

Abstract
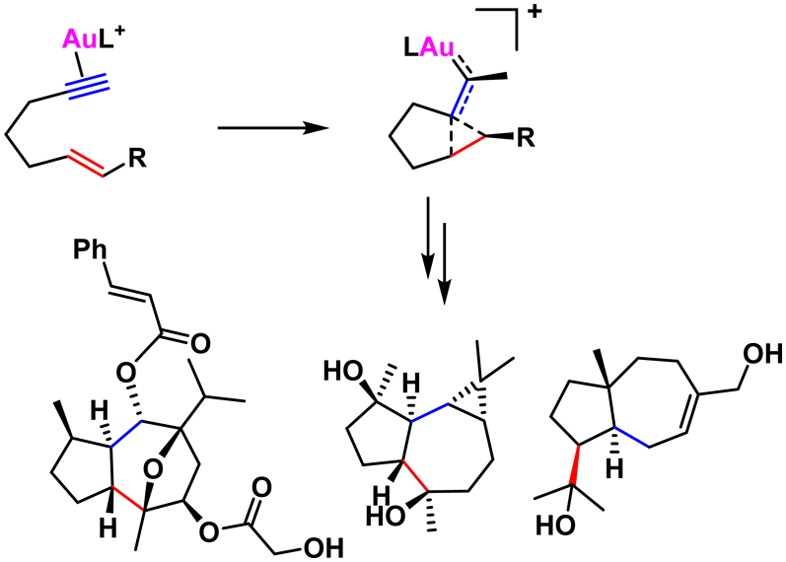
Cycloisomerizations of 1,n-enynes catalyzed by gold(I) proceed via electrophilic species with a highly distorted cyclopropyl gold(I) carbene-like structure, which can react with different nucleophiles to form a wide variety of products by attack at the cyclopropane or the carbene carbons. Particularly important are reactions in which the gold(I) carbene reacts with alkenes to form cyclopropanes either intra- or intermolecularly. In the absence of nucleophiles, 1,n-enynes lead to a variety of cycloisomerized products including those resulting from skeletal rearrangements. Reactions proceeding through cyclopropyl gold(I) carbene-like intermediates are ideally suited for the bioinspired synthesis of terpenoid natural products by the selective activation of the alkyne in highly functionalized enynes or polyenynes.
1. Introduction
Homogeneous gold catalysis is a relatively new field of research that became popular in the early years of this century. Although there were a few scattered papers describing reactions of aromatic compounds with gold salts, the first signals of the synthetic potential of gold in homogeneous catalysis came with the work of Teles1 and Tanaka2 on the addition of alcohols and water to alkynes.
Many reviews have covered different aspects of homogeneous gold catalysis.3−12 Here, we focus on the developments of synthetic methods that were inspired on our initial studies of platinum(II)-catalyzed reactions and, in particular, on the multifaceted nature of cyclopropyl metal carbenes, key electrophilic intermediates formed by the activation of alkynes in functionalized 1,n-enynes and related substrates.
Inspired by the seminal work of the group of Trost on the electrophilic activation of enynes using palladium(II) complexes as catalysts,13 which was followed by related work by Murai and Chatani using ruthenium(II)14,15 and by the synthesis of phenols by intramolecular reactions of furans with alkynes developed by Hashmi,16 we initially examined the intramolecular reaction of allylsilanes and allylstannanes with alkynes, discovering that PtCl2 was the best catalyst, although palladium(II), ruthenium(II), and silver(I) salts and complexes could also be used.17,18 Several other groups had almost concurrently found that platinum(II) also catalyzed the skeletal rearrangement of 1,6-enynes and other cycloisomerizations under relatively mild conditions.19−24 We also found that PtCl2 was an excellent catalyst for the addition of water and alcohols (hydroxy- and alkoxycyclization) to 1,6-enynes such as 1a–d, a process that proceeds stereospecifically (Scheme 1).25 Although these reactions proceed by an exo-dig pathway, the alternative endo-dig cyclization mode was also observed in other cases.26
Scheme 1. Platinum(II)-Catalyzed Methoxycyclization of 1,6-Enynes.

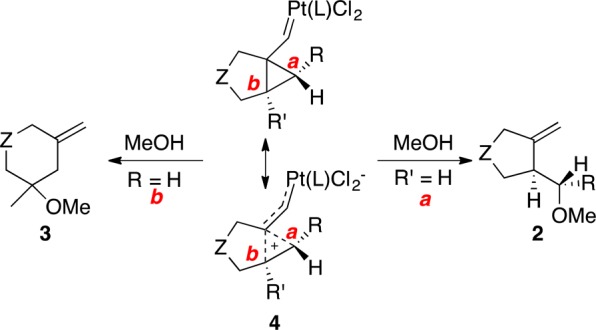
The formation of five- or six-membered ring compounds 2a–c or 3, respectively, was rationalized by the competitive opening of a common intermediate 4 by attack of the nucleophile at the most electrophilic site, which was dictated by the substitution of the alkene following the Markovnikov rule, a hypothesis that was supported by DFT calculations (Scheme 2).27 Intermediates 4 can be described as cyclopropyl platinum(II) carbenes, although the contribution of a charge-separated resonance form corresponding to metal stabilized homoallyl carbocations is also significant. It is important to note that the involvement of very similar reactive intermediates was already suggested in the palladium(II)-13,28 and ruthenium(II)-catalyzed15 reactions of 1,6-enynes.
Scheme 2. Mechanistic Rationale for the Regioselectivity Observed in the Exo-Dig Cyclization of Platinum(II)-Catalyzed Methoxycyclizations of 1,6-Enynes.
In a parallel work, we also examined the synthesis of phenols by cyclization of furans with alkynes using platinum(II) catalysts29,30 instead of gold(III), as originally reported by Hashmi,16 concluding that this transformation is mechanistically related to metal-catalyzed cycloisomerization of enynes in which the furan ring acts as an electron-rich alkene (Scheme 3). According to the DFT studies performed for the platinum(II)-catalyzed reaction, the attack of the furan to the η2-alkyne metal complex 5 leads to a cyclopropyl metal carbene 6, which opens up to form 7. Cyclization to 8, followed by elimination of the metal generates oxepine 9, in tautomeric equilibrium with arene oxide 10, which leads to phenols 11. Although almost all the examples reported concerned with the intramolecular reaction of furanynes, we recently developed an intermolecular version using cationic gold(I) catalysts.31
Scheme 3. Mechanism for the Pt(II)- or Au(I)-Catalyzed Reaction of Furans with Alkynes To Form Phenols.

2. Gold(I)-Catalyzed Cyclizations of 1,n-Enynes
During our initial studies focused on the use of platinum(II) complexes as catalysts, we also found that highly Lewis acidic AuCl3 could be used as a catalyst in some cases.26 However, we soon realized that cationic phosphine gold(I) complexes were more convenient catalysts, actually being the most active and selective for reactions of alkynes, outperforming platinum(II) and all the other metal catalysts that we had examined for the addition of heteronucleophiles, cycloisomerizations of 1,6-enynes, and similar transformations.32,33 First, we generated in situ the reactive cationic gold(I) complexes by protonolysis of neutral complex Ph3PAuMe, which leads to the formation of methane.32 However, this method introduces a strong Brønsted acid into the reaction medium, which may promote unwanted side reactions. More convenient proved to be the use of Ph3PAuCl and a silver(I) salt with a noncoordinating anion such as AgSbF6.32,33 Recently, we found that the mode of activation of the LAuCl precatalysts with silver(I) salts has an important effect in catalysis.34 Thus, if complexes LAuCl are allowed to react with AgX in poorly coordinating solvents and in the absence of the substrate, dinuclear chloride-bridged species [LAuClAuL]+X– are readily formed, regardless of the counterion. These very robust complexes are poor catalysts, so that their formation significantly reduces the reaction rate. The formation of [LAuClAuL]+X– can be minimized by premixing LAuCl with the substrate, followed by addition of silver salts, allowing formation of complexes [LAu(substrate)]+X–, which can directly enter the catalytic cycles. Of course, the formation of dinuclear chloride-bridged complexes can be entirely circumvented by the use of silver-free, cationic [LAuL′]+X–, where L′ is a weakly coordinating ligand, which are often the alternative of choice in gold(I) catalysis.
Along with our initial study in 2004 on the cycloisomerization of 1,6-enynes such as 1e to form 1,3-diene 12a by skeletal rearrangement,32 the groups of Fürstner35 and Toste36 reported related examples of cycloisomerization of 1,5-enynes of type 13a,b to give bicyclo[3.1.0]hex-2-ene derivatives 14a,b, respectively, under almost identical conditions using Ph3PAuCl and a silver salt (Scheme 4). A gold(I)-catalyzed Conia–ene reaction of β-ketoesters with alkynes was also reported by Toste.37
Scheme 4. Gold(I)-Catalyzed Skeletal Rearrangement of 1e(32) and Cycloisomerizations of 1,5-Enynes 13a(35) and 13b(36).

In general, cationic gold(I) complexes activate selectively alkynes3,12 even in the presence of other potentially coordinating functional groups as alkenes.38 It is important to remark that, unlike other transition metals, gold(I) does not promote Alder–ene cycloisomerizations of 1,n-enynes, which require the unfavorable coordination of gold(I) to both the alkyne and the alkene. The oxidative cyclometalation of gold(I) to form a gold(III) metallacycle, is also a very unlikely process.33,39 In the case of 1,6-enynes, gold(I) forms (η2-alkyne)metal complexes 15(40) that react intramolecularly with the alkene to form cyclopropyl gold(I) carbene-like intermediates 16 and/or 17 by 5-exo-dig or 6-endo-dig cyclization, respectively (Scheme 5).32,41−44 Similar pathways are followed by 1,5-,45−47 1,7-,41,48 and higher enynes49−52 in the presence of gold(I) catalysts. According to DFT calculations, intermediates 16 and 17 are even more delocalized cationic structures than their Pt(II) counterparts, although for convenience, we prefer to represent these complexes as cyclopropyl gold(I) carbenes to highlight their propensity to undergo cyclopropanation reactions. However, although the back-donation from gold(I) to the cationic centers in these metal carbene-like structures is low,53−55 it still provides sufficient stabilization56,57 as has recently been shown in a few well-characterized gold carbenes [LAu=CR2]+ and [L2Au=CR2]+.58,59 The bond between Au and C in these gold(I) carbenes has been described as a half-double bond.56 Terminologically, we recommend using the term gold carbenes rather than gold carbenoids for these species57 since genuine gold(I) carbenoids LAu–CH2–X (X = Cl, SPh) have been structurally characterized.60,61 These neutral complexes can be considered as formal precursors of gold methylidene complex [LAu=CH2]+ by an α-elimination of the X group.
Scheme 5. Main Pathways for the Gold(I)-Catalyzed Cycloisomerization of 1,6-Enynes.
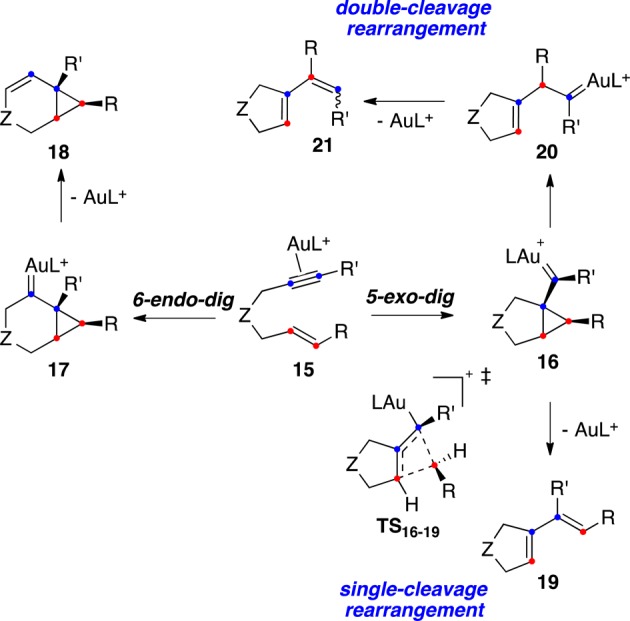
Intermediates 17 usually lead to bicyclo[4.1.0]hept-2-ene derivatives 18 by α-proton elimination (Scheme 5).62−64 On the other hand, intermediates 16 of 5-exo-dig cyclization afford 1,3-dienes 19 by a skeletal rearrangement (single-cleavage) in an interesting process that gives rise directly the η2-coordinated gold(I) diene complex and involves a 1,3-suprafacial migration of the terminal carbon of the alkene via transition state TS16–19.41 Intermediates 16 can also undergo a slightly different migration (double-cleavage rearrangement) by the formal insertion of the terminal alkene carbon into the alkyne carbons that forms a new gold(I) carbene 20. Carbenes 20 then undergo α-proton elimination to give 1,3-dienes 21. Compounds 21 (R = H) with Z configuration usually predominate, although both E- and Z-configured products have been observed.65−67
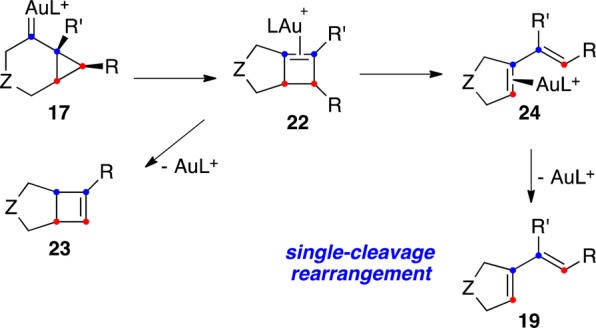
The scenario shown in Scheme 5 is a simplification, as other similar pathways may also be possible depending on the substitution pattern of the substrate. Thus, intermediates 17 of endo-dig cyclization can undergo ring expansion to give (η2-cyclobutene)gold(I) complexes 22, which can evolve to form bicyclo[3.2.0]hept-2-ene derivatives 23 (Scheme 6).62,63,68,69 Highly strained bicyclo[3.2.0]hept-5-enes have only been isolated in a few cases.42,70 Intermediates 22 can also open up to form 24 and then dienes 19 in an overall single-cleavage process.42
Scheme 6. Alternative Mechanism for the Gold(I)-Catalyzed Single-Cleavage Rearrangement of 1,6-Enynes.
Isomeric 1,6-enynes 1f and 1g, which differ in the substitution pattern of the alkyne, react with gold(I) to form the same 1,3-diene 12b by two different mechanisms, single- and double-cleavage rearrangement, respectively (Scheme 7).32,33,41 Other 1,6-enynes bearing terminal alkynes such as 1d and 1h give 1,3-dienes 12c,d by single-cleavage rearrangement.32,33
Scheme 7. Single- and Double-Cleavage Rearrangement of 1,6-Enynes.
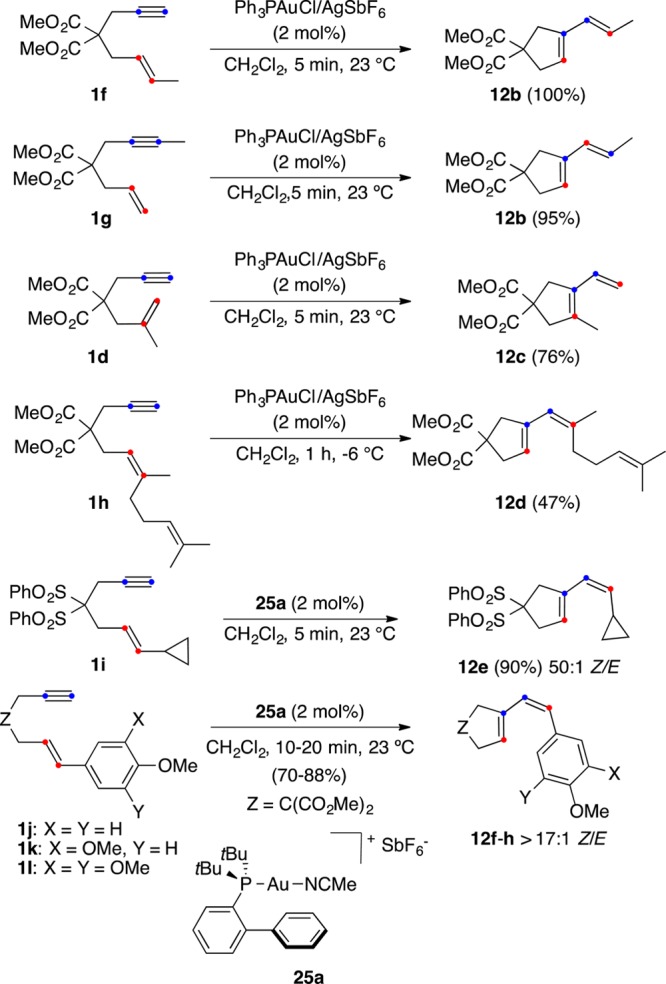
Somewhat surprisingly, E-1,6-enynes such as 1j–l, with strongly electron-donating substituents at the terminal alkene carbon, react with cationic gold(I) catalyst 25a to give Z-configured dienes 12f–h (Scheme 7).71 This Z-selectivity was also found using different electrophilic gold(I) or platinum(II) catalysts. This is rather mechanistically puzzling, since both the platinum(II)- and the gold(I)-catalyzed single-cleavage rearrangements are in the vast majority of cases stereospecific processes in which the configuration of the alkene is retained.32,41,65,72 Complexes of gold(I) bearing commercially available bulky dialkyl biphenyl phosphines such as 25a,73 proved to be particularly useful in homogeneous gold(I) catalysis. Experimental and theoretical studies confirmed that gold(I) does not interact significantly with the closest arene ring of the ligand.74
3. Gold(I)-Catalyzed Intermolecular Reactions of Alkynes with Alkenes
We examined the intermolecular reaction between alkynes and electron-rich alkenes with many different gold(I) catalysts, although only complex mixtures of oligomers were obtained. In addition, electron-rich alkenes are very good ligands for gold(I),38 thus competing with the alkyne for the coordination to the metal. However, by using cationic gold(I) complex 25b with a very bulky phosphine, cyclobutenes 26 were finally obtained regioselectively for a variety of electron-rich alkenes (Scheme 8).75 Interestingly, the isolated yields were improved in most cases by changing the anion from SbF6– to softer BAr4F–, presumably by minimizing the formation of unproductive σ, π-digold(I) alkyne digold(I) complexes, an undesired side reaction in many gold-catalyzed reactions of terminal alkynes.76 The same improvement was observed in the intramolecular [2 + 2] cycloaddition of alkynes with alkynes in substrates of type 27 to form macrocycle 28 (Scheme 9).76,77
Scheme 8. [2 + 2] Cycloaddition of Arylalkynes with Alkenes.
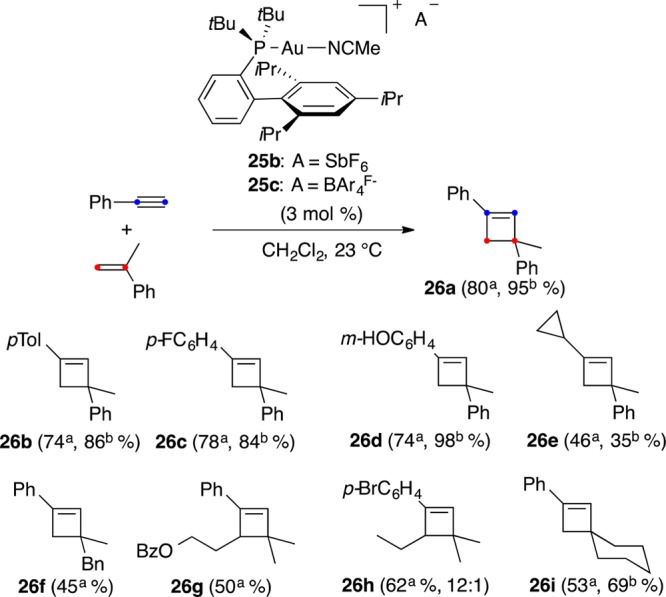
Reaction performed with catalyst 25b.
Reaction performed with catalyst 25c.
Scheme 9. Macrocyclization via [2 + 2] Cycloaddition of Alkynes with Alkenes.

Reaction performed with catalyst 25b.
Reaction performed with catalyst 25c.
The regioselective formation of cyclobutenes by [2 + 2] cycloaddition probably proceeds by electrophilic addition of the (η2-alkyne)gold(I) complex to the alkene via TS29–30 to form a cyclopropyl gold(I) carbene 30 very similar to intermediate 16 involved in the cyclization of enynes (Scheme 10).75 Ring expansion of 30 through TS30–31 would give (η2-cyclobutene)gold(I) complex 31. Intermediate 30 was also trapped intramolecularly with an alkene to form the corresponding cyclopropane.
Scheme 10. Divergent Pathways in the Gold(I)-Catalyzed Intermolecular Reactions of Alkynes with Alkenes.
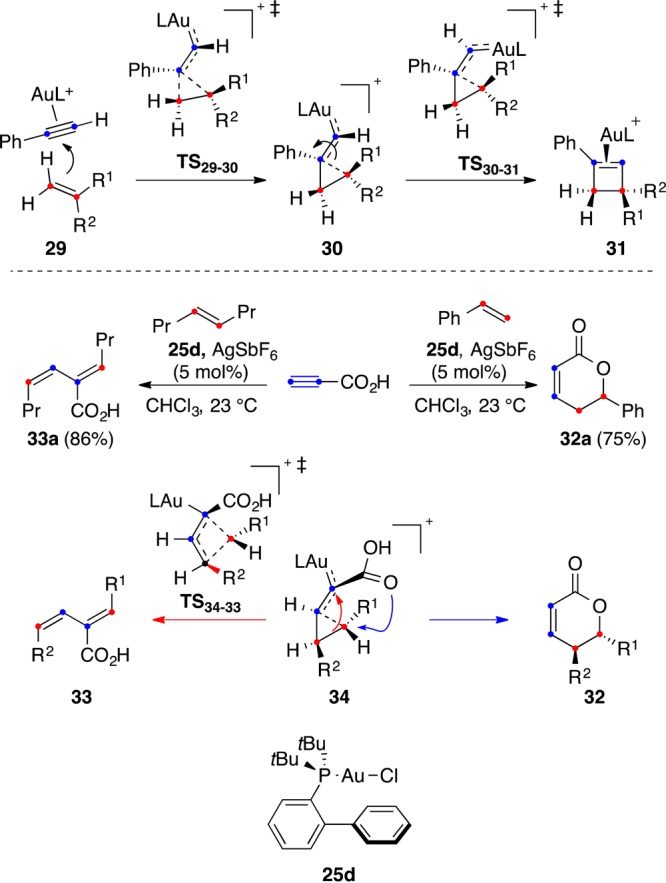
The intermolecular reaction of propiolic acid and related alkynes with strongly electron-withdrawing groups gives dihydropyrones 32 or 1,3-dienes 33 via a similar mechanism, although the initial attack of the alkene to the (η2-alkyne)gold(I) complex occurs with the opposite regiochemistry (Scheme 10).78 Thus, cyclopropyl gold(I) carbene intermediate 34, with gold bonded to the internal carbon of the alkyne, evolves by intramolecular opening to form 32 by attack of the carboxylic acid to the most substituted carbon of the alkene. On the other hand, in the reaction of alkenes with two very similar substituents, intermediate 34 undergoes a 1,3-migration similar to that found in the single-cleavage rearrangement to form stereospecifically 1,3-dienes 33. A transition state similar to TS34–33 has been proposed in the reaction of cyclopropyl gold(I) carbenes generated by retro-Buchner reaction of 7-cyclopropyl 1,3,5-cycloheptatrienes.79
4. Gold(I)-Catalyzed Nucleophilic Additions to Enynes
4.1. Additions of Heteronucleophiles
Gold(I) complexes are able to catalyze the addition of amines, alcohols, or water to 1,n-enynes affording products of amino-, alkoxy-, or hydroxycyclization under much milder conditions than other metal catalysts.26,32,33,62,80−82 These additions are stereospecific, as illustrated by the alkoxycyclization of diastereomeric enynes 1m and 1h, and proceed via opening of cyclopropyl gold(I) carbene intermediates 35 by attack of the nucleophile to the cyclopropane ring (Scheme 11). Thus, the overall process is an anti addition of the alkyne–gold(I) complex and the heteronucleophile to an alkene following the Markovnikov regiochemistry, which is further demonstrated by the reaction of enyne 1d with gold(I) in the presence of MeOH to form six-membered ring 3 via intermediate 36. Analogous additions of heteronucleophiles to 1,5-80,83 and 1,7-enynes48 are also stereospecific.
Scheme 11. Anti-Addition of Nucleophiles to 1,6-Enynes.

1,6-Enynes such as 1n bearing an aryl substituent at the alkyne terminus undergo an analogous 5-exo methoxycyclization to afford 2f in the presence of gold(I) and MeOH (Scheme 12).33 However, similar 1,6-enyne 1o tethered by a benzene ring undergoes gold(I)-catalyzed hydroxy- or alkoxycyclizations preferentially via 6-endo-dig pathway through intermediates 37 giving rise to dihydronaphthalenes 38.84
Scheme 12. Hydroxy- and Alkoxycyclization of 7-Aryl-Substituted 1,6-Enynes.

Amino- and alkoxycyclizations can also take place intramolecularly starting from amino- or hydroxy-1,n-enynes.33,85,86 Thus, 1,6-enyne 1p containing a propargylic alcohol gave 40 quantitatively upon treatment with gold(I) as a result of an intramolecular attack of the hydroxyl group to cyclopropyl gold carbene intermediate 39 (Scheme 13).33 In a mechanistically related transformation, amino- or hydroxy-1,5-enynes 41 afforded in the presence of gold(I) spirofused heterobicyclic compounds 43 through intermediates of type 42.85 In a similar vein, 1,n-enynes bearing nucleophilic moieties such as carboxylic acids,53,87 carbamates,88 or carbonates89 are also prone to undergo gold-catalyzed tandem cyclizations, giving rise to a variety of complex molecular architectures.
Scheme 13. Cyclizations of Amino- And Hydroxy-1,n-enynes.

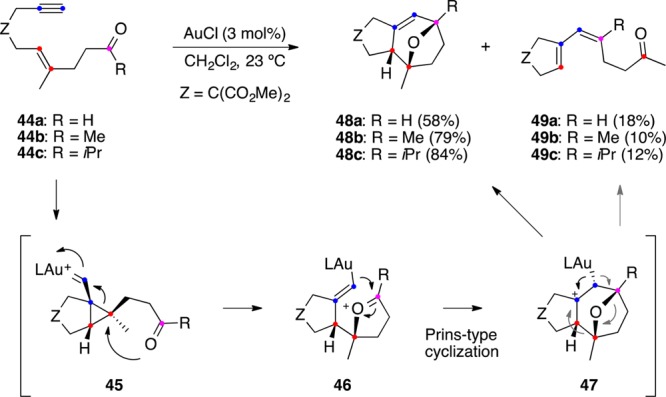
Oxo-1,6-enynes 44 also react in the presence of gold(I) by a formal [2 + 2 + 2] alkyne/alkene/carbonyl cycloaddition to afford predominantly oxatricyclic compounds 48, together with minor amounts of dienes 49 (Scheme 14).90 These reactions proceed via intramolecular attack of the carbonyl to cyclopropyl gold carbenes 45 to form oxonium cations 46, which undergo a Prins-type cyclization to give intermediates 47. Demetalation from 47 forms oxatricycles 48, whereas an alternative elimination with fragmentation of the seven-membered ring leads to dienes 49. Oxo-1,5-enynes also undergo an intramolecular gold(I)-catalyzed reaction to form oxatricyclic adducts.91
Scheme 14. Intramolecular [2 + 2 + 2] Alkyne/Alkene/Carbonyl Cycloaddition of Oxo 1,6-Enynes.
We exploited gold(I)-catalyzed intramolecular [2 + 2 + 2] alkyne/alkene/carbonyl cycloadditions in the synthesis of several oxygen-bridged sesquiterpenoids. Thus, ketoenynes 44d and 44e were respectively converted into oxatricycles 48d and 48e, which are key intermediates in the total syntheses of (+)-orientalol F (50) and pubinernoid B (51) (Scheme 15).92 Oxo-1,6-enyne 44f was analogously cyclized to form 48f, which was subsequently converted into (−)-englerin A (52).93 Another total synthesis of 52 included a very similar gold(I)-catalyzed cyclization as the key step.94 Remarkably, an unprotected aldol subunit could be used in both syntheses as the substrate for the gold-catalyzed reaction.
Scheme 15. Intramolecular [2 + 2 + 2] Cycloadditions of Oxo 1,6-Enynes in Total Synthesis.

1,n-Enynes also react intermolecularly with carbonyl compounds giving rise to a wide range of products depending on the substitution pattern of the alkene and the nature of the carbonyl compound. 1,6-Enynes such as 1q react with aromatic aldehydes in a formal [2 + 2 + 2] cycloaddition to give oxabicyclic adducts of type 55 together with dienes 56, which result from a metathesis-type reaction of the enyne with the aldehyde (Scheme 16).95 The formation of these products can be explained by attack of the aldehyde to the cyclopropyl gold carbene intermediate to form 53a, which undergoes a Prins-type cyclization followed by either demetalation to form 55 or fragmentation to give 56. This mechanism is analogous to that proposed for the intramolecular [2 + 2 + 2] cycloaddition of oxo-1,6-enynes (Scheme 14).90 1,7-Enynes also undergo [2 + 2 + 2] cycloadditions with carbonyl compounds to give analogous products.96 On the other hand, following a related mechanism, 1,6-enyne 1q reacts with cyclopropenones in a ring-expanding spiroannulation to afford spirocyclic cyclopentenones 58 after incorporation of a molecule of water to rearranged intermediate 57.97 1,6-Enynes 1r bearing a monosubstituted alkene react with aldehydes and ketones in a different way to form tricyclic compounds 61.98,99 This transformation presumably proceeds via trapping of the rearranged carbene that results from the enyne to form oxonium cation 59, followed by Prins cyclization and demetalation.
Scheme 16. Intermolecular Cycloadditions of 1,6-Enynes and Carbonyl Compounds.
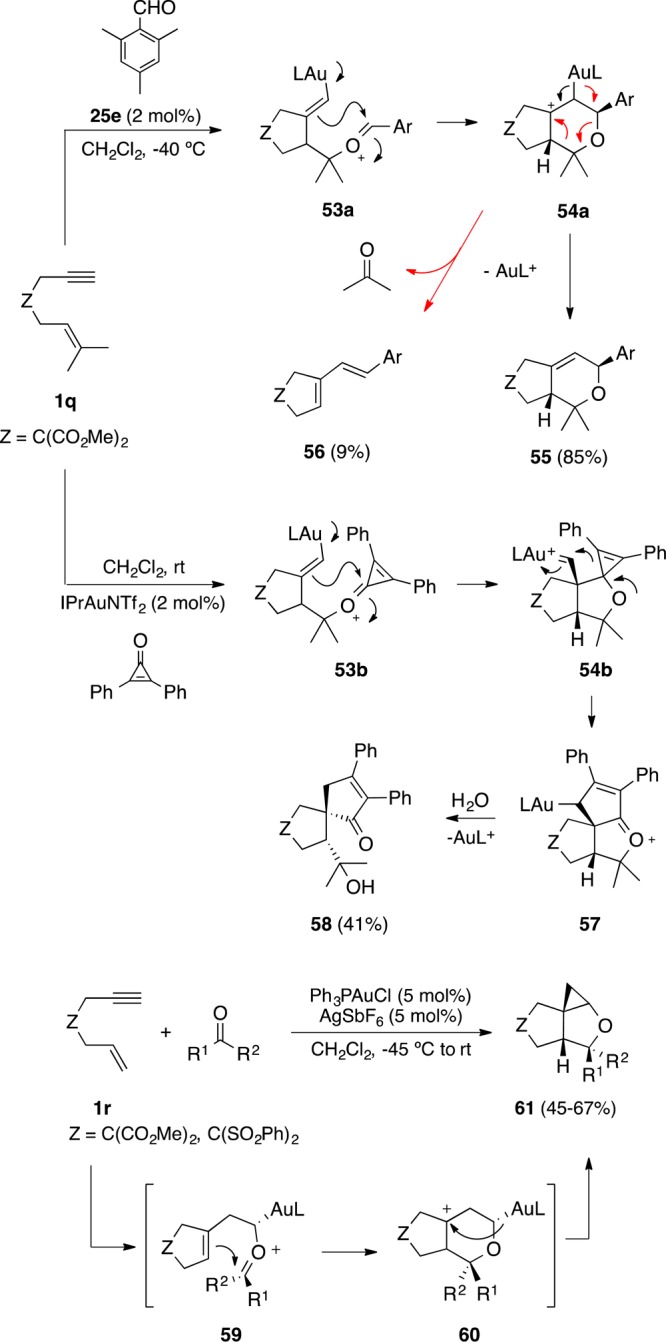
The intermolecular reaction of terminal alkynes with oxoalkenes 62 leads to 8-oxabicyclo[3.2.1]oct-3-enes 64 as a result of a formal [2 + 2 + 2] alkyne/alkene/carbonyl cycloaddition via cyclopropyl gold carbene intermediates 63 (Scheme 17).100
Scheme 17. Intermolecular [2 + 2 + 2] Alkyne/Alkene/Carbonyl Cycloaddition of Terminal Alkynes and Oxoalkenes.
4.2. Additions of Carbonucleophiles
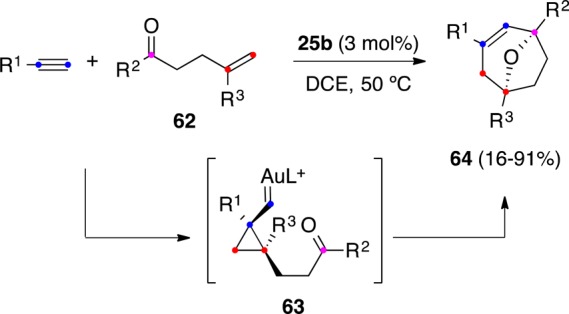
Electron-rich aromatic and heteroaromatic compounds can act as nucleophiles reacting with 1,6-enynes.101−104 Cyclopropyl gold(I) carbenes that result as intermediates in the cyclization of 1,6-enynes can act as bifunctional electrophiles, reacting with arenes either at the carbene or at the cyclopropane.101 Hence, indole reacted with 1,6-enyne 1s in the presence of different gold(I) complexes to give two different adducts 66 and 67 (Scheme 18). We found that the more electrophilic gold(I) complexes with less donating phosphite or phosphine ligands favored the attack to the cyclopropane ring to give 66, whereas complexes with more donating NHC ligands favored the formation of 67 by attack at the metal carbene. An analogous reaction in the presence of a gold(I) catalyst generated in situ from Ph3PAuCl/AgSbF6 was reported to give exclusive formation of products of type 66 by attack to the cyclopropane ring.103
Scheme 18. Regiodivergent Addition of Indole to 1,6-Enynes.
1,3-Dicarbonyl compounds can also add to 1,6-enynes such as 1s acting as carbonucleophiles through their enol tautomer to give compounds 68 (Scheme 19).102 However, some dicarbonyl compounds such as 1,3-cyclohexadione may act as O-nucleophiles to form adduct 69. As in the addition of heteronucleophiles to 1,6-enynes, products derived from the attack at the carbene carbon are favored with more donating ligands, whereas more electrophilic complexes favor the attack to the cyclopropane. Gold(I) also catalyzes the addition of allyl silanes to 1,6-enynes, as well as the addition of a variety of carbonucleophiles to 1,5-enynes.102
Scheme 19. Addition of 1,3-Dicarbonyl Compounds to 1,6-Enynes.

Intramolecular attack of aryl groups in 1,6-enynes of type 1t leads stereospecifically to tricyclic products such as 74 of intramolecular formal [4 + 2] cycloaddition (Scheme 20).62,63,105 This is a very general reaction that proceeds via initial exo-cyclization of the enyne followed by opening of the cyclopropyl gold(I) carbene 70 by a Friedel–Crafts-type reaction. These cycloadditions can be performed enantioselectively in the presence of chiral phosphine106 or phosphite107 gold(I) complexes. The endo-cyclization of the 1,6-enyne also takes place in certain cases, being the preferred pathway for arylalkynes bearing enesulfonamides or enamines such as 72 to afford tricycles 74.108
Scheme 20. Intramolecular Friedel–Crafts-Type Additions to 1,6-Enynes.

Benzyl-substituted 1,5-enynes such as 13c also undergo a formal [4 + 2] cycloaddition via 5-endo-dig cyclization to afford tricyclic product 75.47 In contrast, 1,5-enynes 13d with an aryl substituent at the alkyne react with gold(I) through a different mechanism to form dihydrobenzofluorenes 78 in a formal [3 + 3] cycloaddition.109 This transformation can be explained by a 1,2-H shift in intermediate 77, followed by a Friedel–Crafts alkylation (Scheme 21).
Scheme 21. Intramolecular Friedel–Crafts-Type Additions to 1,5-Enynes.

5. Cyclopropanation Reactions
The carbene-like character of the intermediates formed in metal-catalyzed cycloisomerizations of enynes is more clearly exhibited in intra- and intermolecular cyclopropanations of alkenes.110−112
5.1. Intramolecular Cyclopropanations of Enynes
Formation of tetracyclic compounds 81a,b by cyclization of dienynes 1u,v at room temperature nicely illustrates the high reactivity and stereoselectivity usually exerted by gold catalysts (Scheme 22).112 This reaction, which was first reported using ruthenium(II) complexes under more forcing conditions (80 °C, toluene),15 proceeds by a 5-exo-dig cyclization followed by cyclopropanation of the resulting cyclopropyl gold(I) carbene 80. When the starting dienynes are cyclic substrates these intramolecular cyclopropanations lead to very complex ring systems.113 1,5-Dienynes such as 13e react similarly to afford pentacyclic product 83 through endo-carbene 82.47
Scheme 22. Intramolecular Cyclopropanation of 1,n-Enynes.

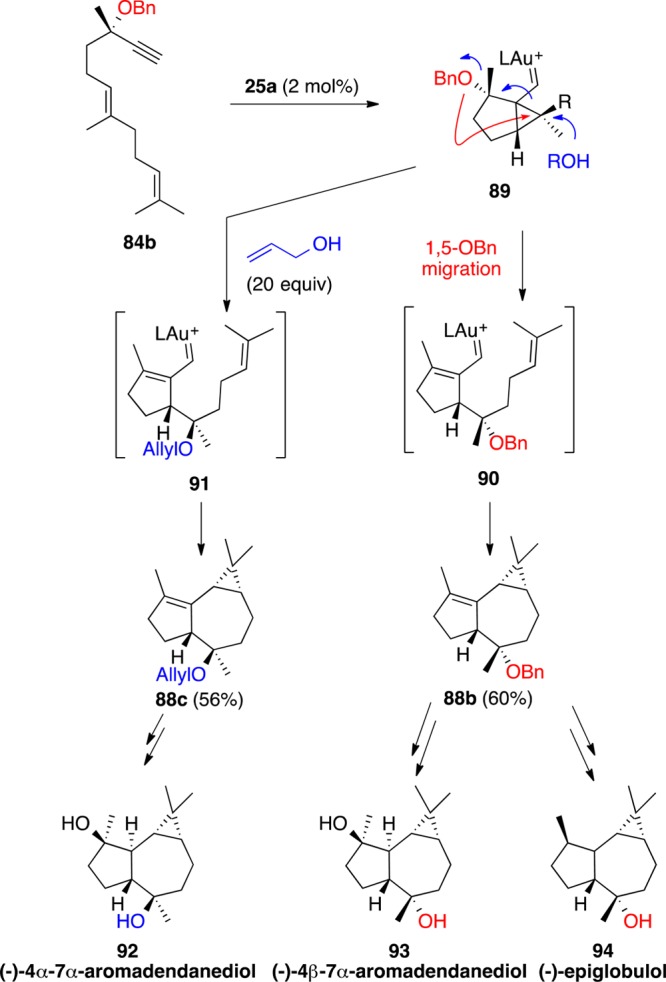
Dienynes bearing a methoxy or other OR group at the propargylic position such as 87a undergo an intramolecular 1,5-OR migration in the presence of gold(I) to form tricyclic compounds 88a (Scheme 23).114 This reaction presumably takes place via cyclopropyl gold(I) carbene intermediate 85, which leads to bridged system 86 by attack of the OR group at the cyclopropane. Opening of 86 results in the formation of α,β-unsaturated gold(I) carbene 87, which is prone to undergo intramolecular cyclopropanation with the alkene at the side chain to form 88a.
Scheme 23. 1,5-Migration of Propargyl OR Groups in Dienynes.

This cascade cyclization process is ideally suited for the total syntheses of sesquiterpenes (−)-4α,7α-aromadendranediol (92), (−)-4β,7α-aromadendranediol (93), and (−)-epiglobulol (94) from a single dienyne 84b by using a stereodivergent gold(I)-catalyzed reaction as the key step (Scheme 24).115 In the absence of any external nucleophile, cyclopropyl gold(I) carbene intermediate 89 undergoes a 1,5-OBn migration followed by intramolecular cyclopropanation to form tricyclic compound 88b, which is the precursor of 93 and 94. On the other hand, by adding an external nucleophile such as allyl alcohol, intermediate 89 leads to 91 and then to 88c after intramolecular cyclopropanation.
Scheme 24. Synthesis of Aromadendrane Sesquiterpenes.
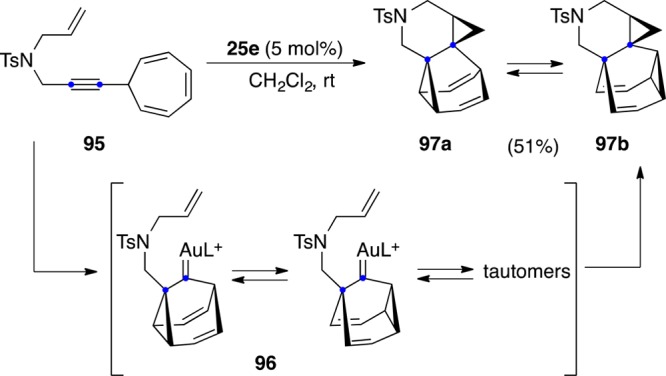
Intramolecular cyclopropanation of a gold(I) carbene can lead to very complex structures starting from 7-alkynylcyclohepta-1,3,5-trienes 95 (Scheme 25).116 This reaction generates highly fluxional barbaralyl gold(I) cations 96 that can be trapped by the pending alkene to form 97a,b, which are in equilibrium by Cope rearrangement both in the solid state and in solution.
Scheme 25. Intramolecular Cyclopropanation from 7-Alkynylcyclohepta-1,3,5-heptatrienes.
5.2. Intermolecular Cyclopropanations of Enynes
Intermediate cyclopropyl gold(I) carbenes such as 65 resulting from the cyclization of 1,6-enynes can also be trapped intermolecularly by cyclic or acyclic alkenes to afford adducts 98 stereoselectively (Scheme 26).117,118 1,6-Enynes such as 1d bearing a terminal alkene react differently yielding 100, probably by rearrangement of cyclopropyl gold(I) carbene 36 to give carbene 99. The involvement of carbene 99 in the trapping by the alkene also supports the involvement of this type of species as intermediates in the double-cleavage rearrangement (see Scheme 5).
Scheme 26. Intermolecular Cyclopropanation of 1,6-Enynes.

In the presence of gold(I), 1,6-enyne 84c undergoes a cyclization followed by 1,5-acetoxy migration to give α,β-unsaturated carbene 103, which reacts intermolecularly with alkene 104 giving rise to 105 with only 5% loss of enantiomeric excess (Scheme 27).119 This transformation has been used as the key step in the total synthesis of antiviral sesquiterpene (+)-schisanwilsonene A (106). It is remarkable that the cyclization/1,5-acetoxy migration is faster than the alternative 1,2-acyloxy migration, which would lead to racemization.
Scheme 27. Synthesis of (+)-Schisawilsonene A.

6. Concluding Remarks
Gold(I) catalysts trigger complex reactions of substituted 1,n-enynes by stabilizing the key reactive cationic intermediates, which can be viewed in a simplified form as cyclopropyl gold(I) carbenes. These intermediates react with diverse nucleophiles inter- or intramolecularly and cyclopropanate alkenes leading to complex structures with total atom economy. This work, along with that of many other groups, has promoted gold from a mere curiosity to the metal of choice for the activation of alkynes in complex molecular settings under homogeneous conditions. The same basic principles uncovered during the mechanistic study of gold(I)-catalyzed reactions of 1,n-enynes can also been extended for reactions of more complex systems as well as for the development of more challenging intermolecular transformations between alkynes and alkenes. In this regard, in order to fulfill the synthetic potential of intermolecular transformations, new types of gold(I) catalysts that selectively activate alkynes in reactions with highly functionalized alkenes are still required, particularly in the arena of enantioselective synthesis.
Acknowledgments
We thank the MINECO (project CTQ2013-42106-P and Severo Ochoa Excellence Accreditation 2014-2018 (SEV-2013-0319), the European Research Council (Advanced Grant No. 321066), the AGAUR (2014 SGR 818), and the ICIQ Foundation for support.
Biographies

Ruth Dorel was born in Zaragoza (Spain) in 1989. She graduated with a degree in Chemistry from the Universidad de Zaragoza in 2012. In 2013, she was awarded the Master of Synthesis and Catalysis Extraordinary Prize at the Universitat Rovira i Virgili (Tarragona, Spain). She has been carrying out her Ph.D. studies at the Institute of Chemical Research of Catalonia (ICIQ) since 2013 under the supervision of Prof. Antonio M. Echavarren, working on the synthesis of natural products and polycyclic aromatic hydrocarbons.

Antonio M. Echavarren was born in Bilbao in 1955 (Basque Country, Spain) and obtained his Ph.D. in 1982 at the Universidad Autónoma de Madrid (UAM, 1982) with Prof. Francisco Fariña. After a postdoctoral stay in Boston College with Prof. T. Ross Kelly, he joined the UAM as an Assistant Professor (1984–1986). Following a two-year period as a NATO fellow in the group of Prof. John K. Stille in Fort Collins (Colorado State University), he joined the Institute of Organic Chemistry of the CSIC in Madrid. In 1992, he returned to the UAM as a Professor of Organic Chemistry. He has also been a Professor of Research of the CSIC since 2004. In 2004, he moved to Tarragona as a Group Leader at the newly created Institute of Chemical Research of Catalonia (ICIQ). He has been Liebig Lecturer (Organic Division, German Chemical Society, 2006), Abbot Lecturer in Organic Chemistry (University of Illinois at Urbana—Campaign, 2009), Schulich Visiting Professor (Technion, Haifa, 2011), Sir Robert Robinson Distinguished Lecturer (University of Liverpool, 2011), and Novartis Lecturer in Organic Chemistry (Massachusetts Institute of Technology, 2015). In 2012, he received a European Research Council Advanced Grant, and in 2014 he was elected president of the 49th EUCHEM Conference on Stereochemistry (Bürgenstock conference). Prof. Echavarren is a member of the International Advisory Board of ChemSusChem (2007−), Organic & Biomolecular Chemistry (2008−), Chemical Society Reviews (2010−), Advanced Synthesis and Catalysis (2011−), and Organic Letters (2014−), member of the Editorial Board of ChemCatChem (2009−) and Chemistry European Journal (2014−), Associate Editor of Chemical Communications (2011−), and Fellow of the Royal Society of Chemistry. He received the 2004 Janssen–Cylag Award in Organic Chemistry and the 2010 Gold Medal of the Royal Spanish Chemical Society. In 2015, he received an Arthur C. Cope Scholar Award from the ACS. His current interests are the discovery of new catalytic methods based on the chemistry of transition metals as well as the synthesis of natural products and polyarenes.
The authors declare no competing financial interest.
References
- Teles J. H.; Brode S.; Chabanas M. Angew. Chem., Int. Ed. 1998, 37, 1415–1418. [DOI] [PubMed] [Google Scholar]
- Mizushima E.; Sato K.; Hayashi T.; Tanaka M. Angew. Chem., Int. Ed. 2002, 41, 4563–4565. [DOI] [PubMed] [Google Scholar]
- Jiménez-Núñez E.; Echavarren A. M. Chem. Rev. 2008, 108, 3326–3350. [DOI] [PubMed] [Google Scholar]
- Gorin D. J.; Sherry B. D.; Toste F. D. Chem. Rev. 2008, 108, 3351–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelet V.; Toullec P. Y.; Genêt J. P. Angew. Chem., Int. Ed. 2008, 47, 4268–4315. [DOI] [PubMed] [Google Scholar]
- Fürstner A. Chem. Soc. Rev. 2009, 38, 3208–3221. [DOI] [PubMed] [Google Scholar]
- Shapiro N. D.; Toste F. D. Synlett 2010, 675–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L.-P.; Hammond G. B. Chem. Soc. Rev. 2012, 41, 3129–3139. [DOI] [PubMed] [Google Scholar]
- Rudolph M.; Hashmi A. S. K. Chem. Soc. Rev. 2012, 41, 2448–2462. [DOI] [PubMed] [Google Scholar]
- Obradors C.; Echavarren A. M. Chem. Commun. 2014, 50, 16–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obradors C.; Echavarren A. M. Acc. Chem. Res. 2014, 47, 902–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorel R.; Echavarren A. M. Chem. Rev. 2015, 10.1021/cr500691k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trost B. M.; Tanoury G. J. J. Am. Chem. Soc. 1988, 110, 1636–1638. [Google Scholar]
- Chatani N.; Morimoto T.; Muto T.; Murai S. J. Am. Chem. Soc. 1994, 116, 6049–6050. [Google Scholar]
- Chatani N.; Kataoka K.; Murai S.; Furukawa N.; Seki Y. J. Am. Chem. Soc. 1998, 120, 9104–9105. [Google Scholar]
- Hashmi A. S. K.; Frost T. M.; Bats J. W. J. Am. Chem. Soc. 2000, 122, 11553–11554. [Google Scholar]
- Fernández-Rivas C.; Méndez M.; Echavarren A. M. J. Am. Chem. Soc. 2000, 122, 1221–1222. [Google Scholar]
- Nevado C.; Charruault L.; Michelet V.; Nieto-Oberhuber C.; Muñoz M. P.; Méndez M.; Rager M.-N.; Genêt J. P.; Echavarren A. M. Eur. J. Org. Chem. 2003, 706–713. [Google Scholar]
- Chatani N.; Furukawa N.; Sakurai H.; Murai S. Organometallics 1996, 15, 901–903. [Google Scholar]
- Oi S.; Tsukamoto I.; Miyano S.; Inoue Y. Organometallics 2001, 20, 3704–3709. [Google Scholar]
- Fürstner A.; Szillat H.; Gabor B.; Mynott R. J. Am. Chem. Soc. 1998, 120, 8305–8314. [Google Scholar]
- Fürstner A.; Szillat H.; Stelzer F. J. Am. Chem. Soc. 2000, 122, 6785–6786. [DOI] [PubMed] [Google Scholar]
- Fürstner A.; Szillat H.; Stelzer F. J. Am. Chem. Soc. 2001, 123, 11863–11869. [DOI] [PubMed] [Google Scholar]
- Mainetti E.; Mouriès V.; Fensterbank L.; Malacria M.; Marco-Contelles J. Angew. Chem., Int. Ed. 2002, 41, 2132–2135. [PubMed] [Google Scholar]
- Méndez M.; Muñoz M. P.; Echavarren A. M. J. Am. Chem. Soc. 2000, 122, 11549–11550. [Google Scholar]
- Nevado C.; Cárdenas D. J.; Echavarren A. M. Chem.—Eur. J. 2003, 9, 2627–2635. [DOI] [PubMed] [Google Scholar]
- Méndez M.; Muñoz M. P.; Nevado C.; Cárdenas D. J.; Echavarren A. M. J. Am. Chem. Soc. 2001, 123, 10511–10520. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Hashmi A. S. K. Angew. Chem., Int. Ed. 1993, 32, 1085–1087. [Google Scholar]
- Martín-Matute B.; Cárdenas D. J.; Echavarren A. M. Angew. Chem., Int. Ed. 2001, 40, 4754–4757. [DOI] [PubMed] [Google Scholar]
- Martín-Matute B.; Nevado C.; Cárdenas D. J.; Echavarren A. M. J. Am. Chem. Soc. 2003, 125, 5757–5766. [DOI] [PubMed] [Google Scholar]
- Huguet N.; Leboeuf D.; Echavarren A. M. Chem.—Eur. J. 2013, 19, 6581–6585. [DOI] [PubMed] [Google Scholar]
- Nieto-Oberhuber C.; Muñoz M. P.; Buñuel E.; Nevado C.; Cárdenas D. J.; Echavarren A. M. Angew. Chem., Int. Ed. 2004, 43, 2402–2406. [DOI] [PubMed] [Google Scholar]
- Nieto-Oberhuber C.; Muñoz M. P.; López S.; Jiménez-Núñez E.; Nevado C.; Herrero-Gómez E.; Raducan M.; Echavarren A. M. Chem.—Eur. J. 2006, 12, 1677–1693. [DOI] [PubMed] [Google Scholar]; Corrigendum: Chem.—Eur. J. 2008, 14, 5096.
- Homs A.; Escofet I.; Echavarren A. M. Org. Lett. 2013, 15, 5782–5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamane V.; Gress T.; Krause H.; Fürstner A. J. Am. Chem. Soc. 2004, 126, 8654–8655. [DOI] [PubMed] [Google Scholar]
- Luzung M. R.; Markham J. P.; Toste F. D. J. Am. Chem. Soc. 2004, 126, 10858–10859. [DOI] [PubMed] [Google Scholar]
- Kennedy-Smith J. J.; Staben S. T.; Toste F. D. J. Am. Chem. Soc. 2004, 126, 4526–4527. [DOI] [PubMed] [Google Scholar]
- Brooner R. E. M.; Widenhoefer R. A. Angew. Chem., Int. Ed. 2013, 52, 11714–11724. [DOI] [PubMed] [Google Scholar]
- Lauterbach T.; Livendahl M.; Rosellón A.; Espinet P.; Echavarren A. M. Org. Lett. 2010, 12, 3006–3009. [DOI] [PubMed] [Google Scholar]
- Brown T. J.; Widenhoefer R. A. Organometallics 2011, 30, 6003–6009. [Google Scholar]
- Nieto-Oberhuber C.; López S.; Muñoz M. P.; Cárdenas D. J.; Buñuel E.; Nevado C.; Echavarren A. M. Angew. Chem., Int. Ed. 2005, 44, 6146–6148. [DOI] [PubMed] [Google Scholar]
- Escribano-Cuesta A.; Pérez-Galán P.; Herrero-Gómez E.; Sekine M.; Braga A. A. C.; Maseras F.; Echavarren A. M. Org. Biomol. Chem. 2012, 10, 6105–6111. [DOI] [PubMed] [Google Scholar]
- Ferrer C.; Raducan M.; Nevado C.; Claverie C. K.; Echavarren A. M. Tetrahedron 2007, 63, 6306–6316. [Google Scholar]
- Soriano E.; Marco-Contelles J. Acc. Chem. Res. 2009, 42, 1026–1036. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Kozmin S. J. Am. Chem. Soc. 2004, 126, 11806–11807. [DOI] [PubMed] [Google Scholar]
- Sun J.; Conley M.; Zhang L.; Kozmin S. J. Am. Chem. Soc. 2006, 128, 9705–9710. [DOI] [PubMed] [Google Scholar]
- López-Carrillo V.; Huguet N.; Mosquera Á.; Echavarren A. M. Chem.—Eur. J. 2011, 17, 10972–10978. [DOI] [PubMed] [Google Scholar]
- Cabello N.; Rodríguez C.; Echavarren A. M. Synlett 2007, 1753–1758. [Google Scholar]
- Böhringer S.; Gagosz F. Adv. Synth. Catal. 2008, 350, 2617–2630. [Google Scholar]
- Odabachian Y.; Gagosz F. Adv. Synth. Catal. 2009, 351, 379–386. [Google Scholar]
- Iwai T.; Okochi H.; Ito H.; Sawamura M. Angew. Chem., Int. Ed. 2013, 52, 4239–4242. [DOI] [PubMed] [Google Scholar]
- Comer E.; Rohan E.; Deng L.; Porco J. A. Org. Lett. 2007, 9, 2123–2126. [DOI] [PubMed] [Google Scholar]
- Fürstner A.; Morency L. Angew. Chem., Int. Ed. 2008, 47, 5030–5033. [DOI] [PubMed] [Google Scholar]
- Hashmi A. S. K. Angew. Chem., Int. Ed. 2008, 47, 6754–6756. [DOI] [PubMed] [Google Scholar]
- Nunes dos Santos; Comprido L.; Klein J. E. M. N.; Knizia G.; Kästner J.; Hashmi A. S. K. Angew. Chem., Int. Ed. 2015, 10.1002/anie.201412401. [DOI] [PubMed] [Google Scholar]
- Benitez D.; Shapiro N. D.; Tkatchouk E.; Wang Y.; Goddard W. A. III; Toste D. F. Nat. Chem. 2009, 1, 482–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Muratore M. E.; Echavarren A. M. Chem.—Eur. J. 2015, 21, 7332–7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussong M. W.; Rominger F.; Krämer P.; Straub B. F. Angew. Chem., Int. Ed. 2014, 53, 9372–9375. [DOI] [PubMed] [Google Scholar]
- Joost M.; Estévez L.; Mallet-Ladeira S.; Miqueu K.; Amgoune A.; Bourissou D. Angew. Chem., Int. Ed. 2014, 53, 14512–14516. [DOI] [PubMed] [Google Scholar]
- Nesmeyanov A. N.; Perevalova E. G.; Smyslova E. I.; Dyadchenko V. P.; Grandberg K. I. Izv. Akad. Nauk SSSR, Ser. Khim. 1977, 2610–2612. [Google Scholar]
- Steinborn D.; Beckea S.; Herzoga R.; Günther M.; Kircheisen R.; Stoeckli-Evans H.; Bruhn C. Z. Anorg. Allg. Chem. 1968, 624, 1303–1307. [Google Scholar]
- Nieto-Oberhuber C.; López S.; Echavarren A. M. J. Am. Chem. Soc. 2005, 127, 6178–6179. [DOI] [PubMed] [Google Scholar]
- Nieto-Oberhuber C.; Pérez-Galán P.; Herrero-Gómez E.; Lauterbach T.; Rodríguez C.; López S.; Bour C.; Rosellón A.; Cárdenas D. J.; Echavarren A. M. J. Am. Chem. Soc. 2008, 130, 269–279. [DOI] [PubMed] [Google Scholar]
- Lee Y. T.; Kang Y. K.; Chung Y. K. J. Org. Chem. 2009, 74, 7922–7934. [DOI] [PubMed] [Google Scholar]
- Ota K.; Chatani N. Chem. Commun. 2008, 2906–2907. [DOI] [PubMed] [Google Scholar]
- Ota K.; Ick Lee S.; Tang J.-M.; Takachi M.; Nakai H.; Morimoto T.; Sakurai H.; Kataoka K.; Chatani N. J. Am. Chem. Soc. 2009, 131, 15203–15211. [DOI] [PubMed] [Google Scholar]
- Lee S. I.; Chatani N. Chem. Commun. 2009, 371–384. [DOI] [PubMed] [Google Scholar]
- Brooner R.; Robertson B. D.; Widenhoefer R. A. Organometallics 2014, 33, 6466–6473. [Google Scholar]
- Brooner R. E. M.; Brown T. J.; Widenhoefer R. A. Angew. Chem. Int. Ed. 2013, 52, 6259–6261. [DOI] [PubMed] [Google Scholar]
- Lee S. I.; Kim S. M.; Choi M. R.; Kim S. Y.; Chung Y. K. J. Org. Chem. 2006, 71, 9366–9372. [DOI] [PubMed] [Google Scholar]
- Jiménez-Núñez E.; Claverie C. K.; Bour C.; Cárdenas D. J.; Echavarren A. M. Angew. Chem., Int. Ed. 2008, 47, 7892–7895. [DOI] [PubMed] [Google Scholar]
- Nieto-Oberhuber C.; López S.; Jiménez-Núñez E.; Echavarren A. M. Chem.—Eur. J. 2006, 12, 5916–5923. [DOI] [PubMed] [Google Scholar]
- Herrero-Gómez E.; Nieto-Oberhuber C.; López S.; Benet-Buchholz J.; Echavarren A. M. Angew. Chem., Int. Ed. 2006, 45, 5455–5459. [DOI] [PubMed] [Google Scholar]
- Pérez-Galán P.; Delpont N.; Herrero-Gómez E.; Maseras F.; Echavarren A. M. Chem.—Eur. J. 2010, 16, 5324–5332. [DOI] [PubMed] [Google Scholar]
- López-Carrillo V.; Echavarren J. Am. Chem. Soc. 2010, 132, 9292–9294. [DOI] [PubMed] [Google Scholar]
- Homs A.; Obradors C.; Leboeuf D.; Echavarren A. M. Adv. Synth. Catal. 2014, 356, 221–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obradors C.; Leboeuf D.; Aydin J.; Echavarren A. M. Org. Lett. 2013, 15, 1576–1579. [DOI] [PubMed] [Google Scholar]
- Yeom H.-S.; Koo J.; Park H.-S.; Wang Y.; Liang Y.; Yu Z.-X.; Shin S. J. Am. Chem. Soc. 2012, 134, 208–211. [DOI] [PubMed] [Google Scholar]
- Solorio-Alvarado C. R.; Wang Y.; Echavarren A. M. J. Am. Chem. Soc. 2011, 133, 11952–11955. [DOI] [PubMed] [Google Scholar]
- Buzas A. K.; Istrate F. M.; Gagosz F. Angew. Chem., Int. Ed. 2007, 46, 1141–1144. [DOI] [PubMed] [Google Scholar]
- Leseurre L.; Toullec P. Y.; Genêt J.-P.; Michelet V. Org. Lett. 2007, 9, 4049–4052. [DOI] [PubMed] [Google Scholar]
- Martínez A.; García–García P.; Fernández–Rodríguez M. A.; Rodríguez F.; Sanz R. Angew. Chem., Int. Ed. 2010, 49, 4633–4637. [DOI] [PubMed] [Google Scholar]
- Böhringer S.; Gagosz F. Adv. Synth. Catal. 2008, 350, 2617–2630. [Google Scholar]
- Sanjuán A. M.; Martínez A.; García-García P.; Fernández-Rodríguez M. A.; Sanz R. Beilstein J. Org. Chem. 2013, 9, 2242–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Kozmin S. A. J. Am. Chem. Soc. 2005, 127, 6962–6963. [DOI] [PubMed] [Google Scholar]
- Pradal A.; Chen Q.; Faudot dit Bel P.; Toullec P. Y.; Michelet V. Synlett 2012, 23, 74–79. [Google Scholar]
- Sethofer S. G.; Mayer T.; Toste F. D. J. Am. Chem. Soc. 2010, 132, 8276–8277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzas A.; Istrate F.; Le Goff X. F.; Odabachian Y.; Gagosz F. J. Organomet. Chem. 2009, 694, 515–519. [Google Scholar]
- Lee Y.; Lim C.; Kim S.; Shin S. Bull. Korean Chem. Soc. 2010, 31, 670–677. [Google Scholar]
- Jiménez-Núñez E.; Claverie C. K.; Nieto-Oberhuber C.; Echavarren A. M. Angew. Chem., Int. Ed. 2006, 45, 5452–5455. [DOI] [PubMed] [Google Scholar]
- Huguet N.; Echavarren A. M. Synlett 2012, 23, 49–53. [Google Scholar]
- Jiménez-Núñez E.; Molawi K.; Echavarren A. M. Chem. Commun. 2009, 7327–7329. [DOI] [PubMed] [Google Scholar]
- Molawi K.; Delpont N.; Echavarren A. M. Angew. Chem., Int. Ed. 2010, 49, 3517–3519. [DOI] [PubMed] [Google Scholar]
- Zhou Q.; Chen X.; Ma D. Angew. Chem., Int. Ed. 2010, 49, 3513–3516. [DOI] [PubMed] [Google Scholar]
- Escribano-Cuesta A.; López-Carrillo V.; Janssen D.; Echavarren A. M. Chem.—Eur. J. 2009, 15, 5646–5650. [DOI] [PubMed] [Google Scholar]
- Huple D. B.; Liu R.-S. Chem. Commun. 2012, 48, 10975–10977. [DOI] [PubMed] [Google Scholar]
- Matsuda T.; Sakurai Y. J. Org. Chem. 2014, 79, 2739–2745. [DOI] [PubMed] [Google Scholar]
- Schelwies M.; Dempwolff A. L.; Rominger F.; Helmchen G. Angew. Chem., Int. Ed. 2007, 46, 5598–5601. [DOI] [PubMed] [Google Scholar]
- Schelwies M.; Moser R.; Dempwolff A. L.; Rominger F.; Helmchen G. Chem.—Eur. J. 2009, 15, 10888–10900. [DOI] [PubMed] [Google Scholar]
- Obradors C.; Echavarren A. M. Chem.—Eur. J. 2013, 19, 3547–3551. [DOI] [PubMed] [Google Scholar]
- Amijs C. H. M.; Ferrer C.; Echavarren A. M. Chem. Commun. 2007, 698–700. [DOI] [PubMed] [Google Scholar]
- Amijs C. H. M.; López-Carrillo V.; Raducan M.; Pérez-Galán P.; Ferrer C.; Echavarren A. M. J. Org. Chem. 2008, 73, 7721–7730. [DOI] [PubMed] [Google Scholar]
- Toullec P. Y.; Genin E.; Leseurre L.; Genêt J.-P.; Michelet V. Angew. Chem., Int. Ed. 2006, 45, 7427–7430. [DOI] [PubMed] [Google Scholar]
- Leseurre L.; Chao C.-M.; Seki T.; Genin E.; Toullec P. Y.; Genêt J.-P.; Michelet V. Tetrahedron 2009, 65, 1911–1918. [Google Scholar]
- Yeh M.-C. P.; Tsao W.-C.; Lee B.-J.; Lin T.-L. Organometallics 2008, 27, 5326–5332. [Google Scholar]
- Chao C. M.; Vitale M. R.; Toullec P. Y.; Genêt J.-P.; Michelet V. Chem.—Eur. J. 2009, 15, 1319–1323. [DOI] [PubMed] [Google Scholar]
- Delpont N.; Escofet I.; Pérez-Galán P.; Spiegl D.; Raducan M.; Bour C.; Sinisi R.; Echavarren A. M. Catal. Sci. Technol. 2013, 3, 3007–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak J. A.; Patrick B. O.; Dake G. R. J. Org. Chem. 2010, 75, 8585–8590. [DOI] [PubMed] [Google Scholar]
- García-García P.; Rashid M. A.; Sanjuán A. M.; Fernández-Rodríguez M. A.; Sanz R. Org. Lett. 2012, 14, 4778–4781. [DOI] [PubMed] [Google Scholar]
- Chatani N.; Kataoka K.; Murai S.; Furuakwa N.; Seki Y. J. Am. Chem. Soc. 1998, 120, 9104–9105. [Google Scholar]
- Harrak Y.; Blaszykowski C.; Bernard M.; Cariou K.; Mainetti E.; Mouriès V.; Dhimane A.-L.; Fensterbank L.; Malacria M. J. Am. Chem. Soc. 2004, 126, 8656–8657. [DOI] [PubMed] [Google Scholar]
- Nieto-Oberhuber C.; López S.; Muñoz M. P.; Jiménez-Núñez E.; Buñuel E.; Cárdenas D. J.; Echavarren A. M. Chem.—Eur. J. 2006, 12, 1694–1702. [DOI] [PubMed] [Google Scholar]
- Kim S. M.; Park J. H.; Choi S. Y.; Chung Y. K. Angew. Chem., Int. Ed. 2007, 46, 6172–6175. [DOI] [PubMed] [Google Scholar]
- Jiménez-Núñez E.; Raducan M.; Lauterbach T.; Molawi K.; Solorio C. R.; Echavarren A. M. Angew. Chem., Int. Ed. 2009, 48, 6152–6155. [DOI] [PubMed] [Google Scholar]
- Carreras J.; Livendahl M.; McGonigal P. R.; Echavarren A. M. Angew. Chem., Int. Ed. 2014, 53, 4896–4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGonigal P. R.; de León C.; Wang Y.; Homs A.; Solorio-Alvarado C. R.; Echavarren A. M. Angew. Chem., Int. Ed. 2012, 51, 13093–13096. [DOI] [PubMed] [Google Scholar]
- López S.; Herrero-Gómez E.; Pérez-Galán P.; Nieto-Oberhuber C.; Echavarren A. M. Angew. Chem., Int. Ed. 2006, 45, 6029–6032. [DOI] [PubMed] [Google Scholar]
- Pérez-Galán P.; Herrero-Gómez H.; Hog D. T.; Martin N. J. A.; Maseras F.; Echavarren A. M. Chem. Sci. 2011, 2, 141–149. [Google Scholar]
- Gaydou M.; Miller R. E.; Delpont N.; Ceccon J.; Echavarren A. M. Angew. Chem., Int. Ed. 2013, 52, 6396–6399. [DOI] [PubMed] [Google Scholar]