

Abstract
Objectives
Given the roles of bcl-2, bax and p53 in apoptosis, we investigated the effect of their expression on the response to cisplatin in order to understand the molecular events of cisplatin-resistance in lung cancers.
Methods
Three parental human lung cancer cell lines (PC9, PC14 and H69) and their in vitro selected cisplatin-resistant sublines were examined. Cells treated with cisplatin were processed for acridine orange and ethidium bromide staining and DNA gel electrophoresis for the morphologic detection of apoptosis. The endogenous levels of bcl-2, bax and p53 protein expression in lung cancer cells were assessed by Western blot analysis and DNA of polymerase chain reaction-amplified exon 5 to 8 of p53 gene was directly sequenced.
Results
H69, which had bcl-2 expression, p53 mutation and decreased expression of p53 and bax, was relatively resistant to cisplatin and delayed and reduced apoptosis. Although apoptosis was markedly reduced in cisplatin-resistant sublines compared to their parental cells, there were no significant differences in the expression of p53, bcl-2 and bax.
Conclusions
Cisplatin-resistance was associated with the reduced cellular susceptibility to apoptosis. Cancer cells with the natural expression of bcl-2 and p53 mutation may be more resistant to cisplatin and less susceptible to apoptosis.
Keywords: Cisplatin-resistance, apoptosis, p53, bcl-2, bax
INTRODUCTION
Cisplatin is one of the most active anticancer agents for the treatment of lung cancer and the cisplatin-based chemotherapeutic regimens have produced a statistically significant and clinically relevant improvement in survival. The long-term survival is directly linked to the degree of clinical response to chemotherapy 1,2). However, more than one-third of patients do not achieve an appreciable clinical response to the cisplatin-based chemotherapy. Therefore, it is important to understand the molecular genetic features that can determine the response or resistance to cisplatin, which could permit the selection of the most suitable patients for the cisplatin-based chemotherapy and enhance the development of an innovative treatment for patients most likely to be refractory to cisplatin. The precise mechanisms responsible for cisplatin-mediated cytotoxicity are not fully understood, but evidence obtained in the last few years indicates that cisplatin inhibits DNA synthesis by double-strand breaks. It reacts readily with the N7 position of purines to form a variety of lethal platinum-DNA adducts, which trigger programmed cell death (apoptosis). Indeed, cells treated with cytotoxic levels of cisplatin display the biochemical and morphologic features of apoptosis3–7). Thus, the resistance to cisplatin can be caused by the loss of the regulation of apoptosis.
p53 is involved in the activation of apoptosis induced by DNA-damage, such as cisplatin. As a transcriptional activator, p53 increases the transcription of a number of genes and the pattern of transcriptional regulation is critical in determining the cellular response to DNA damage. It is known to activate the transcription of death agonist, bax, but to repress the expression of death antagonist, bcl-28–14). Given the roles of bcl-2, bax and p53 in cell death and survival, we examined the expression of these genes in small cell and non-small cell lung cancer cell lines to investigate the effect of their expression on the response to cisplatin in an attempt to understand the molecular events contributing to the development of the cisplatin-resistance in lung cancers.
MATERIALS and METHODS
1. Cell lines
NCI H69 human small cell lung carcinoma, PC9 human lung adenocarcinoma, PC14 human lung adenocarcinoma and their in vitro selected cisplatin-resistant sublines, H69/CDDP, PC9/CDDP, PC14/CDDP, were kindly provided by Dr. N. Saijo (National Cancer Center Research Institute, Tokyo, Japan). All the cisplatin-resistant cells were cultured in cisplatin-free medium for at least 4 weeks before being used for the experiments. Cells were cultured in RPMI-1640 medium supplemented with 2 mM L-glutamine, 10% fetal calf serum, 10 μg/ml penicillin and 10 μg/ml streptomycin and grown in a humidified incubator with 5% CO2 at 37°C. All media and chemical reagents were purchased from Gibco Co.(BRL, Paisley, UK).
2. MTT assay for cytotoxicity
In vitro cisplatin-induced cytotoxicity was determined by the MTT dye reduction assay. Cells were plated out at a density of 3,000–4,000 cells per well into a 96-well microtiter plates and allowed to attach overnight. The next day, cells were treated with 0.1 to 1,000 μg/ml concentrations of cisplatin (Platosin®; Pharmachemie, Haarlem, Netherlands) dissolved in sterile water and incubated for 4 days. After this treatment, 20μl of MTT (3, (4, 4-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide, final concentration of 5 mg/ml, Sigma, MO, USA) were added in each well and incubated at 37°C for 4 hr. After incubation, cells were centrifuged at 200 g for 5 min and the supernatant was aspirated. 200 μl of Dimethyl sulfoxide (DMSO) were added in each well to solubilize the formed formazan crystals. The absorbance was recorded at 540 nm on a Titertek Multiskan® MCC (EFLab, Finland). Wells containing only RPMI-FBS and MTT were used as control. The IC50 (Inhibitory Concentration 50%) values were the drug concentrations inducing 50% reduction in the absorbance. Each experiment was performed using 6 replicate wells for each drug concentration and three independent experiments were carried out with consistent results. The relative resistance to cisplatin was obtained by comparing IC50 values of cisplatin-resistant sublines with those of the parental cell lines.
3. Assay for the detection of apoptotic cell death
3.A. Quantitation of cell viability by acridine orange/ethidium bromide uptake
Approximately 5 × 105 cells were harvested after various times post-treatment with cisplatin (10 μg/ml) and washed with phosphate buffered saline (PBS). Cells were stained with acridine orange (100 μg/ml) and ethidium bromide (100 μg/ml) and viewed immediately under a fluorescence microscope. The staining clearly distinguished between viable cells and cells showing condensed chromatin staining characteristic of apoptosis. A minimum 200 total cells were counted and the percentage of apoptotic cells was calculated as follows: % apoptotic cell=(total number of apoptotic cells/total number of cells counted) × 100
Three independent experiments were performed with consistent results.
3.B. DNA isolation and gel electrophoresis
For the electrophoretic characterization of DNA fragmentation, 3 × 106 cells were harvested after 12, 24, 48 and 72 hr post-treatment with 3.3, 10 and 100 μg/ml concentrations of cisplatin and washed twice with PBS, and the pellet in a 1.5 ml eppendorf tube was resuspended in 100 μl lysis buffer containing 10 mM EDTA (pH 8.0), 10 mM Tris-HCl (pH 7.4), 0.5% Triton-X 100 and lysed at 4°C for 10 min. The suspension was centrifuged at 25,000 g for 20 min and the resulting supernatant was harvested in a new eppendorf tube. Each sample was added with 2 μl RNase (20 μg/ml, Sigma) and incubated at 37°C for 1 hr. Each sample was added with 2 μl Proteinase K (20 μg/ml, Sigma) and incubated at 37°C for 1 hr. Then, 20 μl of 5M NaCl and 120 μl of isopropyl alcohol were added to each sample, which was stored at −20°C overnight. The next day, samples were centrifuged at 25,000 g, 4°C for 15 min. The pellets were dissolved with 20 μl TBE buffer containing 10 mM Tris-HCl (pH 7.4), 1 mM EDTA (pH 8) and electrophoresed through 2% agarose gel after adding 4 μl loading buffer (40 % sucrose, bromophenol blue 0.25%) at 100 volts for 90 min. 100bp DNA markers (Bio-Rad, CA, USA) were run in parallel. The gels were visualized by 1μg/ml ethidium bromide staining under ultraviolet light.
4. p53 Sequencing
Sequencing of exon 5 to 8 of p53 was performed by polymerase chain reaction (PCR) amplification followed by direct automated sequencing of double-stranded DNA using biotinylated terminators.
4.A. PCR amplification
PCR was performed using 200 ng of DNA, 1.5 mg MgCl2, 200 mM/liter of each dNTPs, 1 mM/liter of each primer, 0.4 ml of Taq polymerase enzyme and 5 ml of 10 X Taq buffer ((Promega, WI, USA) in a final volume of 50 ml. The cycling profile comprised 30 cycles of denaturation (95°C for 50 sec), annealing (58°C for 90 sec) and extension (72°C for 90 sec).
4.B. DNA nucleotide primers
Exon 5 to 8 of p53 were amplified using the Human p53 Exons 5–8 Amplifier Panel™ (Clontech, CA, USA) in accordance with the manufacturers instruction. Their sequences were as follows: exon 5 sense strand, 5 CTC TTC CTG CAG TAC TCC CCT GC 3; exon 5 antisense strand, 5 GCC CCA GCT GCT CAC CAT CGC TA 3; exon 6 sense strand, 5 GAT TGC TCT TAG GTC TGG CCC CTG 3; exon 6 antisense strand, GGC CAC TGA CAA CCA CCC TTA ACC 3; exon 7 sense strand, 5 GTG TTG TCT CCT AGG TTG GCT CTG 3; exon 7 antisense strand, 5 CAA GTG GCT CCT GAC CTG GAG TC 3; exon 8 sense strand, 5 ACC TGA TTT CCT TAC TGC CTC TGG C 3; exon 8 antisense strand, 5 GTC CTG CTT GCT TAC CTC GCT TAG T 3.
4.C. Direct DNA sequencing of PCR products
PCR products were primarily purified using 50 ml 7.5 M NH4AC and 300 ml 98% ethanol mix extraction and precipitation with 150 ml 70% ethanol. The same primers used for generating the PCR products were also used for the sequencing reactions. Other reagents were supplied in the GATC-BioCycle Sequencing kit (GATC GmbH, Konstanz, Germany). Both strands were sequenced for each exon. GATC-BioCycle sequencing protocol was followed for cycle sequencing of PCR products. 1 μl of the purified PCR product (approximately 40 ng), 5 pM/ml primer, 2μl reaction-buffer, 2μl dITP-Mix and 5 units of Thermo-Sequenase™ (Amersham, Bucks, UK) were well mixed with 20μl distilled water for extension. 5μl of extension-mix was transferred into a 0.5 ml PCR tube and 1μl ddGTP-Mix was added. G-A-T-C reaction tubes were placed on Thermo Cycler (ERICOM INC. CA, USA) and run the following program: denaturation for 3 min 95°C, 30 cycles, and final 4 min 60°C. 2 ml samples were loaded in a 7% denaturating polyacrylamide gel and the electrophoresis was performed on a GATC 1500 unit. Biotinylated terminator of 55 μl NBT (4-nitro blue tetrazolium chloride) and of 55 μl BCIP (X-phosphate/5-bromo-4-chloro-3-indolyl-phosphate; Boehringer Mannheim) in 20ml reaction buffer was used for visualization of DNA colorimetrically. All PCR products were sequenced in both directions.
5. Western blot analysis of Bcl-2, Bax and p53
Cells were removed from tissue culture flasks using a cell-scraper and centrifuged at 300 X g for 5 min. The pellet was resuspended in ice-cold lysis buffer consisting of 20 mM Tris-HCl buffer (pH 7.4) containing 5 μg/ml aprotinin, 5 μg/ml leupeptin and 1 mM phenylmethylsulphonyl fluoride, and homogenized by the passage through a 26 gauge syringe needle. The suspension was centrifuged at 20,000 rpm for 15 min at 4°C and the supernatant was stored at −70°C until assay. Protein concentration was measured in each cell lysate by the BCA method (Sigma) and equal amounts of total protein were loaded for each blot. Cell lysates were mixed with equal volume of Tris-Glycine SDS-sample buffer and boiled for 2 min. 25 μl of each denatured sample was loaded and electrophoresed through 4–20% Tris-Gylcine gels in Tris-Glycine running buffer (all were Novex, San Diego, USA) for 2 hr at 100 volts. Electrophoresed gels were transferred to nitrocellulose membrane (Bio-Rad) in transfer buffer (12 mM Tris base 1.45 g, 96 mM glycine 7.2 g, methanol 200 ml, distilled water 1,000 ml) for 2.5 hr at 30 volts. Membranes were blocked in 5% skim milk solution overnight and probed with monoclonal mouse anti-human Bcl-2 Ab (1:500, 2 μg/ml of blocking solution, Santa Cruz, CA, USA), polyclonal rabbit anti-human Bax Ab (1:500, 2 μg/ml of blocking solution, Santa Cruz) or monoclonal mouse anti-human p53 Ab (1:500, 2 μg/ml of blocking solution, Zymed, CA, USA) for 90 min at 37°C. After washing with 0.05% Tween-20 three times, membranes were incubated for 45 min with peroxidase-conjugated goat IgG fraction to mouse or rabbit (1:1000, 1 μg/ml of blocking solution, Cappel, NC, USA) at 37°C. After several washings, they were added with 10 ml of tetramethyl-benzidine solution (Zymed) and shook for 10–30 min for visualization of immunoreactive materials. Immunobloting was repeated in all of the cell lines at least three times with consistent results.
6. Statistical analysis
Differences between groups were tested for the significance using student’s two-tailed t-test (SAS Inst. INC, NC, USA), from which P values were calculated.
RESULTS
1. Cisplatin-induced cytotoxicity
The cisplatin-induced cytotoxicity in lung cancer cell lines was evaluated by MTT colorimetric assay (Fig. 1). The calculated IC50 values and the relative resistance to cisplatin are presented in Table 1. In this study, each paired in vitro cisplatin-resistant sublines had 3.1–4.7 fold more resistance to cisplatin than their parental cell lines had (P<0.05). Among the parental cells, H69 was relatively resistant to cisplatin compared to other parental cell lines, in spite of its histologic type, small cell carcinoma (relative resistance 2.1–3.2, P<0.05).
Fig. 1.
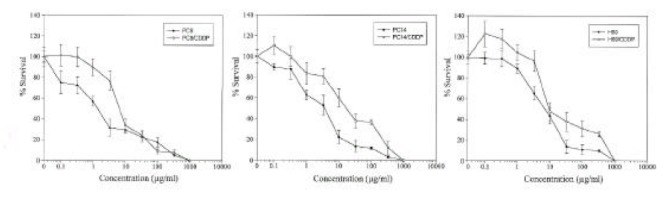
Cytotoxicity induced by cisplatin in PC9 (human lung adenocarcinoma) and PC9/CDDP, PC14 (human lung adenocarcinoma) and PC14/CDDP, and H69 (human small cell lung carcinoma) and H69/CDDP. Points, means of more than three independent experiments, and bars, SD.
Table 1.
IC50 values and the relative resistance of human lung cancer cell lines to cisplatin. IC50 values (mean±SD) were estimated from MTT cytotoxicity assays and the represent mean values of at least three independent experiments. The relative resistance to cisplatin was obtained by comparing the IC50 values of cisplatin-resistant sublines with those of the parental cell lines. a P < 0.05, b P < 0.01
| Cell lines | IC50 (μg/ml) | Relative resistance | |||
|---|---|---|---|---|---|
| PC9 | 2.30 (± 1.39) | – | |||
| PC9/CDDP | 7.95 (± 0.43) | 3.5b | |||
| PC14 | 3.55 (± 0.41) | – | |||
| PC14/CDDP | 16.56 (± 2.09) | 4.7b | |||
| H69 | 7.28 (± 1.89) | – | |||
| H69/CDDP | 22.21 (± 3.80) | 3.1a | |||
|
|
|
||||
| PC9 | 2.30 (± 1.39) | – | |||
| H69 | 7.28 (± 1.89) | 3.2a | |||
|
|
|
||||
| PC14 | 3.55 (± 0.41) | – | |||
| H69 | 7.28 (± 1.89) | 2.1a | |||
2. Cell death induced by cisplatin is due to apoptosis
In order to examine the nature of cell death induced by cisplatin, cells collected at various time points post-treatment with 10 μg/ml of cisplatin were processed for acridine orange and ethidium bromide staining for the detection of condensed or fragmented chromatin and internucleosomal DNA fragmentation, diagnostic of apoptotic cells. Since we could not observe the clear apoptotic features at the concentration of cisplatin near to IC50 (3.3 μg/ml), we examined the cellular response to apoptosis at 10 μg/ml of cisplatin.
Apoptotic patterns (Fig. 2) were observed in PC9 and PC14 cell lines as early as 12 hr after cisplatin exposure, and increased after 24 hr. In H69 cells, the accumulation of acridine orange positive cells in response to cisplatin was slower and lower than that of PC9 or PC14 cell lines. The percentage of apoptotic cells remained below 10% after 24 hr and more than 75% cells were viable even after 72 hr in PC9/CDDP, PC14/CDDP and H69/CDDP (Fig. 3).
Fig. 2.
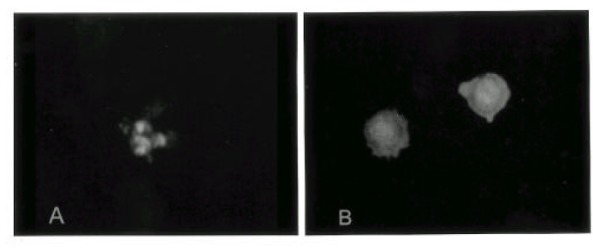
Morphological detection of apoptosis in PC9 cells. Cells were stained with acridine orange and ethidium bromide. Apoptotic cells with nuclear fragmentation into spherical bodies. (A, X100), (B, X200)
Fig. 3.
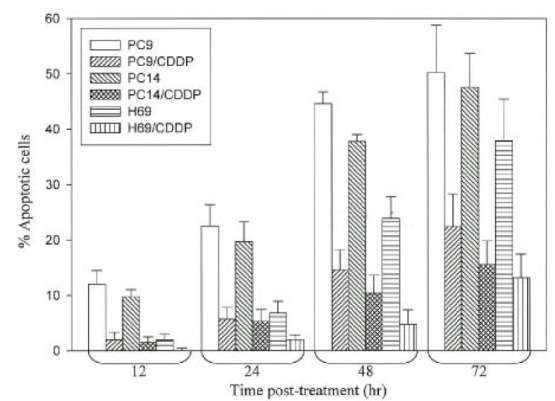
Percentage of apoptotic cells. Cells were harvested after various time post-treatment with 10μg/mL of cisplatin, washed PBS and used directly for staining with acridine orange and ethidium bromide. Results are expressed as mean±SD for three independent experiments.
Examination of internucieosomal DNA fragmentation (DNA ladders) showed the similar pattern of response and DNA ladders were visualized in PC9 and PC14 cells after 24 hr post-treatment, whereas in H69 cells, it was visualized after 48 hr. In cisplatin-resistant sublines, DNA ladders were not visualized after 48 hr (Fig. 4A). However, at higher concentration of cisplatin (100 μg/ml), DNA ladders were observed after 48 hr in all of the cell lines (Fig 4B).
Fig. 4.
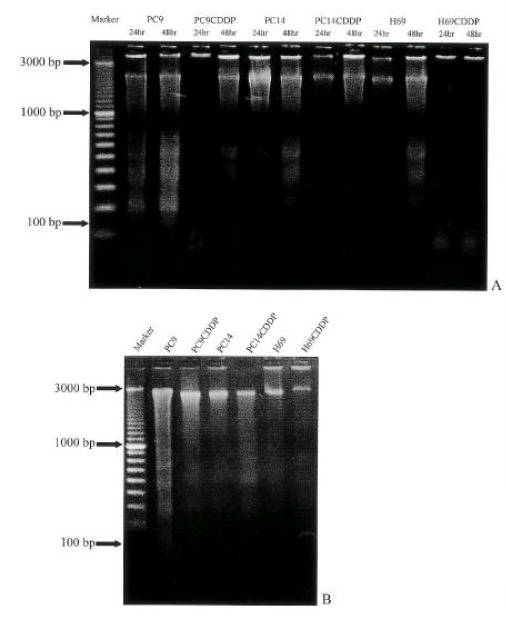
Agarose gel electrophoresis of genomic DNA extraced from lung cancer cells treated with 10μg/mL of cisplatin for 24, 48 hr (A) and 10μg/mL of cisplatin for 48 hr(B).
3. Sequencing analysis of p53 gene
There was no mutation of p53 in PC9, PC14 and their cisplatin-resistant sublines. The same point mutation was detected in H69 and H69/CDDP, localized in exon 5. As shown in Fig. 5, the mutation in these cell lines was nucleotide substitution (transversion, GGA→GTA) at codon 171.
Fig. 5.
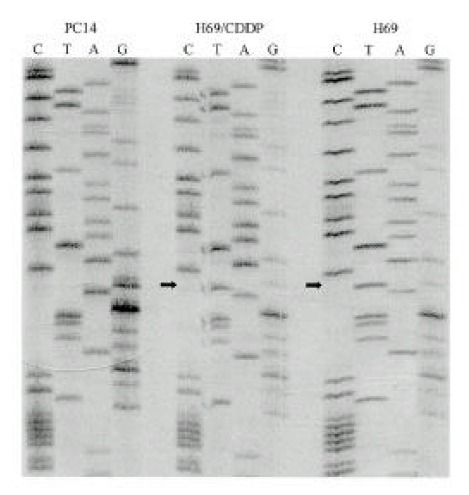
DNA sequence analysis of p53.
The left panel (PC14) represents the normal sequence of p53 exon 5. The arrows show the positions of point mutation. A transversion of G to T at codon 171 is found in H69 and H69/CDDP.
4. Relationship between bcl-2, bax and p53 protein expression
Recent reports have suggested that p53 tumor suppressor gene product may not only regulate down bcl-2 but regulate up bax gene expression8–14). The endogenous levels of bcl-2, bax and p53 protein expression in lung cancer cells were assessed by Western blot analysis. In this study, the expression of p53 was reduced in H69 and H69/CDDP which had the mutation of p53 compared to other cell lines without p53 mutations. Bcl-2 protein had an inverse relationship with p53, which was detectable only in cells with very low expression of p53 protein such as H69 and H69/CDDP, but not detectable in cells with high expression of p53 protein such as PC9, PC9/CDDP, PC14 and PC14/CDDP. Bax protein was expressed in all cells with variable intensities and the relative amount of bax protein was high in cells with high p53 expression (PC9, PC9/CDDP, PC14, PC14/CDDP) comparing cells with very low p53 expression (H69, H69/CDDP)(Fig. 6). This indicates that the reduced expression of p53 protein contributes to up-regulation of bcl-2 as well as down-regulation of bax expression.
Fig. 6.
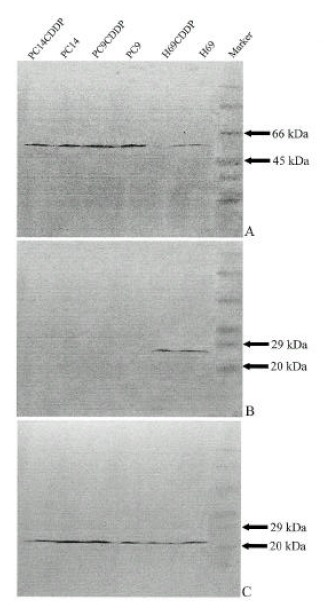
Western blots showing expression of p53 (A), bcl-2(B) and bax (C) in pairs of sensitive and resistant to cisplatin lung cancer cells. The size of these proteins shown were confirmed by simultaneous electrophoresis of molecular weight standards.
5. Regulation of cisplatin-resistance by the expression of p53, bcl-2 and bax
We examined the relationship between bcl-2, bax and p53 expression and cisplatin-resistance. Among parental cell lines, H69 (small cell carcinoma) was more resistan to cisplatin in spite of its histologic subtype and lower apoptosis than other parental cell lines. These findings suggest that its intrinsic resistance to cisplatin and p53 mutation and up-regulation of bcl-2 may confer the intrinsic cisplatin-resistance in this cell line.
In vitro selected cisplatin-resistant sublines had 3.1–4.7 fold resistance to cisplatin (P<0.05) and markedly reduced apoptosis compared to their parental cell lines. However, we observed no difference in levels of bcl-2, bax and p53 proteins between parental cell lines and their resistant sublines. This indicates that the acquired resistance made through cisplatin-exposure is related to the reduced susceptibility to apoptosis, but not to the alteration in the expression of p53, bax and bcl-2 proteins.
DISCUSSION
Lung cancer is one of the most lethal cancers for both men and women in the world. The majority of patients present with advanced, inoperable diseases and overall cure rate for lung cancer approximates 13%. This low rate can be ascribed to the high propensity for metastasis and resistance to chemotherapy5,7).
Cisplatin has a clinical activity against various solid tumors, especially lung cancers. The effectiveness of chemotherapy with cisplatin is restricted by the emergence of resistant cell populations, and defining of the molecular features that determine the resistance to cisplatin has an important clinical implication. The recent evidence suggests that the genetic regulation of apoptosis may affect the cellular response to DNA damages and, therefore, modulate the sensitivity of tumor cells to cisplatin4–6).
p53 tumor suppressor gene is most commonly mutated in human cancers. It is recognized as an important component of the pathway leading from DNA damage to apoptosis and several studies have suggested that p53 is involved in the activation of apoptosis induced by DNA-damaging anticancer agents9, 15, 16). Introduction of DNA strand breaks leads to a post-transcriptional increase in p53 protein levels17,18). However, whether p53 protein activity is altered or the levels are merely increased is not clear. The induced p53 protein is able to transcriptionally activate a number of different genes, including p21, gadd45 and mdm2 and the induction of p21 appears to be the major mechanism of G1 arrest19, 20). However, in some cell types and some physiologic conditions, p53 induction can lead to apoptosis. Although the mechanism remains unclear, the upregulation of bax, death agonist, through transactivation by p53 could play an important role in apoptosis10–13,21,22). As the role in mediating DNA-damage induced cell cycle arrest was elucidated, it became clear that normal p53 function was also required for DNA-damage induced apoptosis23–25). In this study, we examined in vitro selected cisplatin-resistant sublines made by the continuous exposure of cisplatin. We expected the increasement in the level of p53 protein of these resistant sublines by cisplatin induced DNA-damage. However, we were not able to observe a significant difference in the level of p53 protein compared to parental cell lines. Mutation of p53 and a relatively reduced expression of p53 protein were observed in H69 and H69/CDDP, which showed markedly reduced apoptosis. This indicates that p53 mutation results in the functional loss and instabilization of p53 protein. However, other cisplatin-resistant cell lines, such as PC9/CDDP and PC14/CDDP, which had no mutation of p53 and a comparable level of p53 protein, showed markedly reduced apoptosis compared to their parental cell lines. Because the loss of p53 function can be resulted even with the wild type of p53, it is needed to evaluate the functional activity of p53 in these cell lines.
Although p53 is known to transactivate bax, it suppress bcl-2 expression and the magnitude of p53 suppression of bcl-2 expression may be tissue-specific8,16). The recent analysis on breast cancer shows that the reverse relationship between p53 and bcl-2 expression, and the overexpression of mutant p53, can induce down-regulation of bcl-2 both protein and mRNA level8). Immunohistochemical study on non-Hodgkin’s lymphoma shows the significant inverse relationship between p53 and bcl-2 protein expression26). Moreover, in vitro study of overexpression of bcl-2 in cancer cells delays accumulation of p53 and indirectly suppresses bax induction16,27,28). In this study, bcl-2 protein was only detectable in small cell lung cancer cells (H69 and H69/CDDP) which had p53 mutation and the decreased expression of p53, suggesting the reverse relationship between these proteins. Bax protein expression was also reduced in H69 and H69/CDDP, indicating indirectly suppression of induction by bcl-2.
Bcl-2 was originally identified at the chromosomal breakpoint of t(14;18)-bearing B-cell lymphomas. The bcl-2 gene encodes for the p26Bcl-2 protein, which suppresses apoptosis4,27–34). So, it is now known to belong to the growing family of apoptosis-regulatory gene products, which may either be death antagonist (Bcl-2, Bcl-XL, Bcl-w, Bfl-1, Brag-1, Mcl-1, and A1) or death agonists (Bax, Bak, Bcl-Xs, Bad, Bid, Bik and Hrk)30–32). Most of the proteins encoded by the bcl-2 gene family are predominantly localized in the outer mitochondrial membrane and they possess variable amounts of Bcl-2 homology (BH) regions, which determine their capacities to interact with each other or with other unrelated protein. Bax shows extensive amino acid homology with bcl-2 and forms homodimers and heterodimers with bcl-2 in vivo. When bcl-2 protein is expressed at high level in cells, it may form complexes with bax, preventing bax homodimerization and inhibiting cell death29–32). Thus, the ratio of death antagonist (bcl-2, bcl-XL, etc) to agonist (bax, bcl-Xs, bad, etc) is important in determining whether a cell will response to an apoptotic signal29–32, 14, 36, 37). In this study, bcl-2 was expressed in H69 and H69/CDDP with p53 mutation and the level of bax protein was relatively decreased in these cell lines compared to other cell lines without p53 mutation. The delayed and reduced apoptosis by cisplatin in these cell lines may be attributed to bcl-2. Apoptosis in other cisplatin resistant cell lines was markedly reduced in spite of comparable expression of bax. However, the proapoptotic function of bax can be antagonized by other death antagonist, such as bcl-XL rather than bcl-2, which should be further evaluated.
Small cell lung cancer is well known to be more sensitive to chemotherapy than non-small cell lung cancer and the response approximates 90% with cisplatin-based chemotherapy, including 10–50% of complete response1, 2). In this study, H69 is a small cell lung cancer cell. However, it is more resistant to cisplatin than other non-small cell lung cancer cells and it has a reduced and delayed response to apoptosis. Interestingly, p53 mutation and bcl-2 expression were observed in only small cell lung cancer cells (H69 & H69/CDDP). Since our experiment demonstrates that cancer cells with p53 mutation and bcl-2 expression are more resistant to cisplatin and they have a decreased susceptibility to apoptosis, cells naturally expressing bcl-2 and germline mutation of p53 may be intrinsically resistant to cisplatin.
In this study, we could not readily obtain apoptotic features at concentration close to IC50 (3.3 μg/ml) in all of the examined cell lines. Although PC9 and PC14 cell lines had the wild type p53 and a comparable expression of p53 protein in this study, we could not obtain the clearly visualized DNA fragmentations, a morphologic feature of apoptosis at concentration close to IC50 (3.3 μg/ml), but it was relevant at higher concentration of cisplatin (10 μg/ml). This indicates that the examined parental cells were not prone to apoptosis, even with a comparable induction of p53. With respect to the responsiveness of tumors to therapies, it is suggested to be derived from the tendency of the parental cells to undergo rapid apoptosis in response to DNA damage38). For example, the responsive tumor types, such as leukemia, lymphoma or germ cell tumors, are probably derived from cells that primarily use p53 induction as a signal to apoptosis, but the tumors derived from epithelial cells, such as lung carcinoma, do not rapidly signal to apoptosis in response to DNA damage, which use p53 for G1 arrest and which are relatively resistant to current treatments31,41,42). During G1 arrest, interfering with DNA-repair enzyme machinery or additional growth factors in environment may enhance the tolerance to DNA damage, provide a survival signal to cells and render the resistance to chemotherapy3,7,39,40).
In summary, we examined the reduced sensitivity to cisplatin treatment in the examined cell lines paralleled a resistance to apoptosis induction, and cancer cells with endogenous expression of bcl-2 and p53 mutation were more resistant to cisplatin, which might be useful as a factor in tailoring chemotherapy regimens. However, a larger prospective study is needed to confirm the findings of this report. We observed markedly reduced apoptosis in cisplatin-resistant cell lines, but could not find any differences in the expression of apoptosis-regulatory proteins, p53, bcl-2 and bax compared to parental cell lines. It is suggested that the evaluation of functional activity of p53, such as a signal to cell cycle arrest or to apoptosis, is needed to understand the reduced susceptibility to apoptosis in resistant cells.
REFERENCES
- 1.Souquet PJ, Chauvin F, Boissel JP, Cellerino R, Cormier Y, Ganz PA, Kaasa S, Pater JL, Quoix E, Rapp E. Polychemotherapy in advanced non-small cell lung cancer: A meta-analysis. Lancet. 1993;342:19–21. doi: 10.1016/0140-6736(93)91882-m. [DOI] [PubMed] [Google Scholar]
- 2.Bunn PA, Kelly K. New treatment agents for advanced small cell and non-small cell lung cancer. Semin Oncol. 1995;22(Suppl 6):53–63. [PubMed] [Google Scholar]
- 3.Chaney SG, Sancar A. DNA repair: Enzymatic mechanisms and relevance to drug response. J Natl Cancer Inst. 1996;88:1346–1360. doi: 10.1093/jnci/88.19.1346. [DOI] [PubMed] [Google Scholar]
- 4.Hickman JA. Apoptosis and chemotherapy resistance. Eur J Cancer. 1996;32A:921–926. doi: 10.1016/0959-8049(96)00080-9. [DOI] [PubMed] [Google Scholar]
- 5.Roth JA. Modification of tumor suppressor gene expression and induction of apoptosis in non-small cell lung cancer (NSCLC) with an adenovirus vector expressing wildtype p53 and cisplatin. Hum Gene Thera. 1996;7:1013–1030. doi: 10.1089/hum.1996.7.8-1013. [DOI] [PubMed] [Google Scholar]
- 6.Hannun YA. Apoptosis and dilemma of cancer chemotherapy. Blood. 1997;89:1845–1853. [PubMed] [Google Scholar]
- 7.O’Dwyer PJ, Johnson SW, Hamilton TC. Cisplatin and its analogues. In: Devita VT Jr, Heilman S, Rosenberg SA, editors. Cancer: principles and practice of oncology. Philadelphia: Lippincott-Raven; 1997. pp. 418–428.32. [Google Scholar]
- 8.Haldar S, Negrini M, Monne M, Sabbioni S, Croce CM. Down-regulation of bcl-2 by p53 in breast cancer cells. Cancer Res. 1994;54:2095–2097. [PubMed] [Google Scholar]
- 9.Lowe SW, Bodis S, McClatchey A, Remington L, Ruley HE, Fisher DE, Housman DE, Jacks T. p53 status and the efficacy of cancer therapy in vivo. Science. 1994;266:807–810. doi: 10.1126/science.7973635. [DOI] [PubMed] [Google Scholar]
- 10.Miyashita T, Krajewski S, Krajewska M, Wang HG, Lin HK, Liebermann DA, Hoffman B, Reed JC. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene. 1994;9:1799–1805. [PubMed] [Google Scholar]
- 11.Selvakumaran M, Lin HK, Miyashita T, Wang HG, Krajewski S, Redd JC, Hoffman B, Liebermann D. Oncogene. 1994;9:1791–1798. [PubMed] [Google Scholar]
- 12.Zhan Q, Fan S, Bae I, Guillouf C, Lieberman DA, O’Connor PM, Fornace AJ. Oncogene. 1994;9:3743–3751. [PubMed] [Google Scholar]
- 13.Perego P, Giarola M, Righetti SC, Supino R, Caserini C, Delia D, Pierotti MA, Miyashita T, Reed JC, Zunino F. Association between cisplatin resistance and mutation of p53 gene and reduced bax expression in ovarian carcinoma cell systems. Cancer Res. 1996;56:556–562. [PubMed] [Google Scholar]
- 14.Thomas A, Rouby SE, Reed JC, Stanislaw K, Silber R, Potmesil M, Newcomb EW. Drug-induced apoptosis in B-cell chronic lymphocytic leukemia: relationship between p53 gene mutation and bcl-2/bax proteins in drug resistance. Oncogene. 1996;12:1055–1062. [PubMed] [Google Scholar]
- 15.Fan S, EI-Deiry WS, Bae I, Freeman J, Jondle D, Bhatia K, Fornace AJ, Jr, Margrath I, Kohn KW, O’Connor PM. p53 gene mutations are associated with decreased sensitivity of human lymphoma cells to DNA damaging agents. Cancer Res. 1994;54:5824–5830. [PubMed] [Google Scholar]
- 16.Eliopoulos AG, Kerr DJ, Herod J, Hodgkins L, Krajewski S, Reed JC, Young LS. The control of apoptosis and drug resistance in ovarian cancer: influence of p53 and bcl-2. Oncogene. 1995;11:1217–1228. [PubMed] [Google Scholar]
- 17.Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991;51:6304–6311. [PubMed] [Google Scholar]
- 18.Tishler RB, Calderwood SK, Coleman CN, Price BD. Increases in sequence specific DNA binding by p53 following treatment with chemotherapeutic and DNA damaging agents. Cancer Res. 1993;53:2212–2216. [PubMed] [Google Scholar]
- 19.Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature. 1995;377:552–557. doi: 10.1038/377552a0. [DOI] [PubMed] [Google Scholar]
- 20.Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 1995;55:5187–5190. [PubMed] [Google Scholar]
- 21.Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of human bax gene. Cell. 1995;80:293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 22.Kitada S, Krajewski S, Miyashita T, Krajewska M, Reed JC. ϒ-radiation induces upregulation of bax protein and apoptosis in radiosensitive cells in vivo. Oncogene. 1996;12:187–192. [PubMed] [Google Scholar]
- 23.Yonish-Rouach E, Resnitzky D, Lotem J, Sachs L, Kimchi A, Oren M. Wild-type p53 induced apoptosis of myeloid leukemic cells that is inhibited by interleukin-6. Nature. 1991;352:345–347. doi: 10.1038/352345a0. [DOI] [PubMed] [Google Scholar]
- 24.Clarke AR, Purdie CA, Harrison DJ, Morris RG, Bird CC, Hooper ML, Wyllie AH. Thymocyte apoptosis induced by p53-dependent and independent pathway. Nature. 1993;362:849–852. doi: 10.1038/362849a0. [DOI] [PubMed] [Google Scholar]
- 25.Lowe SW, Schmitt SW, Smith SW, Osborne BA, Jacks T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature. 1993;362:847–849. doi: 10.1038/362847a0. [DOI] [PubMed] [Google Scholar]
- 26.Pezzella F, Morrison H, Jones M, Gatler KC, Lane D, Harris AL, Mason DY. Immunohistochemical detection of p53 and bcl-2 proteins in non-Hodgkin’s lymphoma. Histopathology. 1993;22:39–44. doi: 10.1111/j.1365-2559.1993.tb00067.x. [DOI] [PubMed] [Google Scholar]
- 27.Miyashita T, Reed JC. Bcl-2 oncoprotein blocks chemotherapy-induced apoptosis in a human leukemia cell line. Blood. 1993;81:151–157. [PubMed] [Google Scholar]
- 28.Allouche M, Bettaieb A, Vindis C, Rousse A, Grignon C, Laurent G. Influence of bcl-2 overexpression on the ceramide pathway in daunorubicin-induced apoptosis of leukemic cells. Oncogene. 1997;14:1837–1845. doi: 10.1038/sj.onc.1201023. [DOI] [PubMed] [Google Scholar]
- 29.Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 30.Reed JC. Bcl-2 and the regulation of programmed cell death. J Cell Biol. 1994;124:1–6. doi: 10.1083/jcb.124.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sato T, Hanada M, Bodrug S, Irie S, Iwama N, Boise LH, Thompson CB, Golemis E, Fong L, Wang H, Reed JC. Interaction among members of the bcl-2 protein family analyzed with a yeast two-hybrid system. Proc Natl Acad Sci USA. 1994;91:9238–9242. doi: 10.1073/pnas.91.20.9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kroemer G. The proto-oncogene bcl-2 and its role in regulating apoptosis. Nat Med. 1997;3:614–620. doi: 10.1038/nm0697-614. [DOI] [PubMed] [Google Scholar]
- 33.Dole M, Nunez G, Merchant AK, Maybaum J, Rode CK, Bloch CA, Castle VP. Bcl-2 inhibits chemotherapy-induced apoptosis in neuroblastoma. Cancer Res. 1994;54:3253–3259. [PubMed] [Google Scholar]
- 34.Gottschalk AR, Boise LH, Oltvai ZN, Accavitti MA, Korsmeyer SJ, Quintans J, Thompson CB. The ability of bcl-XLand bcl-2 to prevent apoptosis can be differentially regulated. Cell Death & Differ. 1996;3:113–118. [PubMed] [Google Scholar]
- 35.Tu Y, Xu F, Liu J, Vescio R, Berenson J, Fady C, Lichtenstein A. Upregulated expression of bcl-2 in multiple myeloma cells induced by exposure to doxorubicin, etoposide and hydrogen peroxide. Blood. 1996;88:1805–1812. [PubMed] [Google Scholar]
- 36.McConkey DJ, Chandra J, Wright S, Plunkett W, McDonnell TJ, Reed JC, Keating M. Apoptosis sensitivity in chronic lymphocytic leukemia is determined by endogenous endonuclease content and relative expression of bcl-2 and bax. J Immunol. 1996;156:2624–2639. [PubMed] [Google Scholar]
- 37.Pepper C, Bentley P, Hoy T. Regulation of clinical chemoresistance by bcl-2 and bax oncoproteins in B-cell chronic lymphocytic leukaemia. Br J Haematol. 1996;95:513–517. doi: 10.1046/j.1365-2141.1996.d01-1927.x. [DOI] [PubMed] [Google Scholar]
- 38.Slichenmeyer WJ, Nelson WG, Slebos RJ, Kastan MB. Loss of a p53-associated G1 checkpoint does not decrease cell survival following DNA damage. Cancer Res. 1993;53:4164–4168. [PubMed] [Google Scholar]
- 39.Pietras RJ, Fendly B, Chazin VR, Pegram MD, Howell SB, Slamon DJ. Antibody to HER-2/neu receptor blocks DNA repair after cisplatin in human breast and ovarian cancer cells. Oncogene. 1994;9:1829–1838. [PubMed] [Google Scholar]
- 40.Canman CE, Gilmer T, Coutts S, Kastan MB. Growth factor modulation of p53-mediated growth arrest vs. apoptosis. Genes Dev. 1995;9:600–611. doi: 10.1101/gad.9.5.600. [DOI] [PubMed] [Google Scholar]
- 41.Michalovitz D, Halevy O, Oren M. Conditional inhibition of transformation & of cell proliferation by temperature-sensitive mutant of p53. Cell. 1990;62:671–680. doi: 10.1016/0092-8674(90)90113-s. [DOI] [PubMed] [Google Scholar]
- 42.Sabbatini PC, S-K Rao L, White E. Modulation of p53-mediated transcriptional repression & apoptosis by the adenovirus E1B 19K protein. Mol Cell Biol. 1995;15:1060–1070. doi: 10.1128/mcb.15.2.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]