

Abstract
Catalytic assembly of enantiopure aliphatic amines from abundant and readily available precursors has long been recognized as a paramount challenge in synthetic chemistry. Herein, we describe a mild and general copper-catalyzed hydroamination that effectively converts unactivated internal olefins, an important yet unexploited class of abundant feedstock chemicals, into highly enantioenriched α-branched amines (≥ 96% enantiomeric excess) featuring two minimally differentiated aliphatic substituents. This method provides a powerful means to access a broad range of advanced, highly functionalized enantioenriched amines of interest in pharmaceutical research and other areas.
Unactivated internal olefins are manufactured on a very large scale during petroleum processing and thus represent excellent prospective building blocks for chemical synthesis. For example, 2 × 105 metric tons of 2-butene, the simplest member of this family, are produced annually by petroleum cracking and ethylene dimerization (1). In addition, the orthogonal reactivity of internal olefins with respect to carbonyls and other polar functional groups provides an exceptional opportunity for the late-stage diversification of complex molecules. In spite of these attractive attributes, few transition metal-catalyzed enantioselective transformations have been developed to convert unactivated internal olefins into broadly useful products with high levels of enantiocontrol (2, 3). In particular, the catalytic asymmetric addition of a hydrogen atom and a functional group across an unactivated internal olefin (i.e., hydrofunctionalization) constitutes a class of chemical transformations of broad utility yet poses formidable challenges for synthetic chemists (4).
Several factors have impeded the development of efficient asymmetric hydrofunctionalization of unactivated internal olefins using transition metal hydride catalysts. The low binding affinity of internal olefins towards the metal center and the sluggish migratory insertion usually lead to intrinsically demanding hydrometalation; for a handful of well-tailored Co- (5) and Rh- (6, 7) based catalyst systems that are capable of undergoing hydrometalation, the chain-walking mechanism consisting of iterative β-hydride elimination/migratory insertion steps rapidly convert the initially formed unstabilized secondary alkylmetal intermediate into the thermodynamically more stable, yet achiral, primary alkylmetal species. As a result, these processes are typically not amenable to enantioselective catalysis (8, 9).
Enantioenriched amines represent key structural elements in a variety of natural products, pharmaceutical agents, agrochemicals and functional materials. They also constitute an important family of chiral molecular scaffolds that are used stoichiometrically or catalytically to effect a diverse array of asymmetric transformations (10). Thus, the synthesis of optically pure amines has long been recognized as a preeminent goal for organic synthesis. Although enzymatic resolution (11) and stoichiometric chiral auxiliary-based methods (12) can be used to access α-branched aliphatic amines, these methods often require tedious multistep transformations from commercially available materials to arrive at the desired enantiopure amine products. To date, the general and modular catalytic assembly of α-branched aliphatic amines in a highly enantioselective fashion still presents considerable challenge (10). Moreover, catalytic access to such compounds has proven elusive when the two aliphatic substituents are minimally differentiated by their steric and electronic properties (e.g., methyl- vs. ethyl-).
The enantioselective hydroamination of alkenes has been successfully accomplished several times, albeit with a narrow substrate scope and/or moderate levels of enantioselectivity (13, 14). Most notable are intramolecular processes (15, 16) as well as intermolecular reactions of styrenes (17), terminal olefins (18, 19) and norbornene (20, 21). We have recently become interested in stereoselective amine synthesis using copper-catalyzed olefin hydroamination (Fig. 1) (22–27). In this context, we envisioned that the copper-catalyzed enantioselective hydroamination of unactivated internal olefins, if successful, would provide a general and powerful means of accessing enantioenriched α-branched aliphatic amines. The proposed catalytic cycle (Fig. 1C) commences with the enantioselective addition of a copper hydride species I across the double bond of the internal olefin II to provide the secondary alkylcopper intermediate IIIa. Electrophilic interception of the transient alkylcopper species with a hydroxylamine ester IV (28) in turn furnishes the enantioenriched aliphatic amine Va and releases the copper benzoate VI, which is reconverted to I upon reacting with hydrosilane VII (29). However, despite recent advances made in the field of copper(I) hydride-catalyzed asymmetric reduction (30, 31), enantioselective transformations of unactivated internal olefins catalyzed by a copper hydride species have previously not been reported.
Fig. 1. Proposed asymmetric hydroamination of unactivated internal olefins to access enantioenriched branched aliphatic amines.
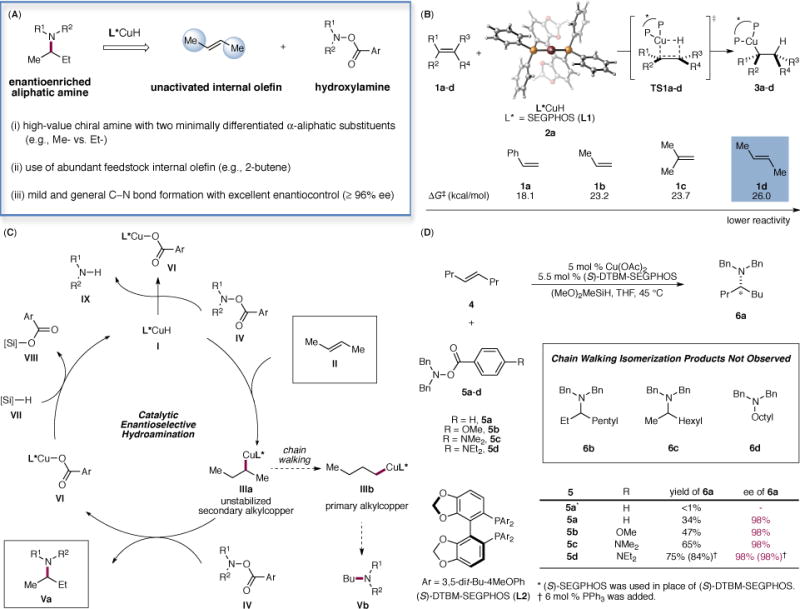
(A) Advantageous properties of the reaction profile. (B): DFT-calculated activation barriers for the hydrocupration of olefins 1a–d. Energies are computed at the M06/SDD-6-311+G(d,p)/SMD(THF) level with geometries optimized at the B3LYP/SDD-6-31G(d) level. (C): Proposed catalytic cycle. (D): Optimization studies. Reactions were performed using 4 (0.60 mmol), 5 (0.20 mmol), (MeO)2MeSiH (0.60 mmol), Cu(OAc)2 (5 mol %), L (5.5 mol %) in THF (1.0 M) at 45 °C for 36 h. Yields were determined by GC analysis using dodecane as the internal standard.
The hydrocupration of unactivated internal olefins is a very challenging process, and the impediments to this hydrocupration were evidenced by computational studies. As described in Fig. 1B, density functional theory (DFT) calculations with a SEGPHOS (L1)-based catalyst (2a) indicate that the hydrocupration of trans-2-butene (1d) is 7.9 and 2.8 kcal/mol more difficult than that of styrene (1a) and 1-propene (1b), respectively. These findings implied that unactivated internal olefin 1d undergoes hydrocupration 6.6×105 times slower than that of styrene 1a and 1.2×102 times slower than terminal olefin 1b. Since competitive and unproductive reduction of the hydroxylamine ester IV is catalyzed by the same copper hydride species I, the sluggish hydrocupration of unactivated internal olefins could be detrimental to the hydroamination process (Fig. 1C). In further computations, we were encouraged to find that the barrier of hydrocupration of 1d is 2.7 kcal/mol lower when 2a is replaced by the DTBM-SEGPHOS (L2, DTBM = 3,5-di-tert-butyl-4-methoxyphenyl)-based catalyst (2b) that has previously shown enhanced capability for asymmetric induction in copper(I) hydride mediated transformations (22, 23, 32). On the basis of these results and our previous work (22, 23), we posited that the use of this catalyst, which is highly active for hydrocupration yet still capable of effectively discerning the small steric bias between the prochiral faces of the internal olefin, would represent the key to the successful implementation of our proposed enantioselective hydroamination.
We began our investigation by exploring our previously developed catalyst systems for the enantioselective hydroamination of terminal olefins using trans-4-octene as the model substrate (Fig. 1C). Although the desired hydroamination product was not observed when (S)-SEGPHOS (L1) was employed, we found that in the presence of 5 mol % (S)-DTBM-SEGPHOS-based catalyst, the desired amine product was formed in 34% yield with excellent enantioselectivity (98% ee). None of the isomeric amine products (6b, 6c and 6d) originating from the chain walking isomerization of the initially formed secondary alkylcopper species was observed (33). In an effort to further improve the catalytic efficiency and suppress the competitive reduction of 5, we reasoned that the use of an electrophilic aminating reagent bearing a more electron-rich benzoyl group that is less prone to reduction yet exhibits comparable or even enhanced reactivity towards the nucleophilic alkylcopper intermediate could serve to address the problem. In addition, we postulated that the copper benzoate VI bearing an electron-rich benzoate would undergo faster σ-bond metathesis with the hydrosilane VII to regenerate the CuH species, thereby accelerating catalyst turnover. Ultimately, fine-tuning of the electronic nature of the benzoyl group led to the identification of 5d as the optimal electrophilic aminating agent. It is practically advantageous that 5d-type electrophilic aminating reagents feature enhanced stability upon storage and could be conveniently prepared from the parent hydroxylamines and commercially available 4-(N,N-diethylamino)benzoic acid on a large scale in a single operation.
Having established the optimal reaction conditions for this copper-catalyzed hydroamination, we focused our effort on the substrate scope of this transformation (Fig. 2). We were particularly interested in the utilization of 2-butene as an alkylating reagent to access branched aliphatic amines of high enantiopurity (Fig. 2(A)). The creation of ‘methyl-ethyl’ stereocenters in a highly enantioselective fashion has widely been considered as a premier challenge in asymmetric catalysis in view of the minimal steric differentiation between the two substituents (34). In particular, catalytic asymmetric synthesis of ‘methyl-ethyl’ α-branched tertiary amines is highly challenging. We found that in the presence of 2-butene, a wide range of hydroxylamine esters could be effectively converted into the corresponding chiral tertiary amines in a highly enantioselective manner. Electron-rich (9a and 9b) and electron-deficient (9c) hydroxylamine esters represented compatible coupling partners. Furthermore, a broad range of sensitive functional groups such as an aniline (9b), an ester (9d), a phenol (9d), an alcohol (9h), an amide (9j), an acetal (9l) and a cyclic trisubstituted olefin (9m) were tolerated under these conditions. Additionally, the successful incorporation of functional group handles such as a boronate (9e) and a chloride (9q and 9r) into the amine fragment provided opportunities for the further transformations of these valuable products using modern cross-coupling techniques (35). Moreover, substrates bearing a variety of privileged heterocyclic motifs that are frequently found in medicinal agents including indole (9f), thiophene (9g), furan (9h), pyridine (9i) and pyrimidine (9n) could be successfully transformed into the corresponding tertiary amines with high levels of enantiocontrol. In addition to acyclic substrates, cyclic aminating reagents could also be accommodated, providing the desired product in excellent enantiomeric excess (9n). Finally, the current catalyst system demonstrated excellent ability to control the diastereoselectivity when chiral electrophilic aminating reagents bearing stereocenters adjacent to the nitrogen atom were applied (9o–9r).
Fig. 2. Substrate scope of the copper-catalyzed enantioselective hydroamination of internal olefins.
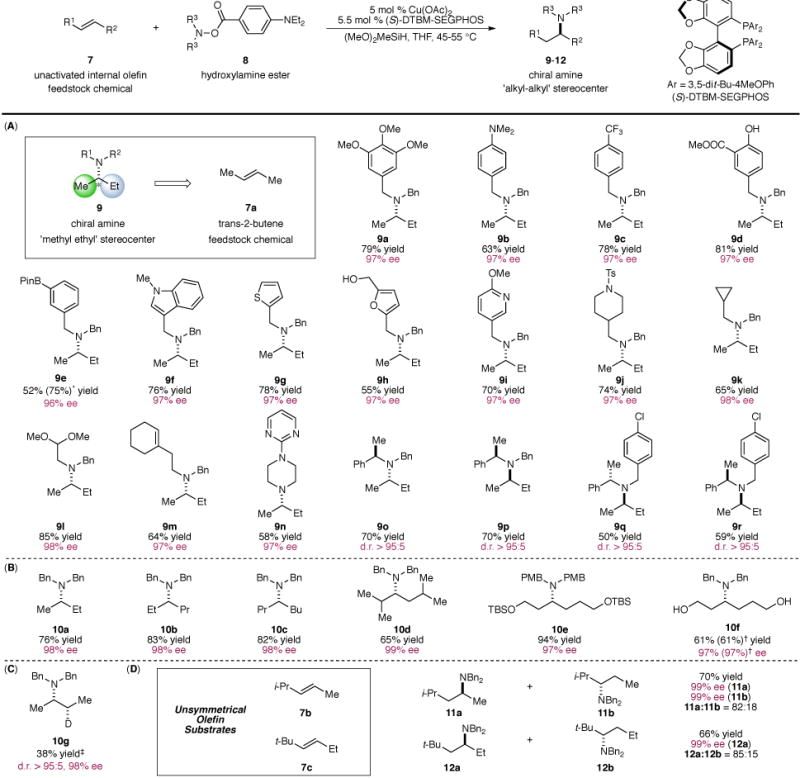
(A): Asymmetric hydroamination of 2-butene with a variety of electrophilic amines. (B): Scope of symmetrical internal olefins. (C): Deuterium Incorporation. (D): Regioselectivity in the hydroamination of unsymmetrical internal olefins. Yields refer to isolated yields on the average of two runs. Enantiomeric excesses were determined by chiral HLPC analysis or using Swager’s protocol (37). * Yield in parentheses was determined by 1H NMR analysis. † Reaction was performed on a 5 mmol scale with 1 mol % Cu(OAc)2, 1.1 mol % (S)-DTBM-SEGPHOS and 2 mol % PPh3. ‡ Ph2SiD2 was used in lieu of (MeO)2SiMeH. d.r. = diastereomeric ratio.
We next surveyed the scope of unactivated internal olefins that could be utilized for the current transformation (Fig. 2(B)). A broad range of abundant and commercially available internal olefins was found to be suitable substrates, affording the corresponding chiral amines featuring two highly similar α-substituents in excellent enantioselectivities (10a–10d). In addition, internal olefins bearing branching secondary alkyl substituents (10d) readily participated in this transformation. Functional groups such as a silyl ether (10e) and a free alcohol (10f) were also compatible under these conditions. Moreover, we were able to perform this transformation on a 5 mmol scale while simultaneously lowering the catalyst loading to 1 mol % without any detrimental effect on the yield and the enantioselectivity (10f), thereby demonstrating the scalability and practicality of this method. Furthermore, by capitalizing on the intrinsic flexibility of the hydrofunctionalization process, pharmacologically relevant β-deuteroamines (10g) (36) could be conveniently accessed in a highly diastereo-and enantioselective manner through the use of a commercially available source of deuteride (Ph2SiD2). Finally, when unsymmetrical internal olefins (7b and 7c) were employed, the hydroamination proceeded in a regioselective manner, affording tertiary amines with uniformly high levels of enantiocontrol (11 and 12).
A central advantage of the current catalytic enantioselective hydroamination is its ability for the late-stage modification of advanced, highly functionalized synthetic intermediates. To demonstrate this strategy, we selected two widely prescribed pharmaceutical drugs, cinacalcet and paroxetine, and subjected the electrophilic amines (13 and 15) derived from these drugs to our reaction conditions (Fig. 3). All of these amine-containing complex molecular scaffolds underwent effective hydroamination with unactivated internal olefins to furnish either of the diastereomeric tertiary amine products in a catalyst-controlled fashion, thereby exemplifying the potential of the current transformation in the rapid and efficient diversification of medicinally relevant molecules.
Fig. 3. Diversification of pharmaceutical agents.
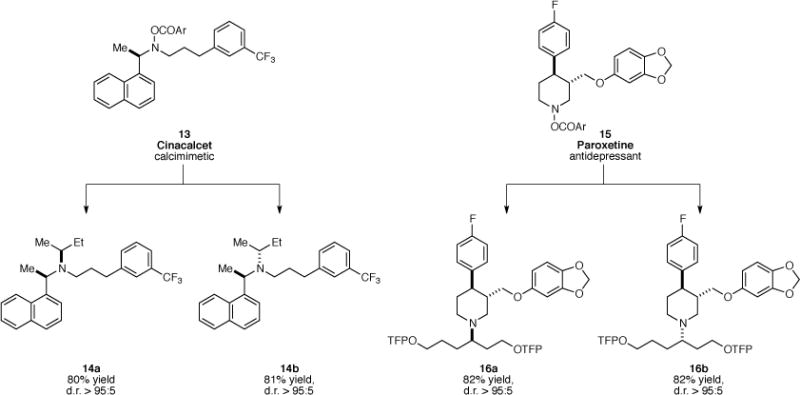
Conditions: a. 5 mol % Cu(OAc)2, 5.5 mol % (S)-DTBM-SEGPHOS, (MeO)2MeSiH, internal olefin, THF, 45 °C. b. (R)-DTBM-SEGPHOS was used instead of (S)-DTBM-SEGPHOS. TFP = 2-(5-trifluoromethyl)pyridyl.
To gain insight into the origin of the high levels of enantiocontrol observed with our system, we performed DFT calculations on the hydrocupration of trans-2-butene that is likely enantiodetermining (Fig. 4). The hydrocupration involves a four-membered transition state where the hydride is delivered to the olefinic carbon while the Cu–C bond is formed simultaneously (38). In this pseudo-four coordinate transition state, the two P atoms on the ligand are perpendicular to the C=C double bond in the substrate. The C2-symmetric bidentate phosphine ligand adopts the axial-equatorial coordination. The equatorial phosphine substituents are in closer contact with the olefin substrate and thus play a dominant role in enantiodiscrimination. In transition state TS2a involving the (Re)-face attack of the internal olefin, the unfavorable steric repulsion of both of the olefin’s aliphatic substituents with the ligand’s equatorial P substituents is minimized, leading to the formation of the preferred (S)-alkylcopper intermediate. The sterically encumbered tert-butyl groups present in the backbone of DTBM-SEGPHOS accentuate the energy differences between the (Re)- and the (Si)-face attack transition states, thereby giving rise to an activation barrier difference (ΔΔG‡) of 3.3 kcal/mol in favor of the (Re)-face attack pathway (TS2a). We expect this enantioselective hydroamination process to find broad application among practitioners of synthetic and pharmaceutical sciences. In addition to the utility of this protocol, we anticipate the distinct reactivity of unactivated internal olefins revealed in the current reaction manifold will inspire the further advances in a range of enantioselective hydrofunctionalization processes utilizing internal olefins.
Fig. 4. Transition state structures of the enantioselectivity-determining hydrocupration step.
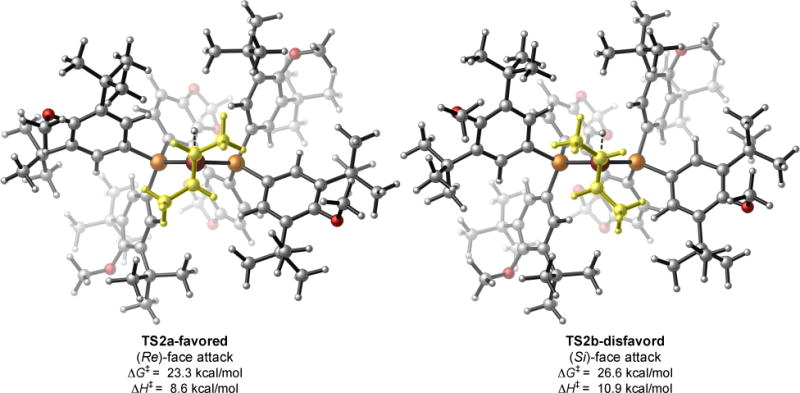
Energies are computed at the M06/SDD-6-311+G(d,p)/SMD(THF) level with geometries optimized at the B3LYP/SDD-6-31G(d) level.
Acknowledgments
We are grateful to the National Institutes of Health (grant GM 58160 for S.L.B.) and the University of Pittsburgh (P.L.) for financial support. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We acknowledge Dr. Yanchuan Zhao (Swager group) for helpful discussions. We thank Dr. Yi-Ming Wang and Dr. Michael T. Pirnot for assistance with the preparation of the manuscript. Calculations were performed at the Center for Simulation and Modeling at the University of Pittsburgh and the Extreme Science and Engineering Discovery Environment (XSEDE) supported by the National Science Foundation.
References
- 1.Geilen FMA, Stochniol G, Peitz S, Schulte-Koerne E. Butenes Ullmann’s Encyclopedia of Industrial Chemistry. 2014:1–13. [Google Scholar]
- 2.Bell S, Wüstenberg B, Kaiser S, Menges F, Netscher T, Pfaltz A. Science. 2006;311:642–644. doi: 10.1126/science.1121977. [DOI] [PubMed] [Google Scholar]
- 3.Mei TS, Patel HH, Sigman MS. Nature. 2014;508:340–344. doi: 10.1038/nature13231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anaikov VP, Tanaka M. Top Organomet Chem. 2013;43 [Google Scholar]
- 5.Obligacion JV, Chirik PJ. J Am Chem Soc. 2013;135:19107–19110. doi: 10.1021/ja4108148. [DOI] [PubMed] [Google Scholar]
- 6.van der Veen LA, Kamer PCJ, van Leeuwen PWNM. Angew Chem Int Ed. 1999;38:336–338. doi: 10.1002/(SICI)1521-3773(19990201)38:3<336::AID-ANIE336>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 7.Pereira S, Srebnik M. J Am Chem Soc. 1996;118:909–910. [Google Scholar]
- 8.Sakai N, Nozaki K, Takaya H. J Chem Soc, Chem Commun. 1994:395–396. [Google Scholar]
- 9.Gadzikwa T, Bellini R, Dekker HL, Reek JNH. J Am Chem Soc. 2012;134:2860–2863. doi: 10.1021/ja211455j. [DOI] [PubMed] [Google Scholar]
- 10.Nugent TC. Chiral Amine Synthesis: Methods, Developments and Applications. Wiley VCH; Weinheim: 2010. [Google Scholar]
- 11.Bornscheuer UT, Kazlauskas RJ. Hydrolases in Organic Synthesis. Wiley VCH; Weinheim: 2006. [Google Scholar]
- 12.Robak MT, Herbage MA, Ellman JA. Chem Rev. 2010;110:3600–3740. doi: 10.1021/cr900382t. [DOI] [PubMed] [Google Scholar]
- 13.Müller TE, Hultzsch KC, Yus M, Foubelo F, Tada M. Chem Rev. 2008;108:3795–3892. doi: 10.1021/cr0306788. [DOI] [PubMed] [Google Scholar]
- 14.Roesky PW, Müller TE. Angew Chem, Int Ed. 2003;42:2708–2710. doi: 10.1002/anie.200301637. [DOI] [PubMed] [Google Scholar]
- 15.Gagné MR, Brard L, Conticello VP, Giardello MA, Stern CL, Marks TJ. Organometallics. 1992;11:2003–2005. [Google Scholar]
- 16.Hong S, Marks TJ. Acc Chem Res. 2004;37:673–686. doi: 10.1021/ar040051r. [DOI] [PubMed] [Google Scholar]
- 17.Kawatsura M, Hartwig JF. J Am Chem Soc. 2000;122:9546–9547. [Google Scholar]
- 18.Zhang Z, Lee SD, Widenhoefer RA. J Am Chem Soc. 2009;131:5372–5373. doi: 10.1021/ja9001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reznichenko AL, Nguyen HN, Hultszch KC. Angew Chem Int Ed. 2010;49:8984–8987. doi: 10.1002/anie.201004570. [DOI] [PubMed] [Google Scholar]
- 20.Dorta R, Egli P, Zürcher F, Togni A. J Am Chem Soc. 1997;119:10857–10858. [Google Scholar]
- 21.Zhou J, Hartwig JF. J Am Chem Soc. 2008;130:12220–12221. doi: 10.1021/ja803523z. [DOI] [PubMed] [Google Scholar]
- 22.Zhu S, Niljianskul N, Buchwald SL. J Am Chem Soc. 2013;135:15746–15749. doi: 10.1021/ja4092819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu S, Buchwald SL. J Am Chem Soc. 2014;136:15913–15916. doi: 10.1021/ja509786v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi SL, Buchwald SL. Nature Chem. 2015;7:38–44. doi: 10.1038/nchem.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Niljianskul N, Zhu S, Buchwald SL. Angew Chem Int Ed. 2015;54:1638–1641. doi: 10.1002/anie.201410326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mikki Y, Hirano K, Satoh T, Miura M. Angew Chem Int Ed. 2013;52:10830–10834. doi: 10.1002/anie.201304365. [DOI] [PubMed] [Google Scholar]
- 27.Rucker RP, Whittaker AM, Dang H, Lalic G. J Am Chem Soc. 2012;134:6571–6574. doi: 10.1021/ja3023829. [DOI] [PubMed] [Google Scholar]
- 28.Berman AM, Johnson JS. J Am Chem Soc. 2004;126:5680–5681. doi: 10.1021/ja049474e. [DOI] [PubMed] [Google Scholar]
- 29.Lee D, Yun J. Tetrahedron Lett. 2004;45:5415–5417. [Google Scholar]
- 30.Apella DH, Moritani Y, Shintani R, Ferreira EM, Buchwald SL. J Am Chem Soc. 1999;121:9473–9474. [Google Scholar]
- 31.Deutsch C, Krause N, Lipshutz BH. Chem Rev. 2008;108:2916–2927. doi: 10.1021/cr0684321. [DOI] [PubMed] [Google Scholar]
- 32.Lipshutz BH, Noson K, Chrisman W, Lower A. J Am Chem Soc. 2003;125:8779–8789. doi: 10.1021/ja021391f. [DOI] [PubMed] [Google Scholar]
- 33.Whitesides GM, Stedronsky ER, Casey CP, San Filippo J., Jr J Am Chem Soc. 1970;92:1426–1427. [Google Scholar]
- 34.Fandrick KR, Fandrick DR, Reeves JT, Gao J, Ma S, Li W, Lee H, Grinberg N, Lu B, Senanayake CH. J Am Chem Soc. 2011;133:10332–10335. doi: 10.1021/ja2028958. [DOI] [PubMed] [Google Scholar]
- 35.de Meijere A, Diederich F. Metal-Catalyzed Cross-Coupling Reactions. Wiley VCH; Weinheim: 2004. [Google Scholar]
- 36.Gant TG. J Med Chem. 2014;57:3595–3611. doi: 10.1021/jm4007998. [DOI] [PubMed] [Google Scholar]
- 37.Zhao Y, Swager TM. J Am Chem Soc. 2015;137:3221–3224. doi: 10.1021/jacs.5b00556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Won J, Noh D, Yun J, Lee JY. J Phys Chem A. 2010;114:12112–12115. doi: 10.1021/jp1081966. [DOI] [PubMed] [Google Scholar]