

Abstract
Background
Neuroinflammation occurs in insulted regions of the brain and may be due to reactive oxygen species (ROS), nitric oxide (NO), cytokines, and chemokines produced by activated glia. Excessive production of neurotoxic molecules causes further neuronal damage. Low levels of vitamin D3 are a risk factor for various brain diseases.
Methods
Using the bacterial endotoxin, lipopolysaccharide (LPS), to induce neuroinflammation in primary cortical neuron-glia cultures, we investigated how 1,25-dihydroxyvitamin D3 (1,25(OH)2D3) affected neuroinflammation.
Results
LPS (100 ng/ml) induced the accumulation of nitrite and the production of ROS, interleukin (IL)-6, and macrophage inflammatory protein (MIP)-2 in time-dependent manners. Inhibition of p38 and extracellular signal-regulated kinase (ERK) but not c-Jun N-terminal kinase (JNK) mitogen-activated protein kinase (MAPK) by 20 μM of SB203580, PD98059, and SP600125, significantly reduced LPS-induced ROS production, NO accumulation, and inducible NO synthase (iNOS) expression, respectively. LPS-induced IL-6 and MIP-2 were significantly attenuated by inhibition of p38, ERK, and JNK MAPK. Cotreatment with 1,25(OH)2D3 attenuated LPS-induced ROS production, NO accumulation, and iNOS expression in concentration-dependent manners. 1,25(OH)2D3 also reduced LPS-induced production of IL-6 and MIP-2. Similarly, iNOS, IL-6, and MIP-2 mRNA expression in cells treated with LPS significantly increased, whereas this effect was attenuated by 1,25(OH)2D3. Moreover, LPS-induced phosphorylation of p38, ERK, and JNK MAPK was significantly inhibited by 1,25(OH)2D3.
Conclusions
Our findings indicate that 1,25(OH)2D3 reduced the LPS-stimulated production of inflammatory molecules in neuron-glia cultures by inhibiting MAPK pathways and the production of downstream inflammatory molecules. We suggest that 1,25(OH)2D3 can be used to alleviate neuroinflammation in various brain injuries.
Introduction
1,25-Dihydroxyvitamin D3 (1,25-(OH)2D3) is a secosteroid hormone, synthesized through a multistep process, which begins in the skin and is completed in the kidneys. Ultraviolet light photocatalyzes conversion of the precursor, 7-dehydrocholesterol, to vitamin D3 or cholecalciferol, which has no biological activity until its conversion to the active form, 1,25-(OH2)D3 [1]. The activated vitamin D metabolite has many roles in regulating homeostasis (e.g., calcium homeostasis and maintenance) throughout the body. 1,25-(OH)2D3 has effects on the classic target organs (e.g., bones, intestines, and kidneys) and stimulates calcium transport from these organs to the blood. A growing body of evidence has demonstrated that 1,25-(OH)2D3 plays an important role in non-classical actions such as regulating immune function [2]. It is known that 1,25-(OH)2D3, as a potent neuromodulator of the immune system, exerts marked effects on neural cells [3]. 1,25-(OH)2D3 was shown to regulate neurotrophic factors in the brain, including nerve growth factors (NGFs) [4], neurotrophin 3 (NT3) [5], and glial cell line-derived neurotrophic factor (GDNF) [6]. Additionally, 1,25-(OH)2D3 increases expressions of microtubule-associated protein-2, growth-associated protein-43 [7], and neurite outgrowth [8] in cultured neurons, indicating that 1,25-(OH)2D3 may also affect neuronal plasticity processes.
Clinical studies suggested that a vitamin D insufficiency is associated with an increased risk of brain insults such as Alzheimer’s disease (AD) [9], Parkinson’s disease [10], and ischemic brain injury [6]. In animal studies, a vitamin D deficiency exacerbated stroke brain injury and dysregulated ischemia-induced inflammation [11], whereas administration of 1,25-(OH)2D3 reduced ischemia-induced brain damage through upregulating GDNF expression [6]. Pretreatment with 1,25-(OH)2D3 attenuated hypokinesia and dopaminergic neurotoxicity induced by 6-OHDA in rats [12]. Moreover, 1,25-(OH)2D3 increased secretion of anti-inflammatory cytokines and reduced secretion of proinflammatory cytokines [4, 5, 13], suggesting that 1,25-(OH)2D3 may be neuroprotective and may regulate neuroinflammation in the brain. However, the underlying mechanisms of vitamin D’s effect on neuroinflammation remain unclear.
Neuroinflammation is a common mechanism and plays a crucial role in the pathogenesis of various nerve diseases. Initiation of a neuroinflammatory response involves a complex interplay of glia. Activated glial cells, mainly astrocytes and microglia, are thus histopathological hallmarks of neurologic diseases. Inflammatory mediators (e.g., nitric oxide (NO), reactive oxygen species (ROS), proinflammatory cytokines, and chemokines) released by activated glia are neurotoxic and can cause neuronal damage [14]. It is known that lipopolysaccharide (LPS), a gram-negative bacterial cell wall endotoxin, can activate glia through Toll-like receptors, triggering downstream signaling, such as mitogen-activated protein kinases (MAPKs). Three major MAPK subfamilies have been described: p38, extracellular signal-regulated kinase (ERK), and c-Jun N-terminal kinase (JNK). Activation of MAPK pathways by LPS initiates neuroinflammatory cascades characterized by activation of glia and increasing production of inflammatory mediators including ROS, NO, cytokines, and chemokines [15–17]. Therefore, controlling activated glia can be a therapeutic strategy for neuroinflammation.
Studying the protective roles of antioxidant compounds in inhibiting the inflammatory response in brain diseases is an important vista for further research and clinical applications. Using cortical neuron-glia cultures, we investigated how 1,25-(OH)2D3 affected LPS-induced neuroinflammatory responses, by exploring whether the effects of 1,25-(OH)2D3 are mediated through MAPK pathways.
Materials and methods
Chemical reagents and antibodies
1,25-(OH)2D3 (SI-D1530) and LPS (L3129) were purchased from Sigma-Aldrich (St. Louis, MO). The p38 MAPK inhibitor, SB203580, ERK inhibitor, PD98059, JNK inhibitor, SP600125, iNOS, and β-actin were purchased from Calbiochem (San Diego, CA). Antibodies against ERK, p38, JNK, phosphorylated (p)-p38, p-ERK (p-p42/p44), and p-JNK (p-p46/p54) were purchased from Cell Signaling Technology (Beverly, MA). Antibodies against microtubule-associated protein-2 (MAP-2) and glial fibrillary acid protein (GFAP) were purchased from Chemicon (Temecula, CA). Antibody against ED1 was purchased from Serotec (Bicester, UK). Antibodies against oligodendrocyte marker 4 (O4), fibronectin 1 (FN1), and rat endothelial cell antigen (RECA-1) were purchased from R&D systems (Minneapolis, MN), Bioworld Technology (MN, USA), and Abcam (Cambridge, MA), respectively.
Primary rat cortical neuron-glia cultures
Primary neuron-glia cultures were prepared from the cerebral cortex of 1-day-old neonatal Sprague–Dawley rats, as previously described [18–31]. All animal procedures were approved by the Institutional Animal Care and Use Committee of Taipei Medical University (Taipei, Taiwan) (permit no.: LAC-101-0249). These procedures were performed according to the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. After the rats were sacrificed, their brains were quickly removed aseptically, and the blood vessels and meninges were discarded. Cerebral cortices were dissected under sterile conditions and kept on ice in Hank’s solution (without Ca2+ or Mg2+). Subsequently, cortical cells were dissociated by trituration using a pipette. Cells were centrifuged (1500 rpm for 5 min) and resuspended in 10 % fetal bovine serum/Dulbecco’s modified Eagle’s medium (Gibco BRL, Grand Island, NY). To each well of 24-well culture plates was seeded 5 × 105 cells in 0.5 ml of culture medium. Cells were incubated at 37 °C and 5 % CO2 at a humidity of 95 % and used for experiments starting on day 14 of cultivation in vitro. The percentage cell composition was determined by immunostaining, followed by cell counting. The neuron-glia cultures consisted of approximately 35 % neurons, 54 % astrocytes, and 6 % microglia. In addition, cultures also consisted of approximately 4 % of fibroblasts and a small percentage (<1 %) of other cells including oligodendrocytes, and endothelial cells (pictures not shown).
Immunocytochemistry
Cultures were fixed in 4 % paraformaldehyde, as previously described [18]. Background staining was reduced by blocking nonspecific binding sites with 10 % goat serum for 1 h at room temperature. After washing with phosphate-buffered saline (PBS), endogenous peroxidase activity was quenched by incubation with a 3 % H2O2 solution in PBS. Cultures were washed again and then incubated overnight with the appropriate primary antibodies (rabbit anti-MAP-2, 1:500, Chemicon; rabbit anti-GFAP, 1:1000, Chemicon; mouse anti-ED1, 1:500, Serotec; mouse anti-O4, 1:300, R&D system; rabbit-anti fibronectin 1, 1:500, Bioworld Technology; mouse-anti RECA-1, 1:300, Abcam) at 4 °C. Cells were washed and visualized using the avidin-biotin peroxidase complex method (ABC Elite kit; Vector Laboratories, Burlingame, CA). Images were viewed on an inverted Olympus IX 70 microscope (Tokyo City, Japan) equipped with a cooled CCD camera and SPOT advanced software (Diagnostic Instruments, Sterling Heights, MI).
Cell-type identification and counting
Types of cells present in the culture were identified by immunocytochemical staining using cell-specific markers (MAP-2 for neurons, GFAP for astrocytes, ED1 for microglia, FN1 for fibroblasts, O4 for oligodendrocytes, and RACE-1 for endothelial cells). Images of immunostained-positive cells in each well (in five randomly selected fields with a calibrated area) were captured as digital micrographs, and cells were counted by an observer who was blinded to our study and confirmed by the experiment operator.
Measurement of ROS
ROS production was detected using the fluorochrome, 2′,7′-dichlorofluorescin diacetate (DCF-DA). Treated cells were incubated with 30 μM DCF-DA in cultured medium for 20 min at 37 °C. After incubation, the culture medium was removed, and then 500 μl of culture medium was added to each culture well. Plates were read with a fluorometric microplate reader at 485/500 nm [23]. Production of ROS was calculated as the maximum DCF fluorescence following incubation (20 min). The value of DCF fluorescence is expressed in arbitrary fluorescence units (AU).
Determination of NO accumulation
Nitrite accumulation was measured using the Griess reaction in culture media. After treatment, culture medium was collected for measurement of nitrite. Briefly, 50 μl of culture medium was mixed with an equal volume of Griess reagent. The absorbance was detected at 540 nm by a microplate reader (Molecular Devices, Menlo Park, CA).
Determination of IL-6 and macrophage inflammatory protein (MIP)-2 release
Levels of IL-6 and MIP-2 secreted into the culture media were measured by enzyme-linked immunosorbent assay (ELISA) kits (BioSource International) according to the manufacturer’s protocol.
Western blotting
Whole cell lysates were prepared from cultured neuronal/glial cells after 24 h of desired treatments. Treated cells were harvested, washed with PBS, and lysed in protein extraction buffer (Mammalian Cell-PE LBTM, Geno Technology) containing protease and phosphatase inhibitors (Complete Mini, Roche Diagnostics, Indianapolis, IN). Cell debris was removed by centrifugation at 12,000 rpm for 15 min at 4 °C, and the supernatant was collected for storage at −80 °C or to perform a Western blot analysis. Equal amounts of protein were separated on 10~15 % sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were electroblotted onto polyvinylidene difluoride membranes for 120 min under 100 V (PerkinElmer Life Sciences). Membranes were blocked with 5 % non-fat milk for 1 h, and then incubated overnight at 4 °C with the indicated antibodies, including those for inducible NO synthase (iNOS), p38, phosphorylated (p)-p38, p42/44, p-p42/44, p46/54, and p-p46/54 (1:1000 dilution) followed by an appropriate secondary antibody (at a 1:2 × 104 dilution) for 1 h at room temperature. Signals were visualized using enhanced chemiluminescent detection reagents (PerkinElmer Life Sciences). The membrane was then stripped and reprobed with an antibody specific for β-actin (at a 1:104 dilution) to ensure the accuracy of each loading. The protein signal intensity was quantified by a BioImaging System (Level Biotechnology) and normalized with the corresponding β-actin intensity.
Real-time reverse-transcription polymerase chain reaction (RT-PCR) assay
Total RNA was extracted from treated cells using the TRIzol® reagent (Invitrogen Life Technologies, Paisley, Scotland) according to the manufacturer’s protocol. Three micrograms of total RNA was reverse-transcribed into complementary (c)DNA using the Rever Tra Ace-α First-strand cDNA Synthesis Kit (TOYOBO Life Science, Japan). The resulting cDNA was incubated with the Rotor-Gene SYBR Green kit (QIAGEN Biosystems, CA) and primers for iNOS (sense, 5′-TTCTTTGCTTCTGTGCTAATGC-3′ and antisense, 5′-ATACTGTTCCATGCAGACAACC-3′); IL-6 (sense, 5′-TTCTTGGGACTGATGTTGTTGAC-3′ and antisense, 5′-AATTAAGCCTCCGACTTGTGAAG-3′); MIP-2 (sense, 5′-AAACTGCACCCAGGAAGCC-3′ and antisense, 5′-ACAGTGAGCTGGCCAATGC-3′); and β-actin (sense, 5′-GACCCAGATCATGTTTGAGACCTTC-3′ and antisense, 5′-GGTGACCGTAACACTACCTGAG-3′). For the semiquantitative analysis, we performed 40 amplification cycles (of denaturation at 95 °C for 5 s and annealing at 60 °C for 10 s) on a Rotor-Gene Q PCR Detection System (QIAGEN Biosystems). A melting curve analysis and sequencing data were used to confirm the specificity of the PCR products. Levels of iNOS, IL-6, and MIP-2 messenger (m)RNAs were normalized to those of β-actin and were then expressed as values relative to the control using the comparative threshold cycle (Ct) method.
3-(4,5-Dimethylthianol-2-yl)-2,5 diphenyl tetrazolium bromide (MTT) reduction assay
To examine cell viability, a colorimetric MTT reduction assay was performed as previously described [24]. After a 2-h treatment, the MTT reagent (0.5 mg/ml MTT in PBS) was added to each culture well, and cultured cells were incubated at 37 °C for 40 min. The culture medium was then aspirated from each well, and cells were dissolved by adding DMSO. The absorbance was measured at a test wavelength of 570 nm and a reference wavelength of 630 nm using a microplate reader (Molecular Devices).
Measurement of lactate dehydrogenase (LDH) release
Cytotoxicity was detected in culture media using an assay of LDH release, as an index of cell injury [18, 25]. LDH activity (units/min) was calculated from the slope of the decrease in the optical density at 340 nm over a 3-min period. The level of LDH was expressed as a percentage of values relative to PBS-treated control (Cont.) cultures.
Data analysis
All values are presented as the mean ± standard error of the mean (SEM). Experimental data were assessed using a one-way analysis of variance (ANOVA) followed by the Newman-Keuls’ test using the SigmaStat program (Jandel Scientific, San Rafael, CA). Significance was set at p < 0.05.
Results
LPS-induced ROS production, nitrite accumulation, and the release of proinflammatory mediators
To examine whether LPS elicited neuroinflammatory responses in cultured neurons/glia, we first examined the time course of LPS-induced ROS production, nitrite accumulation, and the release of IL-6 (a cytokine) and MIP-2 (a chemokine). As shown in Fig. 1a, treating cells with LPS (100 ng/ml) for the indicated periods (3, 6, 9, 12, and 24 h) induced ROS production in a time-dependent manner. ROS production significantly increased in cultured cells exposed to LPS for 6 h (p < 0.05) and continued to increase from 12 to 24 h (p < 0.01 and p < 0.001, respectively). NO production, as reflected by nitrite accumulation, significantly increased in cultured cells exposed to LPS for 12 h (p < 0.01) and continued to increase from 12 to 24 h (p < 0.001; Fig. 1b). Additionally, IL-6 significantly increased in cultured cells exposed to LPS for 6 h (p < 0.05) and remained elevated for up to 24 h (p < 0.001; Fig. 1c). Similarly, we also found that MIP-2 increased in cultured cells exposed to LPS for 9 h (p < 0.05) and remained elevated for up to 24 h (p < 0.001; Fig. 1c). As expected, LPS successfully initiated a neuroinflammatory response in our neuronal/glial coculture system.
Fig. 1.
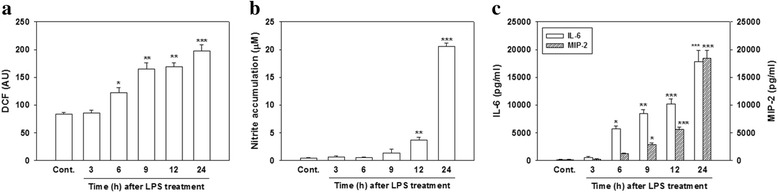
Effect of lipopolysaccharide (LPS) on reactive oxygen species (ROS) production, nitrite accumulation, and interleukin (IL)-6 and macrophage inflammatory protein (MIP)-2 production. Cultured cells were treated with LPS (100 ng/ml) for the indicated time periods. a ROS production by cells was detected by DCF fluorescence. The y-axis of the graph is expressed in DCF fluorescent arbitrary units (AU). b After treatment, the culture medium was collected to analyze levels of nitrite accumulation. c The culture medium was collected to analyze levels of IL-6 and MIP-2. Data are expressed as the mean ± SEM (n = 5 in each group). *p < 0.05, **p < 0.01, ***p < 0.001 vs. the control (Cont.)
Involvement of MAPK signaling pathways in LPS-induced production of proinflammatory mediators
To examine whether MAPKs (e.g., p38, ERK, and JNK) are involved in the production of LPS-initiated proinflammatory mediators, we treated cells with various concentrations (10 and 20 μM) of MAPK inhibitors and observed how these inhibitors affected levels of ROS production, nitrite accumulation, iNOS expression, and IL-6 and MIP2 production. In the absence of LPS, neither SB203580 (a p38 inhibitor), PD98059 (an ERK inhibitor), nor SP600125 (a JNK inhibitor) at 20 μM significantly affected ROS production (Fig. 2a). Cotreatment of 10 or 20 μM SB203580 with LPS for 24 h significantly attenuated LPS-induced ROS production; and 20 μM SB203580 more completely inhibited ROS production compared to 10 μM; however, the difference was non-significant. Cotreatment of 20 μM but not 10 μM PD98059 with LPS produced a significant reduction in ROS (p < 0.05). However, cotreatment of 10 or 20 μM JNK with LPS did not reduce ROS production (Fig. 2a). Figure 2b shows that cells treated with 10 or 20 μM SB203580 and LPS exhibited significantly attenuated nitrite accumulation, and 20 μM more completely inhibited this accumulation compared to 10 μM. Both SB203580 and PD98059 but not SP600125 at 20 μM significantly reduced LPS-induced nitrite accumulation (p < 0.001). Regarding iNOS induction, treatment with 20 μM of each inhibitor (SB203580, PD98059, and SP600125) alone had no effect (data not shown). Western blot analyses indicated that cotreatment with SB203580 or PD98059 and LPS for 24 h significantly attenuated the level of LPS-induced iNOS expression (p < 0.001 and p < 0.01, respectively), whereas cells cotreated with the JNK inhibitor and LPS exhibited no effect (Fig. 2c).
Fig. 2.
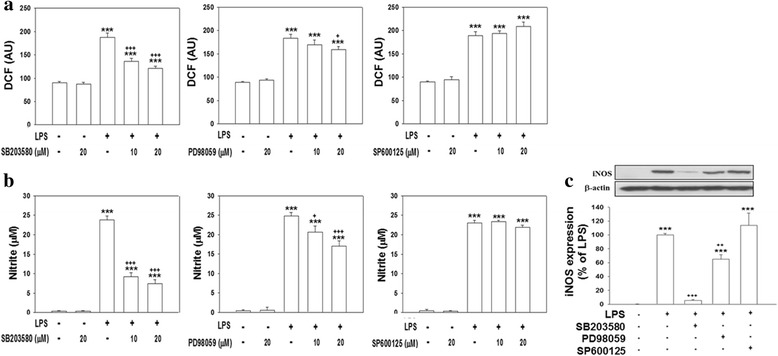
Effect of mitogen-activated protein kinase (MAPK) inhibitors on lipopolysaccharide (LPS)-induced reactive oxygen species (ROS) production, nitrite accumulation, and inducible nitric oxide synthase (iNOS) expression. Cultured cells were treated with various concentrations (10 and 20 μM) of SB203580 (a p38 inhibitor), PD98059 (an extracellular signal-regulated kinase (ERK) inhibitor), or SP600125 (a c-Jun N-terminal kinase (JNK) inhibitor), and then exposed to LPS (100 ng/ml) for 24 h. a ROS production by cells was detected by DCF fluorescence. The y-axis of the graph is expressed in DCF fluorescent arbitrary units (AU). b The culture medium was collected to analyze levels of nitrite accumulation. c After treatment, cells were harvested for a Western blot analysis. Levels of iNOS were normalized to β-actin, and then quantified. The iNOS expression induced by LPS was set to 100 % expression. Data are expressed as the mean ± SEM (n = 4 or 5 in each group). ***p < 0.001 vs. the control (Cont.); + p < 0.05, ++ p < 0.01, +++ p < 0.001 vs. LPS
Additionally, cotreating cells with SB203580, PD98059, or SP600125 and LPS for 24 h significantly attenuated the release of IL-6 (p < 0.001) and MIP-2 (p < 0.001) compared to the release in cultures treated with LPS alone (Fig. 3). Levels of IL-6 and MIP-2 production in cultures treated with SB203580, PD98059, or SP600125 alone did not significantly differ from those produced in control cultures (data not shown). These results suggest that activation of p38, ERK, and JNK MAPKs is involved in LPS-induced ROS production, nitrite accumulation, and proinflammatory mediator release.
Fig. 3.
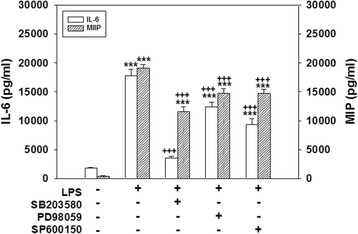
Effect of mitogen-activated protein kinase (MAPK) inhibitors on lipopolysaccharide (LPS)-induced interleukin (IL)-6 and macrophage inflammatory protein (MIP)-2 production. Cultured cells were treated for 24 h with 20 μM SB203580, 20 μM PD98059, or 20 μM SP600125 and were then exposed to LPS (100 ng/ml). The culture medium was collected to analyze levels of IL-6 and MIP-2. Data are expressed as the mean ± SEM (n = 4 in each group). ***p < 0.001 vs. the control (Cont.); +++ p < 0.001 vs. LPS
1,25-(OH)2D3 suppressed LPS-induced ROS production, nitrite accumulation, iNOS expression, and the release of inflammatory mediators without causing cell death
1,25-(OH)2D3, a potent functional hormone, is one of the important nuclear steroid transcription regulators that regulate the transcription of a large number of genes. We examined whether 1,25-(OH)2D3 could attenuate an experimental neuroinflammatory response produced using LPS in a neuron-glia culture model. Exposing cells to LPS for 24 h significantly enhanced ROS production (p < 0.001, Fig. 4a), nitrite accumulation (p < 0.001, Fig. 4b), iNOS expression (p < 0.001, Fig. 4c), and IL-6 and MIP-2 (p < 0.001, Fig. 5) production compared to those of control cultures. 1,25(OH)2D3 (100 nM) alone exerted no effects. Cotreating cells with 10 or 100 nM but not 1 nM of 1,25(OH)2D3 and LPS significantly attenuated levels of ROS production (p < 0.001, Fig. 4a). Similarly, cotreatment with 1, 10, or 100 nM of 1,25(OH)2D3 and LPS for 24 h significantly reduced nitrite accumulation (p < 0.001, Fig. 4b) and levels of iNOS expression (p < 0.001, Fig. 4c) in a concentration-dependent manner. Moreover, cotreating cells with 100 nM of 1,25(OH)2D3 significantly reduced LPS-induced IL-6 and MIP-2 production (p < 0.001, Fig. 5). Regarding protein expressions, levels of iNOS (Fig. 6a), IL-6, and MIP-2 proteins (Fig. 6b) were significantly reduced in cells exposed to 1,25(OH)2D3 plus LPS for 24 h. These results indicated that 1,25(OH)2D3 attenuated LPS-induced iNOS, IL-6, and MIP-2 expressions at both the protein and mRNA levels.
Fig. 4.
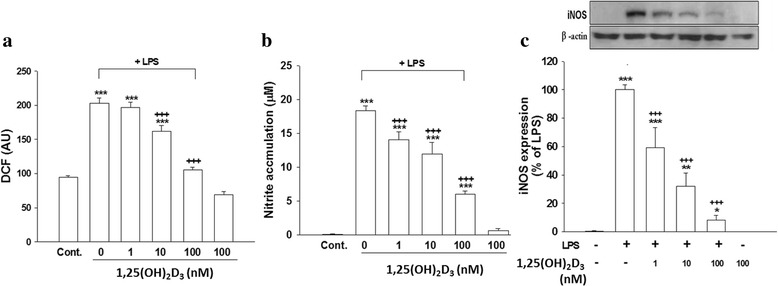
Effects of 1,25(OH)2D3 on levels of lipopolysaccharide (LPS)-induced reactive oxygen species (ROS) production, nitrite accumulation, and inducible nitric oxide synthase (iNOS) expression. Cultured cells were untreated (PBS) or treated with 1,25(OH)2D3 (100 nM), LPS (100 ng/ml), or various concentration of 1,25(OH)2D3 (1, 10, or 100 nM) plus LPS for 24 h. a ROS production by cells was detected by DCF fluorescence. The y-axis of the graph is expressed in DCF fluorescent arbitrary units (AU). b The culture medium was collected to analyze levels of nitrite accumulation. c After treatment, cells were harvested to analyze levels of iNOS protein expression using Western blotting. Levels of iNOS were normalized to β-actin and then quantified. iNOS expression induced by LPS was set to 100 % expression. Data are expressed as the mean ± SEM (n = 5 in each group). *p < 0.05, **p < 0.01, ***p < 0.001 vs. the control (Cont.); +++ p < 0.001 vs. LPS
Fig. 5.
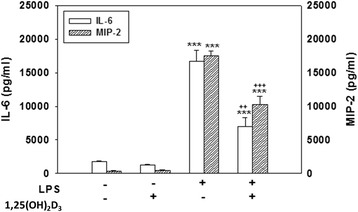
Effect of 1,25(OH)2D3 on lipopolysaccharide (LPS)-induced interleukin (IL)-6 and macrophage inflammatory protein (MIP)-2 release. Cultured cells were untreated (PBS) or treated with 1,25(OH)2D3 (100 nM), LPS (100 ng/ml), or 1,25(OH)2D3 plus LPS for 24 h, and then the culture medium was collected to analyze levels of IL-6 and MIP-2. Data are expressed as the mean ± SEM (n = 4 in each group). ***p < 0.001 vs. the control (Cont.); ++ p < 0.01, +++ p < 0.001 vs. LPS
Fig. 6.
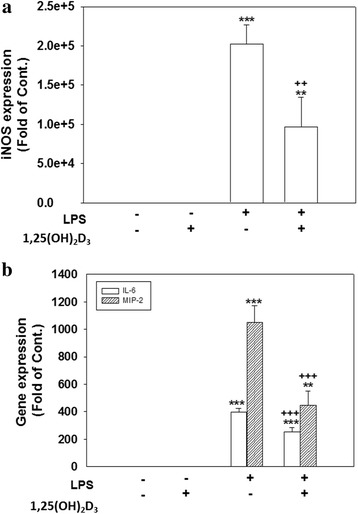
Effect of 1,25(OH)2D3 on lipopolysaccharide (LPS)-induced inducible nitric oxide synthase (iNOS), interleukin (IL)-6, and macrophage inflammatory protein (MIP)-2 gene expressions. Cultured cells were untreated (PBS) or treated with 1,25(OH)2D3 (100 nM), LPS (100 ng/ml), or 1,25(OH)2D3 plus LPS for 24 h. After treatment, cultured cells were harvested to conduct an RT-PCR assay of iNOS (a), IL-6, and MIP-2 (b) mRNA levels. Expression levels of iNOS, IL-6, and MIP-2 mRNAs were normalized to β-actin. Data are expressed as the mean ± SEM (n = 4 in each group). **p < 0.01 and ***p < 0.001 vs. the control (Cont.); ++ p < 0.01, +++ p < 0.001 vs. LPS
We also monitored cell viability using an MTT reduction assay and confirmed it using LDH measurements. Results from both the MTT reduction assay and LDH measurements showed that the viability of cultured cells exposed to 100 nM 1,25(OH)2D3 or LPS alone for 24 h did not significantly differ from that of PBS-treated cells (Fig. 7a). The MTT assay revealed that cotreatment with 1,25(OH)2D3 and LPS non-significantly affected cell viability compared to that of control cultures. Compared to the medium control, no significant differences existed in levels of LDH release (an indicator of cell death) when 1,25(OH)2D3 and LPS were simultaneously added, indicating that the 1,25(OH)2D3 concentration used in this study was not toxic to cells. Determination of LDH release and the MTT reduction test might not be sufficient to study cell damage. Therefore, cytotoxicity was further assessed by immunocytochemical staining, and we subsequently counted the number of positive cells with antibodies against MAP-2 (a neuron marker), GFAP (an astrocyte marker), and ED1 (a microglia marker), respectively. Exposure of cortical neuron-glia cultures to either 1,25(OH)2D3, LPS, or a combination of 1,25(OH)2D3 and LPS for 24 h did not alter the number of MAP-2-positive cells or the morphology of those cells compared to control cultures. Numbers of GFAP-positive and ED1-positive cells did not significantly differ among control culture and cultures treated with 1,25(OH)2D3, LPS, or a combination of 1,25(OH)2D3 and LPS (Fig. 7b, c). Exposure of cells to 1,25(OH)2D3 alone retained both astrocytes and microglia resting morphologies. However, LPS treatment changed the morphologies of astrocytes and microglia from those representing the resting state to those representing the active state. Activated astrocytes had an enlarged size and a massive accumulation of GFAP-positive filaments; ED1-positive microglia changed from the resting state, which exhibits a small soma and long cytoplasmic processes, to the activated form, which is characterized by an amoeboid appearance. A combination of 1,25(OH)2D3 and LPS attenuated the LPS-induced morphological changes in astrocytes and microglia. Taken together, our results suggest that the selected concentration of 1,25(OH)2D3 (100 nM) effectively reduced the LPS-initiated neuroinflammatory response without damaging cells.
Fig. 7.
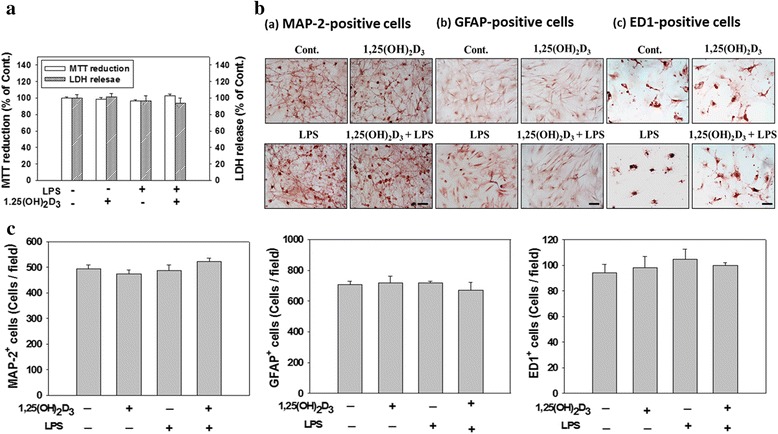
A lack of 1,25(OH)2D3 affected cell survival and cell death. Cultured cells were untreated (PBS) or treated with 1,25(OH)2D3 (100 nM), LPS (100 ng/ml), or 1,25(OH)2D3 plus LPS for 24 h. a After treatment, cultured cells were used for an MTT reduction assay. The culture medium was collected for lactate dehydrogenase (LDH) measurement. Cytotoxicity was assayed based on levels of LDH activity in the culture medium. MTT reduction or LDH release data are expressed as a percentage of values relative to control (Cont.) cultures. Data are expressed as the mean ± SEM (n = 4 in each group). b After treatment, cultured cells were fixed, and then immunocytochemically stained with an antibody against a specific cell marker (MAP-2, GFAP, or ED1). Representative photomicrographs show cultures stained with antibodies to identify neurons (MAP-2-positive cells; a), astrocytes (GFAP-positive cells; b), and microglia (ED1-positive cells; c). Arrows indicate activated glial cells. c Cell counts of MAP-2-, GFAP-, and ED1-positive cells in cultures exposed to either PBS, 1,25(OH)2D3, LPS, or 1,25(OH)2D3 plus LPS for 24 h. Data are the mean ± SEM from three independent experiments
1,25(OH)2D3 inhibited LPS-stimulated activation of MAPK signaling
To examine whether the antioxidant and anti-inflammatory effects of 1,25(OH)2D3 were mediated by inhibition of LPS-induced MAPK activation, we examined the effects of simultaneous treatments with LPS for 180 min using various combinations of the p38 inhibitor, SB203580, ERK inhibitor, PD98059, JNK inhibitor, SP600125, and 1,25(OH)2D3. Western blot results showed that LPS-stimulated phosphorylation of p38, p42/44 (ERK), and p46/54 (JNK) was significantly inhibited by SB203580 (p < 0.01, Fig. 8a), PD98059 (p < 0.001 for p-p42 and p < 0.05 for p-p44; Fig. 8b), and SP600125 (p < 0.001 for both p-p46 and p-p54; Fig. 8c), respectively. Cultured cells cotreated with 1,25(OH)2D3 and LPS exhibited significantly attenuated p-p38 (p < 0.001, Fig. 8a), p-p42/44 (p < 0.05 for both p-p42 and p-p44; Fig. 8b), and p-p46/54 levels (p < 0.001 for both p-p46 and p-p54; Fig. 8c). Activation of the phosphorylation of p38, ERK, and JNK did not significantly differ among control cultures and those treated with MAPK inhibitors or 1,25(OH)2D3 alone (data not shown). These results suggest that 1,25(OH)2D3 suppressed the production of LPS-induced inflammatory mediators by inhibiting the p38, ERK, and JNK MAPK pathways.
Fig. 8.
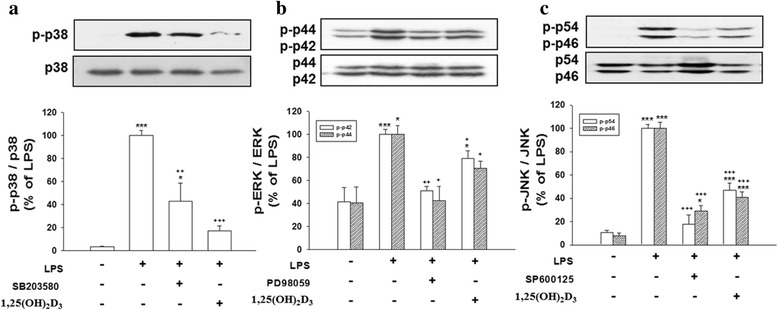
Mitogen-activated protein kinase (MAPK) inhibitors and 1,25(OH)2D3 inhibited lipopolysaccharide (LPS)-induced phosphorylation of p38, extracellular signal-regulated kinase (ERK), and c-Jun N-terminal kinase (JNK). Cultured cells were treated for 180 min with PBS (control; Cont.) alone, or LPS combined with 20 μM of SB203580, PD98059, SP600125, or 100 nM 1,25(OH)2D3, and then harvested to conduct Western blot analyses of phosphorylated (p)-p38 (a), p-ERK (p42/44) (b), and p-JNK (p46/54) (c). Levels of p-p38, p-ERK, and p-JNK were respectively normalized to total levels of p38, ERK, and JNK, and then quantified. Data are expressed as the mean ± SEM (n = 5 in each group). *p < 0.05, ***p < 0.001 vs. the Cont.; + p < 0.05, ++ p < 0.01, +++ p < 0.001 vs. LPS
Discussion
Glial cells, namely astrocytes and microglia, are the most abundant cells in the brain and play pivotal roles in protecting neurons from harmful insults. Activation of glia following infection or neuronal injury causes neuroinflammation; however, excessive glial activation may exacerbate neuronal damage due to the production of inflammatory molecules. In this study, we used LPS as a stimulant in neuron-glia cultures to initiate a neuroinflammatory response. The excessive production of inflammatory molecules was used as an index of activated glia. In this model, LPS significantly and time-dependently induced the production of inflammatory molecules including NO (reflected by nitrite accumulation), ROS, IL-6, and MIP-2 in cortical neuron-glia cultures. Activation of the p38, ERK, and JNK MAPK pathways is involved in LPS-initiated production of inflammatory molecules. 1,25(OH)2D3 attenuated LPS-induced nitrite accumulation, iNOS expression, and ROS production in concentration-dependent manners. LPS-induced production of IL-6 and MIP-2 was also significantly reduced by 1,25(OH)2D3. Furthermore, LPS-elicited phosphorylation of the p38, ERK, and JNK MAPK pathways was decreased by 1,25(OH)2D3. These results suggest that 1,25(OH)2D3 attenuates LPS-initiated neuroinflammation through inhibiting MAPK activation in neuron-glia cultures.
We previously demonstrated that astrocytes and microglia are important early sources of proinflammatory mediators in cerebrospinal fluid and brain tissues during bacterial (Klebsiella pneumoniae) infection of the central nervous system (CNS) [19]. Brain endothelial cells and pericytes which surround endothelial cells not only contribute to maintenance of the barrier function of the blood–brain barrier but also are immunoactive and can respond to LPS [20–22]. It is possible that LPS may stimulate brain immune cells (e.g., astrocyte and microglia) and other brain cells (e.g., pericytes, endothelial cells, and fibroblasts) resulting in a complex cascade of neuroinflammatory responses [20, 21, 26]. We employed cortical neuron-glia cultures as an in vitro model for mimicking the in vivo environment in which neurons and glia can interact. The advantages of our in vitro model were that (1) cultured cells can bypass any possible physiological feedback interactions in vivo to allow direct observation of LPS-induced neuroinflammation in specific cell populations and (2) it provides a simpler system to elucidate cellular mechanisms. However, its limitation is that cell cultures are simply an attempt to provide a simulated microenvironment, and cells are not in their normal physiological or original environment. The cortical neuron-glia cultures in this study consisted of approximately 35 % neurons, 54 % astrocytes, 6 % microglia, 4 % fibroblasts, and a small percentage (<1 %) of other cells including oligodendrocytes, and endothelial cells. The low percentage of other cell types may have been due to the fact that we dissected cerebral cortices and carefully removed blood vessel and meninges during our procedures to prepare the cortical neuron-glia cultures. It is possible that the results of this study can be ascribed to complex interactions between the different cell types found in the cultures. However, considering the relatively poor responses of the small percentage of fibroblasts and other cell types (oligodendrocytes and endothelial cells) to LPS, we propose that glial cells (mainly astrocytes and microglia) were the major responder cells in our culture system.
Glial cells play a role in immune surveillance under normal conditions; however, after a brain injury or exposure to inflammation, glial cells are activated. Under neuroinflammation, glial cells become strongly over-activated through a process that involves phosphorylation of downstream MAPK pathways (e.g., p38, ERK, and JNK) and production of inflammatory molecules [17]. Consistent with our results that treatment of cultured neurons/glia with LPS significantly induced expression of iNOS and the production of NO, ROS, IL-6, and MIP-2, pharmacological inhibition of MAPK pathways by MAPK inhibitors (SB 203580 for p38, PD98059 for ERK, and SP600125 for JNK MAPK) partially attenuated LPS-induced production of inflammatory molecules (ROS, NO, iNOS, IL-6, and MIP-2). A Western blot analysis also confirmed that treatment of cultured neurons-glia with LPS significantly induced phosphorylation of the p38, ERK, and JNK MAPK pathways, indicating that MAPK activation is involved in LPS-induced neuroinflammation. However, sustained overproduction of these inflammatory molecules may further contribute to neuronal damage. Therefore, inhibition of neuroinflammation has become a promising therapeutic target for neuronal injuries.
A dietary vitamin D3 (cholecalciferol) deficiency may lead to oxidative stress and elevation of iNOS expression in the brain and further promote cognitive decline in middle-aged and elderly adults [27]. It was reported that inhibition of iNOS expression can cause improvements in clinical signs observed in rats with experimentally produced allergic encephalomyelitis (EAE) treated with 1,25(OH)2D3 [28]. 1,25(OH)2D3 also effectively attenuated inflammatory cytokine expressions in the spinal cord and ameliorated EAE in rats [29]. In that model, 1,25(OH)2D3 treatment significantly reduced Toll-like receptor 8 (TLR8) expression and TLR8 target gene expressions (TNF-α and IL-1β) [29]. Additionally, 1,25(OH)2D3 provides neuroprotection against 1-methyl-4-1,2,3,6-tetrahydropyridine (MPTP)-induced neuronal injury through inhibition of glial activation and proinflammatory cytokine expression [30]. Those findings suggest that 1,25(OH)2D3 has important roles in the brain. In this study, we found that LPS-induced ROS production, nitrite accumulation, iNOS expression, and IL-6 and MIP-2 production were significantly reduced after treatment with 1,25(OH)2D3. It is known that 1,25(OH)2D3 is one of the important nuclear steroid transcription regulators that can control transcriptions of a large number of genes. 1,25(OH)2D3 is known to exert its biological functions by directly influencing cellular processes and also by influencing gene expressions through vitamin D response elements (VDREs). We also found that levels of iNOS, IL-6, and MIP-2 mRNA expressions were also significantly attenuated by 1,25(OH)2D3, indicating that 1,25(OH)2D3 plays an important role in gene regulation. However, further studies are required to elucidate the role of 1,25(OH)2D3 in LPS-initiated gene expression.
The apparent lack of cell damage induced by LPS in our cortical neuron-glia culture may have been due to regional differences in the susceptibility to the concentration (100 ng/ml or 0.1 μg/ml) of LPS in a shorter time (24 h) period. Differential neurotoxicity in mixed neuron-glia cultures from various brain regions after treatment with LPS was demonstrated in a previous study [31] in which treatment with LPS at an even higher concentration (1 μg/ml) for a longer time (72 h) did not cause neurotoxicity in cortical or hippocampal neuron-glia culture but caused neurotoxicity in cultures derived from the mesencephalon. In another study using mice cortical neuron-glia culture, neurotoxicity was only observed with a longer exposure time (36 h) when cultures were treated with LPS (1 μg/ml) in combination with interferon (IFN)-γ (5 U/ml) [32]. The concentration of 1,25(OH)2D3 we used in this study was similar to that in previous reports [33, 34]. In order to exclude the possibility that 1,25(OH)2D3 attenuated the LPS-induced release of inflammatory molecules because of 1,25(OH)2D3 toxicity, our results confirmed that 100 nM 1,25(OH)2D3 was not toxic to cultured neuron-glia cells as revealed by the MTT and LDH assays as well as immunocytochemistry. Because the concentration of 1,25(OH)2D3 used in our cultures effectively reduced LPS-induced neuroinflammation, we suggest that 1,25(OH)2D3 supplementation may be a candidate for prevention and treatment of neuroinflammation.
1,25(OH)2D3 is a neuro-immunomodulator involved in various neurodegenerative and autoimmune diseases [3]. The neuroprotective effects of 1,25(OH)2D3 have been reported in cultured hippocampal neurons against excitotoxic insults through reduced L-type calcium channel expression [35] and against rotenone-induced neurotoxicity in human neuroblastoma cell line SH-SY5Y cells by enhancing autophagy signaling [36]. Besides the direct neuroprotective effects on neurons, 1,25(OH)2D3 also reduced neurotoxin-induced dopaminergic neuronal loss by inhibiting of microglia activation and proinflammatory cytokine expression [30]. It is known that 1,25(OH)2D3 acts via the vitamin D receptor (VDR), a member of the steroid/thyroid hormone superfamily of transcription factors, and the membrane-associated, rapid-response steroid-binding receptor (MARRS), also known as Erp57/Grp58 [3]. The VDR is expressed by glia [4] and neurons [37] in the brain, and not just those participating in the classic actions of vitamin D. The action of 1,25(OH)2D3 mediates its biological effects by binding to the VDR, which then recruits cofactors to form a transcriptional complex that binds to VDREs in the promoter region of target genes to alter transcriptional cascades within cells [2]. The non-classical action of 1,25(OH)2D3 is that it binds to the MARRS receptor, located on the cell surface, initiating non-genomic effects [3]. A single-nucleotide polymorphism in the VDR gene can influence the affinity of vitamin D to its receptor and thus may be related to neurodegenerative diseases and neuronal damage by altering vitamin D-mediated pathways [38, 39]. β-amyloid (Aβ) disrupts the vitamin D-VDR pathway and results in iNOS expression in cortical neurons, whereas this effect can be prevented by vitamin D [40]. Moreover, treatment with vitamin D in that model protected neurons by preventing cytotoxicity and apoptosis and also by upregulating the VDR [41]. Those results suggest a potential role for vitamin D-VDR-mediated mechanisms in AD. In this study, we were interested in investigating how 1,25(OH)2D3 affected LPS-induced neuroinflammatory responses and exploring whether the effects of 1,25(OH)2D3 were mediated through MAPK signaling pathways. Involvement of MAPK signaling pathways in the production of inflammatory molecules in neurons and glial cells was previously demonstrated [42–44]. The three major MAPK subfamilies of p38, ERK, and JNK are phosphorylated in neurons and glia following LPS treatment [45]. MAPKs are related to LPS signaling in glial cells and lead to iNOS expression and the production of NO and proinflammatory cytokines [45–47]. The current results indicate that the phosphorylation of p38, ERK, and JNK MAPKs occurred after LPS treatment. We demonstrated that 1,25(OH)2D3 decreased the phosphorylation of p38, ERK, and JNK MAPKs as a result of LPS. This is consistent with a previous report that 1,25(OH)2D3 reduced macrophage-induced release of chemokines and cytokines by adipocytes and the chemotaxis of monocytes through inhibiting MAPK signaling pathways [48]. Similarly, 1,25(OH)2D3 diminished LPS-stimulated tumor necrosis factor (TNF)-α and IL-6 release via inhibiting p38 phosphorylation in monocytes/macrophages [49]. Our results suggest that the 1,25(OH)2D3-mediated suppression of the production of neuroinflammatory molecules may occur through inhibition of MAPK pathways. However, further studies are required to better understand the extents of1,25(OH)2D3’s anti-inflammatory effects and its involvement in the progression of neuroinflammation.
Conclusion
Our results suggest that 1,25(OH)2D3 significantly attenuated the LPS-initiated production of inflammatory molecules by inhibiting p38, ERK, and JNK MAPK signaling in neuron-glia cultures. We suggest that 1,25(OH)2D3 might be an innovative approach to ameliorating neuroinflammation resulting from various neuronal injuries.
Acknowledgements
This work was supported in part by grants from the Hsin Sheng Junior College of Medical Care and Management (HSC-103-001) and from Taipei Medical University (03C0720004A) to YNH; and from the Ministry of Science and Technology (MOST-103-2321-B-038-002 and MOST-104-2320-B-038-057-MY3) to JYW, Taiwan.
Abbreviations
- 1,25(OH)2D3
1α,25-dihydroxyvitamin D3
- ERK
extracellular signal-regulated kinase
- GDNF
glial cell line-derived neurotrophic factor
- IL
interleukin
- iNOS
inducible nitric oxide synthase
- JNK
c-Jun N-terminal kinase
- LPS
lipopolysaccharide
- MAPK
mitogen-activated protein kinase
- MARRS
membrane-associated, rapid-response steroid-binding receptor
- MIP
macrophage inflammatory protein
- NGF
nerve growth factor
- NO
nitric oxide
- NT3
neurotrophin 3
- ROS
reactive oxygen species
- VDR
vitamin D receptor
- VDRE
vitamin D response element
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YNH and JYW conceived and designed the experiments and wrote the paper. YNH and YJH performed the experiments. YNH, CCL, and JYW analyzed the data. YNH, CTC, and JYW contributed reagents/materials/analysis tools. All authors read and approved the final version of the manuscript.
Contributor Information
Ya-Ni Huang, Email: yani0106@gmail.com.
Yi-Jung Ho, Email: ejung330@gmail.com.
Chien-Cheng Lai, Email: popula180@gmail.com.
Chien-Tsai Chiu, Email: chientsaichiu@hotmail.com.
Jia-Yi Wang, Phone: 886-2-2736-1661 ext. 3412, Email: jywang2010@tmu.edu.tw.
References
- 1.Lips P. Vitamin D physiology. Prog Biophys Mol Biol. 2006;92:4–8. doi: 10.1016/j.pbiomolbio.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 2.Yang L, Ma J, Zhang X, Fan Y, Wang L. Protective role of the vitamin D receptor. Cell Immunol. 2012;279:160–6. doi: 10.1016/j.cellimm.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 3.Fernandes de Abreu DA, Eyles D, Feron F. Vitamin D, a neuro-immunomodulator: implications for neurodegenerative and autoimmune diseases. Psychoneuroendocrinology. 2009;34(Suppl 1):S265–77. doi: 10.1016/j.psyneuen.2009.05.023. [DOI] [PubMed] [Google Scholar]
- 4.Neveu I, Naveilhan P, Jehan F, Baudet C, Wion D, De Luca HF, et al. 1,25-dihydroxyvitamin D3 regulates the synthesis of nerve growth factor in primary cultures of glial cells. Brain Res Mol Brain Res. 1994;24:70–6. [DOI] [PubMed]
- 5.Neveu I, Naveilhan P, Baudet C, Brachet P, Metsis M. 1,25-dihydroxyvitamin D3 regulates NT-3, NT-4 but not BDNF mRNA in astrocytes. Neuroreport. 1994;6:124–6. doi: 10.1097/00001756-199412300-00032. [DOI] [PubMed] [Google Scholar]
- 6.Wang Y, Chiang YH, Su TP, Hayashi T, Morales M, Hoffer BJ, et al. Vitamin D(3) attenuates cortical infarction induced by middle cerebral arterial ligation in rats. Neuropharmacology. 2000;39:873–80. [DOI] [PubMed]
- 7.Taniura H, Ito M, Sanada N, Kuramoto N, Ohno Y, Nakamichi N, Yoneda Y. Chronic vitamin D3 treatment protects against neurotoxicity by glutamate in association with upregulation of vitamin D receptor mRNA expression in cultured rat cortical neurons. J Neurosci Res. 2006;83:1179–89. doi: 10.1002/jnr.20824. [DOI] [PubMed] [Google Scholar]
- 8.Brown J, Bianco JI, McGrath JJ, Eyles DW. 1,25-dihydroxyvitamin D3 induces nerve growth factor, promotes neurite outgrowth and inhibits mitosis in embryonic rat hippocampal neurons. Neurosci Lett. 2003;343:139–43. doi: 10.1016/S0304-3940(03)00303-3. [DOI] [PubMed] [Google Scholar]
- 9.Yu J, Gattoni-Celli M, Zhu H, Bhat NR, Sambamurti K, Gattoni-Celli S, et al. Vitamin D3-enriched diet correlates with a decrease of amyloid plaques in the brain of AbetaPP transgenic mice. J Alzheimers Dis. 2011;25:295–307. [DOI] [PMC free article] [PubMed]
- 10.Sanchez B, Relova JL, Gallego R, Ben-Batalla I, Perez-Fernandez R. 1,25-Dihydroxyvitamin D3 administration to 6-hydroxydopamine-lesioned rats increases glial cell line-derived neurotrophic factor and partially restores tyrosine hydroxylase expression in substantia nigra and striatum. J Neurosci Res. 2009;87:723–32. doi: 10.1002/jnr.21878. [DOI] [PubMed] [Google Scholar]
- 11.Balden R, Selvamani A, Sohrabji F. Vitamin D deficiency exacerbates experimental stroke injury and dysregulates ischemia-induced inflammation in adult rats. Endocrinology. 2012;153:2420–35. doi: 10.1210/en.2011-1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang JY, Wu JN, Cherng TL, Hoffer BJ, Chen HH, Borlongan CV, et al. Vitamin D(3) attenuates 6-hydroxydopamine-induced neurotoxicity in rats. Brain Res. 2001;904:67–75. [DOI] [PubMed]
- 13.Cantorna MT, Woodward WD, Hayes CE, DeLuca HF. 1,25-dihydroxyvitamin D3 is a positive regulator for the two anti-encephalitogenic cytokines TGF-beta 1 and IL-4. J Immunol. 1998;160:5314–9. [PubMed] [Google Scholar]
- 14.Wang JY, Wen LL, Huang YN, Chen YT, Ku MC. Dual effects of antioxidants in neurodegeneration: direct neuroprotection against oxidative stress and indirect protection via suppression of glia-mediated inflammation. Curr Pharm Des. 2006;12:3521–33. doi: 10.2174/138161206778343109. [DOI] [PubMed] [Google Scholar]
- 15.Korcok J, Wu F, Tyml K, Hammond RR, Wilson JX. Sepsis inhibits reduction of dehydroascorbic acid and accumulation of ascorbate in astroglial cultures: intracellular ascorbate depletion increases nitric oxide synthase induction and glutamate uptake inhibition. J Neurochem. 2002;81:185–93. doi: 10.1046/j.1471-4159.2002.00814.x. [DOI] [PubMed] [Google Scholar]
- 16.Zhang F, Liu J, Shi JS. Anti-inflammatory activities of resveratrol in the brain: role of resveratrol in microglial activation. Eur J Pharmacol. 2010;636:1–7. doi: 10.1016/j.ejphar.2010.03.043. [DOI] [PubMed] [Google Scholar]
- 17.Leow-Dyke S, Allen C, Denes A, Nilsson O, Maysami S, Bowie AG, et al. Neuronal Toll-like receptor 4 signaling induces brain endothelial activation and neutrophil transmigration in vitro. J Neuroinflammation. 2012;9:230. [DOI] [PMC free article] [PubMed]
- 18.Huang YN, Wu CH, Lin TC, Wang JY. Methamphetamine induces heme oxygenase-1 expression in cortical neurons and glia to prevent its toxicity. Toxicol Appl Pharmacol. 2009;240:315–26. doi: 10.1016/j.taap.2009.06.021. [DOI] [PubMed] [Google Scholar]
- 19.Wen LL, Chiu CT, Huang YN, Chang CF, Wang JY. Rapid glia expression and release of proinflammatory cytokines in experimental Klebsiella pneumoniae meningoencephalitis. Exp Neurol. 2007;205:270–8. doi: 10.1016/j.expneurol.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 20.Kacimi R, Giffard RG, Yenari MA. Endotoxin-activated microglia injure brain derived endothelial cells via NF-kappaB, JAK-STAT and JNK stress kinase pathways. J Inflamm (Lond) 2011;8:7. doi: 10.1186/1476-9255-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kovac A, Erickson MA, Banks WA. Brain microvascular pericytes are immunoactive in culture: cytokine, chemokine, nitric oxide, and LRP-1 expression in response to lipopolysaccharide. J Neuroinflammation. 2011;8:139. doi: 10.1186/1742-2094-8-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jansson D, Rustenhoven J, Feng S, Hurley D, Oldfield RL, Bergin PS, et al. A role for human brain pericytes in neuroinflammation. J Neuroinflammation. 2014;11:104. [DOI] [PMC free article] [PubMed]
- 23.Rosenkranz AR, Schmaldienst S, Stuhlmeier KM, Chen W, Knapp W, Zlabinger GJ. A microplate assay for the detection of oxidative products using 2’,7’-dichlorofluorescin-diacetate. J Immunol Methods. 1992;156:39–45. doi: 10.1016/0022-1759(92)90008-H. [DOI] [PubMed] [Google Scholar]
- 24.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 25.Wang JY, Shum AY, Ho YJ. Oxidative neurotoxicity in rat cerebral cortex neurons: synergistic effects of H2O2 and NO on apoptosis involving activation of p38 mitogen-activated protein kinase and caspase-3. J Neurosci Res. 2003;72:508–19. doi: 10.1002/jnr.10597. [DOI] [PubMed] [Google Scholar]
- 26.Wei X, Melemedjian OK, Ahn DD, Weinstein N, Dussor G. Dural fibroblasts play a potential role in headache pathophysiology. Pain. 2014;155:1238–44. doi: 10.1016/j.pain.2014.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keeney JT, Forster S, Sultana R, Brewer LD, Latimer CS, Cai J, et al. Dietary vitamin D deficiency in rats from middle to old age leads to elevated tyrosine nitration and proteomics changes in levels of key proteins in brain: implications for low vitamin D-dependent age-related cognitive decline. Free Radic Biol Med. 2013;65:324–34. [DOI] [PMC free article] [PubMed]
- 28.Garcion E, Nataf S, Berod A, Darcy F, Brachet P. 1,25-Dihydroxyvitamin D3 inhibits the expression of inducible nitric oxide synthase in rat central nervous system during experimental allergic encephalomyelitis. Brain Res Mol Brain Res. 1997;45:255–67. doi: 10.1016/S0169-328X(96)00260-4. [DOI] [PubMed] [Google Scholar]
- 29.Li B, Baylink DJ, Deb C, Zannetti C, Rajaallah F, Xing W, et al. 1,25-Dihydroxyvitamin D3 suppresses TLR8 expression and TLR8-mediated inflammatory responses in monocytes in vitro and experimental autoimmune encephalomyelitis in vivo. PLoS One. 2013;8:e58808. [DOI] [PMC free article] [PubMed]
- 30.Kim JS, Ryu SY, Yun I, Kim WJ, Lee KS, Park JW, et al. 1alpha,25-Dihydroxyvitamin D(3) Protects Dopaminergic Neurons in Rodent Models of Parkinson’s Disease through Inhibition of Microglial Activation. J Clin Neurol. 2006;2:252–7. [DOI] [PMC free article] [PubMed]
- 31.Kim WG, Mohney RP, Wilson B, Jeohn GH, Liu B, Hong JS. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. J Neurosci. 2000;20:6309–16. doi: 10.1523/JNEUROSCI.20-16-06309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jeohn GH, Kim WG, Hong JS. Time dependency of the action of nitric oxide in lipopolysaccharide-interferon-gamma-induced neuronal cell death in murine primary neuron-glia co-cultures. Brain Res. 2000;880:173–7. doi: 10.1016/S0006-8993(00)02737-2. [DOI] [PubMed] [Google Scholar]
- 33.Obradovic D, Gronemeyer H, Lutz B, Rein T. Cross-talk of vitamin D and glucocorticoids in hippocampal cells. J Neurochem. 2006;96:500–9. doi: 10.1111/j.1471-4159.2005.03579.x. [DOI] [PubMed] [Google Scholar]
- 34.Atif F, Sayeed I, Ishrat T, Stein DG. Progesterone with vitamin D affords better neuroprotection against excitotoxicity in cultured cortical neurons than progesterone alone. Mol Med. 2009;15:328–36. doi: 10.2119/molmed.2009.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brewer LD, Thibault V, Chen KC, Langub MC, Landfield PW, Porter NM. Vitamin D hormone confers neuroprotection in parallel with downregulation of L-type calcium channel expression in hippocampal neurons. J Neurosci. 2001;21:98–108. doi: 10.1523/JNEUROSCI.21-01-00098.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jang W, Kim HJ, Li H, Jo KD, Lee MK, Song SH, et al. 1,25-Dyhydroxyvitamin D(3) attenuates rotenone-induced neurotoxicity in SH-SY5Y cells through induction of autophagy. Biochem Biophys Res Commun. 2014;451:142–7. [DOI] [PubMed]
- 37.Gezen-Ak D, Dursun E, Yilmazer S. The effects of vitamin D receptor silencing on the expression of LVSCC-A1C and LVSCC-A1D and the release of NGF in cortical neurons. PLoS One. 2011;6:e17553. doi: 10.1371/journal.pone.0017553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gezen-Ak D, Dursun E, Ertan T, Hanagasi H, Gurvit H, Emre M, et al. Association between vitamin D receptor gene polymorphism and Alzheimer’s disease. Tohoku J Exp Med. 2007;212:275–82. [DOI] [PubMed]
- 39.Butler MW, Burt A, Edwards TL, Zuchner S, Scott WK, Martin ER, et al. Vitamin D receptor gene as a candidate gene for Parkinson disease. Ann Hum Genet. 2011;75:201–10. [DOI] [PMC free article] [PubMed]
- 40.Dursun E, Gezen-Ak D, Yilmazer S. A New Mechanism for Amyloid-beta Induction of iNOS: Vitamin D-VDR Pathway Disruption. J Alzheimers Dis. 2013;36:459–74. doi: 10.3233/JAD-130416. [DOI] [PubMed] [Google Scholar]
- 41.Dursun E, Gezen-Ak D, Yilmazer S. A novel perspective for Alzheimer’s disease: vitamin D receptor suppression by amyloid-beta and preventing the amyloid-beta induced alterations by vitamin D in cortical neurons. J Alzheimers Dis. 2011;23:207–19. doi: 10.3233/JAD-2010-101377. [DOI] [PubMed] [Google Scholar]
- 42.Gorina R, Font-Nieves M, Marquez-Kisinousky L, Santalucia T, Planas AM. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFkappaB signaling, MAPK, and Jak1/Stat1 pathways. Glia. 2011;59:242–55. doi: 10.1002/glia.21094. [DOI] [PubMed] [Google Scholar]
- 43.Zhao S, Zhang L, Lian G, Wang X, Zhang H, Yao X, et al. Sildenafil attenuates LPS-induced pro-inflammatory responses through down-regulation of intracellular ROS-related MAPK/NF-kappaB signaling pathways in N9 microglia. Int Immunopharmacol. 2011;11:468–74. [DOI] [PubMed]
- 44.Kim WI, Ryu HJ, Kim JE, Seo CH, Lee BC, Choi IG, et al. Differential nuclear factor-kappa B phosphorylation induced by lipopolysaccharide in the hippocampus of P2X7 receptor knockout mouse. Neurol Res. 2013;35:369–81. [DOI] [PubMed]
- 45.Xie Z, Smith CJ, Van Eldik LJ. Activated glia induce neuron death via MAP kinase signaling pathways involving JNK and p38. Glia. 2004;45:170–9. doi: 10.1002/glia.10314. [DOI] [PubMed] [Google Scholar]
- 46.Ashwell JD. The many paths to p38 mitogen-activated protein kinase activation in the immune system. Nat Rev Immunol. 2006;6:532–40. doi: 10.1038/nri1865. [DOI] [PubMed] [Google Scholar]
- 47.Kim BW, Koppula S, Hong SS, Jeon SB, Kwon JH, Hwang BY, et al. Regulation of microglia activity by glaucocalyxin-A: attenuation of lipopolysaccharide-stimulated neuroinflammation through NF-kappaB and p38 MAPK signaling pathways. PLoS One. 2013;8:e55792. [DOI] [PMC free article] [PubMed]
- 48.Ding C, Wilding JP, Bing C. 1,25-dihydroxyvitamin D3 protects against macrophage-induced activation of NFkappaB and MAPK signalling and chemokine release in human adipocytes. PLoS One. 2013;8:e61707. doi: 10.1371/journal.pone.0061707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Y, Leung DY, Richers BN, Liu Y, Remigio LK, Riches DW, et al. Vitamin D inhibits monocyte/macrophage proinflammatory cytokine production by targeting MAPK phosphatase-1. J Immunol. 2012;188:2127–35. [DOI] [PMC free article] [PubMed]