

Abstract
Purpose
Diuretics are the primary treatment for the management of chronic heart failure (HF) symptoms and for the improvement of acute HF symptoms. The rate of delivery to the site of action has been suggested to affect diuretic pharmacodynamics. The main objective of this clinical trial was to explore whether a prolonged release tablet formulation of torasemide (torasemide-PR) was more natriuretically efficient in patients with chronic HF compared to immediate-release furosemide (furosemide-IR) after a single-dose administration. Moreover, the pharmacokinetics of torasemide-PR, furosemide-IR, and torasemide-IR were assessed in chronic HF patients as well as urine pharmacodynamics.
Methods
Randomized, open-label, blinded-endpoint, crossover, and single-dose Phase I clinical trial with three experimental periods. Torasemide-PR and furosemide-IR were administered as a single dose in a crossover fashion for the first two periods, and torasemide-IR 10 mg was administered for the third period. Blood and urine samples were collected at fixed timepoints. The primary endpoint was the natriuretic efficiency after administration of torasemide-PR and furosemide-IR, defined as the ratio between the average drug-induced natriuresis and the average drug recovered in urine over 24 hours.
Results
Ten patients were included and nine completed the study. Here, we present the results from nine patients. Torasemide-PR was more natriuretically efficient than furosemide-IR (0.096±0.03 mmol/μg vs 0.015±0.0007 mmol/μg; P<0.0001). Mictional urgency was lower and more delayed with torasemide-PR than with furosemide-IR.
Conclusion
In a study with a limited sample size, our results suggest that 10 mg of torasemide-PR is more natriuretically efficient than 40 mg of furosemide-IR after single-dose administration in patients with chronic HF over a 24-hour collection period. Further studies are necessary to evaluate potential pharmacodynamic differences between torasemide formulations and to assess its impact on clinical therapeutics.
Keywords: torasemide, furosemide, controlled-release preparation, efficiency, heart failure, pharmacodynamics
Introduction
Heart failure (HF) is associated with fluid retention and is one of the leading causes of morbidity and mortality in the world.1,2 Among the wide range of pharmacological treatments available, diuretics continue to play an essential role in the management of fluid overload symptoms.2,3 Torasemide is a high-ceiling loop diuretic that inhibits the Na+–K+–2Cl− reabsorptive pump in the medullary portion of the thick ascending limb of the loop of Henle, resulting in pronounced saluresis and diuresis.2,4 It is rapidly absorbed following oral administration, achieving the peak plasma concentration (Cmax) within the first hour. Metabolism of torasemide involves hepatic biotransformation, with up to 25% of an intravenous dose being excreted unchanged in the urine. Linear pharmacokinetics of torasemide have been observed after intravenous and oral administration of 20–200 mg and oral doses of 50–200 mg to patients with renal failure and congestive heart failure (CHF), respectively.4 On a mole per mole basis, torasemide is more than twice as active compared to furosemide.5
In CHF patients, the bioavailability of torasemide is the same as that observed in healthy volunteers with values approximating 90%.6,7 Vargo et al6 studied the pharmacokinetics of torasemide and furosemide in patients with CHF (classes II and III) and compared them to the pharmacokinetic profiles in healthy subjects.8 Cmax and tmax of torasemide in CHF patients were compared to that reported in healthy subjects, thus indicating that CHF did not affect the rate of absorption of torasemide after oral administration. By contrast, delayed absorption of furosemide was observed. Compared to healthy subjects, patients with CHF showed no pharmacokinetic changes for torasemide, with the exception of decreased chloride and increased Vss. Based on urinary excretion data, the pharmacokinetics of torasemide in this patient population are linear up to doses of at least 200 mg.6,9 As for pharmacodynamic effects, a rapid onset of action for torasemide with a significant increase in diuresis has been observed in healthy volunteers.10 In patients with chronic CHF, 10–20 mg of torasemide appears to produce greater saluresis and diuresis than 40 mg of furosemide; however, urinary potassium excretion does not follow the dose-related increase observed for sodium and chloride excretion after single doses of torasemide of up to 20 mg.4 Some clinical studies propose that torasemide could have a longer duration of action and improved tolerability compared to furosemide.3,11 Furthermore, a greater proportion of patients who received torasemide showed improved functional class (45.8 vs 37.2, P<0.00017) than compared with those who received furosemide.11 Additionally, torasemide could be a cost-saving option compared to furosemide.12
Conventionally, immediate-release (IR) formulations are quickly released into the bloodstream and, after rapid excretion of the drug, plasma concentrations fall sharply to subtherapeutic levels. This can lead to a decrease in drug efficacy depending on the clinical indication. This effect can be minimized by the administration of prolonged-release (PR) formulations. A PR formulation of torasemide (torasemide-PR) has been found to elicit a more physiological response in healthy subjects, resulting in a urinary excretion rate associated with a higher natriuretic efficiency and a more constant diuresis than the IR formulation.13
The main objective of this clinical trial was to explore whether torasemide given as PR tablet (torasemide-PR) was more natriuretically efficient in patients with chronic HF compared to IR furosemide (furosemide-IR) after a single-dose administration. As secondary objectives, the pharmacokinetic and pharmacodynamic profiles of torasemide-PR, torasemide-IR, and furosemide-IR were explored.
Methods
Subjects
Inclusion and exclusion criteria are summarized in Table 1.
Table 1.
Inclusion and exclusion criteria for recruited patients
| Inclusion criteria | |
| Demographic criteria | |
| Sex | Male and female |
| Age | ≥18 years old |
| Population | Diagnosed of HF, NYHA functional classes II–III, with ≥ a 3-month duration at the inclusion. Stable under HF therapy |
| Anthropometric criteria | |
| Body mass index | 19–30 kg/m2 |
| Medical criteria | |
| Laboratory analysis results, vital signs, and ECG were performed to validate that the inclusion criteria were met | |
| Medication criteria | |
| History | Must be under stable drug therapy, defined as no new drug for HF introduced within 4 weeks prior to inclusion |
| Other criteria | |
| Procedures | Informed consent approved by the ethics committee of the HSCSP signed by patients before any intervention |
| Pregnancy | Women not pregnant and assure no risk of pregnancy |
| Exclusion criteria | |
| Medical criteria | |
| Hospitalization | Hospitalization due to HF, any acute cardiac disease or procedure within the previous 6 weeks or any major surgery within the previous 8 weeks |
| Concomitant clinical conditions | Symptoms of angina at rest or with minimal activity (class III or IV, Canadian Cardiovascular Society) Severe aortic or mitral stenosis or clinically significant valvular heart disease that could lead to surgery within 12 months after inclusion Hypertrophic obstructive cardiomyopathy, active myocarditis, constrictive pericarditis, or clinically significant congenital heart disease Hepatitis B, hepatitis C or HIV or AIDS diagnosis Medical history or evidence of drug or alcohol abuse in the 3 months prior to inclusion Concomitant cardiovascular disease that was expected to reduce life expectancy to less than 1 year, or uncontrolled insulin-dependent diabetes SBP >150 mmHg, DBP >95 mmHg, HR in supine ≥100 beats/min after a 5-minute rest or untreated symptomatic bradycardia within 1 month prior to inclusion Total bilirubin ≥1.5 times ULN, or ALT or AST ≥3 times ULN, and estimated GFR ≤30 mL/min/1.73 m2 |
| Ventricular assist devices | To require ventricular assist devices, underwent CRT within 3 months or cardioverter defibrillator in the 4 weeks prior to inclusion |
| Transplants | To have a main organ transplant or a high probability of receiving a heart transplant within 6 months after inclusion |
| Medication criteria | |
| Concomitant therapy | Scheduled routine intravenous infusions for HF (eg, inotropes, vasodilators, diuretics) or routine ultrafiltration Digoxin therapy at a steady state exceeding 1.0 ng/mL at inclusion Current intake of a potent inhibitor of CYP2C9 or within 14 days prior to inclusion Current intake of a potent inducer of CYP2C9 or within 28 days prior to inclusion Chronic therapy with antiepileptic and antiarrhythmic drugs (except beta blockers) Treatment with eplerenone and espironolactone was only permitted after 7 days of washout Amiodarone was only permitted after 14 days of washout Chronic treatment (>7 days) with NSAIDs, except aspirin ≤325 mg dose |
| Other criteria | |
| Participation in other clinical trials within the preceding 60 days, or five half-lives before inclusion Blood donation during 4 weeks prior to inclusion |
|
Abbreviations: CRT, cardiac resynchronization therapy; ECG, electrocardiogram; NSAIDs, nonsteroidal anti-inflammatory drugs; GFR, glomerular filtration rate; HF, heart failure; NYHA, New York Heart Association; HSCSP, Hospital de la Santa Creu i Sant Pau; SBP, systolic blood pressure; DBP, diastolic blood pressure; HR, heart rate; ULN, upper normal limit; ALT, alanine aminotransferase; AST, aspartate aminotransferase.
Study design and treatments
The study was a randomized, open-label, blinded-endpoint, crossover, and single-dose Phase I clinical trial with three experimental periods (EU Clinical Trials Register: registration number 2011-000972-32 [www.clinicaltrialsregister.eu]; Clinical Trials registration number NCT01549158 [www.clinicaltrials.gov]). It was conducted in the Drug Research Center (CIM, IIB Sant Pau), and patients were recruited from the Cardiology Department of the Hospital de la Santa Creu i Sant Pau as well as from three primary care centers (Barcelona, Spain).
Prior to initiation, the study protocol and the informed consent that was signed by the eligible patients, was approved by an independent ethics committee (Comité Ético de Investigación Clínica del Hospital de la Santa Creu i Sant Pau in Barcelona, Spain) and the national competent authority (AEMPS, Spain).
The study included a screening evaluation after a 10-hour fasting period prior to the initiation of any study procedures and an end-of-study evaluation for all patients (physical examination including vital signs, electrocardiogram [ECG], and laboratory analyses).
The experimental phase consisted of three separate periods with a 7-day washout period (Figure 1). The included patients were randomized to receive torasemide-PR (Sutril Neo® 10 mg tablets; Ferrer Internacional S.A., Barcelona, Spain) or furosemide-IR (Seguril® 40 mg, tablets, Sanofi-Aventis SA, Barcelona, Spain) for the first period, and they subsequently received the alternate drug for the second period. All the participants received torasemide-IR (Sutril® 10 mg tablets; Ferrer Internacional SA, Barcelona, Spain) in the third period to minimize the impact of patient withdrawals during the study on the main study objective. Doses for the study were chosen according to clinical practice and authorized indications.
Figure 1.
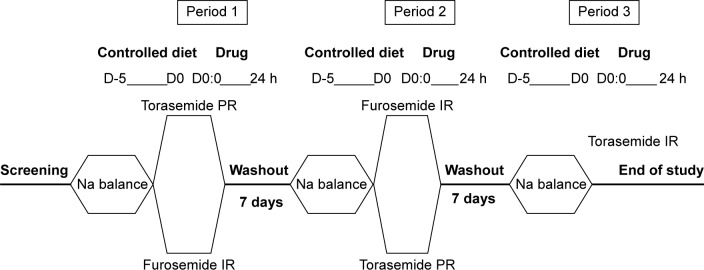
Study schedule.
Notes: The study consisted of three periods separated with a 7-day washout. The first and second period were crossover between torasemide PR and furosemide-IR whereas in the third period the patients taken Torasemide-IR.
Abbreviations: h, hours; PR, prolonged release; IR, immediate release.
Participants were required to follow a controlled diet (30 mEq Na/day, 60 mEq K/day, 1–1.5 g/kg protein/day) during the 5 days prior to dosing in each of the three periods, in order to ensure an appropriate sodium balance. This sodium balance was defined as the maintenance of a stable weight and an absence of sodium excretion fluctuations in the urine over 24 hours. In the morning of the fifth day prior to dosing (D-5), patients were given a bottle to collect urine for 24 hours, until the morning of the fourth day prior to dosing (D-4). Sodium (mEq) was analyzed in urine, and patients were weighed 24 hours before dosing; day-1 (D1) of each study period. In the morning of the study day 1 (D1) of each study period, a single dose of the drug was given with 180 mL of water in fasting conditions. Patients rested in a supine position for the first 8 hours after the administration to avoid the attenuation of the natriuretic response.14 Vital signs were measured using a DINAMAP® sphygmomanometer (Critikon, Inc., Tampa, FL, USA). ECG were recorded using a Pagewrite 300pi ECG recorder (Hewlett Packard, Palo Alto, CA, USA). Hematology, biochemistry, and urine parameters were evaluated before and after each period. No medications were permitted during the study, except for those allowed in the selection criteria.
Study endpoints
The primary endpoint was the natriuretic efficiency after the administration of torasemide-PR and furosemide-IR. The efficiency concept is an alternative way to describe the pharmacological effect, in terms of effect per unit drug concentration, instead of simple effect. However, for loop diuretics, urinary drug concentrations more accurately reflect the effect compared to plasma concentrations.15 In the present study, the total efficiency for the studied diuretics was defined as the ratio between the average drug-induced natriuresis (AeNa) and the average drug recovered in urine (Aedrug) over 24 hours. Secondary endpoints included other pharmacokinetic/pharmacodynamic (PK/PD) parameters of the study drugs, as well as urinary urgency assessed by a visual analog scale (VAS).
PK/PD determinations
For PK parameter evaluations, blood samples were taken for the determination of plasma concentrations of torasemide and furosemide. Urine samples were also taken to assess drug and ion excretion. The following PD parameters were also evaluated: diuretic effect of each study medication, measured as the volume of urine collected during different intervals (drug-induced diuresis) and at 24 hours; the diuresis rate in those intervals; and sodium, potassium, and chloride excretion rate (ERNa, ERK, ERCl, respectively).
Urine samples were collected 12 hours before each study drug administration and 0–0.5 hour, 0.5–1 hour, 1–1.5 hours, 1.5–2 hours, 2–3 hours, 3–4 hours, 4–6 hours, 6–8 hours, 8–12 hours, 12–24 hours postdose intervals. Blood samples were collected at baseline (prior to drug administration), and +0.25 hour, +0.5 hour, +0.75 hour, +1 hour, +1.25 hours, +1.5 hours, +1.75 hours, 2 hours, +2.5 hours, +3.5 hours, +4.5 hours, +5.5 hours, +7 hours, +10 hours, +18 hours, +24 hours, after study drug administration.
Blood samples (5.0 mL per sample, 17 samples per patient per period) were collected into lithium-heparinized tubes for PK analysis. Samples were centrifuged no later than 30 minutes after collection for 10 minutes at 3,000 rpm at 4°C, and the resulting plasma samples were separated into two aliquots of 0.7 mL each that were stored at −80°C.
Cmax and time to reach peak (tmax) were obtained directly from the raw data. The terminal plasma elimination half-life (t1/2) was calculated as t1/2=0.639/ke, where ke represents the first-order elimination rate constant associated with the terminal (log linear) portion of the curve, estimated via linear regression of time versus log concentration. The area under the plasma concentration–time curve (AUC) from 0 to ∞ (AUC0−∞) was calculated as AUC0−∞ = AUC0−tx + Ctx/ke, where tx is the time of the last torasemide or furosemide concentration (Ctx), exceeding the limit of quantification. Partial AUC values with 0 hours and 24 hours as time limits (AUC0−24) were also calculated. All AUC values were calculated by applying the log-trapezoidal method. The apparent volume of distribution (Vz/F) of torasemide and furosemide was calculated as Vz/F=D/(ke *AUC0−∞), where D is dose and F is bioavailability. Total oral clearance (Cl/F) was calculated as D/AUC0−t.
The cumulative amount of torasemide and furosemide excreted up to 24 hours (Ae24) was calculated as the sum of the amount of drug excreted unchanged in each time interval up to 24 hours. The cumulative amount excreted up to infinity (Ae∞) was calculated as the AUC extrapolated to infinity.
The diuretic effect was measured as the volume of urine obtained during the various urine collection intervals and the volume of urine collected at 24 hours and diuresis rate, expressed as milliliters per minute or milliliters per hour.
Calculation of all pharmacokinetic parameters (plasma and urine) and electrolytes in the urine were based on a noncompartmental model using WinNonlin version 2.1 (Pharsight Corporation, St Louis, MO, USA).
Samples analyses
Bioanalytical assays were previously validated (unpublished data, 2011) and performed at the Dr F Echevarne Analytical Laboratory, (Barcelona, Spain) in accordance with Good Laboratory Practices Guidelines.
Plasma and urine concentrations of torasemide and furosemide were determined through the method of liquid chromatography coupled to tandem mass spectrometry (triple quadrupole). Samples containing torasemide and furosemide were thawed at room temperature and centrifuged at 3,000 rpm for 5 minutes. The calibration line ranged from 1 ng/mL to 2,500 ng/mL (plasma samples) and from 1 ng/mL to 1,000 ng/mL (urine samples). The coefficient of variation (CV) at concentrations of 3 ng/mL, 1,000 ng/mL, and 2,000 ng/mL of torasemide in plasma samples was 10.3%, 9.9%, and 5.9%, respectively. The CV at concentrations of 3 ng/mL, 500 ng/mL, and 800 ng/mL of torasemide in urine samples was 10.5%, 5.5%, and 4.8%, respectively. Regarding furosemide, the calibration line ranged from 25 ng/mL to 2,000 ng/mL (plasma samples) and from 25 ng/mL to 5,000 ng/mL (urine samples). The CV at concentrations of 75 ng/mL, 800 ng/mL, and 1,600 ng/mL of furosemide in plasma samples was 4.4%, 4.8%, and 4.5%, respectively. The CV at concentrations of 75 ng/mL, 2,500 ng/mL, and 4,000 ng/mL of furosemide in urine samples was 5.8%, 4.0%, and 3.4%, respectively.
The limit of quantification was 1 ng/mL for torasemide and 25 ng/mL for torasemide. All samples were analyzed within the frozen stability period. Chromatography separation was performed using an analytical column Phenomenex LUNA C18 (150×4.6 mm) 5 μm in all cases.
For pharmacodynamics, urine sodium, chloride, and potassium were measured by indirect potentiometry using ion-selective electrodes (Integra 800; Roche Diagnostics SL, Barcelona, Spain) at the Biochemistry laboratory of the Hospital de la Santa Creu i Sant Pau. Measurements are expressed in millimoles per liter.
Safety and tolerability
Safety was assessed measuring blood pressure, heart rate, respiratory rate, body temperature at baseline and 0.5 hour, 1 hour, 2.5 hours, 3.5 hours, 4.5 hours, 7 hours, 10 hours, 18 hours, and 24 hours after study drug administration. Laboratory analyses were performed before and after each period. Adverse events were recorded throughout the study period. An ECG was performed at the inclusion and 24 hours after drug administration in each period.
Statistical methods
The sample size was calculated assuming a 60% intraindividual variability of furosemide and considering data described by Vargo et al6 for torasemide, on the basis of an estimated difference of 20% in natriuretic efficiency between both drugs. As a result, 30 patients with a maximum sex imbalance of 60%:40% in either direction were planned for study inclusion.
An exploratory analysis of the plasma parameters was carried out by plotting the mean of the raw data for the three treatments obtained in the study. The population for the primary analysis was per protocol population (defined as all randomized patients who received study medication for at least the two first periods and with no major deviations).
The difference in natriuretic efficiency between torasemide-PR and furosemide-IR was formally tested by means of a one-way analysis of variance (ANOVA) (formulation) of repeated measures followed by a post hoc analysis to assess the differences between both formulations. Secondary pharmacokinetic plasma parameters (Cmax, AUC0−t, AUC0−∞, t1/2, Vd/F, Cl/F, tmax, and mean residence time (MRT), pharmacokinetic urine parameters (ERmax, Ae24, and Ae∞), and urine pharmacodynamic variables (diuresis effect, ERNa, ERK, and ERCl) were analyzed for descriptive and comparative purposes. For comparisons between the three formulations (torasemide-PR, furosemide-PR, and torasemide-IR) a one-way ANOVA (formulation) of repeated measures followed by a post hoc analysis was applied to assess the differences by pairs. The test was two sided at the 5% significance level.
All statistical analyses were performed using SPSS/WIN 18 (SPSS Inc., Chicago, IL, USA).
Results
Study subjects
Due to low recruitment rates, only 10 out of the 30 planned patients were included in the study (eight males and two females) and nine patients completed the study. One patient was excluded before taking any study drug on D-1 and, therefore, all pharmacokinetic, pharmacodynamic, and safety analyses were performed on nine patients.
The demographic and baseline characteristics of included patients are shown in Table 2. Treatments were angiotensin converting enzyme inhibitors or angiotensin receptor antagonists (seven patients were treated with enalapril, one with ramipril, and one with valsartan), beta blockers (two patients received carvedilol and seven received bisoprolol), and diuretics (eight patients received diuretics). Three patients were taking mineral corticoid receptor antagonists that were transiently removed and restarted at the end of the study. Patients maintained an appropriate sodium balance, assessed by a stable weight (P=0.152), and no changes in sodium excretion (P=0.141) were observed between D-4 and D-1. A post hoc comparison of baseline (within 12 hours just before drugs administration) electrolytes (Na, K, and Cl) and Cr showed no differences between groups (Na: P=0.477; K: P=0.223; Cl: P=0.515; Cr: P=0.631).
Table 2.
Demographic and baseline characteristics of chronic heart failure patients (N=10)
| Parameter | Mean (SD) | Median (min–max) |
|---|---|---|
| Age (years) | 63.20 (12.44) | 65 (47–81) |
| Body weight (kg) | 80.34 (13.16) | 79 (66–105) |
| Height (cm) | 169.50 (8.63) | 168.5 (153–186) |
| Body mass index (weight [kg]/height [m2]) | 27.60 (3.69) | 27.95 (20.8–34.4) |
| Ejection fraction | 39 (13) | 34 (26–65) |
Abbreviations: max, maximum; min, minimum; SD, standard deviation.
Natriuresis efficiency
The timecourse of each drug excretion rate (Figure 2) had an important influence on the natriuretic efficiency. The sodium excretion rate (ERNa) was found to be similar between each drug administered (Table 3). Mean natriuretic efficiency obtained over the 24 hours collection interval was significantly higher for both torasemide formulations compared to furosemide (P<0.0001). No significant differences were found between PR and IR torasemide formulations (P=0.855; Table 3).
Figure 2.
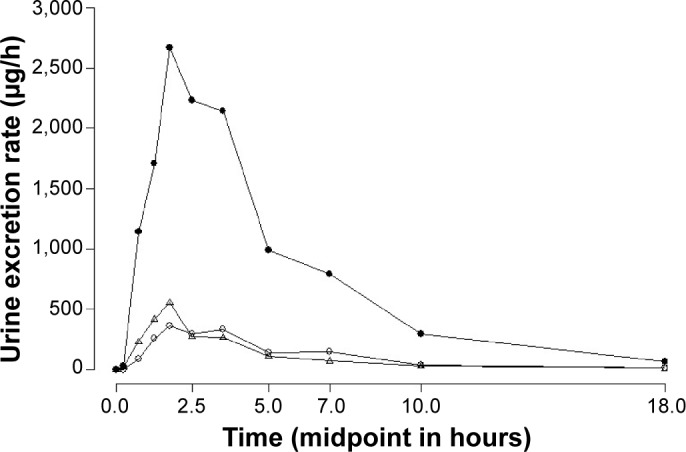
Mean urine excretion rate (μg/h) vs middle point (hours) of 10 mg torasemide-IR (Δ), 10 mg torasemide-PR (○), and 40 mg furosemide-IR (•).
Notes: Data points are presented as the mean of the time-course of each drug excretion rate in urine. The first collected interval starts at 0 hours, after the ingestion of the drugs.
Abbreviations: IR, immediate release; PR, prolonged release.
Table 3.
ERmax, ERNa, and natriuresis efficiency after oral torasemide-IR 10 mg, furosemide-IR 40 mg, and torasemide-PR 10 mg (N=9)
| Parameter | Torasemide-IR | Furosemide-IR | Torasemide-PR |
|---|---|---|---|
| ERmax (μg/h)* | |||
| Arithmetic mean (SD) | 555.24 (205.0) | 3,186.84 (1,410.1) | 404.38 (79.5) |
| Median (min–max) | 533.52 (324.3–913.2) | 2,798.85 (1,091.6–5,316.5) | 416.99 (262.3–503.0) |
| ERNa (mmol/h) | |||
| Arithmetic mean (SD) | 82.61 (28.6) | 81.63 (38.5) | 83.19 (37.1) |
| Median (min–max) | 87.01 (40.0–119.0) | 78.02 (36.5–143.8) | 65.33 (41.5–138.5) |
| Natriuresis efficiency* (mmol/μg) | |||
| Arithmetic mean (SD) | 0.091 (0.024) | 0.015 (0.007) | 0.096 (0.03) |
| Median (min–max) | 0.08 (0.06–0.13) | 0.017 (0.01–0.03) | 0.1 (0.06–0.13) |
Note:
P<0.0001 (both torasemide formulations vs furosemide). Data are the mean (SD) and median (min-max) of the pharmacodynamic parameters obtained for the three formulations.
Abbreviations: ERmax, drug’s excretion rate; ERNa, sodium excretion rate; h, hour; IR, immediate release; max, maximum; min, minimum; PR, prolonged release; SD, standard deviation.
The timecourse of natriuresis efficiency (Figure 3) shows that torasemide-IR induced the highest natriuresis efficiency during the first time interval (basal, 0.5 hour). Afterward, natriuresis efficiency was higher with torasemide-PR. Furosemide showed the lowest natriuretic efficiency from 0.25 hour until the end of the collection interval.
Figure 3.
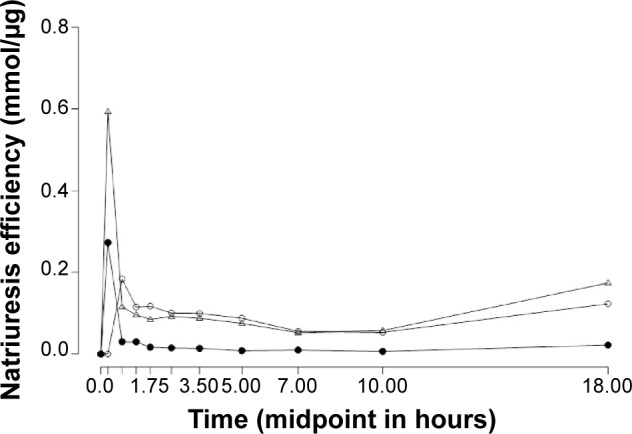
Mean natriuresis efficiency (mmol/μg) following oral administration of 10 mg torasemide-IR (Δ), 10 mg torasemide-PR (○), and 40 mg furosemide-IR (•) plotted against the midpoint (hours) of the entire collection interval.
Notes: Data points are time-course (expressed as midpoint of the entire collected interval) of the mean natriuresis efficiency, calculated as the ratio between drug-induced natriuresis and drug recovered in urine over 24 hours.
Abbreviations: IR, immediate release; PR, prolonged release.
Plasma pharmacokinetics
Measured plasma concentrations ranged from 3.58 ng/mL to 1,781.53 ng/mL for torasemide-IR, from 27.55 ng/mL to 1,903.42 ng/mL for furosemide-IR, and from 3.20 ng/mL to 975.52 ng/mL for torasemide-PR. Plasma concentration–time curves of torasemide-IR, furosemide-IR, and torasemide-PR are shown in Figure 4.
Figure 4.
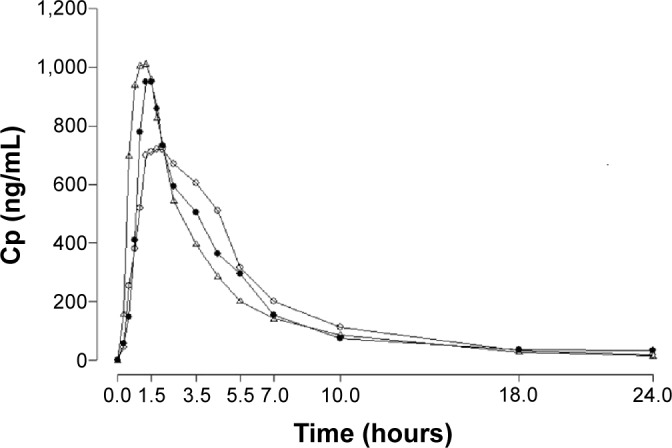
Mean plasma concentration–time curves (ng/mL–hours) of 10 mg torasemide-IR (Δ), 10 mg torasemide-PR (○), and 40 mg furosemide-IR (•).
Abbreviations: IR, immediate release; PR, prolonged release; Cp, plasma concentrations.
The descriptive statistics of the pharmacokinetic plasma parameters are summarized in Table 4. Torasemide-IR was more rapidly absorbed than torasemide-PR and furosemide-IR; achieving Cmax 1±0.4 hours after intake, whereas furosemide tmax was 1.6±0.8 hours and torasemide-PR tmax was 1.8±0.4 hours. The lowest Cmax (807.1±138.8 ng/mL) was observed for torasemide-PR versus furosemide-IR and torasemide-IR (1,039.9±415.4 ng/mL and 1,252.5±299.7 ng/mL, respectively).
Table 4.
Summary of PK parameters in plasma (N=9)
| Parameter | Torasemide-IR | Furosemide-IR | Torasemide-PR |
|---|---|---|---|
| Cmax (ng/mL) | |||
| Arithmetic mean (SD) | 1,252.5 (299.7) | 1,039.9 (415.4) | 807.1 (138.8) |
| Median (min–max) | 1,257.9 (846.1–1781.5) | 922.6 (508.3–1903.4) | 826.7 (558.8–975.5) |
| P-value | |||
| Torasemide-IR | – | 0.262 | 0.002 |
| Furosemide-IR | – | – | 0.313 |
| Torasemide-PR | – | – | – |
| tmax (h) | |||
| Arithmetic mean (SD) | 1.0 (0.4) | 1.6 (0.8) | 1.8 (0.4) |
| Median (min–max) | 1.0 (0.5–1.7) | 1.5 (1–3.5) | 1.7 (1.2–2.5) |
| P-value | |||
| Torasemide-IR | – | 0.028 | 0.012 |
| Furosemide-IR | – | – | 0.246 |
| Torasemide-PR | – | – | – |
| AUC0−t (ng h/mL) | |||
| Arithmetic mean (SD) | 3,976.3 (1277.0) | 3,540.9 (776.6) | 4,362.1 (1386.5) |
| Median (min–max) | 4,050.15 (2131.9–5875.0) | 3,753.2 (2119.5–4528.2) | 4,188.0 (2616.9–6907.7) |
| P-value | |||
| Torasemide-IR | – | 0.321 | 0.031 |
| Furosemide-IR | – | – | 0.091 |
| Torasemide-PR | – | – | – |
| AUC0−∞ (ng h/mL) | |||
| Arithmetic mean (SD) | 4,096.4 (1386.8) | 3,761.1 (852.4) | 4,496.8 (1516.7) |
| Median (min–max) | 4,200.3 (2154.2–6255.3) | 3,896.4 (2244.6–4764.9) | 4,252.5 (2648.4–7396.7) |
| P-value | |||
| Torasemide-IR | – | 0.674 | 0.041 |
| Furosemide-IR | – | – | 0.231 |
| Torasemide-PR | – | – | – |
| Ke (L/h) | |||
| Arithmetic mean (SD) | 0.14 (0.02) | 0.32 (0.13) | 0.15 (0.02) |
| Median (min–max) | 0.15 (0.11–0.17) | 0.34 (0.14–0.52) | 0.16 (0.11–0.17) |
| P-value | |||
| Torasemide-IR | – | 0.014 | 0.425 |
| Furosemide-IR | – | – | 0.021 |
| Torasemide-PR | – | – | – |
| t1/2 (h) | |||
| Arithmetic mean (SD) | 4.92 (0.77) | 2.65 (1.32) | 4.65 (0.80) |
| Median (min–max) | 4.60 (4.01–6.17) | 2.01 (1.32–4.73) | 4.39 (3.94–6.55) |
| P-value | |||
| Torasemide-IR | – | 0.004 | 0.499 |
| Furosemide-IR | – | – | 0.019 |
| Torasemide-PR | – | – | – |
| Vd/F (L) | |||
| Arithmetic mean (SD) | 18.5 (4.8) | 41.4 (19.0) | 15.9 (3.9) |
| Median (min–max) | 16.2 (13.8–28.8) | 42.3 (19.5–77.8) | 15.4 (10.1–22.3) |
| P-value | |||
| Torasemide-IR | – | 0.023 | 0.014 |
| Furosemide-IR | – | – | 0.015 |
| Torasemide-PR | – | – | – |
| Cl/F (L/h) | |||
| Arithmetic mean (SD) | 2.7 (0.98) | 11.2 (3.1) | 2.4 (0.8) |
| Median (min–max) | 2.4 (1.6–4.6) | 10.3 (8.4–17.8) | 2.3 (1.3–3.8) |
| P-value | |||
| Torasemide-IR | – | <0.0001 | 0.046 |
| Furosemide-IR | – | – | <0.0001 |
| Torasemide-PR | – | – | – |
| MRT (h) | |||
| Arithmetic mean (SD) | 4.5 (1.0) | 3.8 (1.1) | 5.4 (0.9) |
| Median (min–max) | 4.5 (3.2–6.3) | 3.6 (2.5–5.9) | 5.5 (4.2–6.8) |
| P-value | |||
| Torasemide-IR | – | 0.243 | <0.0001 |
| Furosemide-IR | – | – | 0.008 |
| Torasemide-PR | – | – | – |
Notes: Data are mean (SD) and median (min-max) for every pharmacokinetic parameter. Significance for parameters was tested by means of a one-way analysis of variance (ANOVA) followed by a post hoc analysis to assess differences by pairs. Bold values indicate statistical significance.
Abbreviations: AUC, area under the curve; Cmax, maximum plasma concentration; Cl/F, clearance; h, hours; Ke, elimination rate constant; max, maximum; min, minimum; MRT, mean residence time; PK, pharmacokinetic; IR, immediate-release; PR, prolonged release; tmax, time to reach Cmax; t1/2, elimination half-life; Vd/F, volume of distribution; SD, standard deviation.
Urine pharmacokinetics/pharmacodynamics
Table 5 shows the summarized urine pharmacokinetics. The cumulative amount of torasemide, both IR and PR formulations, was lower than that of furosemide-IR. Potassium and chlorine excretion rates were numerically slightly higher and lower for furosemide, respectively, compared to either formulation of torasemide.
Table 5.
PK/PD urinary parameters (N=9)
| Parameter | Torasemide-IR | Furosemide-IR | Torasemide-PR |
|---|---|---|---|
| Ae24 (μg) | |||
| Arithmetic mean (SD) | 1,805.14 (502.8) | 13,586.26 (4,061.2) | 2,020.50 (757.4) |
| Median (min–max) | 1,700.29 (1,145.7–2,961.4) | 15,346.48 (5,620.4–18,586.0) | 2,146.61 (1,052.6–3,688.1) |
| P-value | |||
| Torasemide-IR | – | <0.0001 | 0.214 |
| Furosemide-IR | – | – | <0.0001 |
| Torasemide-PR | – | – | – |
| Ae∞ (μg) | |||
| Arithmetic mean (SD) | 1,869.75 (512.8) | 13,953.82 (3,887.4) | 2,109.94 (765.6) |
| Median (min–max) | 1,831.38 (1,160.2–3,024.3) | 15,672.18 (6,347.8–18,757.1) | 2,223.9 (1,085.02–3,768.1) |
| P-value | |||
| Torasemide-IR | – | <0.0001 | 0.155 |
| Furosemide-IR | – | – | <0.0001 |
| Torasemide-PR | – | – | – |
| ERK (mmol/h) | |||
| Arithmetic mean (SD) | 29.75 (9.6) | 33.51 (8.6) | 29.08 (10.1) |
| Median (min–max) | 31.98 (16.7–45.5) | 32.21 (22.81–44.40) | 26.45 (18.2–50.8) |
| P-value | |||
| Torasemide-IR | – | 0.588 | 0.996 |
| Furosemide-IR | – | – | 0.435 |
| Torasemide-PR | – | – | – |
| ERCl (mmol/h) | |||
| Arithmetic mean (SD) | 98.69 (30.0) | 93.46 (40.0) | 95.06 (38.4) |
| Median (min–max) | 110.96 (53.0–131.0) | 101.55 (36.4–147.0) | 74.46 (54.3–152.2) |
| P-value | |||
| Torasemide-IR | – | 0.678 | 0.939 |
| Furosemide-IR | – | – | 0.994 |
| Torasemide-PR | – | – | – |
Notes: Data are mean (SD) and median (min-max) for every pharmacokinetic and pharmacodynamic parameter. Significance for parameters was tested by means of a one-way analysis of variance (ANOVA) followed by a post hoc analysis to assess differences by pairs. Bold values indicate statistical significance.
Abbreviations: Ae24, cumulative amount of drug excreted up to 24 hours; Ae∞, cumulative amount of drug excreted up to infinity; ERK, potassium excretion rate; ERCl, chlorine excretion rate; h, hour; max, maximum; min, minimum; PK/PD, pharmacokinetic/pharmacodynamic; IR, immediate-release; PR, prolonged release; SD, standard deviation.
Diuretic effect
The 0–24 hours diuretic effect for all evaluated drugs was similar. The total volume of urine collected after torasemide-PR was 2,335.1 mL at a diuresis rate of 97.3 mL/h. After torasemide-IR, the total volume of urine collected was 2,422.7 mL at a diuresis rate of 100.9 mL/h and after furosemide-IR, the total volume of urine collected was 2,478.7 mL at a diuresis rate of 103.3 mL/h.
The urine volume–time curves (Figure 5) show that in the interval from administration to +1.75 hours, torasemide-PR treatment resulted in a smaller quantity of excreted urine compared to both IR drugs. By contrast, in the interval from +3.5 hours to +10 hours torasemide-PR induced a larger volume of urine excretion compared to the other two IR formulations. In short, furosemide-IR and torasemide-IR induced urine excretion sooner than torasemide-PR.
Figure 5.
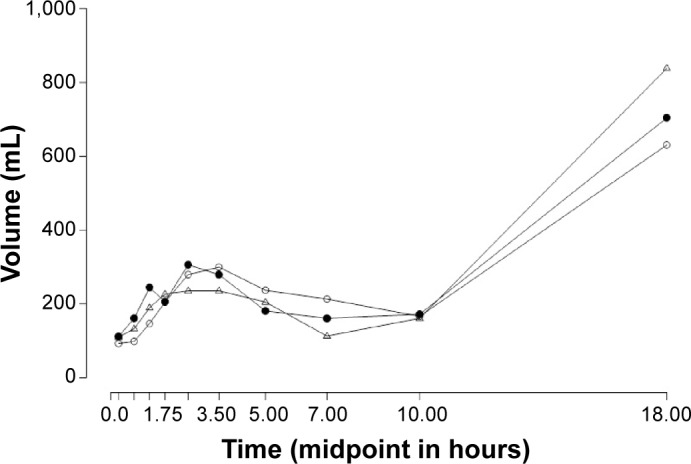
Mean urine volume excreted (mL) after 10 mg torasemide-IR (Δ), 10 mg torasemide-PR (○), and 40 mg furosemide-IR (•) plotted against the midpoint (hours) of the entire collection interval.
Notes: The entire collection interval comprise 10 different intervals after the drug ingestion from 0 h to 24 h.
Abbreviations: IR, immediate release; PR, prolonged release.
Subjective urinary urgency
The higher mictional urgency (41.78 mm), based on a VAS, was reported with furosemide-IR at the 1–1.5 hours interval after administration. Highest mictional urgency reported with torasemide-IR and torasemide-PR was 33.1 mm at the interval 1.5–2 hours postdose and 34.1 mm at the interval 3–4 hours postdose, respectively.
Safety and tolerability
No serious adverse events were recorded during the study. Three patients reported a total of nine adverse events, seven of which were considered to be related to study medication and were transient. Among these, two adverse events were evaluated as mild (abdominal distension, asthenia) and five were moderate (three reports of increased urea in one patient, two reports of increased triglycerides in another one patient). Other deviations in laboratory findings were not considered clinically relevant. There were no clinically relevant deviations on cardiologic parameters, that is, blood pressure, heart rate, and ECG. There were no withdrawals related to adverse events.
Discussion
Loop diuretics inhibit the sodium–potassium–chloride transporter in the thick ascending limb of the loop of Henle, resulting in a decreased reabsorption of sodium, chloride, and water.16 Furosemide was the first loop diuretic approved by the Food and Drug Administration in 1966, and torasemide is the most recently approved loop diuretic in 1993.16 With the exception of the actual drug, the type of release of a drug’s formulation has been revealed to be an important factor for not only pharmacokinetics but also pharmacodynamics.13,17,18
Regarding diuretics, a key pharmacodynamic endpoint is efficiency, a parameter that describes the effect of a drug in terms of effect per unit of concentration, and particularly useful for those drugs following concentration–effect relationships according to maximum effect or sigmoid maximum effect models.15 It is known that with identically administered doses of certain diuretics, such as furosemide in healthy volunteers19,20 or bumetanide in chronic renal insufficiency21 and in CHF (classes II–III),22 constant inputs induce higher total effects than rapid inputs, meaning that sustained drug concentrations elicit higher effect than oscillating concentrations with large differences between peaks and troughs. The present study was designed to compare natriuretic efficiency of equivalent doses of PR torasemide and IR furosemide. Previous results showed that a PR formulation of torasemide was associated with higher natriuretic efficiency than an IR torasemide formulation in healthy volunteers.13 Results from our study show that the amount of sodium excreted per molecule by the loop of Henle is higher with torasemide-PR than with furosemide-IR. Therefore, a higher natriuretic efficiency was reached with torasemide-PR. Similarly, torasemide-IR was also shown in our study population to enhance natriuretic efficiency compared to furosemide-IR. Nevertheless, no significant differences were found between both formulations of torasemide, which may be the result of, or influenced by, an insufficient sample size. The natriuresis efficiency results obtained in our study for furosemide-IR are very similar to the results obtained for furosemide by Alván et al.18 Alván et al investigated the bioavailability and diuretic effect of two oral controlled release formulations and a plain tablet of furosemide and they found that natriuretic efficiency after controlled release formulations of furosemide were higher than after the plain formulation. These findings are in agreement with our results obtained with the torasemide-PR formulation investigated.
The pharmacokinetic results of the present study are in accordance with the known kinetic characteristics of the three study medications. Torasemide-PR shows a typical PR profile, reaching a lower maximum concentration (P=0.002) over a longer period of time (P=0.012) and with a more gradual decrease in plasmatic concentrations compared to the IR formulation. In addition, the pharmacokinetic parameters of PR and IR torasemide formulations were in general comparable to those obtained in a previous study in healthy subjects.13 The time to maximum concentration was significantly longer with the PR formulation than with the IR in healthy volunteers (1.5 hours vs 0.7 hours)13 and at a similar magnitude of difference compared to this study (1.7 hours vs 1.0 hours). In a previous study of patients with HF,6 a similar tmax mean (1.1 hours) was already shown with a 10 mg dose of torasemide-IR; these results are similar to the results observed in our study. Results obtained by Barbanoj et al13 also showed that in healthy volunteers the Cmax achieved with a PR torasemide formulation was 32% lower than with the IR formulation. In our study, despite obtaining slightly lower absolute concentrations, the Cmax reached with the PR formulation was 36% lower than with the IR formulation. It is known that low cardiac output with reduced intestinal perfusion and decreased intestinal motility affect drugs absorption and may lead to lower Cmax and delayed tmax in HF patients compared to healthy subjects.7 Despite this, the relative difference due to formulation was maintained regardless the clinical condition of the patients with HF.
Results from the present study suggest that treatment with torasemide is related with less urinary urgency. This is in agreement with the results from a clinical trial conducted to evaluate efficacy and quality of life in patients with HF NYHA II–IV who received torasemide or furosemide over nine months.3 Quality-of-life was defined in the trial as a compound endpoint, with urinary urgency being one of its items. The number of mictions was also lower in the torasemide group compared to furosemide, and the authors concluded that torasemide was associated with less functional and social limitations in patients with CHF under treatment.3 Although urinary urgency was measured through a different method in the present study, our results show numeric differences in the same direction than the mentioned clinical trial.
For both formulations of torasemide, similar results were observed regarding diuretic effect, urine volume–time, and urinary urgency. Considering data from urine volume–time curves and times for highest VAS scoring between PR and IR torasemide formulations, the rate of drug release may have a role in improving symptoms by delaying urinary urgencies, which may potentially impact patients’ quality of life.3
Safety data from the present study confirm that torasemide-PR is a safe and well-tolerated drug, in accordance with the known safety profile of this drug.3,6,13,23
One limitation of the study was the low number of patients enrolled, so the study could not reach the expected sample size, limiting the possibility to detect further differences between the studied formulations. Despite this study not assessing clinical endpoints, our findings suggest that torasemide-PR may be a preferred option for chronic stable HF patients, due to its better tolerability and natriuretic efficiency. In this sense and due to the limited sample size, it would be desirable to perform additional studies to provide further information about clinical endpoints (eg, decompensation or hospital readmissions) in chronic HF patients treated with PR formulations of loop diuretics.
In conclusion, the findings from this study suggest that torasemide-PR was more natriuretically efficient than furosemide-IR after a single-dose administration in patients with chronic HF, over the 24-hour collection period evaluated.
Acknowledgments
The study results were presented in abstract form at the 35th Congress of the Spanish Society of Pharmacology; September 24–September 26, 2014, Madrid, Spain.
The authors thank all the patients who participated in their study and the entire staff of the Drug Research Center (CIM), specially the physician Joan Martínez, the nurses team Mireia Gonzalez, Mayte Garrido, David Martínez, and Maribel Martínez, the laboratory technician Judith Claramunt and also to Susanna Clos for their valuable dedication in the study. Special thanks to Dr Xavier Cos (CAP Sant Martí de Provençals, BCN), Dr Albert Boada (CAP Maragall, Barcelona), and Dr Carles Brotons (EAP Sardenya, Barcelona) for patient recruitment.
The authors also thank Elena Roldán and Valle Madueño (TFS Develop, Spain) for clinical trial monitoring, Marta Muñoz-Tudurí (TFS Develop) for editing the manuscript and for editorial assistance, and Jonathan Mackinnon (TFS Develop) for the language review. Finally, we would like to thank Natalia Oudovenko (Clinical Development Department, Ferrer Internacional SA) and Ester Fernández (Medical Department, Ferrer Internacional SA). This study was sponsored by Ferrer Internacional SA (Barcelona, Spain).
Footnotes
Author contribution
MRB, ER, JD, RMA, and BS designed the trial. ER was the study cardiologist and supervised the patient’s recruitment. MP and RMA were the study physicians. MRB and MP collected the data. IG and MRB performed the analyses. BS developed and implemented the simulation model, managed the bioanalytical part and supervised the pharmacokinetic results. MRB and RMA drafted the manuscript. MRB prepared the figures. All authors interpreted the data, wrote the paper, and have seen and approved the final version.
Disclosure
JD is employee of Ferrer International S.A (Barcelona, Spain) and BS was an employee of Ferrer International S.A (Barcelona, Spain) when the study was conducted. The authors report no other conflicts of interest in this work.
References
- 1.MacIntyre K, Capewell S, Stewart S, et al. Evidence of improving prognosis in heart failure: trends in case fatality in 66 547 patients hospitalized between 1986 and 1995. Circulation. 2000;102(10):1126–1131. doi: 10.1161/01.cir.102.10.1126. [DOI] [PubMed] [Google Scholar]
- 2.Patterson JH, Adams KF, Applefeld MM, Corder CN, Masse BR. Oral torsemide in patients with chronic congestive heart failure: effects on body weight, edema, and electrolyte excretion. Torsemide Investigators Group. Pharmacotherapy. 1994;14(5):514–521. [PubMed] [Google Scholar]
- 3.Müller K, Gamba G, Jaquet F, Hess B. Torasemide vs furosemide in primary care patients with chronic heart failure NYHA II to IV – efficacy and quality of life. Eur J Heart Fail. 2003;5(6):793–801. doi: 10.1016/s1388-9842(03)00150-8. [DOI] [PubMed] [Google Scholar]
- 4.Dunn CJ, Fitton A, Brogden RN. Torasemide. An update of its pharmacological properties and therapeutic efficacy. Drugs. 1995;49(1):121–142. doi: 10.2165/00003495-199549010-00009. [DOI] [PubMed] [Google Scholar]
- 5.Lesne M. Comparison of the pharmacokinetics and pharmacodynamics of torasemide and furosemide in healthy volunteers. Arzneimittel for Schung. 1988;38(1A):160–163. [PubMed] [Google Scholar]
- 6.Vargo DL, Kramer WG, Black PK, Smith WB, Serpas T, Brater DC. Bioavailability, pharmacokinetics, and pharmacodynamics of torsemide and furosemide in patients with congestive heart failure. Clin Pharmacol Ther. 1995;57(6):601–609. doi: 10.1016/0009-9236(95)90222-8. [DOI] [PubMed] [Google Scholar]
- 7.Kramer WG. Pharmacokinetics and pharmacodynamics of torasemide in congestive heart failure. Cardiology. 1994;84(suppl 2):108–114. doi: 10.1159/000176463. [DOI] [PubMed] [Google Scholar]
- 8.Brater DC, Rudy DR, Voelker JR, Greene PK, Gehr T, Sica DA. Torasemide dose-proportionality of pharmacokinetics and pharmacodynamics. Prog Pharmacol Clin Pharmacol. 1990;8:29–37. [Google Scholar]
- 9.Vargo D, Kramer WG, Black PK, Smith WB, Serpas T, Brater DC. The pharmacodynamics of torsemide in patients with congestive heart failure. Clin Pharmacol Ther. 1994;56(1):48–54. doi: 10.1038/clpt.1994.100. [DOI] [PubMed] [Google Scholar]
- 10.Lambe R, Kennedy O, Kenny M, Darragh A. Study of the tolerance and diuretic properties of torasemide following oral or intravenous administration to healthy volunteers. Eur J Clin Pharmacol. 1986;31(suppl):9–14. doi: 10.1007/BF00541461. [DOI] [PubMed] [Google Scholar]
- 11.Cosín J, Díez J, TORIC investigators Torasemide in chronic heart failure: results of the TORIC study. Eur J Heart Fail. 2002;4(4):507–513. doi: 10.1016/s1388-9842(02)00122-8. [DOI] [PubMed] [Google Scholar]
- 12.Stroupe KT, Forthofer MM, Brater DC, Murray MD. Healthcare costs of patients with heart failure treated with torasemide or furosemide. Pharmacoeconomics. 2000;17(5):429–440. doi: 10.2165/00019053-200017050-00002. [DOI] [PubMed] [Google Scholar]
- 13.Barbanoj MJ, Ballester MR, Antonijoan RM, et al. A bioavailability/bioequivalence and pharmacokinetic study of two oral doses of torasemide (5 and 10 mg): prolonged-release versus the conventional formulation. Clin Exp Pharmacol Physiol. 2009;36(5–6):469–477. doi: 10.1111/j.1440-1681.2008.05089.x. [DOI] [PubMed] [Google Scholar]
- 14.Ring-Larsen H, Henriksen JH, Wilken C, Clausen J, Pals H, Christensen NJ. Diuretic treatment in decompensated cirrhosis and congestive heart failure: effect of posture. Br Med J Clin Res Ed. 1986;292(6532):1351–1353. doi: 10.1136/bmj.292.6532.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alván G, Paintaud G, Wakelkamp M. The efficiency concept in pharmacodynamics. Clin Pharmacokinet. 1999;36(5):375–389. doi: 10.2165/00003088-199936050-00005. [DOI] [PubMed] [Google Scholar]
- 16.Wargo KA, Banta WM. A comprehensive review of the loop diuretics: should furosemide be first line? Ann Pharmacother. 2009;43(11):1836–1847. doi: 10.1345/aph.1M177. [DOI] [PubMed] [Google Scholar]
- 17.Banerjee PS, Robinson JR. Novel drug delivery systems. An overview of their impact on clinical pharmacokinetic studies. Clin Pharmacokinet. 1991;20(1):1–14. doi: 10.2165/00003088-199120010-00001. [DOI] [PubMed] [Google Scholar]
- 18.Alván G, Paintaud G, Eckernäs SA, Grahnén A. Discrepancy between bioavailability as estimated from urinary recovery of frusemide and total diuretic effect. Br J Clin Pharmacol. 1992;34(1):47–52. doi: 10.1111/j.1365-2125.1992.tb04106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wakelkamp M, Blechert A, Eriksson M, Gjellan K, Graffner C. The influence of frusemide formulation on diuretic effect and efficiency. Br J Clin Pharmacol. 1999;48(3):361–366. doi: 10.1046/j.1365-2125.1999.00015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Meyel JJ, Smits P, Russel FG, Gerlag PG, Tan Y, Gribnau FW. Diuretic efficiency of furosemide during continuous administration versus bolus injection in healthy volunteers. Clin Pharmacol Ther. 1992;51(4):440–444. doi: 10.1038/clpt.1992.44. [DOI] [PubMed] [Google Scholar]
- 21.Rudy DW, Voelker JR, Greene PK, Esparza FA, Brater DC. Loop diuretics for chronic renal insufficiency: a continuous infusion is more efficacious than bolus therapy. Ann Intern Med. 1991;115(5):360–366. doi: 10.7326/0003-4819-115-5-360. [DOI] [PubMed] [Google Scholar]
- 22.Ferguson JA, Sundblad KJ, Becker PK, Gorski JC, Rudy DW, Brater DC. Role of duration of diuretic effect in preventing sodium retention. Clin Pharmacol Ther. 1997;62(2):203–208. doi: 10.1016/S0009-9236(97)90069-2. [DOI] [PubMed] [Google Scholar]
- 23.Knauf H, Mutschler E. Clinical pharmacokinetics and pharmacodynamics of torasemide. Clin Pharmacokinet. 1998;34(1):1–24. doi: 10.2165/00003088-199834010-00001. [DOI] [PubMed] [Google Scholar]