

Abstract
Alcohol-related diseases of the nervous system are caused by excessive exposures to alcohol, with or without co-existing nutritional or vitamin deficiencies. Toxic and metabolic effects of alcohol (ethanol) vary with brain region, age/developmental stage, dose, and duration of exposures. In the mature brain, heavy chronic or binge alcohol exposures can cause severe debilitating diseases of the central and peripheral nervous systems, and skeletal muscle. Most commonly, long-standing heavy alcohol abuse leads to disproportionate loss of cerebral white matter and impairments in executive function. The cerebellum (especially the vermis), cortical-limbic circuits, skeletal muscle, and peripheral nerves are also important targets of chronic alcohol-related metabolic injury and degeneration. Although all cell types within the nervous system are vulnerable to the toxic, metabolic, and degenerative effects of alcohol, astrocytes, oligodendrocytes, and synaptic terminals are major targets, accounting for the white matter atrophy, neural inflammation and toxicity, and impairments in synaptogenesis. Besides chronic degenerative neuropathology, alcoholics are predisposed to develop severe potentially life-threatening acute or subacute symmetrical hemorrhagic injury in the diencephalon and brainstem due to thiamine deficiency, which exerts toxic/metabolic effects on glia, myelin, and the microvasculature. Alcohol also has devastating neurotoxic and teratogenic effects on the developing brain in association with fetal alcohol spectrum disorder/fetal alcohol syndrome. Alcohol impairs function of neurons and glia, disrupting a broad array of functions including neuronal survival, cell migration, and glial cell (astrocytes and oligodendrocytes) differentiation. Further progress is needed to better understand the pathophysiology of this exposure-related constellation of nervous system diseases and better correlate the underlying pathology with in vivo imaging and biochemical lesions.
Overview: alcohol use guidelines, abuse, metabolism and toxicity, public health problems and established limits
After tobacco and obesity, alcohol abuse is the third leading preventable cause of death in the United States. Furthermore, the alcohol abuse death rate is nearly doubled by including the premature deaths that are alcohol-related, e.g., motor vehicle accidents. Heavy drinking worsens morbidity from chronic disease as it exacerbates the effects of hypertension, diabetes mellitus, and hepatitis, and interferes with the metabolism and therapeutic actions of various medications. Societal costs of alcohol abuse are extremely high due to increased rates of severe injury, accidental deaths, lost income, over use of healthcare resources, and disruption of the family life [17]. Since disease-related effects of alcohol can occur with either chronic or binge drinking, the National Institutes of Alcohol Abuse and Alcoholism (NIAAA) established guidelines for (non-disease risk) acceptable upper limits of alcohol intake by adults. For men aged 21–65 years, the NIAAA recommends a maximum of 14 standard drinks per week and four drinks on any given day, whereas for women in the same age bracket, and men over 65, the recommended upper limits are seven standard drinks per week and three drinks on any given day.
Standard drinks all contain the same quantity of alcohol, although the definition of a standard drink and the recommended upper limits of alcohol intake vary by country. In the USA, one standard drink equals 14 grams of pure alcohol which is contained in 12 oz (355 ml) of beer or cooler (5 % alcohol), 5 oz (148 ml) of wine (12 % alcohol), 1.5 oz (44 ml) of 80-proof spirits (40 % alcohol), 8 oz (237 ml) of malt liquor (7 % alcohol), or 3 oz (89 ml) of fortified wine (http://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/standard-drink). In Australia and New Zealand, a standard drink is 10 g ethanol and upper limits of 4 drinks per day and 14 per week are recommended (http://www.drinkwise.org.au/you-alcohol/alcohol-facts/what-is-a-standard-drink/). In Japan, a standard drink contains 19.75 g alcohol, whereas in the United Kingdom, a standard drink has 8 g alcohol. In the European Union, the alcohol content in a standard drink varies by country, ranging from 6 to 17 g (http://www.icap.org/PolicyIssues/DrinkingGuidelines/StandardDrinks/KeyFactsandIssues/tabid/209/Default.aspx). Most guidelines recommend abstinence for pregnant women or those breastfeeding, and reduced intake in the elderly or persons on medications.
Alcohol abuse
The rates of heavy chronic and binge drinking are highest among 18–25 year olds. With increasing age, alcohol abuse rates decline and are 50–60 % lower among individuals who are 26 years and older compared with the 18–25-year-old bracket. On the other hand, the soaring rates of heavy drinking among teens and even younger minors are disconcerting, particularly because both long- and short-term consequences of extreme under-age drinking threaten physical health, mental health, and socioeconomic well-being. Correspondingly, in adolescents and young adults, chronic heavy and binge drinking increase for subsequently meeting DSM-IV criteria for alcohol dependence, and subsequently developing neurocognitive impairment and neurodegeneration with deficits in learning, memory, and executive functions. This article reviews the nature of acute and chronic alcohol-mediated neuropathologic lesions, including vulnerable targets of injury in the nervous system.
Alcohol metabolism and toxins
Alcohol (ethanol) is absorbed in the upper gastrointestinal tract by diffusion, and then rapidly distributes to all organs. Alcohol is eliminated primarily by oxidation in the liver where it is degraded to acetaldehyde followed by acetate, and then CO2 + H2O. There are three major pathways of alcohol metabolism: (1) alcohol dehydrogenase–aldehyde dehydrogenase; (2) microsomal ethanol oxidizing system (MEOS); and (3) catalase. With low levels of alcohol, aldehyde dehydrogenase is the principal enzyme utilized for metabolism. With high levels of alcohol, MEOS is recruited into action. The same enzymatic pathways exist in the brain. However, in the liver, different isoforms of alcohol and acetaldehyde dehydrogenases may be preferentially utilized. However, more research is needed to understand the consequences and limitations of the brain’s differential use of selected alcohol-metabolizing enzyme pathways.
Alcohol-induced brain injury: indirect effects
Growing evidence suggests that alcohol-mediated brain injury may be partly consequential to liver injury via a liver–brain axis [34]. Therefore, it is highly relevant that neuropathologists and neuroscientists understand the structural and functional effects of alcohol on the liver. Alcohol-mediated injury compromises the liver’s capacity to detoxify ethanol and thereby protect the brain and other organs from the toxic effects of ethanol and acetaldehyde. In addition, chronic liver injury leads to production of toxic, metabolic, and inflammatory mediators that injure the brain [28, 34]. In brief, alcohol-induced liver injury can be divided into 3 stages. Stage 1 is fatty liver disease (hepatic steatosis), which is relatively benign, generally reversible, and associated with hepatocellular accumulations of lipids, principally triglycerides. Stage 2, steatohepatitis, develops in the setting of hepatic steatosis and is associated with organelle dysfunction, oxidative stress, hepatocellular injury, and inflammation. Steatohepatitis is a disease state that is triggered by complex interactive hepatotoxic effects of acetaldehyde, NADH, and reactive oxygen species (ROS), together with lipotoxicity, endoplasmic reticulum (ER) stress, proinflammatory cytokine activation, and/or gut endotoxin-mediated injury [40, 88, 122, 151]. Stage 3 reflects progression of steatohepatitis from a predominantly inflammatory and injury state to a stage where hepatocellular regeneration and repair are severely compromised and fibrogenesis begins to dominate, further limiting metabolic and homeostatic functions of the liver [5, 48]. Chronic, progressive alcohol-induced liver injury increases risk for cirrhosis and hepatocellular carcinoma [51].
Mediators of acute alcohol-related encephalopathy
Alcohol abuse has acute and chronic adverse effects on brain structure and function. Secondary injuries such as contusions and stroke are not considered in this review. Alcohol’s toxic/metabolic effects on the brain are mediated by hepatic encephalopathy, as well as other complex factors that are not entirely understood, despite decades of research in this field. Nonetheless, the recurring theme is that alcohol-related metabolic encephalopathy is mediated by a self-perpetuating injury loop initiated by astrocyte swelling, which leads to oxidative and nitrosative stress, impaired intracellular signaling, and modifications of protein and gene expression. Ammonia toxicity is still regarded as a causal factor, and therefore resolving hyperammonemia is one of the principal targets of care. However, emerging data suggest that other agents such as aromatic amino acids and toxic lipids may be responsible for the impairments in neuronal activity in hepatic encephalopathy. In addition, cofactors such as deficiencies in folate, thiamine, pyridoxine and zinc may play critical roles [147]. Correspondingly, meta-analysis of alcoholic liver disease (ALD) studies showed that nutritional supplementation ameliorates symptoms of hepatic encephalopathy [9].
Mediators of chronic alcohol-related encephalopathy: neurotoxicity versus nutritional deficiency
Neurotoxic injury
In humans, chronic heavy alcohol abuse can be associated with significant structural and functional injury to the brain, as well as cognitive impairment with deficits in executive function. Correspondingly, experimental model data also show that heavy alcohol exposures lead to neurodegeneration and cognitive impairment [30, 71, 92, 154]. Although, to some extent, cognitive impairment in humans can be reversed by abstinence, detoxification reverses alcohol-related deficits in learning, memory, and executive function in less than half the cases. Moreover, postmortem studies showed that the vast majority (75 %) of chronic alcoholics have significant brain damage/degeneration. Therefore, the factors that govern the development of neurotoxic injury and degeneration, reversibility of injury, and long-term consequences with respect to brain function have not yet been determined. Attempts to address these points have focused on determining if chronic encephalopathy and neurodegeneration among alcoholics are mediated by the neurotoxic effects of chronic heavy or repeated binge alcohol exposures, nutritional deficiencies, or both.
Cortical and subcortical nuclear degeneration
The age-old controversy about the relative roles of alcohol toxicity and thiamine or other nutritional deficiencies in the pathogenesis of chronic alcohol-related brain diseases remains unresolved, probably due to overlap in their effects, combined with the fact that alcohol impairs absorption and utilization of thiamine. Conceivably, nutritional intake and peripheral blood levels of thiamine could be normal, while endorgan utilization of thiamine is deficient. In adult humans, the diencephalon, cerebral cortex, hippocampus, and white matter are susceptible to the neurotoxic effects of alcohol and metabolic insults caused by thiamine deficiency. Nonetheless, the potential contributions of thiamine deficiency in relation to chronic alcoholic brain disease have been partly resolved by experimental data that illustrate the distinct adverse effects of thiamine deficiency with some additive effects on chronic alcohol exposure [63, 162]. With regard to the selectively vulnerable gray matter targets of degeneration in alcoholic brain disease, thiamine deficiency alone reduces neurotrophic protein levels in the thalamus, and neurotransmitter levels in the hippocampus and cerebral cortex. Repeated bouts of thiamine deficiency cause severe and permanent deficits in spatial memory and increased perseverative behavior. Regarding its additive effects, thiamine deficiency, together with binge or chronic ethanol exposures, causes progressive cognitive dysfunction and loss of neural plasticity due to reduced GABAergic inhibition and increased glutamatergic excitation. This suggests that thiamine deficiency helps drive the cascade of alcohol-related neurodegeneration. Indeed, long-term consequences of both insults include cell loss in the cerebral cortex, and reduced expression neurotrophins that are needed for neuronal survival and plasticity [162].
White matter degeneration
White matter (myelin) is another major target of alcohol toxicity in the CNS [19, 26, 33, 78, 106]. Alcohol abuse and thiamine deficiency together cause greater reductions in white matter volume compared with thiamine deficiency alone [82, 162]. Since the neurodegenerative effects of thiamine deficiency and alcohol abuse overlap and often co-exist, it is unlikely that alcohol-related neurological disorders or brain atrophy would be entirely reversed by thiamine repletion therapy. This concept is supported by the finding that strategies designed to reverse thiamine deficiency do not fully restore cognitive and behavioral function. At the present time, there is no clear way to fully weigh the relative contributions of chronic alcohol neurotoxicity and nutritional deficiencies in relation to neurodegeneration. Neurologists, neuropathologists, and neuroradiologists must remain cognizant of the current limitations in our ability to distinguish between the direct neurotoxic and metabolic effects of alcohol and its metabolites, and the adverse effects of thiamine and other nutritional deficiency states.
Neuroimaging the neuropathology of alcoholic brain disease
Alcoholic brain diseases have been interrogated through the use of cranial computerized tomography (CT), magnetic resonance imaging (MRI), voxel-based morphometry, deformation-based morphometry, diffusion tensor MRI, diffusion-weighted MRI, and functional imaging via magnetic resonance spectroscopy (MRS), positron emission tomography (PET), single photon emission computed tomography (SPECT), and functional MRI. The value of these approaches is that they could potentially detect neuropathologic effects of chronic alcohol abuse and/or thiamine deficiency and thereby aid in diagnosis and treatment management. For example, these approaches have been used to quantify volume reductions in gray and white matter, and detect microstructural disruption of white matter tracts [78, 87]. Moreover, these tools could be used to evaluate reversibility of structural lesions [49] and assess activity in the specific brain regions. For instance, fMRI has been used to demonstrate that alcohol relapse tendencies could be predicted by increased activity in the mesocorticolimbic system [19].
Brain pathology in alcoholics: the big picture
It is very difficult to fully understand the spectrum of alcohol-related brain diseases in humans because accurate clinical histories are often lacking, the clinical course is frequently complicated by other substance abuses, e.g., illicit drugs, licit drugs, tobacco, systemic diseases, and nutritional deficiencies. Moreover, in contrast to experimental models in which alcohol dosing and timing of exposure are regulated, these parameters vary widely among individuals and over the individual’s lifespan. Matters are further complicated by evidence that underlying genetic susceptibility factors influence behavior, alcohol consumption, and the consequences of excessive alcohol intake [41, 104]. To help resolve these matters, the National Institutes of Health (NIH) funded a Tissue Resource Center in New South Wales, Australia that focuses on the systematic study of human alcoholic brains [136]. The relatively high rates of alcohol abuse, low rates of other substance abuses, and willingness of subjects to participate have enabled successful banking of brains and characterization of human alcoholic brain diseases [58, 136].
Alcohol-related metabolic brain injury and disease
Acute effects of alcohol on the CNS are mainly caused by alcohol poisoning or hepatic dysfunction leading to encephalopathy and myelopathy. Subacute and chronic alcohol-related CNS diseases are generally associated with subacute and chronic liver disease. Neurons, glia, myelin, brain microvessels, and all levels of the neuraxis from cerebrum to spinal cord, and including peripheral nerves and skeletal muscle are targets of alcohol-related metabolic dysfunction and disease. The degree to which the injury is mediated by acute metabolic insults may determine its reversibility.
Alcohol poisoning
Acute alcohol poisoning can be fatal due to hemorrhage in the ventral diencephalon, mesencephalon, and basal ganglia, and severe white matter edema in the cerebral hemispheres and pontine and medullary tegmenta. These histopathologic features reflect metabolic dysfunction, and overlap with lesions seen in Leigh’s disease (mitochondrial) and Wernicke’s encephalopathy [99]. Vasculopathy with secondary ischemic injury is the most prominent lesion. Like Leigh’s and Wernicke’s, glial-vascular pathology with relative preservation of neuronal cell bodies occurs in the brainstem, whereas acute neuronal necrosis occurs more commonly in the cerebral cortex, diencephalon (thalamus), and cerebellum [142]. It is unclear whether the neuronal necrosis reflects secondary hypoxic-ischemic injury, or a primary response to neurotoxic effects of alcohol. Acute alcohol poisoning is mimicked experimentally by binge ethanol exposures which result in marked brain edema [146]. The pathogenesis of the brain edema is not fully understood. However, evidence suggests that the brain edema is a consequence of acute alcohol withdrawal [15] with attendant impairments in autoregulation of brain blood flow [15, 70].
Hepatic encephalopathy (HE)
Acute HE is a neuropsychiatric disorder that is clinically manifested by confusion, delirium, coma, asterixis, loss of fine motor coordination, hyper-reflexia, slowed speech and mild cognitive impairment. However, acute HE can be covert or subtle in that afflicted individuals may have deficits in psychometric performance, ability to work, and capacity to carry out activities of daily living in the absence of overt encephalopathy. This phenomenon could account for the discordances between clinical and pathological findings at postmortem exam.
Although acute HE is reversible, survival can be as low as 50 % within the first year, and further reduced to 25 % within 3 years of diagnosis. Acute HE occurs in the setting of severe liver dysfunction or portosystemic shunting, but the majority (50–80 %) are associated with cirrhosis and hepatic dysfunction caused by continued alcohol abuse [69]. High levels of serum ammonia mediated by reduced hepatic clearance correlate with severity of acute HE [20, 23]; however, other toxins derived from the gut, e.g., short- and medium-chain length fatty acids, benzodiazepine-like substances, phenols, and mercaptans also contribute to this disease process [22, 161, 170].
Neuroimaging studies generally reveal non-specific or non-diagnostic results. Abnormalities in brain structure, including loss of gray and white matter volumes in cirrhotics, are detectable by advanced cerebral MRI and voxel-based morphometrics, but such structural changes can occur independent of acute HE. Moreover, since neuroimaging abnormalities, including brain atrophy, persist after liver transplantation [53], most likely they reflect irreversible cirrhosis-associated brain degeneration rather than effects of acute HE. On the other hand, recent data suggest that using MR spectroscopy, high apparent diffusion coefficients, with or without increased glutamine or reduced choline and myo-inositol, which correspond to shifts in brain water distribution, do correlate with acute HE [27].
Postmortem studies demonstrated that the principal abnormality in acute HE is non-inflammatory diffuse brain edema. With persistence and progression of liver failure, and development of hyperammonemia, which causes direct and indirect neurotoxic injury to the brain [23, 62], metabolic encephalopathy ensues. Acute HE is characterized by increased abundance and prominence of Alzheimer type II astrocytes in gray matter structures, particularly deep cerebral nuclei and the cerebral cortex. Alzheimer type II astrocytes have characteristic large pale nuclei with peripherally distributed chromatin and scant cytoplasm [21]. In more chronic stages of HE, pseudolaminar spongy degeneration can occur in deep layers of the cerebral cortex [164]. In addition to metabolic encephalopathy with Alzheimer Type II astrocytosis, consequence of cirrhosis and acute HE include brain acidosis and attendant RNA degradation [140]. Therefore, it should be noted that molecular and biochemical studies of alcoholic brain disease must consider tissue pH and intactness of RNA to avoid mis-attributing any reductions in gene expression to the neurotoxic and neurodegenerative effects of chronic alcohol abuse.
Hepatic myelopathy
Although the spinal cord is usually not considered a target of alcohol neurotoxicity, hepatic myelopathy is a subacute syndrome associated with liver failure and liver-brain shunting [32, 79, 145]. Clinically, the patients exhibit bilateral progressive and irreversible spastic paraparesis, hyper-reflexia of the legs, and extensor plantar responses. MRI with FLAIR can show abnormalities in subcortical and corticospinal tract white matter [135]. Postmortem findings include symmetrical demyelination of the lateral columns with variable degrees of axonal loss in the corticospinal tracts [68, 84, 166]. In addition, progressive myelopathy has been reported in alcoholics without portacaval shunting or hepatic dysfunction. Since the clinical signs and symptoms of distal extremity paresthesias, spastic paraparesis, and evidence of dorsal and lateral column involvement are indistinguishable from the entity linked to liver disease [132], myelopathy could be mediated by direct neurotoxic effects of alcohol.
Acquired hepatocerebral degeneration (AHD)
Wilson’s disease (not discussed here) is the hereditary form of hepatocerebral degeneration. AHD is a chronic, largely irreversible neuropsychiatric syndrome that arises in the setting of chronic ALD, as well as in other forms of cirrhosis [44]. The clinical course of AHD can be modulated by either worsening or improving hepatic function. AHD causes depression, dementia, dysarthria, gait ataxia, intention tremor, and choreathetosis. Since AHD is often preceded by bouts of HE, AHD could represent a chronic, irreversible form of HE. AHD commonly overlaps with orobuccolingual dyskinesia, Parkinsonism, and postural or kinetic tremors that respond poorly to dopaminergic drugs. Under these circumstances, the brains will not exhibit pathologic lesions of Parkinson’s disease or striatonigral degeneration.
AHD pathology is mainly centered in the diencephalon and cerebellum, although basal ganglia and spinal cord (overlapping with hepatic myelopathy) can be involved. The fact that cirrhosis and end-stage liver disease from other causes can also result in AHD suggests that the pathology of AHD is not specific for ALD. Human MRI with T1 weighted imaging studies of AHD show hyper-intensities in the globus pallidus, as well as overlap with other lesions of alcoholic brain disease. For example, the mammillary bodies and thalamic nuclei, which are characteristically affected in Wernicke’s encephalopathy, can be involved in AHD. In addition, one report of AHD demonstrated Wernicke’s encephalopathy-type pathology with demyelination, capillary and glial proliferation in subcortical white matter, and relative preservation of cortical neurons in the frontal lobe [16]. Alternatively, AHD can be associated with neuronal loss, foci of necrosis, and variable degrees of Alzheimer’s type II astrocytosis [81] in the cerebral and cerebellar cortex and basal ganglia. A limited number of ultrastructural studies have revealed that cortical pathology in AHD is associated with dilated and decompacted rough endoplasmic reticulum (RER) with formation of cisternae, abundant loose ribosomes, degeneration of mitochondria with disorganization of cristae, intra-mitochondrial dense-body inclusions, thinning and fragmentation/splitting of myelin sheaths, and swollen dendritic processes at synaptic terminals [143].
Mechanistically, alcoholic cirrhosis and AHD are associated with upregulation of the isoprenoid pathway and elevated digoxin secretion from the hypothalamus. The isoprenoid pathway produces three major metabolites: digoxin, dolichol, and ubiquinone. Digoxin modulates tryptophan/tyrosine transport. Dolichol mediates N-glycosylation of proteins. Ubiquinone is a free radical scavenger. In alcoholics, diencephalic levels of digoxin synthesis, dolichol, glycoconjugate, and free radicals are elevated, while ubiquinone levels are decreased [83]. Consequences could include increased NMDA/glutamate excitotoxicity and dysregulated lipid metabolism. The latter could exert neurotoxic effects that promote further metabolic dysfunction and myelin degradation [4].
Alcohol-related dementias
Overview
Chronic alcohol abuse can have long-lasting adverse effects on brain function and produce deficits ranging from mild cognitive impairment (MCI) to dementia. Unlike Alzheimer’s and cerebrovascular diseases which account for the vast majority of dementia cases in older age groups, alcohol-related cognitive impairment is more common in middle-aged people [94]. Since alcohol-related MCI and dementia can persist after cessation of drinking, these conditions can be difficult to diagnose and they could also confound the course of other neurodegenerative diseases, including Alzheimer’s and vascular dementia. Alcohol-related cognitive impairment is associated with brain atrophy with sulcal widening and dilatation of the ventricles, and can be accompanied by other alcohol-related diseases such as peripheral neuropathy and cerebellar atrophy [56]. Neuroimaging studies can be helpful for detecting alcohol-related atrophy of the frontal lobes, cerebellum, and medial temporal structures, including hippocampi [94]. Postmortem examination of alcoholics’ brains often yields a spectrum of abnormalities that curiously damage ventromedial and periventricular structures as illustrated in Fig. 1.
Fig. 1.

Mid-sagittal view of the brain with major targets of alcohol-mediated injury and degeneration circled. Structures included are the cingulate gyrus, corpus callosum, hypothalamus, periventricular thalamus, cerebellar vermis, basal ganglia, medial temporal structures, and periventricular white matter
The spectrum of dementias includes, Wernicke-Korsakoff syndrome (WKS), Marchiafava–Bignami, and acquired hepatocerebral degeneration [26, 163]. In addition, alcoholics often sustain traumatic brain injuries, and can secondarily develop debilitating or fatal CNS diseases due to hepatic encephalopathy, central pontine myelinolysis, or nutritional deficiencies pellagrous encephalopathy (due to niacin deficiency), [26].
Pellagrous encephalopathy is characterized by the clinical triad of dementia (delirium), dermatitis, and diarrhea. The neuropathology of pellagrous encephalopathy is manifested by central chromatolysis of neurons in the brainstem (mainly basis pons), dentate nuclei of the cerebellum, cranial nerve nuclei III, VI, VII, and VIII, arcuate nuclei, reticular nuclei, and spinal cord posterior horn cells. Less often, neurons in the central gray of the midbrain, inferior and superior colliculi, midbrain interpeduncular nuclei, cranial nerve nuclei X and XII, gracile and cuneate nuclei, and anterior horn cells are injured [61], Central chromatolysis is a pre-apoptotic stage of cellular injury in which neuronal chromatin/Nissl bodies dissolve and nuclei with prominent nucleoli are pushed to the side in response to axonal disconnection, ischemia, toxic injury, or pellagra.
Although postmortem assessments of alcohol-related neurodegeneration can be limited by the scarceness of clinical information, autopsy series can be informative and help to further evaluate the spectra of diseases. For example, in an older study in which alcoholism was present in 8.4 % of the cases, the incident rates (among the 8.4 %) of Wernicke’s encephalopathy, cerebellar atrophy, and central pontine myelinolysis were 18, 12, and 7 %, respectively [124].
Wernicke’s encephalopathy (WE)
WE is an acute neuropsychiatric disorder caused by thiamine (vitamin B1) deficiency, and associated with altered mental status, ataxia, and ophthalmoplegia. The increased incidence of WE among alcoholics is due to inadequate nutritional intake, together with alcohol’s inhibitory effects on thiamine absorption through the gastrointestinal tract, and on thiamine activation via phosphorylation [155]. WE is life threatening. Undiagnosed and untreated, WE can progress to a chronic neuropsychiatric disease termed, Korsakoff Syndrome (KS). It is noteworthy that WE caused by nutritional deficiency alone, i.e., in the absence of alcohol dependence and abuse, generally does not progress to KS. This suggests that the toxic effects of alcohol contribute to the WE-to-KS transition, and thereby mediate progressive neurodegeneration. Mechanistically, alcohol compromises thiamine transport into tissues, including across the blood–brain barrier, and it damages apoenzymes, increasing the levels of thiamine needed for metabolism. Since thiamine diphosphate is a cofactor for several key thiamine-dependent enzymes, thiamine deficiency impairs mitochondrial activity, oxidative metabolism, energy status, and neuronal viability. Alcohol-mediated impairments in thiamine uptake, tissue delivery and utilization compromise therapeutic efforts, particularly oral administration [153]. Consequently, effective therapeutic intervention for WE may require up to 1 g/day of i.v. thiamine.
The rates of WE vary among countries [156], but they have declined due to increased awareness, use of neuroimaging, rapid administration of therapy, and convenient public health interventions [86]. In Australia where the incident rate of WE was once 2.8 % and highest in the world (prior to 1995) [59], a national food thiamine supplementation program significantly reduced the rates of WE (1998) [60]. In more affluent societies, increased rates of WE can occur in complex debilitating states, such as malignancy [109, 117], and increasingly following bariatric surgery for obesity [29, 65, 134]. However, in drastically poor populations such as South African Blacks, the rates of WE remain stubbornly very high (6.6 % of autopsies) due to under-detection and failure to diagnose rather than the lack of treatment measures. Consequently, WE contributes substantially to alcohol-related morbidity and mortality [138]. Moreover, WE and KS can lead to sudden unexpected death due to injury in cardiorespiratory centers of the brainstem.
In over 80 % of cases, a diagnosis of WE is rendered based on the constellation of ophthalmoplegia, nystagmus, ataxia, and mental confusion, together with reduced blood levels of thiamine, and neuroimaging evidence of cytotoxic and vasogenic edema, and bilateral symmetric hyperintensity alterations around the third ventricle, aqueduct, mammillary bodies, and midbrain tectum on T2-weighted MR [89]. However, incomplete clinical presentations together with a normal MRI vis-à-vis inadequate clinical suspicion lead to under-detection and under-treatment of WE. Once diagnosed, besides repletion of systemic thiamine stores, the clinical management of WE could include neuroimaging to monitor therapeutic responses in the brain [89, 169].
Korsakoff syndrome (KS)
KS is a persistent neuropsychiatric syndrome associated with amnesia and disorientation, and caused by combined effects of thiamine deficiency and excessive alcohol consumption [94]. KS can be regarded as the chronic version of WE. Although KS can develop after a single bout of WE, it usually occurs in the setting of chronic alcoholism and multiple bouts of WE, and therefore is often referred to as Wernicke–Korsakoff syndrome (WKS). KS is characteristically associated with deficits in forming memory and in the implementation of executive functions due to disruption of neuronal circuits in the thalamus, mammillary bodies, hippocampus, frontal lobes and cerebellum [72]. WKS neuroimaging reveals bilateral macro-hemorrhages in the anterior thalami and fornix [101]. However, with chronicity, KS may be more accurately diagnosed with 3-D MRI with voxel-based morphometry to demonstrate parahippocampal, hippocampal, and thalamic atrophy with enlargement of the third ventricle [94].
Neuropathology of WE and KS
The neuropathologic hallmarks of acute WE include symmetrical hemorrhagic lesions in mammillary bodies, hypothalamus, thalamus, brainstem, and cerebellum. Histopathological sections characteristically reveal recent peri-capillary petechial hemorrhages in the mammillary bodies, periventricular zones around the third and fourth ventricles and aqueduct, together with proliferated and dilated capillaries, spongiosis (edema with vacuolation), demyelination, and relative preservation of neuronal cell bodies. The consistent feature of perivascular micro-hemorrhages reflects loss of vessel wall integrity. In acute WE (Fig. 2), the medial thalamic nuclei and inferior olives are frequently damaged by spongiosis, demyelination, and gliosis [54]. Axonal fragmentation, irregular swellings, and disruption can be seen by silver staining. The modest degrees of neuronal cell body loss help to distinguish these metabolic lesions from ischemic injury. Inflammation is not a standard feature of acute WE or WKS. It is noteworthy that with the exception of the petechial hemorrhages, the pathology of WE resembles Leigh’s disease, which is a metabolic disorder mediated by mitochondrial dysfunction and/or gene mutation [25, 95]. WE is often accompanied by hepatic encephalopathy with prominence and proliferation of Alzheimer type II astrocytes in basal forebrain structures and the cerebral cortex [16].
Fig. 2.

Acute Wernicke’s encephalopathy. a Mammillary body with darkened area of hemorrhage. b Acute perivascular hemorrhage in hypothalamus. c Prominent reactive proliferation of vascular endothelial cells, d normal vessel, e, f hypothalamus with e acute or f chronic Wernicke’s encephaopathy. Note neuronal cytoplasmic eosinophilia and mild microvesicular as well a pericellular edema in e compared with gliosis, abundant hemosiderin-laden macrophages (see inset), and prominent spongiosis due to macrovesicular and microvesicular edema in f
In the subacute phases of WE, the mammillary bodies, anterior and mediodorsal nuclei of the thalamus are most prominently affected. Endothelial cell nuclei become prominent and proliferative, resulting in narrowing of vascular lumens. Despite extensive parenchymal injury, neurons tend to be relatively spared. However, in some severe cases, the dominant abnormality is acute ischemia-like cellular injury with loss of neurons.
In the chronic phases of WKS, the long-standing nature of injury results in atrophy and brownish discoloration of the mammillary bodies, anterior nuclei, and medial-dorsal nuclei of the thalamus (Figs. 2a, 3a). This distribution of lesions is responsible for the amnestic disorders in WKS. Histopathologic studies reveal focal hemosiderin deposits and hemosiderin-laden macrophages, spongy or cystic degeneration, proliferation of small, thin-walled vessels, mild neuronal loss and prominently increased reactive astrocytes in affected diencephalic structures. In addition, white matter fibers and tracts exhibit reduced myelin staining which corresponds more to dysmyelination rather than unequivocal demyelination.
Fig. 3.

Chronic alcoholic neurodegeneration. The brain was from a middle-aged alcoholic man with documented Wernicke–Korsakoff psychosis. a Coronal section through the cerebral hemispheres at the level of the hypothalamus and foramen of Munro demonstrating atrophy of the fornix and mammillary bodies. b Closer image of the atrophic hypothalamus, mammillary bodies, and fornix. The atrophy is extreme yet the typical rust discoloration associated with WKS was not observed. c Central demyelination in the mid-portion of the corpus callosum (region circled in panel b), with features suggestive of Marchiafava–Bignami
Chronic WKS can be associated with global brain atrophy with disproportionate loss of white matter volume. In severe cases, cortical necrosis with neuronal loss can occur, particularly in superior frontal regions. Cerebellar degeneration marked by loss of Purkinje cells, is another neuropathologic finding in chronic WKS. Cerebellar degeneration contributes to motor dysfunction, including loss of balance and coordination, and neurocognitive deficits in WKS.
Altogether, CNS degenerative metabolic pathology in WKS includes glial-vascular injury with relative preservation of neuronal cell bodies in the diencephalon and brainstem, white matter myelin loss and/or impaired maintenance, and cerebellar atrophy. The glial-vascular degenerative lesions are caused by thiamine deficiency and impaired thiamine utilization in the brain. The link to alcohol abuse stems from the poor nutritional status of many alcoholics, as well as alcohol’s inhibitory effects on thiamine uptake and utilization. Myelin-associated pathology is linked to combined effects of thiamine deficiency, impaired thiamine utilization, and heavy alcohol abuse. Although the direct and proportional contributions of alcohol on CNS dysmyelination have not been determined, the lower propensity for WE to progress to KS in the absence of alcohol abuse suggests that the neurotoxic effects of alcohol have a causal role. Whether cerebellar degeneration in WKS reflects long-standing effects of alcohol neurotoxicity, thiamine deficiency, or both has not been fully resolved, although evidence suggests that the cerebellar pathology occurs mainly in the setting of thiamine deficiency [11], and that cerebellar dysfunction can be reversed by thiamine treatment [148].
White matter toxic, metabolic, and degenerative brain pathology
Central pontine myelinolysis (CPM)
CPM is an osmotic demyelinating disease that principally results in damage to bundles of myelinated fibers that intercalate among gray matter in the basis pontis. However, extrapontine myelinolysis can occur in the basal ganglia, thalamus, deep layers of the cerebral cortex, at cortical-white matter junctions, and in the tips of cerebellar folia. CPM is associated with cytotoxic edema, whereas in extra-pontine myelinolysis, the edema seems to be mainly vasogenic in origin [7]. Although osmotic demyelination is typically precipitated by rapid shifts in blood osmolality [111], CPM has been reported to occur in states of thiamine deficiency [75, 171]. In alcoholics, CPM and extrapontine myelinolysis occur with alcohol withdrawal, even in the absence of rapid correction of hyponatremia. CPM symptoms include dysarthria, dysphagia, pseudobulbar palsy, and gait disturbances. Diffusion tensor imaging (DTI) studies detect high signal intensities in the pons, thalamus, and basal ganglia in cases of CPM. Due to severe toxic and metabolic derangements in critical diencephalic and brainstem structures, mortality rates are high and can approach 30 %, while survivors often are left with severe motor disabilities [90].
CPM primarily occurs in the central basis pontis rather than the tegmentum. Extrapontine myelinolysis occurs when lesions of osmotic demyelination extend rostrally into the midbrain and lateral geniculate body, or caudally into the medulla (uncommon). By macroscopic examination, CPM is manifested by an irregularly bordered area of granular darkening or grayish discoloration in the basis pontis [66]. Histological sections stained with a dye that detects myelin, e.g., Luxol fast blue, demonstrates demyelination and degeneration of myelin, and relative preservation of axons and neuronal cell bodies (pontine nuclei) (Fig. 4). Immunohistochemical studies demonstrate reduced immunoreactivity to myelin basic protein, myelin-associated glycoprotein carbonic anhydrase C, and transferrin, and increased immunoreactivity to glial fibrillary acidic protein (GFAP; Fig. 5a, b) and S-100 protein in the lesions [50]. In addition, immunohistochemical staining of both human [128] and experimental models [152] of CPM demonstrated abundant microglial cell infiltration (Fig. 5c, d) with proinflammatory cytokine activation in active lesions. Ultrastructure studies revealed intramyelinic edema with distended myelin sheaths that eventually rupture, as well as axonal spheroids which reflect secondary axonal injury [118].
Fig. 4.
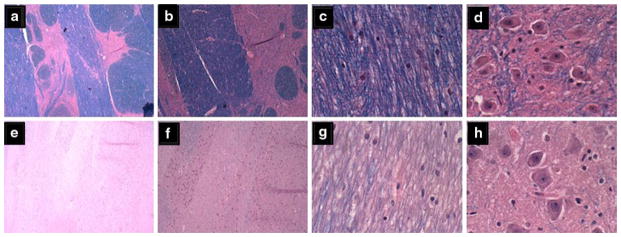
Central pontine myelinolysis. a–d Control basis pontis showing a–c intact myelin, a, b clear delineations between gray and white matter structures, and d abundant pontine neurons. e–h Alcoholic basis pontis (same as Fig. 7a) showing e–g almost complete loss of myelin, e, f virtually no delineation between gray and white mater structures, and b reduced abundance of glial cells, yet h preservation of neurons in pontine neurons (Luxol Fast Blue, Hematoxylin and Eosin stain)
Fig. 5.

Central pontine myelinolysis. Sections of pons from alcoholic patient in Fig. 4 were immunostained for a, b GFAP to detect reactive astrocytosis and gliosis, or c, d CD68 for macrophages and microglia. Examples of labeled cells are marked with arrows. Immunoreactivity was revealed with diaminobenzidine (brown) and sections were counterstained with hematoxylin (a, c, d 200× and b 400× original magnification)
Marchiafava–Bignami
Marchiafava–Bignami is a very rare, severe, and typically fatal disease that was originally described in middle-aged men living in the Chianti region of Italy. Initially, its association with the consumption of “rough red wine” suggested that the Marchiafava–Bignami was mediated by alcohol or possibly contaminating toxins. However, the occurrences of Marchiafava–Bignami in people who had no evidence of alcohol abuse [80, 85], and instead were malnourished, including from thiamine and niacin (alcoholic pellagra) deficiencies [139], cast doubt on the concept that alcohol was the sole cause of this disease. Moreover, the successful treatment of Marchiafava–Bignami with intravenous thiamine [113], points towards thiamine and other deficiencies as cofactors in this disease.
Brain CT, MRI, and diffusion-weighted imaging (DWI) can be used to detect the characteristic corpus callosum lesions of Marchiafava–Bignami [12]. By DWI, Marchia-fava–Bignami callosal lesions exhibit high T2 and FLAIR signals [2]. More extensive white matter involvement is manifested by large bilaterally symmetrical hypodense areas in subcortical white matter. Corresponding with the favorable clinical responses to thiamine [113], Marchi-afava–Bignami associated with T2 and FLAIR signals in the corpus callosum decrease after vitamin B complex treatments [2]. Postmortem studies revealed that Marchi-afava–Bignami is associated with cystic necrosis or cavitation, demyelination, and edema in the corpus callosum (see Fig. 3b, c), with variable degrees of centrum ovale and anterior commissure involvement [18, 64, 141]. Extensive damage to central and decussating white matter bundles results in inter-hemispheric disconnection with inability of one hemisphere to respond to visual or somaesthetic stimuli projected to the other. Besides white matter injury and degeneration, Marchiafava–Bignami can be associated with subcortical infarcts, cortical laminar sclerosis, and lacunae in the basal ganglia and pons.
Cerebral white matter degeneration
White matter atrophy is a well-established feature of chronic alcoholic brain disease [33], and readily documented by MRI and postmortem studies. Although white matter atrophy and degeneration occur throughout the cerebrum, neuroimaging and neurobehavioral studies tend to focus on abnormalities in the genu and body of the corpus callosum, including fibers that interconnect prefrontal brain structures. Severity of white matter atrophy increases with level of alcohol abuse. For example, among males 70+ years or age, reductions in the premotor frontal corpus callosum volume correlate with amounts of alcohol regularly consumed [73].
The clinical correlates of white matter atrophy vary widely from having no detectible signs or symptoms, to exhibiting overt cognitive impairment [114]. Even more challenging is that despite atrophy, specific, alcohol-related abnormalities have not been delineated by routine histopathologic studies. In this regard, high resolution MRI coupled with generalized fractional anisotropy could be used to assess micro- and macro-structural and metabolic abnormalities in white matter. In diffusion imaging, fractional anisotropy is a measure reflecting white matter fiber density, axonal diameter, and myelination. For example, this approach helped to demonstrate that males who fulfilled DSM-IV criteria for alcohol dependence had significant reductions in the microstructural integrity of fibers in all segments of the corpus callosum, particularly those that interconnect the orbitofrontal regions [87]. Severity of microstructural alterations in brain white matter correlates with impaired performance on non-verbal reasoning, attention tests, and psychomotor speed tests [78, 115], behavioral impulsivity, reduced bilateral processing, and deficits in pathways that mediate visual-motor integration [137]. In addition, the same neuroimaging approaches demonstrated that abstinence from alcohol could partially reverse alcohol-related macro-structural, micro-structural, and metabolic abnormalities in white matter [49].
Altogether, neuroimaging has provided significant insight into the nature and consequences of white matter degeneration in alcoholic brains due to the ability to link macro- and micro-structural abnormalities with deficits in function. Postmortem studies have the potential to enhance our understanding of alcoholic leukoencephalopathy and degeneration by characterizing the pathological, molecular, and biochemical lesions that mediate disease and correlate with neuroimaging results. Image analysis of postmortem brains demonstrated global atrophy of cerebral white matter with prominent involvement of the frontal lobes in alcoholics [33, 58]. White matter atrophy is associated with pallor of myelin staining (Fig. 6) and gliosis marked by increased glial fibrillary acidic protein immunoreactivity in enlarged activated astrocytes distributed in central and subcortical white matter. Ultrastructural studies revealed striking disturbances in cerebral white matter integrity characterized by swelling and fragmentation of myelin, irregular loss of axons, and degeneration of oligodendrocytes [143]. These findings, together with results of neuroimaging studies indicate that white matter is a major target of alcoholic brain disease. However, additional research is needed to better understand the pathophysiology of white matter degeneration, its correlations with neuroimaging and behavioral abnormalities, and parameters that define treatable or reversible stages of disease.
Fig. 6.

Alcoholic white matter degeneration. a Periventricular frontal white matter from the anterior frontal region of a non-alcoholic middle-aged man. Other regions of brain showed similar degrees of myelin staining. b–d Cerebral white matter degeneration is present in the brain of an alcoholic middle-aged man with dementia. White matter from the b anterior frontal, c periventricular frontal, and d periventricular region at the level of the hypothalamus with variable degrees of myelin pallor, vacuolation, and gliosis relative to normal
Cortical and subcortical gray matter toxic, metabolic, and degenerative brain pathology
Limbic circuitry
The hippocampus has a critical role in establishing memory and is the target of neurodegeneration in a number of diseases, including Alzheimer’s and alcoholism. Limbic circuitry interconnects the hippocampus with other medial temporal lobe structures, the diencephalon, and cingulate gyrus. Disruption of limbic circuitry in alcoholics is associated with reduced blood flow to the callosomarginal region, pericallosal region, thalamus, hippocampus, parahippocampal gyrus, amygdala, and anterior and middle cingulate regions [150], and atrophy of corresponding structures. As noted with respect to the cerebral cortex, functional abnormalities and atrophy of limbic structures are frequently not associated with loss of neuronal cell bodies, and instead mark white matter fiber degeneration and loss of synaptic connections [57].
Cortical degeneration
The most prevalent brain abnormalities in alcoholics include generalized atrophy of the cerebral cortex and white matter. The clinical manifestations range from subtle or undetectable to cognitive impairment with deficits in executive functions. High resolution T1-weighted MRI studies demonstrated reduced cortical thickness in the superior frontal, precentral, postcentral, middle frontal, middle and superior temporal, and lateral occipital gyri in alcoholics, and correlations between degree of cortical atrophy and severity of alcohol abuse [45]. Furthermore, alcohol-associated cerebral atrophy was linked to functional deficits manifested by reductions in regional blood flow to the frontal, temporal, parietal and occipital cortices, putamen, thalamus, and corpus callosum [110]. Although brain atrophy can be detected by postmortem examination, routine histopathologic studies have not revealed specific or overt lesions [46]. Instead, more refined and targeted assessments of disease seem to be required. For example, in alcoholics’ brains, the densities of cholinergic muscarinic receptors in the frontal cortex [47], and benzodiazepine receptors in the hippocampus and frontal cortex [46] are significantly reduced. In addition, ultrastructural studies demonstrated severe loss of dendritic spines, shrinkage of neuronal cell bodies, axonal damage, and astrocyte injury in the prefrontal cortex, striatum, and substantia nigra of alcoholics [143]. Together, these findings suggest that cortical atrophy in alcoholics is associated with loss of synapses and impaired synaptic function.
Subcortical pathology
Alcoholics’ brains exhibit atrophy (volume reductions) in the diencephalon, including thalamus and hypothalamus, neostriatum (caudate and putamen), and ventral forebrain, including the nucleus basalis of Meynert in the substantia innominata. The causes and behavioral consequences of subcortical nuclear injury and degeneration vary. Atrophy of gray and white matter structures in the medial thalamic and hypothalamic (mammillary bodies) nuclei correlate with cognitive-behavioral deficits in KS, the most severe form of alcoholic dementia [116]. Atrophy of mesocorticolimbic reward centers predicts tendency to relapse after a period of abstinence [24]. Loss of cholinergic neurons in the nucleus basalis of Meynert accounts for some aspects of cognitive impairment [3]. Apart from the hemorrhagic, glial-vascular pathology seen in WKS, alcoholic dementias manifest rather few specific changes in subcortical nuclei that are detectable by routine histopathologic examination. One exception is that striatal atrophy in alcoholics is associated with intra-neuronal acidophilic crystalline inclusion bodies. Ultrastructural analysis demonstrated that the inclusions correspond to filamentous substances associated with RER cisterns, similar to Malory’s hyaline in alcoholic liver disease [105].
Cerebellar degeneration
Cerebellar Purkinje cells, granule cells, and white matter fibers are major targets of neurodegeneration in alcoholics. The anterior mid-portion of the vermis exhibits the most conspicuous atrophy by macroscopic examination, although significant neuronal loss is not always associated with macroscopic evidence of atrophy [74]. In an autopsy series from Japan, 10.9 % of alcoholics had cerebellar degeneration with more prominent atrophy of the anterior and superior portions of the vermis compared with adjacent regions, and severity of cerebellar degeneration was greater in symptomatic compared with asymptomatic cases [167]. Cerebellar degeneration is associated with loss of Purkinje and granule cells, thinning of the molecular layer, and narrowing of white matter cores (Fig. 7). The severity of Purkinje cell loss increases with alcohol dose and duration. For example, 20–30 years of regular alcohol consumption (41–80 g/day) produces a 15 % reduction in the Purkinje cell population, whereas consumption of 81–180 g/day produces a 33.4 % loss of Purkinje cells. Purkinje cell degeneration and white matter loss in the vermis correlate with ataxia, whereas Purkinje cell loss in the lateral lobes correlates with cognitive dysfunction [11].
Fig. 7.
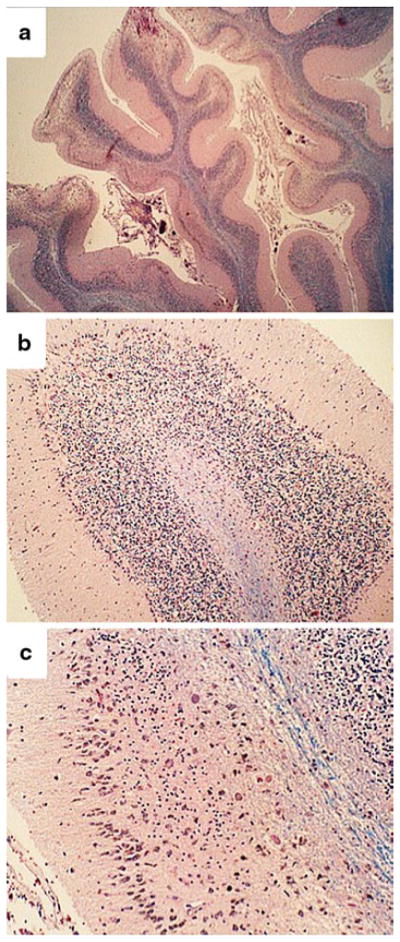
Alcoholic cerebellar degeneration. a Pronounced atrophy of cerebellar folia in the anterior vermis marked by widening of fissures and thinning of cortical and white matter structures. b Higher magnification of the cortex showing attenuation of cells in the inner granule cell (igc) layer, and proliferation of Bergmann’s glia (band-like; arrowheads) with loss of Purkinje cells. c A higher magnification image depicting severe degeneration of the cortex with subtotal depletion of neurons in the granule and Purkinje cell layers, and fiber loss in white matter cores (blue, center right). At the far right, the granule cell population is reduced but better preserved compared with the left side of the same folium
The causes of cerebellar atrophy and degeneration in alcoholics are still debated with regard to the roles of alcohol neurotoxicity and thiamine deficiency. Experimental data support the concept that alcohol exposure is sufficient to cause cerebellar degeneration [30], but in humans, thiamine deficiency could be either a cofactor or possibly a pathogenic agent. This controversy is kept alive by the findings in two independent studies in which opposite results were obtained using unbiased stereology to quantify and map the distributions of neuronal loss. One study showed that alcoholics without WE had significant reductions in Purkinje cell density and neuronal perikarya size [8], while another showed that significant neuronal loss in alcoholic cerebella only occurred in the presence of WE [11]. The latter study was corroborated by the experimental finding that chronic thiamine-deficient states cause cerebellar degeneration with reductions in the population of Purkinje cells and volumes of the molecular layer and white matter [31, 112].
Brainstem
As discussed earlier, brainstem pathology in alcoholics can result from rapid shifts in blood osmolality, leading to the development of central pontine myelinolysis. In addition, alcohol can exert neurotoxic effects on the brainstem. For example, alcohol poisoning causes acute injury and swelling of catecholaminergic and dopaminergic neurons in the mesocorticolimbic dopaminergic and ventral mesencephalic tegmentum [38]. Chronic injury and degeneration reduce the areas occupied by neuronal cell processes in the dorsal raphe nucleus of serotonergic neurons, reflecting impairment of serotonergic transmission to the cortex [158].
Neuromuscular diseases in alcoholics
Peripheral neuropathy
Alcohol-related peripheral neuropathies are chronic poly-neuropathies that involve sensory, autonomic, and motor nerves [91, 107, 108]. Alcohol-related neuropathies are mainly caused by axonal degeneration [6], but also can be mediated by demyelination, combinations of axonal degeneration and demyelination, or possibly functional impairments with minimal structural pathology [77, 96]. Early sensory signs and symptoms are caused by degeneration of distal sensory nerves of the lower extremities. Over time and with continued heavy drinking, sensory, motor, and autonomic nerves undergo progressive degeneration [6]. The relative contributions of alcohol versus B vitamin deficiencies, e.g., B1 (thiamine), B6 (niacin) and B12 (cobalamin) to the pathogenesis of alcohol-related neuropathies are still debated because nutritional deficiencies complicate the pathophysiology of alcoholism, independently cause peripheral neuropathy, and can exacerbate alcoholic neuropathy [77].
Evidence that alcoholic neuropathy is axonal in nature was demonstrated by biopsy-proven loss of mainly large myelinated fibers, followed by medium size myelinated fibers, and then unmyelinated fibers, with a distal predominance of lesions, suggesting a dying back process of neurodegeneration [133]. Ultrastructure and quantitative morphometric studies of alcohol-related polyneuropathies demonstrated Wallerian degeneration of myelinated and unmyelinated fibers, active but inadequate fiber regeneration, and a shift to smaller size myelinated and unmyelinated fibers [157]. However, alcoholic polyneuropathies can be demyelinating in nature and affect sensory, motor or autonomic nerves [55, 76]. In one study, Schwann cell injury with evidence of demyelination and remyelination was demonstrated in sural nerve biopsies from symptomatic subjects [76]. More recently, we conducted a pilot study and examined intra-epidermal nerve fiber degeneration in skin punch biopsies to detect Ubiquitin 9.5 to detect distal polyneuropathies in alcoholics [97]. Those investigations demonstrated striking attenuation of intra-epithelial nerve fibers in alcoholics relative to normal controls (Fig. 8). These preliminary results suggest that the neuropathology or alcohol-related peripheral neuropathy could be investigated more thoroughly and routinely through the use of minimally invasive skin biopsies. This approach could help improve our understanding of disease pathogenesis and response to treatment.
Fig. 8.

Alcoholic small fiber neuropathy detected by skin biopsy. Proximal (a, b) and distal (c, d) 8-mm punch biopsies of full-thickness skin were obtained from the medial thigh and lateral lower calf region from control (a, c) and alcoholic (b, d) patients. The tissues were fixed in 4 % paraformaldehyde, paraffin-embedded, and immunostained with antibodies to Ubiquitin 9.5. Immunoreactivity was detected with nickle-diaminobenzidine. Black linear staining corresponds to intra-epidermal nerve fibers Note the complexity of nerve fiber networks in the epidermis of control versus alcoholic specimens
Alcoholic myopathy
Alcoholic myopathy is the most prevalent skeletal muscle disorder in the Western Hemisphere, as it occurs in 40–60 % of alcohol abusers [1, 119]. Since alcoholic myopathy arises in the presence or absence of polyneuropathy [98], the two disease processes should be evaluated independently. Alcoholic myopathy is a progressive disease that impairs strength due to loss of lean tissue and worsens with duration and level of alcohol abuse [129, 131, 159, 160]. Myofiber atrophy can result in 30 % loss in muscle mass [119]. Severity of myofiber atrophy is linked to levels and duration of alcohol misuse [43] and not necessarily nutritional deficiencies [103], suggesting that it could be mediated by the toxic effects of alcohol [98]. Nonetheless, the pathogenesis of alcoholic myopathy is likely to be multifactorial because the histopathologic changes are only partly reversed by cessation of drinking [42], and disease is more prevalent in alcoholics who have cirrhosis [121]. A potential role for malnutrition is suggested by the finding that B vitamin deficiencies that occur in alcoholics cause myopathy. However, since there is clear evidence that alcohol exposure is sufficient to cause myopathy [39, 43], nutritional deficiencies may be cofactors in the pathogenesis and complicate clinical diagnosis and treatment of chronic myopathies in alcoholics.
The dominant histopathologic features of alcoholic myopathy include selective atrophy of Type II (anaerobic/glycolytic, white) fibers, and either sparing or mild atrophy of Type I (aerobic/red oxidative) fibers [93, 120]. Ultrastructure studies demonstrated that Type II fiber atrophy was associated with tubular aggregates in the sarcoplasmic reticulum [36]. However, acute, repeated high-dose administration of alcohol induces skeletal muscle injury in the absence of nutritional deficiency [144]. Corresponding ultrastructure studies revealed intracellular edema, enlarged and distorted mitochondria, dilated sarcoplasmic reticulum, increased fat and glycogen [130], and increased lipid droplets localized subjacent to the sarcolemma and between fibrils [149]. These abnormalities are reminiscent of the findings in acute alcoholic liver disease, except that giant mitochondria were not observed [35].
Fetal alcohol spectrum disorder
In 1973, Jones and Smith described teratogenic effects of alcohol in humans and termed the constellation of craniofacial abnormalities, “Fetal alcohol syndrome” (FAS). Subsequent studies correlated FAS with major CNS abnormalities and long-lasting cognitive, motor, and neurobehavioral abnormalities ranging from attention deficit hyperactivity disorder to mental retardation. However, it soon became evident that the phenotypic features were broad and heterogeneous, causing the syndrome to be re-named, ‘fetal alcohol spectrum disorder’ (FASD), with the diagnosis of FAS reserved for the most severe form of FASD. It is now recognized that FASD, which has incidence rates of 1–7/1,000 live births, is the most common preventable cause of neurodevelopmental defects including mental retardation [37]. In general, the severity of FASD is linked to alcohol dose, duration, and timing of exposure during pregnancy.
Neuroimaging studies are able to detect many of the macroscopic abnormalities originally described in FASD [127]. For example, MRI has been used to demonstrate overall reductions in brain volume, prominent abnormalities, including displacements in the corpus callosum, disproportionate reductions in frontal lobe volumes, structural abnormalities in the cerebellum, caudate, and hippocampus [102, 106], and functional/perfusion abnormalities in the temporal lobes [13, 126]. Diffusion tensor imaging studies identified probable microstructural abnormalities in white matter bundles of the corpus callosum, cerebellum, and corticospinal tracts in which the extent of injury correlated with severity of FASD phenotype and neurocognitive deficits [165].
Postmortem macroscopic brain pathology in FASD includes microcephaly, hypogenesis of the corpus callosum, structural abnormalities in basal ganglia and hippocampus, and cerebellar hypotrophy/hypoplasia. Morphometric studies demonstrated significant reductions in the volumes of the intracranial vault (7.6 %), cortical gray matter (7.8 %), white matter (8.6 %), and deep gray nuclei (13.1 %). Although all subcortical nuclei were found to be affected, the most prominent hypotrophy occurred in the hippocampus, thalamus, and globus pallidus [100]. By routine neuropathologic examination, microcephaly can be associated with small frontal and parietal lobes [10], reduced cerebral white matter volume, particularly in the posterior cingulate region [14], small hippocampi, basal ganglia and cerebellum (particularly anterior vermis) [125], and volume reductions with displacements of the corpus callosum. In addition, FASD may be associated with hypogenesis or agenesis of the corpus callosum.
Histopathologic studies of confirmed fetal cases revealed a broad range of abnormalities. In severe cases, microcephaly was associated with leptomeningeal heterotopias, thinning, laminar disorganization, and evidence of neuronal and glial cell migration disorder in the cerebral cortex, agenesis, and hypogenesis or thinning of the corpus callosum and anterior commissure, dysgenesis of the cerebellum and occasionally the brainstem, heterotopias in the cerebellum, hypoplasia or even agenesis of the cerebellar vermis and hydrocephalus [52, 127]. In addition, reported abnormalities include, congenital absence of olfactory bulbs and tracts, hippocampal hypoplasia, malformation of the basal ganglia, and in rare cases, cerebral dysgenesis including variable degrees of holoprosencephaly.
In the early stages of embryogenesis, ethanol has profound negative effects on survival and proliferation of progenitor cells, which could account for the microcephaly in FAS. From 7 to 20 weeks gestation when neuroepithelial cell migration and proliferation peak, ethanol exposures alter patterns of neuronal migration and cell fate. Consequences of the latter include reductions in neuronal and glial populations in the neocortex, hippocampus and sensory nuclei [52]. Later in development in the third trimester of pregnancy, ethanol exposures disrupt the brain growth spurt late in gestation and thereby interfere with astrocyte and oligodendrocyte development, synaptogenesis and cerebellar development. In addition, ethanol exposures during this highly vulnerable period can result in substantial apoptosis of brain cells throughout the cerebrum [168].
Conclusions
In this review of the human alcohol-related neuropathology including major disease processes in mature and developing brains, peripheral nerve, and skeletal muscle, the over-arching theme is that ethanol causes direct metabolic and toxic injury to neurons and glial cells. Major consequences include impaired function of astrocytes and oligodendrocytes, leading to neuronal pathology including reduced synaptogenesis, synapse maintenance, and cell survival. White matter pathology ranges from dysmyelination, to demyelination to myelin degeneration, and it occurs in all forms of alcohol-related CNS pathology. The nature, severity, and distribution of white matter lesions vary with age, timing of exposure, and ethanol dose. The consequences of ethanol-mediated neuropathology are frequently complicated by thiamine and other nutritional deficiencies. Thiamine deficiency occurs because alcohol inhibits its absorption and physiological actions. While thiamine deficiency alone is sufficient to cause Wernicke’s encephalopathy and peripheral neuropathy, many aspects of the adult CNS and neuromuscular alcohol-related diseases seem to be caused by combined effects of both thiamine deficiency and alcohol toxicity. Little is known about the role of thiamine deficiency in FASD. Despite decades of awareness of the neurological and neuropathological effects of heavy alcohol abuse, we are still lagging in our understanding of disease spectra and mechanisms, in part due to inadequate clinical data, and overlap between alcohol-related and other metabolic diseases. However, with growing availability of long-lasting biochemical markers of alcohol exposure such as fatty acid ethyl ester [123] and phosphatidylethanol [67], future studies will be better equipped to characterize ethanol’s effects in both developing and mature nervous systems.
Acknowledgments
Supported by AA-11431, AA-12908 and AA-12725 from the National Institutes of Health.
Contributor Information
Suzanne M. de la Monte, Email: Suzanne_DeLaMonte_MD@Brown.edu, Departments of Pathology (Neuropathology), Neurology, and Medicine, Rhode Island Hospital and the Warren Alpert Medical School of Brown University, 55 Claverick Street, Room 419, Providence, RI 02903, USA
Jillian J. Kril, Email: jillian.kril@sydney.edu.au, Disciplines of Medicine and Pathology, Sydney Medical School, The University of Sydney, Sydney, NSW 2006, Australia
References
- 1.Adachi J, Asano M, Ueno Y, et al. Alcoholic muscle disease and biomembrane perturbations (review) J Nutr Biochem. 2003;14(11):616–625. doi: 10.1016/s0955-2863(03)00114-1. [DOI] [PubMed] [Google Scholar]
- 2.Aggunlu L, Oner Y, Kocer B, Akpek S. The value of diffusion-weighted imaging in the diagnosis of Marchiafava–Bignami disease: apropos of a case. J Neuroimaging: Off J Am Soc Neuroimaging. 2008;18(2):188–190. doi: 10.1111/j.1552-6569.2007.00202.x. [DOI] [PubMed] [Google Scholar]
- 3.Akai J, Akai K. Neuropathological study of the nucleus basalis of meynert in alcoholic dementia. Arukoru kenkyu to yakubutsu izon = Jpn J Alcohol Stud Drug Depend. 1989;24(2):80–88. [PubMed] [Google Scholar]
- 4.Albrecht J, Sidoryk-Wegrzynowicz M, Zielinska M, Aschner M. Roles of glutamine in neurotransmission. Neuron Glia Biol. 2010;6(4):263–276. doi: 10.1017/S1740925X11000093. [DOI] [PubMed] [Google Scholar]
- 5.Altamirano J, Bataller R. Alcoholic liver disease: pathogenesis and new targets for therapy. Nat Rev Gastroenterol Hepatol. 2011;8(9):491–501. doi: 10.1038/nrgastro.2011.134. [DOI] [PubMed] [Google Scholar]
- 6.Ammendola A, Tata MR, Aurilio C, et al. Peripheral neuropathy in chronic alcoholism: a retrospective cross-sectional study in 76 subjects. Alcohol. 2001;36(3):271–275. doi: 10.1093/alcalc/36.3.271. [DOI] [PubMed] [Google Scholar]
- 7.An JY, Park SK, Han SR, Song IU. Central pontine and extrapontine myelinolysis that developed during alcohol withdrawal, without hyponatremia, in a chronic alcoholic. Intern Med. 2010;49(6):615–618. doi: 10.2169/internalmedicine.49.3069. [DOI] [PubMed] [Google Scholar]
- 8.Andersen BB. Reduction of Purkinje cell volume in cerebellum of alcoholics. Brain Res. 2004;1007(1–2):10–18. doi: 10.1016/j.brainres.2004.01.058. [DOI] [PubMed] [Google Scholar]
- 9.Antar R, Wong P, Ghali P. A meta-analysis of nutritional supplementation for management of hospitalized alcoholic hepatitis. Can J Gastroenterol = J Can de Gastroenterologie. 2012;26(7):463–467. doi: 10.1155/2012/945707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Archibald SL, Fennema-Notestine C, Gamst A, Riley EP, Mattson SN, Jernigan TL. Brain dysmorphology in individuals with severe prenatal alcohol exposure. Dev Med Child Neurol. 2001;43(3):148–154. [PubMed] [Google Scholar]
- 11.Baker KG, Harding AJ, Halliday GM, Kril JJ, Harper CG. Neuronal loss in functional zones of the cerebellum of chronic alcoholics with and without Wernicke’s encephalopathy. Neuroscience. 1999;91(2):429–438. doi: 10.1016/s0306-4522(98)90664-9. [DOI] [PubMed] [Google Scholar]
- 12.Baron R, Heuser K, Marioth G. Marchiafava–Bignami disease with recovery diagnosed by CT and MRI: demyelination affects several CNS structures. J Neurol. 1989;236(6):364–366. doi: 10.1007/BF00314384. [DOI] [PubMed] [Google Scholar]
- 13.Bhatara VS, Lovrein F, Kirkeby J, Swayze V, 2nd, Unruh E, Johnson V. Brain function in fetal alcohol syndrome assessed by single photon emission computed tomography. S D J Med. 2002;55(2):59–62. [PubMed] [Google Scholar]
- 14.Bjorkquist OA, Fryer SL, Reiss AL, Mattson SN, Riley EP. Cingulate gyrus morphology in children and adolescents with fetal alcohol spectrum disorders. Psychiatry Res. 2010;181(2):101–107. doi: 10.1016/j.pscychresns.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blaha M, Aaslid R, Douville CM, Correra R, Newell DW. Cerebral blood flow and dynamic cerebral autoregulation during ethanol intoxication and hypercapnia. J Clin Neurosci: Off J Neurosurg Soc Aust. 2003;10(2):195–198. doi: 10.1016/s0967-5868(02)00126-1. [DOI] [PubMed] [Google Scholar]
- 16.Blanco-Munez O, Suarez-Gauthier A, Martin-Garcia H, Diaz-Konrad V, San Antonio-Roman V, Cabello A. Unusual cortical compromise in a case of Wernicke’s encephalopathy. Revis Neurol. 2006;42(10):596–599. [PubMed] [Google Scholar]
- 17.Bouchery EE, Harwood HJ, Sacks JJ, Simon CJ, Brewer RD. Economic costs of excessive alcohol consumption in the US, 2006. Am J Prev Med. 2011;41(5):516–524. doi: 10.1016/j.amepre.2011.06.045. [DOI] [PubMed] [Google Scholar]
- 18.Bourrat C, Tommasi M, Bochu M, Kopp N, Malsch S. X-ray scanning in Marchiafava–Bignami disease. Rev Neurol. 1984;140(6–7):426–431. [PubMed] [Google Scholar]
- 19.Buhler M, Mann K. Alcohol and the human brain: a systematic review of different neuroimaging methods. Alcohol Clin Exp Res. 2011;35(10):1771–1793. doi: 10.1111/j.1530-0277.2011.01540.x. [DOI] [PubMed] [Google Scholar]
- 20.Butterworth RF. Portal-systemic encephalopathy: a disorder of neuron-astrocytic metabolic trafficking. Dev Neurosci. 1993;15(3–5):313–319. doi: 10.1159/000111350. [DOI] [PubMed] [Google Scholar]
- 21.Butterworth RF. Cerebral dysfunction in chronic alcoholism: role of alcoholic liver disease. Alcohol Suppl. 1994;2:259–265. [PubMed] [Google Scholar]
- 22.Butterworth RF. Role of circulating neurotoxins in the pathogenesis of hepatic encephalopathy: potential for improvement following their removal by liver assist devices. Liver Int: Off J Int Assoc Study Liver. 2003;23(Suppl 3):5–9. doi: 10.1034/j.1478-3231.23.s.3.1.x. [DOI] [PubMed] [Google Scholar]
- 23.Butterworth RF, Giguere JF, Michaud J, Lavoie J, Layrargues GP. Ammonia: key factor in the pathogenesis of hepatic encephalopathy. Neurochem Pathol. 1987;6(1–2):1–12. doi: 10.1007/BF02833598. [DOI] [PubMed] [Google Scholar]
- 24.Cardenas VA, Durazzo TC, Gazdzinski S, Mon A, Studholme C, Meyerhoff DJ. Brain morphology at entry into treatment for alcohol dependence is related to relapse propensity. Biol Psychiatry. 2011;70(6):561–567. doi: 10.1016/j.biopsych.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cavanagh JB. Selective vulnerability in acute energy deprivation syndromes. Neuropathol Appl Neurobiol. 1993;19(6):461–470. doi: 10.1111/j.1365-2990.1993.tb00474.x. [DOI] [PubMed] [Google Scholar]
- 26.Charness ME. Brain lesions in alcoholics. Alcohol Clin Exp Res. 1993;17(1):2–11. doi: 10.1111/j.1530-0277.1993.tb00718.x. [DOI] [PubMed] [Google Scholar]
- 27.Chavarria L, Alonso J, Garcia-Martinez R, et al. Brain magnetic resonance spectroscopy in episodic hepatic encephalopathy. J Cereb Blood Flow Metab: Off J Int Soc Cereb Blood Flow Metab. 2013;33(2):272–277. doi: 10.1038/jcbfm.2012.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen CH, Walker J, Momenan R, Rawlings R, Heilig M, Hommer DW. Relationship between liver function and brain shrinkage in patients with alcohol dependence. Alcohol Clin Exp Res. 2012;36(4):625–632. doi: 10.1111/j.1530-0277.2011.01662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clements RH, Katasani VG, Palepu R, et al. Incidence of vitamin deficiency after laparoscopic Roux-en-Y gastric bypass in a university hospital setting. Am Surg. 2006;72(12):1196–1202. doi: 10.1177/000313480607201209. (discussion 1203–1194) [DOI] [PubMed] [Google Scholar]
- 30.Cohen AC, Tong M, Wands JR, de la Monte SM. Insulin and insulin-like growth factor resistance with neurodegeneration in an adult chronic ethanol exposure model. Alcohol Clin Exp Res. 2007;31(9):1558–1573. doi: 10.1111/j.1530-0277.2007.00450.x. [DOI] [PubMed] [Google Scholar]
- 31.Collins GH, Converse WK. Cerebellar degeneration in thiamine-deficient rats. Am J Pathol. 1970;58(2):219–233. [PMC free article] [PubMed] [Google Scholar]
- 32.Conn HO, Rossle M, Levy L, Glocker FX. Portosystemic myelopathy: spastic paraparesis after portosystemic shunting. Scand J Gastroenterol. 2006;41(5):619–625. doi: 10.1080/00365520500318932. [DOI] [PubMed] [Google Scholar]
- 33.de la Monte SM. Disproportionate atrophy of cerebral white matter in chronic alcoholics. Arch Neurol. 1988;45(9):990–992. doi: 10.1001/archneur.1988.00520330076013. [DOI] [PubMed] [Google Scholar]
- 34.de la Monte SM, Longato L, Tong M, DeNucci S, Wands JR. The liver-brain axis of alcohol-mediated neurodegeneration: role of toxic lipids. Int J Environ Res Public Health. 2009;6(7):2055–2075. doi: 10.3390/ijerph6072055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Del Villar Negro A, Merino Angulo J, Rivera-Pomar JM. Skeletal muscle changes in chronic alcoholic patients. A conventional, histochemical, ultrastructural and morphometric study. Acta Neurol Scand. 1984;70(3):185–196. doi: 10.1111/j.1600-0404.1984.tb00818.x. [DOI] [PubMed] [Google Scholar]
- 36.del Villar Negro A, Merino Angulo J, Rivera Pomar JM, Aguirre Errasti C. Tubular aggregates in skeletal muscle of chronic alcoholic patients. Acta Neuropathol. 1982;56(4):250–254. doi: 10.1007/BF00691255. [DOI] [PubMed] [Google Scholar]
- 37.Disabilities AAoPCoSAaCoCW. Fetal alcohol syndrome and alcohol-related neurodevelopmental disorders. Pediatrics. 2000;106(2 Pt 1):358–361. [PubMed] [Google Scholar]
- 38.Droblenkov AV. Morphological signs of ethanol poisoning, alcohol abstinence and chronic alcoholic intoxication in the mesocorticolimbic dopaminergic system. Sud Med Ekspert. 2011;54(5):11–17. [PubMed] [Google Scholar]
- 39.Duane P, Peters TJ. Nutritional status in alcoholics with and without chronic skeletal muscle myopathy. Alcohol. 1988;23(4):271–277. [PubMed] [Google Scholar]
- 40.Duddempudi AT. Immunology in alcoholic liver disease. Clin Liver Dis. 2012;16(4):687–698. doi: 10.1016/j.cld.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 41.Enoch MA. The influence of gene-environment interactions on the development of alcoholism and drug dependence. Curr Psychiatry Rep. 2012;14(2):150–158. doi: 10.1007/s11920-011-0252-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Estruch R, Sacanella E, Fernandez-Sola J, Nicolas JM, Rubin E, Urbano-Marquez A. Natural history of alcoholic myopathy: a 5-year study. Alcohol Clin Exp Res. 1998;22(9):2023–2028. [PubMed] [Google Scholar]
- 43.Fernandez-Sola J, Sacanella E, Estruch R, Nicolas JM, Grau JM, Urbano-Marquez A. Significance of type II fiber atrophy in chronic alcoholic myopathy. J Neurol Sci. 1995;130(1):69–76. doi: 10.1016/0022-510x(95)00005-m. [DOI] [PubMed] [Google Scholar]
- 44.Finlayson M. Observations on the pathogenesis of hepatocerebral degeneration in cirrhosis. Bol Estud Med Biol. 1982;32(1–2):3–11. [PubMed] [Google Scholar]
- 45.Fortier CB, Leritz EC, Salat DH, et al. Reduced cortical thickness in abstinent alcoholics and association with alcoholic behavior. Alcohol Clin Exp Res. 2011;35(12):2193–2201. doi: 10.1111/j.1530-0277.2011.01576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Freund G, Ballinger WE. Loss of synaptic receptors can precede morphologic changes induced by alcoholism. Alcohol Suppl. 1991;1:385–391. [PubMed] [Google Scholar]
- 47.Freund G, Ballinger WE., Jr Loss of cholinergic muscarinic receptors in the frontal cortex of alcohol abusers. Alcohol Clin Exp Res. 1988;12(5):630–638. doi: 10.1111/j.1530-0277.1988.tb00255.x. [DOI] [PubMed] [Google Scholar]
- 48.Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology. 2011;141(5):1572–1585. doi: 10.1053/j.gastro.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gazdzinski S, Durazzo TC, Mon A, Yeh PH, Meyerhoff DJ. Cerebral white matter recovery in abstinent alcoholics—a multimodality magnetic resonance study. Brain: J Neurol. 2010;133(Pt 4):1043–1053. doi: 10.1093/brain/awp343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gocht A, Lohler J. Changes in glial cell markers in recent and old demyelinated lesions in central pontine myelinolysis. Acta Neuropathol. 1990;80(1):46–58. doi: 10.1007/BF00294221. [DOI] [PubMed] [Google Scholar]
- 51.Grewal P, Viswanathen VA. Liver cancer and alcohol. Clin Liver Dis. 2012;16(4):839–850. doi: 10.1016/j.cld.2012.08.011. [DOI] [PubMed] [Google Scholar]
- 52.Guerri C, Bazinet A, Riley EP. Foetal alcohol spectrum disorders and alterations in brain and behaviour. Alcohol. 2009;44(2):108–114. doi: 10.1093/alcalc/agn105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guevara M, Baccaro ME, Gomez-Anson B, et al. Cerebral magnetic resonance imaging reveals marked abnormalities of brain tissue density in patients with cirrhosis without overt hepatic encephalopathy. J Hepatol. 2011;55(3):564–573. doi: 10.1016/j.jhep.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 54.Gui QP, Zhao WQ, Wang LN. Wernicke’s encephalopathy in nonalcoholic patients: clinical and pathologic features of three cases and literature reviewed. Neuropathol: Off J Jpn Soc Neuropathol. 2006;26(3):231–235. doi: 10.1111/j.1440-1789.2006.00665.x. [DOI] [PubMed] [Google Scholar]
- 55.Guo YP, McLeod JG. Pathological changes in the vagus nerves in chronic alcoholism. Zhonghua shen jing jing shen ke za zhi = Chin J Neurol Psychiatry. 1990;23(4):234–236. 255. [PubMed] [Google Scholar]
- 56.Harada T, Ishizaki F, Horie N, et al. Alcohol-induced persistent mild cognitive impairment with successful withdrawal from alcohol dependence—a case report. Hiroshima J Med Sci. 2011;60(1):11–13. [PubMed] [Google Scholar]
- 57.Harding AJ, Wong A, Svoboda M, Kril JJ, Halliday GM. Chronic alcohol consumption does not cause hippocampal neuron loss in humans. Hippocampus. 1997;7(1):78–87. doi: 10.1002/(SICI)1098-1063(1997)7:1<78:AID-HIPO8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 58.Harper C. The neurotoxicity of alcohol. Hum Exp Toxicol. 2007;26(3):251–257. doi: 10.1177/0960327107070499. [DOI] [PubMed] [Google Scholar]
- 59.Harper C, Fornes P, Duyckaerts C, Lecomte D, Hauw JJ. An international perspective on the prevalence of the Wernicke–Korsakoff syndrome. Metab Brain Dis. 1995;10(1):17–24. doi: 10.1007/BF01991779. [DOI] [PubMed] [Google Scholar]
- 60.Harper CG, Sheedy DL, Lara AI, Garrick TM, Hilton JM, Raisanen J. Prevalence of Wernicke–Korsakoff syndrome in Australia: has thiamine fortification made a difference? Med J Aust. 1998;168(11):542–545. doi: 10.5694/j.1326-5377.1998.tb139081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hauw JJ, De Baecque C, Hausser-Hauw C, Serdaru M. Chromatolysis in alcoholic encephalopathies. Pellagra-like changes in 22 cases. Brain J Neurol. 1988;111(Pt 4):843–857. doi: 10.1093/brain/111.4.843. [DOI] [PubMed] [Google Scholar]
- 62.Hazell AS, Butterworth RF. Hepatic encephalopathy: an update of pathophysiologic mechanisms. Proc Soc Exp Biol Med. 1999;222(2):99–112. doi: 10.1046/j.1525-1373.1999.d01-120.x. [DOI] [PubMed] [Google Scholar]
- 63.He X, Sullivan EV, Stankovic RK, Harper CG, Pfefferbaum A. Interaction of thiamine deficiency and voluntary alcohol consumption disrupts rat corpus callosum ultrastructure. Neuropsychopharmacol: Off Pub Am Coll Neuropsychopharmacol. 2007;32(10):2207–2216. doi: 10.1038/sj.npp.1301332. [DOI] [PubMed] [Google Scholar]
- 64.Heepe P, Nemeth L, Brune F, Grant JW, Kleihues P. Marchiafava–Bignami disease. A correlative computed tomography and morphological study. Eur Arch Psychiatry Neurol Sci. 1988;237(2):74–79. doi: 10.1007/BF00382370. [DOI] [PubMed] [Google Scholar]
- 65.Iannelli A, Addeo P, Novellas S, Gugenheim J. Wernicke’s encephalopathy after laparoscopic Roux-en-Y gastric bypass: a misdiagnosed complication. Obes Surg. 2010;20(11):1594–1596. doi: 10.1007/s11695-010-0116-0. [DOI] [PubMed] [Google Scholar]
- 66.Ibrahim NB. Central pontine myelinolysis. Postgrad Med J. 1981;57(665):178–180. doi: 10.1136/pgmj.57.665.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Isaksson A, Walther L, Hansson T, Andersson A, Alling C. Phosphatidylethanol in blood (B-PEth): a marker for alcohol use and abuse. Drug Test Anal. 2011;3(4):195–200. doi: 10.1002/dta.278. [DOI] [PubMed] [Google Scholar]
- 68.Iwabuchi K, Yagishita S, Itoh Y, Amano N, Saitoh A. An autopsied case of alcoholic cerebellar degeneration with spastic paraplegia and neuropathy. No to shinkei = Brain Nerve. 1990;42(5):489–496. [PubMed] [Google Scholar]
- 69.Jepsen P, Ott P, Andersen PK, Vilstrup H. The clinical course of alcoholic cirrhosis: effects of hepatic metabolic capacity, alcohol consumption, and hyponatremia—a historical cohort study. BMC Res Notes. 2012;5:509. doi: 10.1186/1756-0500-5-509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jochum T, Reinhard M, Boettger MK, Piater M, Bar KJ. Impaired cerebral autoregulation during acute alcohol withdrawal. Drug Alcohol Depend. 2010;110(3):240–246. doi: 10.1016/j.drugalcdep.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 71.Johnsen-Soriano S, Bosch-Morell F, Miranda M, et al. Ebselen prevents chronic alcohol-induced rat hippocampal stress and functional impairment. Alcohol Clin Exp Res. 2007;31(3):486–492. doi: 10.1111/j.1530-0277.2006.00329.x. [DOI] [PubMed] [Google Scholar]
- 72.Jung YC, Chanraud S, Sullivan EV. Neuroimaging of Wernicke’s encephalopathy and Korsakoff’s syndrome. Neuropsychol Rev. 2012;22(2):170–180. doi: 10.1007/s11065-012-9203-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kapogiannis D, Kisser J, Davatzikos C, Ferrucci L, Metter J, Resnick SM. Alcohol consumption and premotor corpus callosum in older adults. Eur Neuropsychopharmacol: J Eur Coll Neuropsychopharmacol. 2012;22(10):704–710. doi: 10.1016/j.euroneuro.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Karhunen PJ, Erkinjuntti T, Laippala P. Moderate alcohol consumption and loss of cerebellar Purkinje cells. BMJ. 1994;308(6945):1663–1667. doi: 10.1136/bmj.308.6945.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kishimoto Y, Ikeda K, Murata K, Kawabe K, Hirayama T, Iwasaki Y. Rapid development of central pontine myelinolysis after recovery from Wernicke encephalopathy: a non-alcoholic case without hyponatremia. Intern Med. 2012;51(12):1599–1603. doi: 10.2169/internalmedicine.51.7498. [DOI] [PubMed] [Google Scholar]
- 76.Knill-Jones RP, Goodwill CJ, Dayan AD, Williams R. Peripheral neuropathy in chronic liver disease: clinical, electrodiagnostic, and nerve biopsy findings. J Neurol Neurosurg Psychiatry. 1972;35(1):22–30. doi: 10.1136/jnnp.35.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Koike H, Iijima M, Sugiura M, et al. Alcoholic neuropathy is clinicopathologically distinct from thiamine-deficiency neuropathy. Ann Neurol. 2003;54(1):19–29. doi: 10.1002/ana.10550. [DOI] [PubMed] [Google Scholar]
- 78.Konrad A, Vucurevic G, Lorscheider M, et al. Broad disruption of brain white matter microstructure and relationship with neuropsychological performance in male patients with severe alcohol dependence. Alcohol. 2012;47(2):118–126. doi: 10.1093/alcalc/agr157. [DOI] [PubMed] [Google Scholar]
- 79.Koo JE, Lim YS, Myung SJ, et al. Hepatic myelopathy as a presenting neurological complication in patients with cirrhosis and spontaneous splenorenal shunt. Korean J Hepatol. 2008;14(1):89–96. doi: 10.3350/kjhep.2008.14.1.89. [DOI] [PubMed] [Google Scholar]
- 80.Kosaka K, Aoki M, Kawasaki N, Adachi Y, Konuma I, Iizuka R. A non-alcoholic Japanese patient with Wernicke’s encephalopathy and Marchiafava–Bignami disease. Clin Neuropathol. 1984;3(6):231–236. [PubMed] [Google Scholar]
- 81.Kril JJ, Butterworth RF. Diencephalic and cerebellar pathology in alcoholic and nonalcoholic patients with end-stage liver disease. Hepatology. 1997;26(4):837–841. doi: 10.1002/hep.510260405. [DOI] [PubMed] [Google Scholar]
- 82.Kril JJ, Halliday GM, Svoboda MD, Cartwright H. The cerebral cortex is damaged in chronic alcoholics. Neuroscience. 1997;79(4):983–998. doi: 10.1016/s0306-4522(97)00083-3. [DOI] [PubMed] [Google Scholar]
- 83.Kurup RK, Kurup PA. Hypothalamic digoxin and hemispheric chemical dominance: relation to alcoholic addiction, alcoholic cirrhosis, and acquired hepatocerebral degeneration. Int J Neurosci. 2003;113(8):1105–1125. doi: 10.1080/00207450390212023. [DOI] [PubMed] [Google Scholar]
- 84.Lefer LG, Vogel FS. Encephalomyelopathy with hepatic cirrhosis following portosystemic venous shunts. Arch Pathol. 1972;93(2):91–97. [PubMed] [Google Scholar]
- 85.Leong AS. Marchiafava–Bignami disease in a non-alcoholic Indian male. Pathology. 1979;11(2):241–249. doi: 10.3109/00313027909061950. [DOI] [PubMed] [Google Scholar]
- 86.Lindboe CF, Loberg EM. The frequency of brain lesions in alcoholics. Comparison between the 5-year periods 1975–1979 and 1983–1987. J Neurol Sci. 1988;88(1–3):107–113. doi: 10.1016/0022-510x(88)90209-2. [DOI] [PubMed] [Google Scholar]
- 87.Liu IC, Chiu CH, Chen CJ, Kuo LW, Lo YC, Tseng WY. The microstructural integrity of the corpus callosum and associated impulsivity in alcohol dependence: a tractography-based segmentation study using diffusion spectrum imaging. Psychiatry Res. 2010;184(2):128–134. doi: 10.1016/j.pscychresns.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 88.Longato L, Ripp K, Setshedi M, et al. Insulin resistance, ceramide accumulation, and endoplasmic reticulum stress in human chronic alcohol-related liver disease. Oxid Med Cell Long. 2012;2012:479348. doi: 10.1155/2012/479348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lough ME. Wernicke’s encephalopathy: expanding the diagnostic toolbox. Neuropsychol Rev. 2012;22(2):181–194. doi: 10.1007/s11065-012-9200-7. [DOI] [PubMed] [Google Scholar]
- 90.Louis G, Megarbane B, Lavoue S, et al. Long-term outcome of patients hospitalized in intensive care units with central or extrapontine myelinolysis. Crit Care Med. 2012;40(3):970–972. doi: 10.1097/CCM.0b013e318236f152. [DOI] [PubMed] [Google Scholar]
- 91.Low PA, Walsh JC, Huang CY, McLeod JG. The sympathetic nervous system in alcoholic neuropathy. A clinical and pathological study. Brain: J Neurol. 1975;98(3):357–364. doi: 10.1093/brain/98.3.357. [DOI] [PubMed] [Google Scholar]
- 92.Lukoyanov NV, Pereira PA, Paula-Barbosa MM, Cadete-Leite A. Nerve growth factor improves spatial learning and restores hippocampal cholinergic fibers in rats withdrawn from chronic treatment with ethanol. Exp Brain Res Exp Hirnforschung Exp Cereb. 2003;148(1):88–94. doi: 10.1007/s00221-002-1290-7. [DOI] [PubMed] [Google Scholar]
- 93.Martin FC, Slavin G, Levi AJ, Peters TJ. Investigation of the organelle pathology of skeletal muscle in chronic alcoholism. J Clin Pathol. 1984;37(4):448–454. doi: 10.1136/jcp.37.4.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Matsui T, Sakurai H, Toyama T, Yoshimura A, Matsushita S, Higuchi S. Clinical application of neuroimaging to alcohol-related dementia. Nihon Arukoru Yakubutsu Igakkai zasshi = Jpn J Alcohol Stud Drug Depend. 2012;47(3):125–134. [PubMed] [Google Scholar]
- 95.Matthews PM, Marchington DR, Squier M, Land J, Brown RM, Brown GK. Molecular genetic characterization of an X-linked form of Leigh’s syndrome. Ann Neurol. 1993;33(6):652–655. doi: 10.1002/ana.410330616. [DOI] [PubMed] [Google Scholar]
- 96.Mellion M, Gilchrist JM, de la Monte S. Alcohol-related peripheral neuropathy: nutritional, toxic, or both? Muscle Nerve. 2011;43(3):309–316. doi: 10.1002/mus.21946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mellion ML, Silbermann E, Gilchrist JM, Leggio L, de la Monte SM. Small fiber involvement in alcohol related peripheral neuropathy (ALN) Ann Neurol. 2013;74(Suppl 17):S100. [Google Scholar]
- 98.Mills KR, Ward K, Martin F, Peters TJ. Peripheral neuropathy and myopathy in chronic alcoholism. Alcohol. 1986;21(4):357–362. [PubMed] [Google Scholar]
- 99.Mizuguchi M, Tomonaga M. Acute encephalopathy with symmetrical, widespread, edematous and necrotic lesions–an autopsy case report. No to shinkei = Brain Nerve. 1989;41(8):789–794. [PubMed] [Google Scholar]
- 100.Nardelli A, Lebel C, Rasmussen C, Andrew G, Beaulieu C. Extensive deep gray matter volume reductions in children and adolescents with fetal alcohol spectrum disorders. Alcohol Clin Exp Res. 2011;35(8):1404–1417. doi: 10.1111/j.1530-0277.2011.01476.x. [DOI] [PubMed] [Google Scholar]
- 101.Nazarov B, Jeannin S, Mejdoubi M, Signate A, Smadja D. Teaching neuroimages: bilateral anterior thalami and fornix macrohemorrhage in Wernicke–Korsakoff syndrome. Neurology. 2011;77(22):e129. doi: 10.1212/WNL.0b013e31823a0ca5. [DOI] [PubMed] [Google Scholar]
- 102.Niccols A. Fetal alcohol syndrome and the developing socio-emotional brain. Brain Cogn. 2007;65(1):135–142. doi: 10.1016/j.bandc.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 103.Nicolas JM, Garcia G, Fatjo F, et al. Influence of nutritional status on alcoholic myopathy. Am J Clin Nutr. 2003;78(2):326–333. doi: 10.1093/ajcn/78.2.326. [DOI] [PubMed] [Google Scholar]
- 104.Nixon K, Morris SA, Liput DJ, Kelso ML. Roles of neural stem cells and adult neurogenesis in adolescent alcohol use disorders. Alcohol. 2010;44(1):39–56. doi: 10.1016/j.alcohol.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nonaka H. Acidophilic crystalline inclusion body in the caudate nerve cells. Acta Pathol Jpn. 1983;33(5):855–861. doi: 10.1111/j.1440-1827.1983.tb02133.x. [DOI] [PubMed] [Google Scholar]
- 106.Norman AL, Crocker N, Mattson SN, Riley EP. Neuroimaging and fetal alcohol spectrum disorders. Dev Disabil Res Rev. 2009;15(3):209–217. doi: 10.1002/ddrr.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Novak DJ, Victor M. Affection of the vagus and sympathetic nerves in alcoholic polyneuropathy. Trans Am Neurol Assoc. 1973;98:63–67. [PubMed] [Google Scholar]
- 108.Novak DJ, Victor M. The vagus and sympathetic nerves in alcoholic polyneuropathy. Arch Neurol. 1974;30(4):273–284. doi: 10.1001/archneur.1974.00490340001001. [DOI] [PubMed] [Google Scholar]
- 109.Ogershok PR, Rahman A, Nestor S, Brick J. Wernicke encephalopathy in nonalcoholic patients. Am J Med Sci. 2002;323(2):107–111. doi: 10.1097/00000441-200202000-00010. [DOI] [PubMed] [Google Scholar]
- 110.Oishi M, Mochizuki Y, Shikata E. Corpus callosum atrophy and cerebral blood flow in chronic alcoholics. J Neurol Sci. 1999;162(1):51–55. doi: 10.1016/s0022-510x(98)00279-2. [DOI] [PubMed] [Google Scholar]
- 111.Okeda R, Kitano M, Sawabe M, Yamada I, Yamada M. Distribution of demyelinating lesions in pontine and extrapontine myelinolysis—three autopsy cases including one case devoid of central pontine myelinolysis. Acta Neuropathol. 1986;69(3–4):259–266. doi: 10.1007/BF00688302. [DOI] [PubMed] [Google Scholar]
- 112.Oliveira FA, Galan DT, Ribeiro AM, Santos Cruz J. Thiamine deficiency during pregnancy leads to cerebellar neuronal death in rat offspring: role of voltage-dependent K+ channels. Brain Res. 2007;1134(1):79–86. doi: 10.1016/j.brainres.2006.11.064. [DOI] [PubMed] [Google Scholar]
- 113.Pasutharnchat N, Phanthumchinda K. Marchiafava–Bignami disease: a case report. J Med Assoc Thailand = Chotmaihet Thangphaet. 2002;85(6):742–746. [PubMed] [Google Scholar]
- 114.Pfefferbaum A, Lim KO, Desmond JE, Sullivan EV. Thinning of the corpus callosum in older alcoholic men: a magnetic resonance imaging study. Alcohol Clin Exp Res. 1996;20(4):752–757. doi: 10.1111/j.1530-0277.1996.tb01682.x. [DOI] [PubMed] [Google Scholar]
- 115.Pitel AL, Chanraud S, Sullivan EV, Pfefferbaum A. Callosal microstructural abnormalities in Alzheimer’s disease and alcoholism: same phenotype, different mechanisms. Psychiatry Res. 2010;184(1):49–56. doi: 10.1016/j.pscychresns.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pitel AL, Chetelat G, Le Berre AP, Desgranges B, Eustache F, Beaunieux H. Macrostructural abnormalities in Korsakoff syndrome compared with uncomplicated alcoholism. Neurology. 2012;78(17):1330–1333. doi: 10.1212/WNL.0b013e318251834e. [DOI] [PubMed] [Google Scholar]
- 117.Pittella JE, de Castro LP. Wernicke’s encephalopathy manifested as Korsakoff’s syndrome in a patient with promyelocytic leukemia. South Med J. 1990;83(5):570–573. doi: 10.1097/00007611-199005000-00024. [DOI] [PubMed] [Google Scholar]
- 118.Powers JM, McKeever PE. Central pontine myelinolysis. An ultrastructural and elemental study. J Neurol Sci. 1976;29(1):65–81. doi: 10.1016/0022-510x(76)90081-2. [DOI] [PubMed] [Google Scholar]
- 119.Preedy VR, Adachi J, Ueno Y, et al. Alcoholic skeletal muscle myopathy: definitions, features, contribution of neuropathy, impact and diagnosis. Eur J Neurol: Off J Eur Fed Neurol Soc. 2001;8(6):677–687. doi: 10.1046/j.1468-1331.2001.00303.x. [DOI] [PubMed] [Google Scholar]
- 120.Preedy VR, Crabb DW, Farres J, Emery PW. Alcoholic myopathy and acetaldehyde. Nov Found Symp. 2007;285:158–177. doi: 10.1002/9780470511848.ch12. (discussion 177–182, 198–159) [DOI] [PubMed] [Google Scholar]
- 121.Preedy VR, Ohlendieck K, Adachi J, et al. The importance of alcohol-induced muscle disease. J Muscle Res Cell Motil. 2003;24(1):55–63. doi: 10.1023/a:1024842817060. [DOI] [PubMed] [Google Scholar]
- 122.Ramirez T, Longato L, Dostalek M, Tong M, Wands JR, de la Monte SM. Insulin resistance, ceramide accumulation and endoplasmic reticulum stress in experimental chronic alcohol-induced steatohepatitis. Alcohol. 2013;48(1):39–52. doi: 10.1093/alcalc/ags106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Refaai MA, Nguyen PN, Cluette-Brown JE, Laposata M. Ethyl arachidonate is the predominant fatty acid ethyl ester in the brains of alcohol-intoxicated subjects at autopsy. Lipids. 2003;38(3):269–273. doi: 10.1007/s11745-003-1060-6. [DOI] [PubMed] [Google Scholar]
- 124.Riethdorf L, Warzok R, Schwesinger G. Alcoholic encephalopathies in autopsy material. Zentralblatt fur Pathologie. 1991;137(1):48–56. [PubMed] [Google Scholar]
- 125.Riley EP, McGee CL. Fetal alcohol spectrum disorders: an overview with emphasis on changes in brain and behavior. Exp Biol Med (Maywood) 2005;230(6):357–365. doi: 10.1177/15353702-0323006-03. [DOI] [PubMed] [Google Scholar]
- 126.Riley EP, McGee CL, Sowell ER. Teratogenic effects of alcohol: a decade of brain imaging. Am J Medical Genet Part C Semin Med Genet. 2004;127C(1):35–41. doi: 10.1002/ajmg.c.30014. [DOI] [PubMed] [Google Scholar]
- 127.Roebuck TM, Mattson SN, Riley EP. A review of the neuroanatomical findings in children with fetal alcohol syndrome or prenatal exposure to alcohol. Alcohol Clin Exp Res. 1998;22(2):339–344. doi: 10.1111/j.1530-0277.1998.tb03658.x. [DOI] [PubMed] [Google Scholar]
- 128.Roh JK, Nam H, Lee MC. A case of central pontine and extrapontine myelinolysis with early hypermetabolism on 18FDG-PET scan. J Korean Med Sci. 1998;13(1):99–102. doi: 10.3346/jkms.1998.13.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Rubin E. Alcoholic myopathy in heart and skeletal muscle. New Engl J Med. 1979;301(1):28–33. doi: 10.1056/NEJM197907053010107. [DOI] [PubMed] [Google Scholar]
- 130.Rubin E, Katz AM, Lieber CS, Stein EP, Puszkin S. Muscle damage produced by chronic alcohol consumption. Am J Pathol. 1976;83(3):499–516. [PMC free article] [PubMed] [Google Scholar]
- 131.Sacanella E, Fernandez-Sola J, Cofan M, et al. Chronic alcoholic myopathy: diagnostic clues and relationship with other ethanol-related diseases. QJM: Month J Assoc Phys. 1995;88(11):811–817. [PubMed] [Google Scholar]
- 132.Sage JI, Van Uitert RL, Lepore FE. Alcoholic myelopathy without substantial liver disease. A syndrome of progressive dorsal and lateral column dysfunction. Arch Neurol. 1984;41(9):999–1001. doi: 10.1001/archneur.1984.04050200109030. [DOI] [PubMed] [Google Scholar]
- 133.Said G. A clinicopathologic study of acrodystrophic neuropathies. Muscle Nerve. 1980;3(6):491–501. doi: 10.1002/mus.880030606. [DOI] [PubMed] [Google Scholar]
- 134.Salas-Salvado J, Garcia-Lorda P, Cuatrecasas G, et al. Wernicke’s syndrome after bariatric surgery. Clin Nutr. 2000;19(5):371–373. doi: 10.1054/clnu2000.0138. [DOI] [PubMed] [Google Scholar]
- 135.Santos-Garcia D, Arias-Rivas S, Dapena D, Arias M. Past hepatitis B virus infection and demyelinating multiphasic disease: casual or causal relationship? Neurologia. 2007;22(8):542–546. [PubMed] [Google Scholar]
- 136.Sarris M, Garrick TM, Sheedy D, Harper CG. Banking for the future: an Australian experience in brain banking. Pathology. 2002;34(3):225–229. doi: 10.1080/00313020220131260. [DOI] [PubMed] [Google Scholar]
- 137.Schulte T, Muller-Oehring EM, Rohlfing T, Pfefferbaum A, Sullivan EV. White matter fiber degradation attenuates hemispheric asymmetry when integrating visuomotor information. J Neurosci: Off J Soc Neurosci. 2010;30(36):12168–12178. doi: 10.1523/JNEUROSCI.2160-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sechi G, Serra A. Wernicke’s encephalopathy: new clinical settings and recent advances in diagnosis and management. Lancet Neurol. 2007;6(5):442–455. doi: 10.1016/S1474-4422(07)70104-7. [DOI] [PubMed] [Google Scholar]
- 139.Serdaru M, Hausser-Hauw C, Laplane D, et al. The clinical spectrum of alcoholic pellagra encephalopathy. A retrospective analysis of 22 cases studied pathologically. Brain. J Neurol. 1988;111(Pt 4):829–842. doi: 10.1093/brain/111.4.829. [DOI] [PubMed] [Google Scholar]
- 140.Sheedy D, Say M, Stevens J, Harper CG, Kril JJ. Influence of liver pathology on markers of postmortem brain tissue quality. Alcohol Clin Exp Res. 2012;36(1):55–60. doi: 10.1111/j.1530-0277.2011.01580.x. [DOI] [PubMed] [Google Scholar]
- 141.Shiota JY, Nakano I, Kawamura M, Hirayama K. An autopsy case of Marchiafava–Bignami disease with peculiar chronological CT changes in the corpus callosum: neuroradiopathological correlations. J Neurol Sci. 1996;136(1–2):90–93. doi: 10.1016/0022-510x(95)00300-q. [DOI] [PubMed] [Google Scholar]
- 142.Shormanov SV, Shormanova NS. Histomorphometric characteristic of human brain in acute alcoholic intoxication. Sud Med Ekspert. 2005;48(2):13–16. [PubMed] [Google Scholar]
- 143.Skuja S, Groma V, Smane L. Alcoholism and cellular vulnerability in different brain regions. Ultrastruct Pathol. 2012;36(1):40–47. doi: 10.3109/01913123.2011.629770. doi:10(3109/01913123)2011629770. [DOI] [PubMed] [Google Scholar]
- 144.Song SK, Rubin E. Ethanol produces muscle damage in human volunteers. Science. 1972;175(4019):327–328. doi: 10.1126/science.175.4019.327. [DOI] [PubMed] [Google Scholar]
- 145.Spencer DC, Forno LS. February 2000: Dementia with motor dysfunction in a patient with liver disease. Brain Pathol. 2000;10(2):315–316. 319. doi: 10.1111/j.1750-3639.2000.tb00205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Sripathirathan K, Brown J, 3rd, Neafsey EJ, Collins MA. Linking binge alcohol-induced neurodamage to brain edema and potential aquaporin-4 upregulation: evidence in rat organotypic brain slice cultures and in vivo. J Neurotrauma. 2009;26(2):261–273. doi: 10.1089/neu2008.0682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Strohle A, Wolters M, Hahn A. Alcohol intake—a two-edged sword. Part 1: metabolism and pathogenic effects of alcohol. Medizinische Monatsschrift fur Pharmazeuten. 2012;35(8):281–292. (quiz 293–284) [PubMed] [Google Scholar]
- 148.Sullivan EV, Pfefferbaum A. Neuroimaging of the Wernicke–Korsakoff syndrome. Alcohol. 2009;44(2):155–165. doi: 10.1093/alcalc/agn103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sunnasy D, Cairns SR, Martin F, Slavin G, Peters TJ. Chronic alcoholic skeletal muscle myopathy: a clinical, histological and biochemical assessment of muscle lipid. J Clin Pathol. 1983;36(7):778–784. doi: 10.1136/jcp.36.7.778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Suzuki Y, Oishi M, Ogawa K, Mizutani T. Atrophy of the parahippocampal gyrus and regional cerebral blood flow in the limbic system in chronic alcoholic patients. Alcohol. 2010;44(5):439–445. doi: 10.1016/j.alcohol.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 151.Szabo G, Petrasek J, Bala S. Innate immunity and alcoholic liver disease. Dig Dis. 2012;30(Suppl 1):55–60. doi: 10.1159/000341126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Takefuji S, Murase T, Sugimura Y, et al. Role of microglia in the pathogenesis of osmotic-induced demyelination. Exp Neurol. 2007;204(1):88–94. doi: 10.1016/j.expneurol.2006.09.025. [DOI] [PubMed] [Google Scholar]
- 153.Thomson AD, Guerrini I, Marshall EJ. The evolution and treatment of Korsakoff’s syndrome: out of sight, out of mind? Neuropsychol Rev. 2012;22(2):81–92. doi: 10.1007/s11065-012-9196-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tiwari V, Chopra K. Resveratrol abrogates alcohol-induced cognitive deficits by attenuating oxidative-nitrosative stress and inflammatory cascade in the adult rat brain. Neurochem Int. 2013;62(6):861–869. doi: 10.1016/j.neuint.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 155.Todd KG, Hazell AS, Butterworth RF. Alcohol-thiamine interactions: an update on the pathogenesis of Wernicke encephalopathy. Addict Biol. 1999;4(3):261–272. doi: 10.1080/13556219971470. [DOI] [PubMed] [Google Scholar]
- 156.Torvik A. Wernicke’s encephalopathy—prevalence and clinical spectrum. Alcohol. 1991;(Suppl1):381–384. [PubMed] [Google Scholar]
- 157.Tredici G, Minazzi M. Alcoholic neuropathy. An electron-microscopic study. J Neurol Sci. 1975;25(3):333–346. doi: 10.1016/0022-510x(75)90155-0. [DOI] [PubMed] [Google Scholar]
- 158.Underwood MD, Mann JJ, Arango V. Morphometry of dorsal raphe nucleus serotonergic neurons in alcoholism. Alcohol Clin Exp Res. 2007;31(5):837–845. doi: 10.1111/j.1530-0277.2007.00365.x. [DOI] [PubMed] [Google Scholar]
- 159.Urbano-Marquez A, Estruch R, Fernandez-Sola J, Nicolas JM, Pare JC, Rubin E. The greater risk of alcoholic cardiomyopathy and myopathy in women compared with men. JAMA J Am Med Assoc. 1995;274(2):149–154. doi: 10.1001/jama.1995.03530020067034. [DOI] [PubMed] [Google Scholar]
- 160.Urbano-Marquez A, Estruch R, Navarro-Lopez F, Grau JM, Mont L, Rubin E. The effects of alcoholism on skeletal and cardiac muscle. New Engl J Med. 1989;320(7):409–415. doi: 10.1056/NEJM198902163200701. [DOI] [PubMed] [Google Scholar]
- 161.Van Thiel DH, Gavaler JS, Tarter R, Schade RR, Stone BG. Hepatic encephalopathy and the neural regulation of hypothalamic pituitary function. Semin Liver Dis. 1985;5(1):70–83. doi: 10.1055/s-2008-1041759. [DOI] [PubMed] [Google Scholar]
- 162.Vetreno RP, Hall JM, Savage LM. Alcohol-related amnesia and dementia: animal models have revealed the contributions of different etiological factors on neuropathology, neurochemical dysfunction and cognitive impairment. Neurobiol Learn Mem. 2011;96(4):596–608. doi: 10.1016/j.nlm.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Victor M. Alcoholic dementia. Can J Neurol Sci Le J Can des Sci Neurol. 1994;21(2):88–99. doi: 10.1017/s031716710004899x. [DOI] [PubMed] [Google Scholar]
- 164.Victor M, Adams RD, Cole M. The acquired (non-Wilsonian) type of chronic hepatocerebral degeneration. Medicine. 1965;44(5):345–396. doi: 10.1097/00005792-196509000-00001. [DOI] [PubMed] [Google Scholar]
- 165.Wozniak JR, Muetzel RL. What does diffusion tensor imaging reveal about the brain and cognition in fetal alcohol spectrum disorders? Neuropsychol Rev. 2011;21(2):133–147. doi: 10.1007/s11065-011-9162-1. [DOI] [PubMed] [Google Scholar]
- 166.Yengue P, Adler M, Bouhdid H, Mavroudakis N, Gelin M, Bourgeois N. Hepatic myelopathy after splenorenal shunting: report of one case and review of the literature. Acta Gastroenterol Belgica. 2001;64(2):231–233. [PubMed] [Google Scholar]
- 167.Yokota O, Tsuchiya K, Terada S, et al. Frequency and clinicopathological characteristics of alcoholic cerebellar degeneration in Japan: a cross-sectional study of 1,509 postmortems. Acta Neuropathol. 2006;112(1):43–51. doi: 10.1007/s00401-006-0059-7. [DOI] [PubMed] [Google Scholar]
- 168.Young C, Roth KA, Klocke BJ, et al. Role of caspase-3 in ethanol-induced developmental neurodegeneration. Neurobiol Dis. 2005;20(2):608–614. doi: 10.1016/j.nbd.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 169.Zahr NM, Kaufman KL, Harper CG. Clinical and pathological features of alcohol-related brain damage. Nat Rev Neurol. 2011;7(5):284–294. doi: 10.1038/nrneurol.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zaki AE, Wardle EN, Canalese J, Ede RJ, Williams R. Potential toxins of acute liver failure and their effects on blood-brain barrier permeability. Experientia. 1983;39(9):988–991. doi: 10.1007/BF01989765. [DOI] [PubMed] [Google Scholar]
- 171.Zara G, Codemo V, Palmieri A, et al. Neurological complications in hyperemesis gravidarum. Neurol Sci: Off J Italian Neurol Soc Italian Soc Clin Neurophysiol. 2012;33(1):133–135. doi: 10.1007/s10072-011-0660-y. [DOI] [PubMed] [Google Scholar]