 5), suggesting a proliferative priming to enter the cell cycle in the absence of G0S2. To further confirm increased proliferative capacity using a different approach, T cells were labeled with CFSE and activated in vitro on plates coated with anti-CD3 and in the presence of anti-CD28. Loss of G0S2 showed increased dilution of CFSE dye in CD8+ T cells (Figure 6c), indicative of augmented proliferation, with no significant differences in the activation marker CD25 or the capacity to produce IL-2 (Figure 6d).
Figure 6. G0S2 restricts the proliferation of CD8+ T cells.

(a) Dual detection of the proliferation marker Ki67 and DNA content in naïve CD8+ T cells freshly isolated from the spleen of wild type and G0s2−/− mice. (b) Expression of the proliferation marker Ki67 and DNA content in CD8+ T cells cultured on plates coated with anti-CD3 antibody and anti-CD28 in the media. (c) Cell division of purified CD8+ T cells activated with anti-CD3/anti-CD28 was examined by dilution of the CFSE dye. Corresponding proliferation indexes are plotted as bar graphs (mean ± SD, n=3). (d) Cell surface expression of CD25 and intracellular IL-2 content in wild type and G0s2−/− CD8+ T cells activated in vitro.
We next evaluated the proliferation of CD8+ T cells in vivo by systemic infection with Listeria monocytogenes expressing OVA (Lm-OVA). Naïve CD44low CD8+ T cells from G0s2−/− OT-I and wild-type OT-I mice were adoptively transferred into congenic recipient hosts. The next day, mice were infected with Lm-OVA and the immune response of donor CD8+ T cells was monitored in the peripheral blood by flow cytometry (Figure 7a). Overall, we did not detect significant differences in the kinetics of expansion and contraction despite an increase in G0s2−/− OT-I CD8+ T cells at the peak of the response (Figure 7b). Because G0s2−/− mice displayed increased levels of free fatty acid in circulation, an important substrate for energy production, we transferred wild type OT-I CD8+ T cells into wild type or G0s2−/− recipient mice (Figure 7c). No significant differences were observed in the kinetics of the immune response of wild type CD8+ T cells in recipient mice with targeted deletion of the G0s2 gene. Next, we re-challenged mice with Lm-OVA to test the role of G0S2 in the function of memory CD8+ T cells generated in the primary infection. Again, no differences were observed in the capacity of memory G0s2−/− CD8+ T cells to expand in a recall response (Figure 7d). Finally, we studied the differentiation of CD8+ T cells during infection by measuring the expression of the adhesion molecule CD62L (L-selectin). In naïve T cells, CD62L protein is shed from the cell surface by proteolysis during activation whereas the expression of the CD62L gene is silenced or activated during differentiation of central and effector memory T cells, respectively. Loss of G0S2 did not affect the expression of CD62L throughout the progression from naïve to effector and memory CD8+ T cells (Figure 7e). Altogether, our findings show that G0S2 modulates the proliferation of naive CD8+ T cells in vitro but not following in vivo Lm-OVA infection.
Figure 7. Redundant role of G0S2 in CD8+ T cells during immune response to infection.

(a) Diagram depicting the adoptive transfer approach to study immune response of wild type or knockout CD8+ T cells in wild type or knock out recipient mice. (b) To study the immune response of G0s2−/− CD8+ T cells, wild type B6.SJL (CD45.1+) mice adoptively transferred with either OT-I or OT-I G0s2−/− naïve CD8+ T cells were infected with Listeria monocytogenes-OVA (Lm-OVA) (n = 5). (c) To study the effect of the G0s2−/− environment on the activation of CD8+ T cells, G0s2−/− and wild-type mice adoptively transferred with CD8+ T cells purified from the spleens of OT-I.B6.SJL (CD45.1+) mice were infected with Lm-OVA (n = 5). (d) The recall response was evaluated in mice 30 days after primary infection with Lm-OVA. The percentage of donor (CD45.2+) CD8+ T cells is shown. Primary (1°) and secondary (2°) infections are indicated with arrows. (e) Distribution of CD62L positive cells throughout primary and secondary infection with Lm-OVA. Data represent three independent experiments. *, P < 0.05 , ***, P < 0.001 (two-tailed Student’s t-test).
Loss of G0S2 leads to increased homeostatic proliferation of CD8+ T cells
In addition to TCR-driven proliferation of CD8+ T cells either by TCR crosslinking in vitro or by dendritic cell-mediated activation during infection with intracellular pathogens, naïve CD8+ T cells undergo homeostatic proliferation to maintain the pool of circulating T cells. Likewise, it is possible that the abundance of nutrients and therefore the usage of energy sources may be different at sites of pathogen-driven versus homeostatic T cell activation. To study the homeostatic proliferation of CD8+ T cells, we used the well-established model of lymphopenic mice, in which the proliferation of naïve CD8+ T cells is driven predominantly by weak interactions with self peptide-MHC complexes and availability of IL-7 34. We adoptively transferred CD44low CD8+ T cells purified from wild type or G0s2−/− mice that were labeled with CFSE into lymphopenic mice (Figure 8a). Nine days after adoptive transfer, the recovery of donor G0s2−/− CD8+ T cells in the spleen was greater than that of wild type CD8+ T cells (Figure 8b). Furthermore, the CFSE dilution indicated an increased percentage of G0s2−/− cells undergoing more than 5-6 divisions, which is reflected by a higher proliferation index (Figure 8c). Collectively, our data suggest that G0S2 modulates proliferation of CD8+ T cells in greater extent during homeostasis than in response to infection with an intracellular pathogen.
Figure 8. G0S2 modulates the homeostatic proliferation of CD8+ T cells.

(a) Naïve CD8+ T cells purified from the spleens of wild type and G0s2−/− mice (CD45.2+) were labeled with CFSE dye and injected into B6.SJL mice (CD45.1+) that were made lymphopenic by sub-lethal radiation. Cell division was determined by flow cytometric analysis of CFSE dilution at 9 days. (b) Donor-derived (CD45.2+) CD8+ T cells in the spleens of recipient lymphopenic mice (n = 5) is shown on the top and statistical analysis in the bottom. (c) CFSE profile of CD45.2+ CD8+ T cells is shown on the top and proliferation index in the bottom. Data represent three independent experiments. **, P < 0.01 (two-tailed Student’s t-test).
DISCUSSION
The non-transcriptional mechanisms regulating the maintenance of T cell quiescence and their exit into cell division have not been well studied. Naïve T cells utilize OXPHOS to generate just the amount of ATP that is required during a dormant quiescent state 31. Conversely, antigen-activated T cells switch to an anabolic metabolism, aerobic glycolysis, to satisfy the high demand for energy and biomolecules during clonal expansion. In this study, we show that G0S2 expression is repressed upon T cell receptor activation, and homozygous deletion of the G0s2 gene leads to increased mitochondrial respiratory capacity by increasing AMPK activity without increasing mitochondrial biogenesis. G0S2 controls the proliferation induced by anti-CD3/anti-CD28 TCR activation, and more significantly, by homeostatic signals, but not that induced by dendritic cell-mediated activation of naïve T cells.
We previously reported that the ectopic expression of G0S2 induces quiescence in hematopoietic stem cells 10. In CD8+ T cells, the observed G0S2 repression downstream of TCR signaling suggested a putative role in promoting exit from quiescence. Embryonic deletion of the G0s2 gene was viable, although heterozygous females must be used to generate G0s2−/− live pups, suggesting a defect in milk production, as recently reported by another group 25. Consistent with a role for G0S2 as an inhibitor of adipocyte lipolysis 11, 12, 35, we found elevated levels of free fatty acids in adult G0s2−/− mice maintained on a normal chow diet. In addition, we described a new function for G0S2 in the energy metabolism of CD8+ T cells, primarily in the inhibition of OXPHOS by preventing the activation of AMPK in naïve and activated T cells. In contrast to our findings, G0S2 was recently described as a positive regulator of OXPHOS activity via a direct interaction with the F0F1-ATP synthase in cardiomyocytes 14. This differential regulation of OXPHOS may be caused by different energetic requirements between cardiomyocytes and CD8+ T cells and how they cope with a fluctuating availability of oxygen and nutrients at steady state. Similar to cancer cells, CD8+ T cells undergo a metabolic switch from predominantly OXPHOS in resting cells to aerobic glycolysis in activated effector CD8+ T cells, a process known as the Warburg effect 36. It is possible that G0S2 modulates the transition from OXPHOS to glycolysis in CD8+ T cells by restricting AMPK activation. Strikingly, memory CD8+ T cells showed a similar increase in SRC compared to effector and naïve T cells 37, indicating that G0s2−/− CD8+ T cells are metabolically primed as memory T cells. Although G0S2 can be localized to the mitochondria, we ruled out the possibility that the increased OXPHOS is mediated by increased mitochondrial biogenesis by measuring mitochondrial mass by flow cytometry. Instead, immunoblot analysis revealed increased phosphorylation of AMPKα, which drives cells to OXPHOS, with a moderate increased phosphorylation of the mTOR target S6K at later stages of activation. This deregulated activation could be caused by an increase in the activity of LKB1, accelerated cellular deprivation of ATP, or increased activation of AMPK by calcium downstream of the TCR.
Surprisingly, loss of G0S2 did not result in a greater impact on T cell proliferation in vivo given the increased basal and spare respiratory capacity. It is possible that the requirement for G0S2 in the release of T cell quiescence may depend not only on TCR signals but also on the type and strength of co-stimulation. For example, in vitro TCR crosslinking and CD28 co-stimulation, a potent mitogenic stimulus, led to a significant increase in the proliferation rate of naïve CD8+ T cells. Paradoxically, despite increased AMPK activation, naive OT-I G0s2−/− CD8+ T cells were able to expand and differentiate into effector and memory T cells as well as the wild type controls in conditions with maximized co-stimulatory and cytokine signals, such as infection with Listeria monocytogenes. Furthermore, re-challenge of OVA-specific memory G0s2−/− CD8+ T cells resulted in expansion similar to the controls. Notably, the activation of wild-type OVA-specific CD8+ T cells in a G0s2-null environment did not affect T cell expansion upon infection, even though G0s2−/− mice, with elevated levels of circulating free fatty acids, would have an increased probability of generating acetyl-coenzyme A for further metabolism in the tricarboxylic acid (TCA) cycle. However, naïve G0s2−/− CD8+ T cells in leukopenic hosts, driven by a “relative abundance” of IL-7 in addition to weak interactions with self-peptide in the context of self-MHC 4, 34, 38-40, showed a significant increase in homeostatic expansion. Although AMPKα1 is not essential for T cell homeostasis in loss-of-function mouse models 41, G0S2 mediated regulation of AMPK activity may modulate bursts of T cell expansion at steady state. AMPK is a sensor of small variations in energy by limiting ATP consumption while promoting ATP production via OXPHOS during peripheral T cell homeostasis 32. Therefore, it is possible that increased basal respiratory capacity offers a proliferative advantage of G0s2−/− CD8+ T cells at steady state, whereas the spare respiratory capacity, a maximal energy available to CD8+ T cells, may not be utilized in the infection model and may require further stress, such as chronic infections or aging.
Our work highlights a novel function for G0S2 in the control of energy homeostasis in CD8+ T cells by promoting catabolic pathways of ATP production during peripheral T cell homeostasis but not when T cells encounter foreign antigens presented by APCs with full co-stimulatory signals. Therefore, G0S2 potentially acts as a rheostat by modulating the bioenergetic requirements between homeostasis and antigen-driven proliferation and differentiation.
METHODS
Mice
G0s2−/− mice were generated in our laboratory using embryonic stem cells with targeted deletion of the G0S2 gene (VelociGene, Regeneron), purchased from the Knockout Mouse Project (KOMP) repository. The embryonic stem cell line VGB6 (C57BL/6NTac strain) contains the ZEN-Ub1 cassette inserted in the G0s2 gene. Embryonic stem cells were injected into the blastocoels of 3.5 d.p.c. C57BL/6 albino mouse embryos in the Genetically Engineered Mouse Core at Baylor College of Medicine. Chimeras were crossed to C57BL/6 albino mice to assess germline transmission, and then heterozygous mice were backcrossed to C57BL/6 and intercrossed to establish a colony of G0s2−/− mice. For the infection model, we bred mice with OT-I transgenic mice to generate OT-I G0s2—/— mice. Mice were genotyped by PCR using tail genomic DNA. Heterozygous females and homozygous males were used for the generation of null mice because G0s2−/− females failed to nourish newborn pups. C57BL/6 (B6) and B6.SJL mice were purchased from The Jackson Laboratory (Bar Harbor, Maine, U.S.A.). Mice were maintained under specific pathogen-free conditions at the Baylor College of Medicine. All experiments were performed with the approval of the Institutional Animal Care and Usage Committee of Baylor College of Medicine.
Flow cytometry
The following antibodies were used: allophycocyanin (APC)-anti-CD45.1, phycoerythrin (PE)-anti-CD19, PE-anti-CD11b, APC-anti-Gr-1, biotin-anti-CD3e, PE-Cy7-anti-Sca-1, fluorescein isothiocyanate-anti-CD4, APC-anti-CD8, PE-anti-Ter119 and APC-Cy7-anti-c-kit. All antibodies and streptavidin-APC were from BD Biosciences (San Jose, California, U.S.A.). The mitochondria content was evaluated using MitoTracker Green MF (LifeTechnologies, Grand Island, New York, U.S.A.). The mitochondrial membrane potential was determined using mitoscreen (JC-1) kit (BD Biosciences, San Jose, California, U.S.A.). Apoptosis was estimated using the Annexin V apoptosis detection kit (BD Biosciences, San Jose, California, U.S.A.). For cell cycle analysis, cells were fixed in 70% ethanol and stained with anti-Ki67 and 7-AAD (BD Biosciences, San Jose, California, U.S.A.). Samples were analyzed by flow cytometry using a FACSCanto flow cytometer (BD Biosciences, San Jose, California, U.S.A.). Cell surface and intracellular staining profiles were analyzed using FlowJo software (TreeStar, Ashland, Oregon, U.S.A.).
T cell activation and proliferation
Naïve CD44low CD8+ T cells were purified from the spleen by negative selection using the BD-IMag system (BD Biosciences, San Jose, California, U.S.A.). To measure G0S2 expression, CD8+ T cells were activated in 96-well plates coated with 10 μg/ml anti-CD3 (BD Biosciences, San Jose, California, U.S.A.) in the absence or presence of rapamycin (Sigma-Aldrich), LY294002 (Sigma-Aldrich, St Louis, Missouri, U.S.A), cyclosporin A (Millipore, Billerica, Massachusetts, U.S.A.), or PD98059 Millipore, Billerica, Massachusetts, U.S.A.) for 24 hours.
For in vitro stimulation, CD44low CD8+ T cells labeled with 4 μM CFSE (LifeTechnologies, Grand Island, New York, U.S.A.) or 5 μM eFluor 670 (eBioscience, San Diego, California, U.S.A.) were cultured for 2 or 3 days at a density of 1×105 cells per well in RPMI 1640 medium containing 10% (vol/vol) FBS in 96-well plates coated with 10 μg/ml anti-CD3 (BD Biosciences, San Jose, California, U.S.A.) in the presence of anti-CD28 (2 μg/ml; BD Biosciences). For homeostatic proliferation, CFSE-labeled CD44low CD8+ T cells from CD45.2+ WT or G0s2−/− mice were transferred into sub-lethally irradiated B6.SJL (CD45.1+) mice. Dilution of CFSE dye was analyzed by flow cytometry and the proliferation index (PI) was calculated using FlowJo software (TreeStar, Ashland, Oregon, U.S.A.).
Metabolism studies
CD44low CD8+ T cells purified from WT and G0s2−/− mice were activated in 96-well plates with plate-bound anti-CD3 (10 μg/ml) and 2 μg/ml anti-CD28 (BD Biosciences, San Jose, California, U.S.A.) for 36 hours and then transferred to a 24-well plate coated with BD Cell-Tak Adhesive reagent (BD Biosciences, San Jose, California, U.S.A.). Oxygen consumption rates (OCR) and extracellular acidification (ECAR) were measured using the XF24 Extracellular Flux Analyzer (Seahorse Bioscience, Massachusetts, U.S.A.). The XF assay media contained 25 mM glucose, 2 mM L-glutamine, 1 mM sodium pyruvate, 1 μM oligomycin, 1.8 μM fluorocarbonyl cyanide phenyl-hydrazone (FCCP), 1 μM rotenone, 10 mM glucose, and 100 mM 2-deoxyglucose (2-DG) (Sigma-Aldrich, St Louis, Missouri, U.S.A). The MTT assay was performed using a CellTiter-Glo kit (Promega, Madison, Wisconsin, U.S.A.). ATP content was measured using an ATP determination kit, according to the manufacturer’s instructions (LifeTechnologies, Grand Island, New York, U.S.A.). NAD/NADH was measured using a NAD+/NADH Quantification Kit (BioVision, Milpitas, California, U.S.A.). To quantify mitochondrial content, genomic DNA was extracted from CD8+ T cells using a Blood and Cell Culture DNA mini kit (QIAGEN, Valencia, California, U.S.A.), and the ratio of mitochondrial DNA (mtDNA) to nuclear DNA (nDNA) was determined as previously described 42. To study expression levels of enzymes involved in glucose metabolism, we analyzed RNA extracted from naïve and activated CD8+ T cells using the RT2 Profiler PCR assay (QIAGEN, Valencia, California, U.S.A.).
Infection with Listeria monocytogenes
Naive CD8+ T cells were purified from the spleens of OT-I (CD45.1+) and OT-I G0s2—/— (CD45.2+) mice using the BD-Imag magnetic separation system (BD Biosciences, San Jose, California, U.S.A.), following protocol of manufacturer, with the addition of biotinylated anti-CD44 antibody (1-2 μg per spleen). Then, naïve CD8+ T cells (approximately 95% purity) were adoptively transferred to B6.SJL (CD45.1+) recipients (1,000 cells/mouse i.v. to approach the physiological frequency). Recipient mice were infected 24 h later with recombinant OVA-expressing Listeria monocytogenes (Lm-OVA) strain (4×103 CFU, i.v.). For recall responses, mice were challenged 30 days after the first infection with a lethal dose of Lm-OVA (200×103 CFU).
Immunoblot analysis
CD8+ T cells were lysed with RIPA buffer (50mM Tris-HCl, pH 8.0, 1% NP-40, 0.5% Sodium deoxycholate, and 150mM NaCl) with addition of Halt Protease & Phosphatase Inhibitor Cocktail (Thermo Scientific, Waltham, Massachusetts, U.S.A.) and protein concentration was determined using the Bio-Rad protein assay (Hercules, California, U.S.A.). Proteins were resuspended in LDS Sample Buffer, loaded (15 μg/lane) onto NuPAGE Bis-Tris gels, and transferred to polyvinylidene difluoride (PVDF) membranes using iBlot2 system (LifeTechnologies, Grand Island, New York, U.S.A.). Proteins were detected using rabbit anti-S6K, phospho-S6K, AMPKα, phospho-AMPKα, Acetyl-CoA carboxylase (ACC), phospho-ACC, cyclin E, cdk2, phospho-Rb (Ser780), tubulin, and anti-rabbit IgG HRP-linked antibody from Cell Signaling Technology (Danvers, Massachusetts, U.S.A.). Primary antibodies were used at a 1:1,000 dilution whereas secondary antibody was used at a 1:50,000 dilution. PVDF membranes were developed using ECL Western Blotting Substrate (Thermo Scientific, Waltham, Massachusetts, U.S.A.) and exposed to Amersham hyperfilm ECL (GE Healthcare Life Sciences, Piscataway, New Jersey, U.S.A.)
Quantitative real-time PCR
Total RNA was extracted from CD8+ T cells using the RNeasy Mini kit (QIAGEN, Valencia, California, U.S.A.) and cDNA was synthesized from 100-500 ng RNA using random hexamer primers and the SuperScript III kit (LifeTechnologies, Grand Island, New York, U.S.A.). Quantitative real time PCR was performed using LightCycler FastStart DNA Master SYBR Green I (Roche, Nutley, New Jersey, U.S.A.) as specified by the manufacturer. Primer sequences for PCR were as follows: mouse β-actin forward, 5’-GTGGGCCGCTCTAGGCACCA-3’ and reverse, 5’-CGGTTGGCCTTAGGGTTCAGGGG-3’; mouse G0S2 forward, 5’-GTGCTCGGCCTAGTTGAGAC-3’ and reverse, 5’-CACCTGGGTCATGATCTGTG-3’; mouse cyclin E2 forward, 5’- AGGAATCAGCCCTTGCATTATC -3’ and reverse, 5’- CCCAGCTTAAATCTGGCAGAG -3’; mouse Cpt1a forward, 5’-CCAGGCTACAGTGGGACATT-3’ and reverse, 5’-GAACTTGCCCATGTCCTTGT-3’. Thermocycler conditions consisted of an initial step at 95°C for 10 min, followed by 35 cycles of a 3-step PCR program consisting of 95°C for 15 sec, 55°C for 30 sec, and 72°C for 30 sec. The relative expression for each gene was calculated by normalizing to β-actin.
Immunofluorescence detection
CD8+ T cells transduced with MIGR1-G0S2-V5 were cytospun onto glass slides (Thermo Scientific, Waltham, Massachusetts, U.S.A.) and immediately fixed with 1% (vol/vol) paraformaldehyde. Fixed cells were permeabilized with 0.1% (vol/vol) Triton X-100. Slides were stained with mouse anti-V5 and a mouse-specific secondary antibody conjugated to Alexa Fluor 555 (LifeTechnologies, Grand Island, New York, U.S.A.). Mitochondrial localization was detected with rabbit anti-COX IV (Abcam, Cambridge, Massachusetts, U.S.A.) and a rabbit-specific secondary antibody conjugated to Alexa Fluor 488 (LifeTechnologies, Grand Island, New York, U.S.A.). The slides were mounted with a mounting solution containing 4’,6-diamidino-2-phenylindole (DAPI) (LifeTechnologies, Grand Island, New York, U.S.A.) to stain the nuclei and analyzed on an Eclipse 90i microscope using the NIS Elements imaging software (Nikon Instruments Inc., Melville, New York, U.S.A.).
Retroviral expression of G0S2
V5-tagged G0S2 cDNA was cloned into the retroviral vector MIGR1 to generate CD8+ T cells with V5-G0S2 expression. 293T cells were co-transfected with a plasmid containing ψ-eco and MIGR1-V5-G0S2 vector. Bone marrow cells were isolated from wild type mice treated with 5-FU (150 mg/Kg) 4 days earlier and then cultured for two days in the presence of SCF (100 ng/ml), IL-3 (10 ng/ml), and IL-6 (6 ng/ml) from R&D Systems (Minneapolis, Minnesota, U.S.A.) in X-vivo 15 medium (Lonza, Houston, Texas, U.S.A.) 10. Bone marrow cells were transduced by spinoculation (60 min at 1,400 rpm) in the presence of Polybrene (8 μg/ml). Transduced bone marrow cells were then injected into the lateral tail veins of cytoablated mice (950 Rad). After 5 months of transplantation, GFP+ CD8+ T cells were purified from the spleen by cell sorting using a dual-laser MoFlo cell sorter (Beckman Coulter, Brea, California, U.S.A.).
Statistical analysis
Statistical differences were determined by paired two-tailed Student’s t-test using Prism (GraphPad Software Inc., La Jolla, California, U.S.A.). A p value <0.05 was considered significant. Statistics are indicated in each figure legend.
Supplementary Material
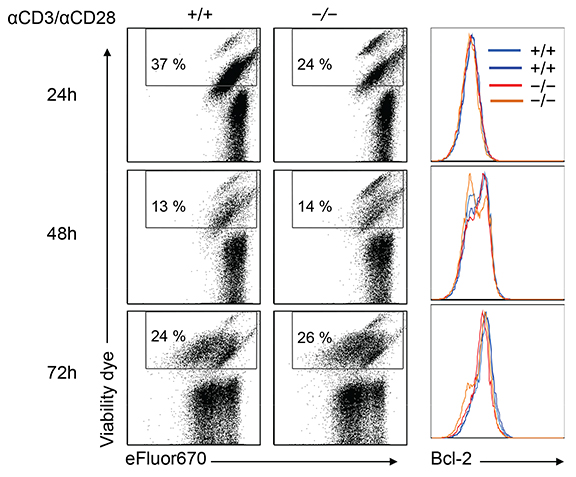
Supplementary Figure 1: Loss of G0S2 does not affect viability of CD8+ T cells. CD8+ T cells isolated from wild type and G0s2—/— mice were activated in vitro with plate-bound anti-CD3 and anti-CD28 for 24, 48, and 72 hours. T cells were analyzed by flow cytometry for cell division (dye eFluor670), viability (fixable viability dye eFluor® 780), and intracellular levels of Bcl-2.
{kind=link}
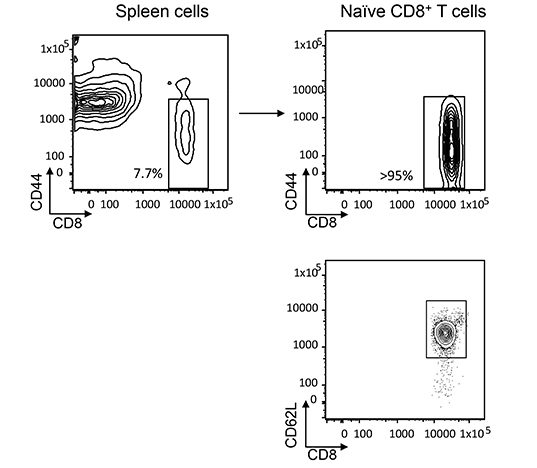
Supplementary Figure 2: Purification of naïve CD8+ T cells from the spleen. CD44lo CD8+ T cells were isolated from wild type and G0s2—/— spleen as described in Methods (95% purity). Naïve T cells were homogenously positive for CD62L.
{kind=link}
Supplementary Figure 3: Immunophenotype of activated CD8+ T cells. Naïve CD44lo CD8+ T cells were activated in plate-bound anti-CD3 and anti-CD28 for 12, 24, and 30 hours. Cells were analyzed for the expression of the activation markers CD69, CD44, and CD25 by flow cytometry.
{kind=link}
ACKNOWLEDGMENTS
The authors would like to thank Tatiana Goltsova for cell sorting, Karen Prince for formatting figures, and the assistance provided by the Genetically Engineered Mouse (GEM) Shared Resource (NIH/NCI 5P30CA125123) and the Cytometry and Cell Sorting Core (P30 AI036211, P30 CA125123, and S10 RR024574) at Baylor College of Medicine. This work was supported by the National Institutes of Health Grants R01-AI077536 (to H.D.L), R01-AI077536-02S1 (to H.D.L), and the Gabrielle’s Angel Foundation for Cancer Research (to H.D.L.).
Footnotes
DISCLOSURES
The authors have no financial conflict of interest.
REFERENCES
- 1.Hamilton SE, Jameson SC. CD8 T cell quiescence revisited. Trends Immunol. 2012;33(5):224–30. doi: 10.1016/j.it.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gerriets VA, Rathmell JC. Metabolic pathways in T cell fate and function. Trends Immunol. 2012;33(4):168–73. doi: 10.1016/j.it.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342(6155):1242454. doi: 10.1126/science.1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Surh CD, Sprent J. Homeostasis of naive and memory T cells. Immunity. 2008;29(6):848–62. doi: 10.1016/j.immuni.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 5.Rocha B, Tanchot C. The Tower of Babel of CD8+ T-cell memory: known facts, deserted roads, muddy waters, and possible dead ends. Immunol Rev. 2006;211:182–96. doi: 10.1111/j.0105-2896.2006.00378.x. [DOI] [PubMed] [Google Scholar]
- 6.Yang K, Shrestha S, Zeng H, Karmaus PW, Neale G, Vogel P, et al. T cell exit from quiescence and differentiation into Th2 cells depend on Raptor-mTORC1-mediated metabolic reprogramming. Immunity. 2013;39(6):1043–56. doi: 10.1016/j.immuni.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yao S, Buzo BF, Pham D, Jiang L, Taparowsky EJ, Kaplan MH, et al. Interferon regulatory factor 4 sustains CD8(+) T cell expansion and effector differentiation. Immunity. 2013;39(5):833–45. doi: 10.1016/j.immuni.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Russell L, Forsdyke DR. A human putative lymphocyte G0/G1 switch gene containing a CpG-rich island encodes a small basic protein with the potential to be phosphorylated. DNA Cell Biol. 1991;10(8):581–91. doi: 10.1089/dna.1991.10.581. [DOI] [PubMed] [Google Scholar]
- 9.Yamada T, Park CS, Shen Y, Rabin KR, Lacorazza HD. G0S2 inhibits the proliferation of K562 cells by interacting with nucleolin in the cytosol. Leuk Res. 2014;38(2):210–7. doi: 10.1016/j.leukres.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamada T, Park CS, Burns A, Nakada D, Lacorazza HD. The Cytosolic Protein G0S2 Maintains Quiescence in Hematopoietic Stem Cells. PLoS One. 2012;7(5):e38280. doi: 10.1371/journal.pone.0038280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heckmann BL, Zhang X, Xie X, Saarinen A, Lu X, Yang X, et al. Defective adipose lipolysis and altered global energy metabolism in mice with adipose overexpression of the lipolytic inhibitor G0/G1 switch gene 2 (G0S2) J Biol Chem. 2014;289(4):1905–16. doi: 10.1074/jbc.M113.522011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang X, Lu X, Lombes M, Rha GB, Chi YI, Guerin TM, et al. The G(0)/G(1) switch gene 2 regulates adipose lipolysis through association with adipose triglyceride lipase. Cell Metab. 2010;11(3):194–205. doi: 10.1016/j.cmet.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Welch C, Santra MK, El-Assaad W, Zhu X, Huber WE, Keys RA, et al. Identification of a protein, G0S2, that lacks Bcl-2 homology domains and interacts with and antagonizes Bcl-2. Cancer Res. 2009;69(17):6782–9. doi: 10.1158/0008-5472.CAN-09-0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kioka H, Kato H, Fujikawa M, Tsukamoto O, Suzuki T, Imamura H, et al. Evaluation of intramitochondrial ATP levels identifies G0/G1 switch gene 2 as a positive regulator of oxidative phosphorylation. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(1):273–8. doi: 10.1073/pnas.1318547111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tokumaru Y, Yamashita K, Osada M, Nomoto S, Sun DI, Xiao Y, et al. Inverse correlation between cyclin A1 hypermethylation and p53 mutation in head and neck cancer identified by reversal of epigenetic silencing. Cancer Res. 2004;64(17):5982–7. doi: 10.1158/0008-5472.CAN-04-0993. [DOI] [PubMed] [Google Scholar]
- 16.Kusakabe M, Watanabe K, Emoto N, Aki N, Kage H, Nagase T, et al. Impact of DNA demethylation of the G0S2 gene on the transcription of G0S2 in squamous lung cancer cell lines with or without nuclear receptor agonists. Biochem Biophys Res Commun. 2009;390(4):1283–7. doi: 10.1016/j.bbrc.2009.10.137. [DOI] [PubMed] [Google Scholar]
- 17.Faber K, Bullinger L, Ragu C, Garding A, Mertens D, Miller C, et al. CDX2-driven leukemogenesis involves KLF4 repression and deregulated PPARgamma signaling. J Clin Invest. 2013;123(1):299–314. doi: 10.1172/JCI64745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kao LC, Germeyer A, Tulac S, Lobo S, Yang JP, Taylor RN, et al. Expression profiling of endometrium from women with endometriosis reveals candidate genes for disease-based implantation failure and infertility. Endocrinology. 2003;144(7):2870–81. doi: 10.1210/en.2003-0043. [DOI] [PubMed] [Google Scholar]
- 19.Nakamura N, Shimaoka Y, Tougan T, Onda H, Okuzaki D, Zhao H, et al. Isolation and expression profiling of genes upregulated in bone marrow-derived mononuclear cells of rheumatoid arthritis patients. DNA Res. 2006;13(4):169–83. doi: 10.1093/dnares/dsl006. [DOI] [PubMed] [Google Scholar]
- 20.Kobayashi S, Ito A, Okuzaki D, Onda H, Yabuta N, Nagamori I, et al. Expression profiling of PBMC-based diagnostic gene markers isolated from vasculitis patients. DNA Res. 2008;15(4):253–65. doi: 10.1093/dnares/dsn014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koczan D, Guthke R, Thiesen HJ, Ibrahim SM, Kundt G, Krentz H, et al. Gene expression profiling of peripheral blood mononuclear leukocytes from psoriasis patients identifies new immune regulatory molecules. Eur J Dermatol. 2005;15(4):251–7. [PubMed] [Google Scholar]
- 22.Yamada T, Park CS, Shen Y, Rabin KR, Lacorazza HD. G0S2 inhibits the proliferation of K562 cells by interacting with nucleolin in the cytosol. Leuk Res. 2013 doi: 10.1016/j.leukres.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamada T, Park CS, Mamonkin M, Lacorazza HD. Transcription factor ELF4 controls the proliferation and homing of CD8+ T cells via the Kruppel-like factors KLF4 and KLF2. Nat Immunol. 2009;10(6):618–26. doi: 10.1038/ni.1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cristillo AD, Heximer SP, Russell L, Forsdyke DR. Cyclosporin A inhibits early mRNA expression of G0/G1 switch gene 2 (G0S2) in cultured human blood mononuclear cells. DNA Cell Biol. 1997;16(12):1449–58. doi: 10.1089/dna.1997.16.1449. [DOI] [PubMed] [Google Scholar]
- 25.Ma T, Lopez-Aguiar AG, Li A, Lu Y, Sekula D, Nattie EE, et al. Mice lacking G0S2 are lean and cold-tolerant. Cancer Biol Ther. 2014;15(5) doi: 10.4161/cbt.28251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu X, Yang X, Liu J. Differential control of ATGL-mediated lipid droplet degradation by CGI-58 and G0S2. Cell Cycle. 2010;9(14):2719–25. doi: 10.4161/cc.9.14.12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460(7251):103–7. doi: 10.1038/nature08097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pearce EL. Metabolism in T cell activation and differentiation. Curr Opin Immunol. 2010;22(3):314–20. doi: 10.1016/j.coi.2010.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, et al. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460(7251):108–12. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang R, Green DR. Metabolic checkpoints in activated T cells. Nat Immunol. 2012;13(10):907–15. doi: 10.1038/ni.2386. [DOI] [PubMed] [Google Scholar]
- 31.Yang K, Chi H. mTOR and metabolic pathways in T cell quiescence and functional activation. Semin Immunol. 2012;24(6):421–8. doi: 10.1016/j.smim.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blagih J, Krawczyk CM, Jones RG. LKB1 and AMPK: central regulators of lymphocyte metabolism and function. Immunol Rev. 2012;249(1):59–71. doi: 10.1111/j.1600-065X.2012.01157.x. [DOI] [PubMed] [Google Scholar]
- 33.Jones RG, Thompson CB. Revving the engine: signal transduction fuels T cell activation. Immunity. 2007;27(2):173–8. doi: 10.1016/j.immuni.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 34.Sprent J, Surh CD. Cytokines and T cell homeostasis. Immunol Lett. 2003;85(2):145–9. doi: 10.1016/s0165-2478(02)00221-3. [DOI] [PubMed] [Google Scholar]
- 35.Forsdyke DR. An alternative way of thinking about stem-loops in DNA. A case study of the human G0S2 gene. J Theor Biol. 1998;192(4):489–504. doi: 10.1006/jtbi.1998.0674. [DOI] [PubMed] [Google Scholar]
- 36.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124(3215):269–70. [PubMed] [Google Scholar]
- 37.van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36(1):68–78. doi: 10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Surh CD, Boyman O, Purton JF, Sprent J. Homeostasis of memory T cells. Immunol Rev. 2006;211:154–63. doi: 10.1111/j.0105-2896.2006.00401.x. [DOI] [PubMed] [Google Scholar]
- 39.Surh CD, Sprent J. Regulation of mature T cell homeostasis. Semin Immunol. 2005;17(3):183–91. doi: 10.1016/j.smim.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 40.Tan JT, Dudl E, LeRoy E, Murray R, Sprent J, Weinberg KI, et al. IL-7 is critical for homeostatic proliferation and survival of naive T cells. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(15):8732–7. doi: 10.1073/pnas.161126098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mayer A, Denanglaire S, Viollet B, Leo O, Andris F. AMP-activated protein kinase regulates lymphocyte responses to metabolic stress but is largely dispensable for immune cell development and function. Eur J Immunol. 2008;38(4):948–56. doi: 10.1002/eji.200738045. [DOI] [PubMed] [Google Scholar]
- 42.Guo W, Jiang L, Bhasin S, Khan SM, Swerdlow RH. DNA extraction procedures meaningfully influence qPCR-based mtDNA copy number determination. Mitochondrion. 2009;9(4):261–5. doi: 10.1016/j.mito.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Loss of G0S2 does not affect viability of CD8+ T cells. CD8+ T cells isolated from wild type and G0s2—/— mice were activated in vitro with plate-bound anti-CD3 and anti-CD28 for 24, 48, and 72 hours. T cells were analyzed by flow cytometry for cell division (dye eFluor670), viability (fixable viability dye eFluor® 780), and intracellular levels of Bcl-2.
Supplementary Figure 2: Purification of naïve CD8+ T cells from the spleen. CD44lo CD8+ T cells were isolated from wild type and G0s2—/— spleen as described in Methods (95% purity). Naïve T cells were homogenously positive for CD62L.
Supplementary Figure 3: Immunophenotype of activated CD8+ T cells. Naïve CD44lo CD8+ T cells were activated in plate-bound anti-CD3 and anti-CD28 for 12, 24, and 30 hours. Cells were analyzed for the expression of the activation markers CD69, CD44, and CD25 by flow cytometry.