

Abstract
Nucleotide excision repair (NER) removes DNA helix-distorting lesions induced by UV light and various chemotherapeutic agents such as cisplatin. These lesions efficiently block the elongation of transcription and need to be rapidly removed by transcription-coupled NER (TC-NER) to avoid the induction of apoptosis. Twenty-nine genes have been classified to code for proteins participating in nucleotide excision repair (NER) in human cells. Here we explored the transcriptional and post-transcriptional regulation of these NER genes across 13 human cell lines using Bru-seq and BruChase-seq, respectively. Many NER genes are relatively large in size and therefore will be easily inactivated by UV-induced transcription-blocking lesions. Furthermore, many of these genes produce transcripts that are rather unstable. Thus, these genes are expected to rapidly lose expression leading to a diminished function of NER. One such gene is ERCC6 that codes for the CSB protein critical for TC-NER. Due to its large gene size and high RNA turnover rate, the ERCC6 gene may act as dosimeter of DNA damage so that at high levels of damage, ERCC6 RNA levels would be diminished leading to the loss of CSB expression, inhibition of TC-NER and the promotion of cell death.
1. Introduction
Ultraviolet (UV) light is a strong mutagen and has promoted natural selection among organisms throughout evolutionary time. Nucleotide excision repair (NER) evolved to safeguard the DNA from the deleterious effects of UV light and can be found in all species of life from bacteria and plants to mammals [1]. NER also protects cells from cyclopurines formed by endogenous reactive oxygen species (ROS) and cancer cells use NER to repair damage induced by certain chemotherapeutic agents such as cisplatin. A better understanding of the regulation of expression of NER genes could aid in predicting the sensitivity of cells to UV light and chemotherapeutic agents and may promote the development of new therapeutic regiments.
Global genomic NER (GG-NER) deals with lesions in the whole genome. The lesion recognition complex in GG-NER, consisting of XPC, RAD23A, RAD23B, CETN2, DDB1 and DDB2, acts by recognizing the DNA lesions in chromatin and recruits the core NER complex to sites of damage (Fig. 1) [1–3]. A specialized damage recognition step of NER has evolved to aid in the removal of bulky lesions that block transcription elongation [4]. This sub-pathway of NERis called transcription-coupled NER (TC-NER) where Cockeyes’ syndrome factors A (CSA) and B (CSB), XAB2 and UVSSA factors orchestrate the recruitment of the NER core complexes to sites of transcription-stalling lesions. Following damage recognition, which is the rate-limiting step, the pre-incision complex consisting of XPA and RPA verifies the presence of the lesion followed by the DNA unwinding by the TFIIH complex. The damaged strand is then incised by the incision enzymes XPG, ERCC1 and XPF, DNA polymerases re-synthesize the DNA in the excised gap and DNA ligase 1 seals the newly synthesized strand with existing strand (Fig. 1).
Figure 1.
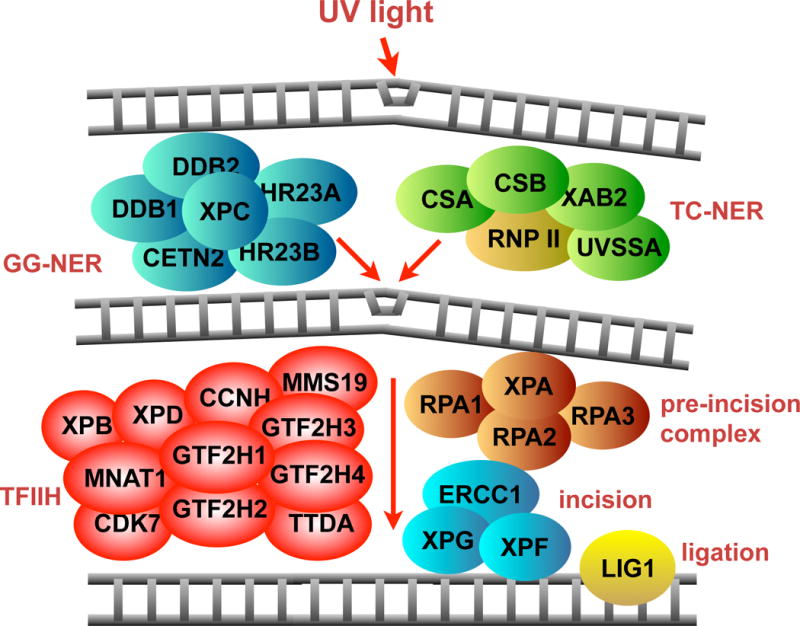
The 29 gene products of NER that were examined in this study and their roles in NER.
Mutations in core components of NER leads to the human disorders xeroderma pigmentosum and trichothiodystrophy [5] while defects in the factors responsible for TC-NER give rise to the Cockayne’s and UV-sensitive syndromes [6]. Polymorphisms in NER genes have been linked to reduced repair capacity and cancer predisposition [7]. Furthermore, Inactivating somatic mutations of the NER genes ERCC2, ERCC3, ERCC4, ERRC5, XPA, XPC and DDB2 promote cancer and therefore these genes are known as cancer predisposition genes [8]. Many studies have also found a correlation between the expression level of DNA repair genes in cancer cells and their sensitivity to cisplatin [7]. Interestingly, a low level of expression or a defect in the GG-NER factors XPC and DDB2 does not sensitize cells to cisplatin or UV light while reduced expression or defects in the TC-NER factors CSA and CSB results in a marked sensitivity [9, 10].
The expression of NER genes have been previously analyzed in cell lines using total cellular RNA, which reports on the steady-state level of RNA but does not distinguish between the contribution of synthesis and turnover of RNA to RNA homeostasis. In this study, we used Bru-seq and BruChase-seq [11, 12] to specifically examine the rate of RNA synthesis and turnover of NER transcripts across 13 human cell lines. These techniques are based on the pulse-labeling of nascent RNA with bromouridine (Bru) followed by either immediate harvest (Bru-seq) or harvest after a 6-hour chase in uridine (BruChase-seq). The Bru-labeled RNA is then isolated using anti-BrdU antibodies conjugated to magnetic beads, converted into a cDNA library and deep sequenced. Surprisingly, our results show that many critical NER genes produce fairly unstable transcripts that would be expected to quickly vanish following induction of transcription-blocking lesions by UV light and bulky chemical adducts – the very adducts NER was designed to remove.
2. Materials and Methods
2.1. Cell lines
The cell lines used were HF1 (gift from Mary Davis, University of Michigan), HPNE, Panc1, MiaPaCa2, BxPC3, UM16, UM28, UM59 and HEK293 (gifts from Diane Simeone University of Michigan), HeLa, GM12878 and K562 (ATCC) and LN428 (gift from Rob Sobol, University of Pittsburgh). The media used were MEM with 10% FBS and antibiotics (HF1), DMEM with 10% FBS and Pen/Strep (Panc1, MiaPaCa2, UM16, UM28, UM59, HEK293), RPMI1640 with 10% FBS and antibiotics (BxPC3), F12K with 10% FBS and Pen/Strep (HeLa), RPMI1640 with 15% FBS (GM12878), IMDM with 10% FBS (K562), and MEMalpha with 10% FBS, antibiotics, gentamycin and puromycin (LN428). The HPNE cells were grown in 75% DMEM without glucose (Sigma D-5030) containing 2 mM L-glutamine (Sigma G7513), 1.5g/L sodium bicarbonate (Sigma S 4019), 25% Medium M3 Base (Incell Corp, M300F-500) with the additives 10 ng/mL human recombinant EGF (BD Sciences 354052), 5.5 mM D-glucose (1g/L) (Sigma G8644) and 750 ng/mL puromycin dihydrochloride (Invitrogen A11138-02).
2.2. Bru-seq and BruChase-seq
For detailed descriptions of these techniques, please see [11, 12]. In short: the labeling of nascent RNA was performed for 30 min at 37°C with 2 mM bromouridine in conditioned medium. For the BruChase-seq experiments, the bromouridine-containing medium was removed after 30-min labeling, the plates were rinsed twice in PBS and then conditioned medium containing 20 mM uridine was added. The cells were then incubated for 6 hours at 37°C. At the completion of the labeling +/− chase, the cells were lysed in TRIzol and the Bru-containing RNA was isolated using anti-BrdU antibodies conjugated to magnetic beads. The isolated RNA was converted into cDNA libraries using the Illumina True-seq library kit followed by deep sequencing to around 50 million single-end 50 nucleotide reads.
2.3. Analysis
Bru-seq and BruChase-seq gene expression was measured in transcripts per million reads (TPM) in order to compare gene expression across cell lines. The TPM of each gene can be thought of as the percent of expression associated with a gene in relation to the total expression of the genome [13]. The TPM formula was slightly modified and implemented as:
Where transcript densities are defined as:
The natural logarithm of TPM measurements corresponding to the 29 NER genes were used in the analysis. When plotting Bru-seq and BruChase-seq data, genes with expression values equal to zero were represented in black. The stability measurement of each transcript is calculated by dividing the exonic reads at 6 hours by the reads across the entirety of the gene at 0 hours. Since it is impossible to calculate a stability value for a gene with no synthesis expression, we left such cases displayed as white boxes in Figure 4B.
Figure 4.
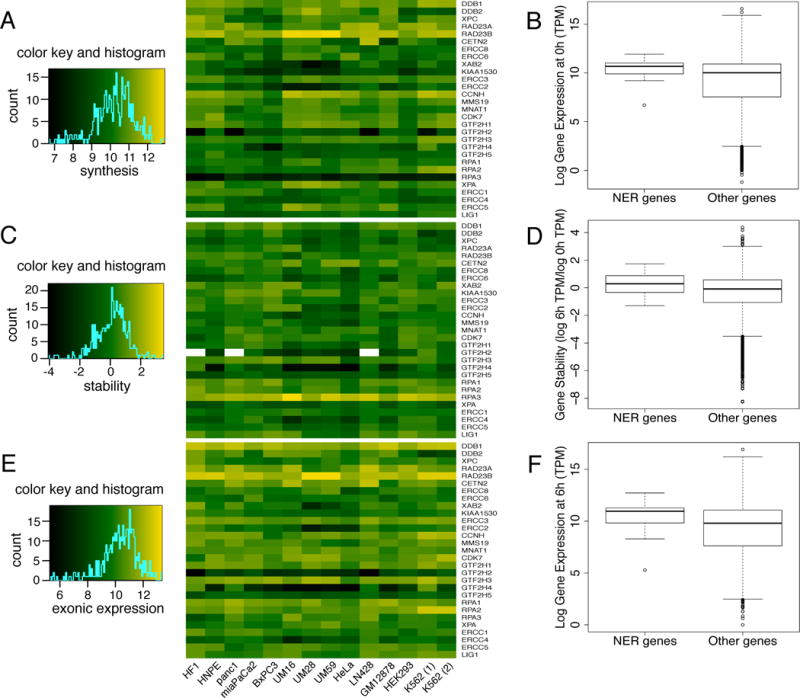
Expression of the 29 NER genes in 13 human cell lines. (A) Relative rates of RNA synthesis of NER genes as determined by Bru-seq. (B) Boxplots showing the comparisons of RNA synthesis between NER genes and all other expressed genes in cell line HF1. (C) Relative RNA stability of NER transcripts as determined by BruChase-seq. (D) Boxplots showing the comparisons of relative RNA stability between NER genes and all other expressed genes in cell line HF1. (E) Exonic reads of 6-hour old NER RNAs. (F) Boxplots showing the comparisons of exonic reads of 6-hour old RNA between NER genes and all other expressed genes in cell line HF1. The boxplots are used to demonstrate the distribution of the data where the horizontal line inside the box indicates the median (50th percentile). The edges of the boxes indicate the 25th and 75th percentile and the spaces between them are called interquartile ranges. Whiskers extend from the box edges up to the most extreme point that is not more than 1.5 times the interquartile range. Individual data points more extreme than the extension of the whiskers are indicated with circles.
3. Results
The total, steady-state level of RNA in cells is a reflection of the equilibrium between synthesis and degradation of RNA. We previously showed that the expression of different human genes appear to have their unique ratios of RNA synthesis and degradation suggesting that both of these processes are coordinately regulated by cells to obtain defined, homeostatic, levels of RNA [12]. The particular settings of the ratios of synthesis and degradation of RNA from NER genes are not known. Since many of the adducts that NER has been designed to remove from DNA have the capacity to block the elongation of transcription, the ratio of synthesis and stability of a particular RNA may have a profound effect on the steady-state level of its expression following DNA insult. For example, a setting of high synthesis coupled with low stability would rapidly lead to the depletion of this RNA while a gene with the strategy of low synthesis and high RNA stability would fare much better at times of exposure to transcription-blocking DNA damage. Here we present the signatures of RNA synthesis and stability of 29 NER genes using Bru-seq and BruChase-seq across 13 human cell lines.
3.1. Differences in NER gene regulation across cell lines
To obtain estimates of the relative stability of the 29 NER transcripts using BruChase-seq, we compared the amount of sequencing reads from all of the exons of a particular gene after a 6-hour chase with the amount of sequencing reads from the whole gene immediately after Bru-labeling. If a particular transcript is stable this ratio will be high while for an unstable transcript this ratio will be low. We observed that the stability of some NER transcripts was differentially regulated in the different cell lines. For example, the XPA transcript was synthesized to similar levels in the pancreatic cancer cell lines BxPC3 and UM59 but this transcript was unstable in BxPC3 cells but stable in UM59 cells (compare the heights of the exonic peaks in red) (Fig. 2A). The XPC transcript was much more stable in human fibroblasts compared to UM28 cells (Fig. 2B) and the ERCC6 transcript was synthesized at a lower level in GM12878 B-cells than in MiaPaCa2 pancreatic cancer cells while showing relatively low stability in both cell lines (Fig. 2C).
Figure 2.
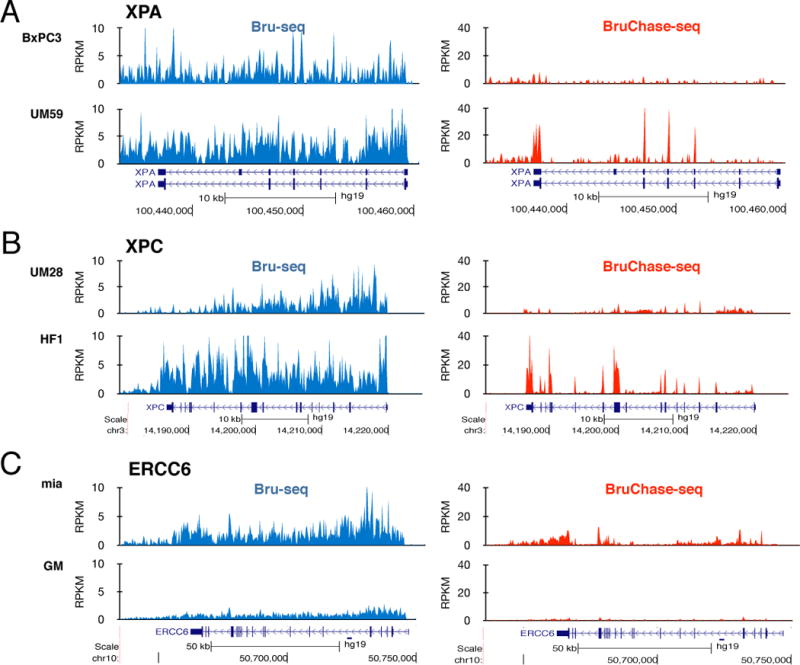
Differential regulation of relative synthesis and stability of key NER transcripts. (A) The relative synthesis of XPA RNA in the pancreatic cancer cell lines BxPC3 and UM59 (blue trace, left, expressed as reads per thousand base pairs per million reads or RPKM) is similar but the stabilities of the XPA transcript differ dramatically between these two cell lines (red trace, right). (B) Transcription appears to diminish at the 3′-end of the XPC gene in UM28 cells while the XPC gene is transcribed evenly in the skin fibroblasts HF1 (left). The stability of the XPC transcript is much higher in the HF1 cells than in the UM28 cells (right). (C) Differential rates of RNA synthesis of the ERCC6 gene in MiaPaCa2 cells and GM12878 cells (left). Low stability of the ERCC6 transcript (ratio of 6-h exonic reads to full gene reads at 0h) in both cell types (right).
The RAD23A and RAD23B genes encode functionally redundant proteins acting as ubiquitin receptors and they interact with the XPC protein to promote damage recognition for GG-NER [3]. Using Bru-seq we found that both RAD23A and RAD23B genes were highly transcribed across the cell lines. In the three primary pancreatic cell lines UM16, UM28 and UM59, both genes were similarly transcribed while the relative stabilities of these transcript showed large variations (Fig. 3). Interestingly, in the UM16 cells the RAD23A transcript was very stable while the RAD23B transcript was not. Conversely, in UM28 and UM59 cells, the RAD23A transcript was unstable while the RAD23B transcript was stable. Thus, each cell line arrived at a desired level of either RAD23A or RAD23B by post-transcriptional regulation involving RNA stability.
Figure 3.
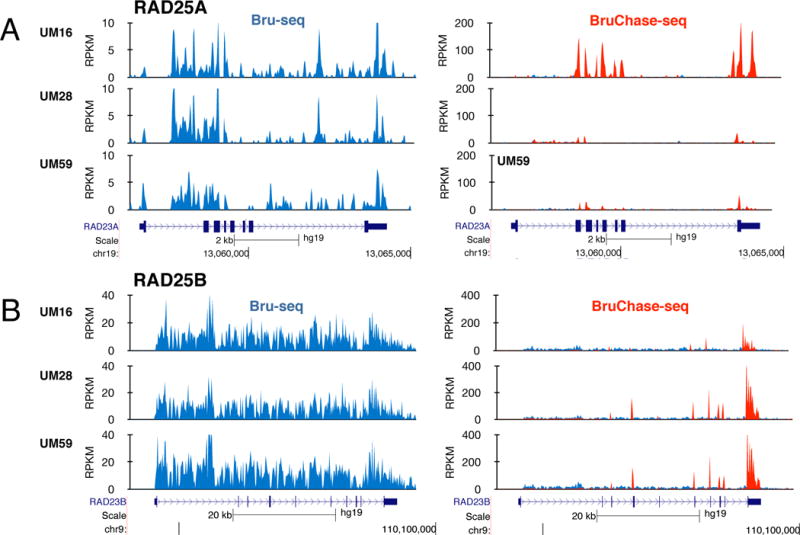
Post-transcriptional regulation of RAD23A and RAD23B genes in pancreatic cancer cell lines. (A) The relative synthesis of RAD25A RNA is very similar across three pancreatic cancer cell lines while the relative stability of the RAD25A transcript is high in UM16 cells but low in UM28 and UM59 cells. (B) Similar relative synthesis of RAD25B across the three UM cell lines while higher stability of the RAD25B transcript in UM28 and UM59 cells.
3.2. Relative rates of RNA synthesis of NER genes
To enable direct comparisons of RNA synthesis between samples, transcript intensities were measured in transcripts per million reads (TPM). It was found that the 29 NER genes were synthesized at a spectrum of different rates (Fig. 4A). These rates differed between genes and between different cell lines for the same gene. To explore whether the origin of this variability was due to different biology of the cell lines or from technical variation between experiments, we compared the relative RNA synthesis between two biological samples prepared at different occasions from the same cell line (K562) and found them to be highly correlated (r2=0.952) (Supplemental Fig. 1A). Thus, technical variability did not appear to be the main cause of the variability seen in the rate of RNA synthesis of the NER genes across the cell lines. We next compared the synthesis rates of the 29 NER genes to the synthesis rates of all expressed genes in the same cell line (HF1) and found that NER genes are synthesized at a median rate that is comparable to the median rate of all expressed genes (Fig. 4B).
3.3. Relative stability of NER transcripts
The relative stabilities of transcripts were estimated by assessing the ratio of transcription reads obtained from the exons of the different genes in the 6-hour sample (BruChase-seq) with the reads throughout the corresponding genes in the 0-hour sample (Bru-seq). The relative stabilities of the 29 NER transcripts are shown in Figure 4C with the correlation between the two K562 cell samples shown in Supplemental Figure 1B. In Figure 4E, the exonic reads from the different genes in the 6-hour samples are shown with the correlation between the K562 cell samples shown in Supplemental Figure 1C. The densities of reads across the exons of genes after a 6-hour chase in uridine are approximations of the steady-state levels of RNA. Taken together, it can be seen that there was large differences in transcript stability between different genes and also across the 13 cell lines.
3.4. Gene and cell type-specific regulation of NER gene expression
We observed differential regulation of gene expression at the synthesis, stability and mature RNA content both at the gene and at the cell type level. For a given variable we used the gene’s median (across all cell types) in order to calculate a gene regulation score (Fig. 5A–C). In order to understand regulation of NER genes in individual cell lines, we calculated overall scores for the cell lines. For a given variable we calculated the percent difference for a given variable of a given gene from the median of that gene calculated across all cell lines. This allowed us to calculate the overall distance of individual cell lines from the median signal observed across all cell lines (Fig. 5D–F).
Figure 5.
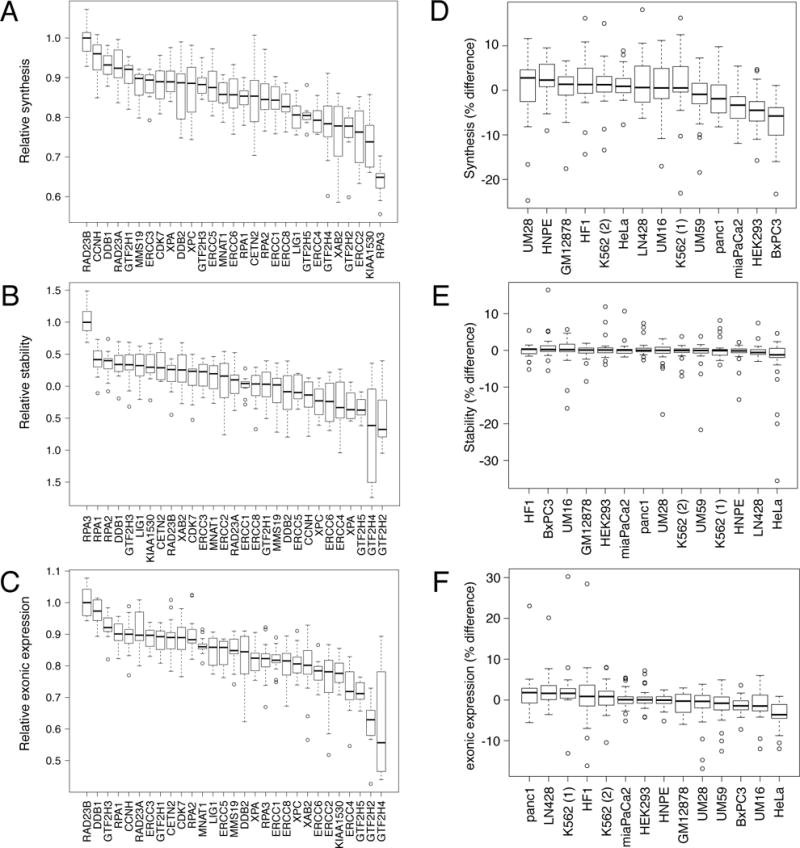
Comparative analysis of transcription regulation across genes and cell lines. Relative transcript synthesis (A), stability (B), and exonic signal in 6-hour old RNA (C). Data is grouped by gene to indicate genes with highest and lowest values. Transcript percent difference from median values (calculated across cell lines) in synthesis (D), stability (E), and exonic signal in 6-hour old RNA (F). Data is grouped by cell lines to indicate cell lines with the greatest discrepancies from the median expression of each gene. Boxplots are designed as described in Fig. 4.
Our analyses of the synthesis and stability of the RNA data showed that the NER genes showed different patterns of gene regulation. For example, it can be seen that the RAD23B was the highest transcribed gene across the cell lines with RPA3 being the lowest (Fig. 5A). Among the cell lines, the primary pancreatic cancer line UM28 and the normal pancreatic epithelial cell line HPNE showed the highest rates of transcription across the 13 cell lines while the established pancreatic cancer cell lines Panc1, MiaPaCa2 and BxPC3, together with HEK293 cells, had the lowest rates of transcription (Fig. 5D). When we tabulated the median relative stability of the different NER transcript we got a very different picture than that of the synthesis data. The most striking difference was seen for the RPA3 transcript, which had the overall lowest median rate of transcription (Fig. 5A) but had by far the highest median relative stability of all tested NER transcripts (Fig. 5B). Similarly, the BxPC3 cell line had the lowest median relative synthesis rate of the NER genes among the 13 cell lines (Fig. 5D) while the transcripts produced scored near the top for median relative stability (Fig. 5E). The transcripts generated by HeLa cells showed the overall lowest median relative stability. The exonic values of the 6-hour samples are the products of the RNA synthesis and RNA stability scores and represent the mature population of RNA resembling steady-state RNA. The NER transcript with the highest median relative abundance at 6 hours after labeling was RAD23B with DDB1 as close second (Fig. 5C). The cell lines with the highest median relative levels of NER transcripts at 6 hours were the pancreatic cancer cell line panc1 and the glioblastoma cell line LN428 while HeLa cells showed the lowest level (Fig. 5F).
4. Discussion
Our study provides a comprehensive view of the complex regulation of synthesis and stability of transcripts of the 29 genes of the NER pathway across 13 cell lines. NER is thought to have evolved primarily to deal with DNA lesions induced by UV light but this repair pathway is also needed to remove cyclopyrine adducts induced by endogenously produced reactive oxygen species (ROS) [3]. Therefore all cells need NER in some capacity to deal with a set of lesions that could pose threats to transcription and replication. Indeed, in our survey of synthesis and stability of the NER genes across both normal and cancerous tissues, no cell line was found to lack expression of the NER genes. However, there were large differences in the expression signatures of the NER genes among the cell lines. Moreover, the balance between synthesis and turnover of the different transcripts showed unique patterns. For example, the RPA1 gene generated very little nascent RNA, but this RNA showed a very high stability allowing this transcript to have fairly high relative abundance at 6 hours after its synthesis. On the other hand, the ERCC6 gene coding for the CSB protein involved in TC-NER, was synthesized at fairly high rates in many cell lines but this transcript was rather unstable. An interesting pattern of post-transcriptional regulation was also observed for the RAD23A and RAD23B transcripts in the cell lines UM16, UM28 and UM59 (Fig. 3). The gene products of these two genes are thought to be functionally equivalent and we found that each cell line synthesized them at similar rates but that they retained one or the other by post-transcriptional regulation.
Our approach of exploring transcript synthesis and stability across cancer cells lines could potentially be used to predict susceptibility to particular chemotherapeutic agents. In this test case of examining the NER pathway we calculated NER gene regulation scores for each cell line and ordered the cell lines according to their median expression (Fig. 5D–F). Functional repair and survival assays would be needed to discern whether the median expression levels of the NER genes in the different cell lines are predictive of the sensitivity to UV light or cisplatin for example. Alternatively, the “outlier” genes with very low expression may be used as predictors of treatment outcomes. A future refinement of this scoring system would be to assign each gene in a particular pathway different “weight” according to whether they are expressed at rate-limiting levels and how important they are for the function of the pathway.
Transcription acts as a sensor of DNA damage and is linked to p53 induction via RPA and ATR signaling [14]. Pro-apoptotic genes induced by p53 are in general more compact in size than anti-apoptotic genes, a situation that ensures a dose-dependent induction of apoptosis as larger survival genes become preferentially blocked by DNA lesions [15]. Furthermore, apoptosis may be induced by transcription-blocking lesions as a result diminished levels of critical gene products, such as MCL-1 [9, 16, 17]. Finally, proliferating cells entering S-phase before resolving stalled RNA polymerase II complexes, will have lethal encounters between replication and transcription machineries [18]. Thus, it is critical that NER, and especially the TC-NER sub-pathway, remains operational following damage exposure to promote survival. To remain operational even after induction of transcription-blocking lesions, it would be plausible to hypothesize that NER genes may be under a selective pressure to reduce their sizes. However, when examining the gene sizes of NER genes we found that they are not generally smaller than the median size of human genes (Fig. 6A).
Figure 6.
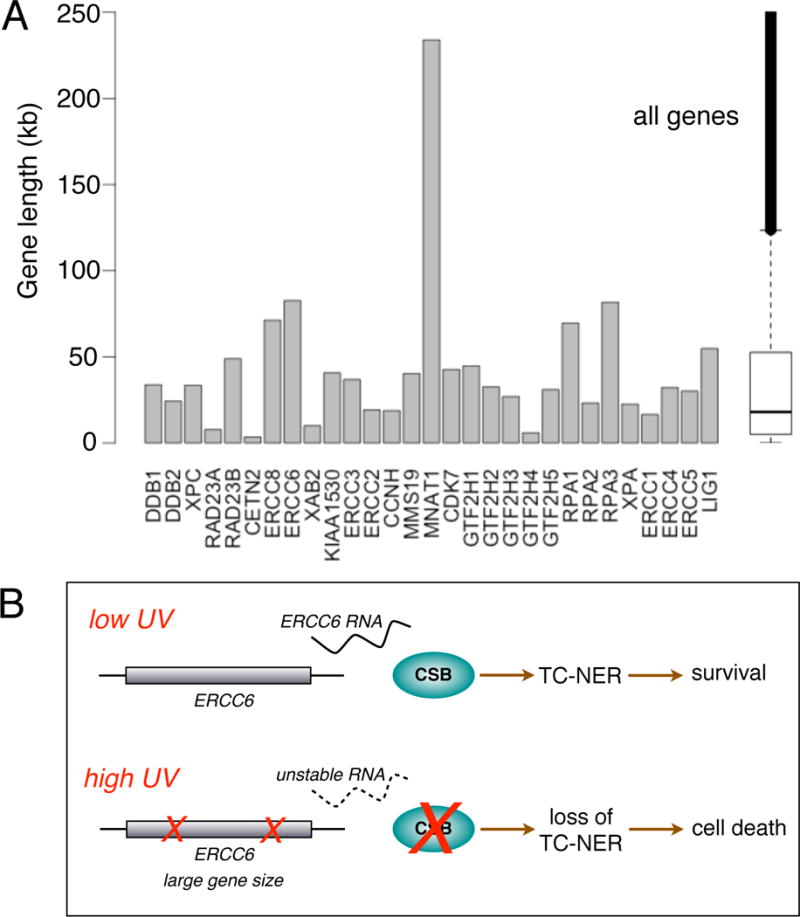
Genomic lengths of human NER genes. (A) The lengths of the 29 human NER genes are expressed in kb. As can be seen, the MNAT1 is the longest gene followed by ERCC6, RPA3 and ERCC8. The median length of all human genes is 18 kb. (B) Model of how ERCC6/CSB may act as a dosimeter of DNA damage to ensure an appropriate level of cell death as a function of UV dose. At lower doses of UV light, cells are able to transcribe sufficient amounts of RNA from the ERCC6 gene to sustain CSB protein levels for proficient TC-NER. At higher levels of UV exposure, transcription-blocking lesions have a high likelihood of forming in the large ERCC6 gene resulting in the inhibition of new synthesis of ERCC6 RNA. Since the ERCC6 transcript is very unstable, it is expected that the levels of ERCC6 RNA will rapidly diminish as transcription of the gene is prohibited, resulting in a time-dependent loss of the CSB protein, loss of TC-NER and induction of cell death.
One of the key factors in TC-NER is the CSB protein, which promotes the recruitment of NER to sites of stalled RNA polymerase II complexes [3, 4]. The CSB protein is synthesized from the ERCC6 gene that spans about 87 kb, a size about three times larger than the median size of all human protein-coding genes, placing it second in size out of the 29 NER genes (Fig. 6A&B). Due to its large size, it represents a rather large target for inactivation by UV light and other agents that are capable of inducing transcription-blocking lesions. Moreover, the ERCC6 transcript was found to be quite unstable across the cell lines so it is expected that the levels of ERCC6 RNA will rapidly decline in the cells following a challenge with high doses of a transcription-blocking agent, such as UV light, eventually leading to diminished levels of CSB protein (Fig. 6C). Since CSB is critical for the recovery of RNA synthesis and survival following exposure to UV light or other agents inducing transcription-blocking lesions, a time-dependent decline in the level of CSB proteins after high doses paints the cells into a corner by running out of the one factor designed to save them from these types of insults. The findings of the unstable nature of the ERCC6 transcript, and its rather large gene size, suggests to us that ERCC6/CSB may act as a dosimeter of DNA damage where above a certain threshold of DNA damage the ERCC6 transcript is depleted to such a degree that it can no longer sustain the translation of the CSB protein resulting in deficient TC-NER and promotion of cell death.
Supplementary Material
Acknowledgments
We would like to thank Michelle Paulsen for excellent technical assistance and the other members of the Ljungman lab for valuable input to this project. We are also grateful for the assistance by Manhong Dai and Fan Meng for administration and maintenance of the University of Michigan Molecular and Behavioral Neuroscience Institute (MBNI) computing cluster and by the personnel at the University of Michigan Sequencing Core. This work has been supported by funds from The University of Michigan Pancreatic Cancer Center, The Translational Oncology Program, University of Michigan Bioinformatics Program, National Institute of Environmental Sciences (1R21ES020946) and National Human Genome Research Institute (1R01HG006786).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement:
None of the authors have any conflicts of interests regarding this manuscript to declare.
References
- 1.Friedberg E, Walker G, Siede W, Wood R, Schultz R, Ellenberger T. DNA Repair and Mutagenesis. ASM Press; Washington, D.C.: 2006. [Google Scholar]
- 2.Wood RD, Mitchell M, Sgouros J, Lindahl T. Human DNA repair genes. Science. 2001;291:1284–1289. doi: 10.1126/science.1056154. [DOI] [PubMed] [Google Scholar]
- 3.Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JH. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol. 2014;15:465–81. doi: 10.1038/nrm3822. [DOI] [PubMed] [Google Scholar]
- 4.Hanawalt PC, Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol. 2008;9:958–70. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- 5.Offman J, Jina N, Theron T, Pallas J, Hubank M, Lehmann A. Transcriptional changes in trichothiodystrophy cells. DNA Repair (Amst) 2008;7:1364–71. doi: 10.1016/j.dnarep.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 6.Spivak G, Itoh T, Matsunaga T, Nikaido O, Hanawalt P, Yamaizumi M. Ultraviolet-sensitive syndrome cells are defective in transcription-coupled repair of cyclobutane pyrimidine dimers. DNA Repair. 2002;1:629–643. doi: 10.1016/s1568-7864(02)00056-3. [DOI] [PubMed] [Google Scholar]
- 7.Bowden NA. Nucleotide excision repair: why is it not used to predict response to platinum-based chemotherapy? Cancer Lett. 2014;346:163–71. doi: 10.1016/j.canlet.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 8.Rahman N. Realizing the promise of cancer predisposition genes. Nature. 2014;505:302–8. doi: 10.1038/nature12981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ljungman M, Zhang F. Blockage of RNA polymerase as a possible trigger for u.v. light-induced apoptosis. Oncogene. 1996;13:823–31. [PubMed] [Google Scholar]
- 10.McKay BC, Becerril C, Ljungman M. P53 plays a protective role against UV- and cisplatin-induced apoptosis in transcription-coupled repair proficient fibroblasts. Oncogene. 2001;20:6805–6808. doi: 10.1038/sj.onc.1204901. [DOI] [PubMed] [Google Scholar]
- 11.Paulsen MT, Veloso A, Prasad J, Bedi K, Ljungman EA, Magnuson B, Wilson TE, Ljungman M. Use of Bru-Seq and BruChase-Seq for genome-wide assessment of the synthesis and stability of RNA. Methods. 2014;67:45–54. doi: 10.1016/j.ymeth.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paulsen MT, Veloso A, Prasad J, Bedi K, Ljungman EA, Tsan YC, Chang CW, Tarrier B, Washburn JG, Lyons R, Robinson DR, Kumar-Sinha C, Wilson TE, Ljungman M. Coordinated regulation of synthesis and stability of RNA during the acute TNF-induced proinflammatory response. Proc Natl Acad Sci U S A. 2013;110:2240–5. doi: 10.1073/pnas.1219192110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wagner GP, Kin K, Lynch VJ. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012;131:281–5. doi: 10.1007/s12064-012-0162-3. [DOI] [PubMed] [Google Scholar]
- 14.Derheimer FA, O’Hagan HM, Krueger HM, Hanasoge S, Paulsen MT, Ljungman M. RPA and ATR link transcriptional stress to p53. Proc Natl Acad Sci USA. 2007;104:12778–83. doi: 10.1073/pnas.0705317104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKay BC, Stubbert LJ, Fowler CC, Smith JM, Cardamore RA, Spronck JC. Regulation of ultraviolet light-induced gene expression by gene size. Proc Natl Acad Sci U S A. 2004;101:6582–6586. doi: 10.1073/pnas.0308181101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ljungman M, Lane DP. Transcription – guarding the genome by sensing DNA damage. Nat Rev Cancer. 2004;4:727–37. doi: 10.1038/nrc1435. [DOI] [PubMed] [Google Scholar]
- 17.Nijhawan D, Fang M, Traer E, Zhong Q, Gao W, Du F, Wang X. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 2003;17:1475–86. doi: 10.1101/gad.1093903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKay B, Becerril C, Spronck J, Ljungman M. Ultraviolet light-induced apoptosis is associated with S-phase in primary human fibroblasts. DNA Repair. 2002;1:811–820. doi: 10.1016/s1568-7864(02)00109-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.