

Abstract
Increased vascular resistance and reduced fetoplacental blood flow are putative aetiologies in the pathogenesis of fetal growth restriction (FGR); however, the regulating sites and mechanisms remain unclear. We hypothesised that placental vessels dictate fetoplacental resistance and in FGR exhibit endothelial dysfunction and reduced flow-mediated vasodilatation (FMVD). Resistance was measured in normal pregnancies (n = 10) and FGR (n = 10) both in vivo by umbilical artery Doppler velocimetry and ex vivo by dual placental perfusion. Ex vivo FMVD is the reduction in fetal-side inflow hydrostatic pressure (FIHP) following increased flow rate. Results demonstrated a significant correlation between vascular resistance measured in vivo and ex vivo in normal pregnancy, but not in FGR. In perfused FGR placentas, vascular resistance was significantly elevated compared to normal placentas (58 ± 7.7 mmHg and 36.8 ± 4.5 mmHg, respectively; 8 ml min−1; means ± SEM; P < 0.0001) and FMVD was severely reduced (3.9 ± 1.3% and 9.1 ± 1.2%, respectively). In normal pregnancies only, the highest level of ex vivo FMVD was associated with the lowest in vivo resistance. Inhibition of NO synthesis during perfusion (100 μm l-NNA) moderately elevated FIHP in the normal group, but substantially in the FGR group. Human placenta artery endothelial cells from FGR groups exhibited increased shear stress-induced NO generation, iNOS expression and eNOS expression compared with normal groups. In conclusion, fetoplacental resistance is determined by placental vessels, and is increased in FGR. The latter also exhibit reduced FMVD, but with a partial compensatory increased NO generation capacity. The data support our hypothesis, which highlights the importance of FMVD regulation in normal and dysfunctional placentation.
Key points
A correlation was found between in vivo umbilical artery Doppler velocimetry and resistance to fetal-side flow in the human ex vivo dually perfused placenta, highlighting that the fetoplacental vascular bed is a key site of resistance to umbilico-placental flow in pregnancy.
We discovered high resistance and poor flow-mediated vasodilatory responses in placentas from pregnancies associated with fetal growth restriction (FGR).
Endothelial cells isolated from the FGR placentas and grown in static and flow culture showed a dysregulated phenotype, with biochemical signalling demonstrating a failed compensatory response to high blood-flow resistance.
Introduction
Fetal growth restriction (FGR) is a serious pregnancy complication, which affects 3–8% of pregnancies (Alberry & Soothill, 2007) and is associated with a significant increase in perinatal morbidity, mortality and stillbirth (Yanney & Marlow, 2004; Figueroa-Diesel et al. 2007). Correlations between low birth weight and increased risk of cardiovascular disease in adulthood have also been well documented (Barker et al. 1989; Barker et al. 1995; Demicheva & Crispi, 2013), with growing evidence that programming during prenatal life can predispose to adult cardiovascular and other diseases (Palinski & Napoli, 2008; Thornburg et al. 2010). Currently there are no proven therapies available to prevent or treat FGR; the only option available is delivery, often preterm, which is associated with its own risks (Lawn et al. 2005).
Abnormal placentation, with reduced fetoplacental blood flow, is a major cause of FGR (Chaddha et al. 2004; Sibley et al. 2005; Kiserud et al. 2006). In healthy pregnancies, placental vascular resistance changes dramatically in the third trimester resulting in a high flow, low resistance circulation, achieved through vascular adaptations including angiogenesis and vasodilatation (Sala et al. 1995; Chaddha et al. 2004). In severe cases of FGR, these adaptations fail to occur properly (Baschat, 2004) and vascular resistance remains high, observed clinically as increased pulsatility and resistance indices (PI and RI) in umbilical artery Doppler assessment (Acharya et al. 2005). The severity of fetal compromise in pregnancies affected by FGR is reflected by the degree of abnormality of the Doppler waveform, with absent or reversed flow at the end of diastole (Ghosh & Gudmundsson, 2009; Vergani et al. 2010). In these cases the flow in the umbilical artery is often intermittent, with a rapid progression of demise, suggesting that increased vascular resistance in FGR cannot solely be attributed to the structural constraints of an altered vascular architecture; dysregulation of vascular tone is also likely to play an important role (Turan et al. 2008).
Vascular tone in the fetoplacental circulation is principally regulated by endocrine or mechanical stimuli since it lacks autonomic innervation (Marzioni et al. 2004). Vasodilators responsible for relaxing smooth muscle and maintaining low vascular resistance include nitric oxide (NO), prostacyclin (PGI2), and endothelial derived hyperpolarising factor (EDHF) (Poston, 1997) with possible roles for carbon monoxide (CO) (Lyall et al. 2000; Bainbridge et al. 2002) and hydrogen sulphide (H2S2) (Holwerda et al. 2012; Cindrova-Davies et al. 2013; Wang et al. 2013). NO is the most studied of the vasodilators in the fetoplacental circulation, and evidence suggests that it plays a crucial role in the pregnancy induced vascular adaptations involved in maintaining low resistance (Baylis et al. 1992; Myatt, 1992; Magness et al. 1997; Sladek et al. 1997; Kulandavelu et al. 2012).
In many vascular beds one of the most potent stimulators of NO synthesis and vasodilatation is shear stress, the frictional force between flowing blood and the endothelium, calculated from the flow rate, viscosity of blood and radius of the vessel (Sprague et al. 2010). In the placenta, it has been shown that inhibition of NO during placental perfusion significantly increases pressure in the fetal circulation when shear stress is elevated through increased flow rate or increased fluid viscosity (Wieczorek et al. 1995). Shear stress has also been shown to increase eNOS expression and activation in ovine fetoplacental endothelial cells (Li et al. 2004). Endothelial dysfunction and an inability to respond appropriately to shear stress and increased blood flow would therefore result in increased vascular resistance similar to that seen in the placenta of pregnancies affected by severe FGR.
In the current study we tested the hypotheses that placental vessels dictate fetoplacental resistance and that in FGR these vessels exhibit endothelial dysfunction and reduced flow-mediated vasodilatation (FMVD).
Using human ex vivo dual placental perfusion we measured vascular resistance and FMVD in the fetoplacental vasculature of normal and FGR pregnancies, and correlated these with resistance data obtained in vivo via umbilical artery Doppler velocimetry. We also directly investigated endothelial responses to shear stress using human placental chorionic plate artery endothelial cells (HPAECs) from normal and FGR pregnancy in an in vitro laminar flow system. In addition the role of NO in placental endothelial responses to shear stress in normal and FGR pregnancy was investigated.
Methods
Ethical approval
Procedures were followed in accordance with institutional guidelines and conformed to the standards set by the latest revision of the Declaration of Helsinki. All tissue was acquired with written consent from fully informed women attending St Mary’s Hospital, Manchester for their antenatal care. Tissues were collected following informed consent, under ethical approval by the North West Haydock Park Research Ethics Committee (08/H1010/05) and the University of Manchester Ethics Committee.
Human placentas from healthy control pregnancies and FGR pregnancies were collected within 30 min of delivery by Caesarean section or vaginal delivery. Volunteer demographics are given in Table1. In this study, idiopathic FGR was defined as an individualised birthweight centile (IBC) at or below the fifth centile, using an algorithm (GROW) accounting for maternal height, weight at booking, parity, gestational age, ethnicity, fetal sex and birth weight (Gardosi, 2006). Only singleton pregnancies were recruited and patients were excluded if they had pre-existing hypertension, pre-eclampsia, diabetes, renal disease, or a body mass index <18.5 or >30.
Table 1.
Clinical and outcome characteristics
| Perfusion study | Normal (n = 10) | FGR (n = 10) | P |
|---|---|---|---|
| Maternal age (years) | 29.5 [25.75–33] | 29 [22.75–34.75] | 0.91 |
| BMI | 22.9 [22.4–27.5] | 25 [22.75–27.50] | 0.649 |
| Parity | 1 [0–1.25] | 1 [0.75–2.25] | 0.362 |
| Gestation (weeks) | 39.2 [38.9–40.6] | 37.5 [35.63–39.18] | < 0.05* |
| Delivery (V/CS)† | 2/8 | 4/6 | N.A |
| PI‡ | 0.88 [0.80–0.93] | 1.09 [0.908–1.783] | < 0.05* |
| RI‡ | 0.565 [0.528–0.603] | 0.670 [0.612–0.800] | < 0.005*** |
| Birth weight (g) | 3517 [3359–3704] | 2189 [1518–2483] | < 0.001*** |
| IBR (centile) | 61 [22–84.3] | 1 [0–2.5] | < 0.001*** |
| Offspring sex# | 6M, 4F | 4M, 6F | N.A |
| Placenta disc weight (g) | 499 [438–642] | 279 [225–372] | < 0.001*** |
| Perfused lobule weight (g) | 49.75 [42–76] | 43.75 [24–72] | 0.623 |
| HPAEC study | Normal (n = 7) | FGR (n = 7) | P |
|---|---|---|---|
| Maternal age (years) | 26 [23–29] | 26 [23–33] | 0.364 |
| BMI | 25 [23–26] | 23 [21–25] | 0.174 |
| Parity | 1 [0–2] | 1[0–2] | 0.947 |
| Gestation (weeks) | 39 [38.1–39.0] | 38 [36–39.4] | 0.653 |
| Delivery (V/CS)† | 0/7 | 6/1 | N.A. |
| Birth weight (g) | 3816 [3560–3860] | 2440 [1980–2778] | < 0.001*** |
| IBR (centile) | 83 [75–88] | 3 [1–5] | < 0.005** |
| Offspring sex# | 3M, 4F | 4M, 3F | N.A. |
| Placenta disc weight (g) | 588 [532–660] | 432 [294–455] | < 0.005** |
Values are shown as median [interquartile range];
statistical analysis was performed using a Mann–Whitney U test.
BMI, body mass index; IBR, individualised birth weight ratio.
Vaginal (V)/Caesarean section (CS).
Pulsitility index (PI) and resistance index (RI) values represent n = 8 for the FGR group since there was absent end diastolic flow in two of the pregnancies.
Male (M), female (F).
Doppler ultrasound measurements of the umbilical artery
Doppler velocimetry measurements of the umbilical artery free loop were obtained within 1 week of delivery using a Voluson E6 with RA4B 3D 4–8 MHz curvilinear probe (GE Healthcare, UK). The PI and RI were then calculated. PI = (S – D)/A and RI = (S – D)/S, where S is the systolic peak, D is the end diastolic flow and A is the temporal average frequency.
Ex vivo dual perfusion of human placental cotyledons
The fetal sex of placentas used in perfusions is given in Table1. Perfusion of one or more grouped placental cotyledons was established as previously described (Brownbill & Sibley, 2006). Briefly, experiments were performed in a humidified cabinet at 37°C. The perfusate was modified Earle’s bicarbonate buffer (EBB; 117 mm NaCl, 10.7 mm KCl, 5.6 mm d-glucose, 3.6 mm CaCl2, 1.8 mm NaH2PO4, 13.6 mm NaHCO3, 0.04 mm l-arginine, 0.8 mm MgSO4, 3.5% (w/v) dextran, 0.1% (w/v) bovine serum albumin, 5000 IU l–1 heparin sodium) equilibrated with 95% O2–5% CO2 to pH 7.4 and warmed to 37°C. The fetal circulation was initially perfused at a standard circulation flow rate of 6 ml min−1. Fetal-side inflow hydrostatic pressure (FIHP) was recorded continuously throughout the experiments as a measure of fetoplacental vascular resistance and vasodilatation in response to altered flow rates (maternal and fetal hydrostatic pressure transducers: Medex, Digitimer, Welwyn Garden City, UK; Nanologger and associated software, Gaeltec, Isle of Skye, Scotland). FIHP was measured as an indication of vascular resistance at steady state following incremental increases in fetal-side inflow rate. The maternal circulation was perfused at a constant flow rate of 14 ml min−1 via perfusion manifold (Harvard Apparatus, Model MPP-5 Perfusion Manifold, 5 Inputs). n = 10 lobules from normal pregnancies and n = 10 FGR pregnancies were studied. Two FGR cases had absent end-diastolic flow in the umbilical artery, preventing their inclusion in some data sets.
Upon the establishment of dual-sided perfusion in open-circuit, placentas were left to equilibrate for 20 min. The fetal-inflow rate was then ramped rapidly between 2 ml min−1 and 12 ml min−1 until the pressure at 12 ml min−1 remained constant to ensure that all available vessels within the cotyledon were open. For experimental observations, fetal-inflow rate was incrementally increased from 2 ml min−1 through to 12 ml min−1 (2, 4, 6, 8, 10 and 12 ml min−1), with 7 min intervals at each flow rate to allow a steady state FIHP to be achieved. The ramping process was performed twice and all experimental values were taken during a second ramping stage to ensure adequate perfusion efficiency and accuracy of the response. In experiments where NO synthase was blocked Nω-nitro-l-arginine (l-NNA; 100 μm) or vehicle control (0.025% (v/v) HCl) was added to the EBB for 40 min at a standard fetal-inflow rate (6 ml min−1) and then maintained prior to repeating the two sets of fetal-side flow ramps. Vascular resistance was taken as the mean steady state FIHP, measured between 6 and 7 min following alteration of flow. FMVD was measured as the percentage reduction in FIHP from peak FIHP following alteration of flow, to the mean steady state FIHP, measured as described above.
Isolation and culture of HPAECs
HPAECs were isolated from chorionic placental vessels of normal and FGR pregnancies using a method previously described (Lang et al. 2008). The sex of the cell lines is given in Table1. Cells were maintained in microvascular endothelial growth medium (EGM-MV; Lonza, UK) supplemented with 10 ng ml−1 fibroblast growth factor 2 (FGF-2; R&D Systems, UK) and 6.7 ng ml−1 vascular endothelial growth factor-165 (VEGF-165; Peprotech, UK) at 37°C in a humidified atmosphere with 5% CO2. Medium was changed every 2–3 days and cells were passaged when they achieved 70% confluency.
A human placenta was obtained within 30 min of delivery, the amnion removed and two to four chorionic plate arteries dissected off the chorionic plate (intermediate diameter: 1–2 mm; minimum length: 3 cm without side branching). Each artery was placed into a 50 ml capped tube containing 20 ml sterile Hanks’ balanced salt solution (HBSS) (with calcium and magnesium, Invitrogen, UK), agitated to release excess blood and transferred to a new tubes with fresh 20 ml HBSS.
In turn, each artery was catheterised and the endothelial cells digested and flushed out of the vessel within a biological safety cabinet: the plastic sheath of an 18G catheter (Venflon; NuCare, UK) was cut with sterile scissors to a bevelled form. The washed artery was placed on a sterile chromatography glass plate in the biological safety cabinet and cannulated. The metal needle was withdrawn and a 15 cm length of fine fishing line was used to tie the artery onto the cannula. Twenty millilitres of collagenase/dispase enzyme solution was drawn up into a 20 ml syringe. The syringe was Luer-locked attached to the catheter, the assembly was held vertically with the vessel dangling, and the enzyme solution (0.1 U ml−1 collagenase, 0.8 U ml−1 dispase, Roche Diagnostics, cat no. 11 097 113 01; prepared in Ca2+- and Mg2+-free HBSS, Gibco, supplied by Invitrogen, UK) was slowly eluted through the vessel lumen across a 7 min period, with the first three bloody drops going to waste. The cell digest solution emanating from the distal cut-end of the vessel was caught into a 5 ml quench solution of 100% (v/v) heat-inactivated fetal bovine serum (FBS; Gibco from Invitrogen, UK).
The cell digest and calf serum were mixed thoroughly and centrifuged at 200 g for 10 min at ambient room temperature. The cell pellet was not visible due to a low cell count, so the supernatant was aspirated to waste leaving 200 μl, which was re-suspended into 3 ml of supplemented microvascular endothelial basal growth medium (EBM-MV bullet kit, Lonza, Belgium, supplied supplementary bullets: gentamycin sulphate/amphotericin-B, bovine brain extract, hydrocortisone rhEGF, FBS; the prepared medium was additionally supplemented with 88 pmol l–1 rhVEGF-A165, Peprotec, UK and 590 pmol l–1 rhFGF-2, R&D Systems, UK). The resuspension from each vessel was added to one well of the coated 24-well plate (attachment factor protein from Gibco, supplied by Invitrogen, UK) and incubated (37°C, 5% CO2, ambient O2). After 1 h of incubation, the medium was replaced with 1ml fresh EBM-MV and incubated for 1–2 weeks, changing the medium every day for the first three days and every three days thereafter. HPAECs had a classic, cobblestone morphology when confluent, preceded by an elongated form, with pseudopodia appearing to make contact between cells when sparse. At between 2 and 4 weeks of culture, sufficient cells (ca 2000) were present as colonies growing from the well periphery, which were then trypsinised (trypsin-EDTA; Lonza, Belgium) and passaged into a well of a six-well plate (uncoated here on in). Subsequent passaging occurred at 70–80% confluency. Trypsinisation was preceded with a Ca2+- and Mg2+-free HBSS wash. The second passage was into 1 × T25 flask, the third into 1 × T75 flask, the fourth into 4 × T75 flasks and the fifth into 4–6 × T175 flasks. From this point, cells were available for experimental use, characterisation and cryogenic storage (storage medium: supplemented EGM-MV + 10% dimethyl sulphoxide; Sigma-Aldrich, cell culture grade).
Characterisation of HPAECs
HPAECs were seeded (5 × 104−1 × 105 cells) onto sterile 12 mm glass coverslips in a 24-well plate. At confluency they were fixed in 100% cold methanol (30 min) and stored under sterile PBS for use in immunohistochemistry. They were stained for endothelium-specific markers: mouse monoclonal anti-human CD31 (CD31; Dako, cat no. M0823, 1 in 50), VEGF receptor 2 (VEGFR2; anti-VEGF R2/KDR fluorescein conjugated mouse IgG; R&D Systems, cat no. FAB357F, 1 in 10), von Willebrand factor (polyclonal rabbit anti-human, Dako, cat no. A0082, stock concentration 3.1 mg ml−1, 1 in 250), and Ulex europaeus agglutinin 1 lectin (direct fluorescent conjugate; Vector Laboratories, 1 in 25). Smooth muscle α-actin was used as a negative control (mouse monoclonal anti-human; Dako, cat no. M085, 1 in 100). Vectashield mounting medium was used with 4’,6-diamidino-2-phenylindole (DAPI). Secondary antibodies, fluorescently labelled with fluorescein isothiocyanate, were used accordingly: (i) polyclonal rabbit anti-mouse (Dako, cat no. F0232, 1 in 50); and (ii) polyclonal swine anti-rabbit IgG (Dako, cat no. F0205, 1 in 50).
Shear stress studies on HPAECs
Details on the preparation of materials and loading of microslides (0.4 mm Luer, T/C treated; Thistle Scientific, UK) can be found at: http://ibidi.com/xtproducts/en/ibidi-Labware/Flow-Chambers/m-Slide-I-Luer-Family. Unidirectional laminar shear-stress experiments on HPAECs were performed using Ibidi fluidic units (Thistle Scientific, UK), on HPAECs between passages 5 and 8 to assess the role of the NO synthase systems between normal and FGR groups. Cells were plated onto Ibidi μ-slides (0.4 Luer T/C treated; Thistle Scientific, UK) at a density of 1×105 cells per slide and cultured overnight to achieve a confluent monolayer. Medium was replaced and four μ-slides were attached in series to a single fluidic unit. Cell culture medium flowing across the attached cells was driven by the fluidic unit coupled to an air pump, delivering software-generated flow rates equating to a pre-set shear stress for 48 h. Several fluidic units were used in parallel for different treatments, or at different shear-stresses (5, 10 or 20 dyn cm–2; n = 6 for each of the normal and FGR groups; n = 5 for each group in the presence and absence of l-NNA, paired analysis). In a modification to the system, for the addition of l-NNA or vehicle and the removal of samples from the reservoirs, a sterile silicone injection port was built into the Ibidi fluidic unit (Thistle Scientific, UK) between one of the syringe barrel reservoirs and the perfusion set. Injections and withdrawals (25G needle and 1 ml syringe) were limited to 0.5 ml (isotonic diluent) and done whilst the associated barrel was in filling mode, so as not to influence the shear rate across the cells during the procedure. At the end of the shear stress run, cell lysates were recovered from Ibidi slides by placing the slides directly onto ice and eluting with 125 μl ice-cold radio-immunoprecipitation assay (RIPA) buffer. Lysates were merged from all four slides and flow-conditioned medium was collected from the perfusion sets and stored at –80°C. Lysate protein levels were determined using a micro-BCA protein detection kit (Pierce, UK). Culture conditioned medium was sampled via the perfusion set ports at 0, 1, 4, 24 and 48 h and stored at –80°C for the later determination of levels of nitrite, the stable breakdown product of NO (Greiss reaction assay kit; Life Technologies, UK).
Samples of medium were taken via a needle port at 1, 4, 24 and 48 h for nitrite analysis, using the Griess reaction following the manufacturer’s instructions (Life Technologies, UK). Immediately on cessation of flow, cell lysates were recovered at the end of the shear stress experiments by eluting the μ-slide chambers with ice-cold RIPA buffer containing protease inhibitor cocktail. Protein concentration was established using a micro-BCA kit (Pierce, UK) and lysates stored at –80°C prior to Western blotting and phosphokinase array assay.
Western blotting
eNOS, phosphoserine1177-eNOS (P-Ser1177-eNOS) and iNOS expression were compared in HPAECs between groups in static culture (n = 3, 7 and 7 from normal pregnancy and n = 4, 7 and 7 from FGR pregnancy; cells harvested at T = 48 h). In a separate study using normal HPACEs, eNOS and P-Ser1177-eNOS expression were analysed between static and shear stress culture (20 dyn cm–2; n = 7 per environment; cells harvested at T = 48 hours). Proteins were separated using SDS–PAGE (7.5% resolving gel) and transferred to Immobilon-P polyvinylidene difluoride membranes (Millipore, UK). Membranes were probed with rabbit polyclonal anti-eNOS (1:1000), anti-phospho-eNOS (Ser1177) (1:1000), anti-β-actin (1:2000) (all from New England Biolabs, UK) or anti-iNOS (1:200) (Abcam, UK) antibodies overnight at 4°C. The following day membranes were washed (×3) in Tris-buffered saline–Tween (50 mmol l–1 Tris-HCl, 150 mmol l–1 NaCl, 0.02% (v/v) Tween, pH 7.4), and incubated with horseradish peroxidase-conjugated secondary antibody (Dako, UK) for 1 h at room temperature. Proteins were detected used enhanced chemiluminescence (Fisher Scientific, UK) and visualised using X-ray film (Kodak, Biomax MR-1).
Phosphokinase array
A proteome profiler human phosphokinase array (R&D Systems, Inc., UK) was performed on pooled normal and pooled FGR HPAEC lysates (n = 4 cell lines for each group) following the manufacturer’s instructions.
Statistical analysis
Data are presented as means ± SEM. Statistical analysis was performed using a two-way ANOVA and Bonferroni post hoc test. Correlations were performed using the Pearson correlation coefficient and comparisons between two groups were performed using Student’s t test for paired or unpaired data, or Sidak’s multiple comparison test. Statistical significance was taken as P < 0.05. Data analysis was performed using GraphPad Prism version 5.
Results
Fetal vascular resistance measured in vivo by umbilical artery Doppler velocimetry reflects vascular resistance in the fetoplacental circulation
Correlation of ex vivo FIHP measured during perfusion with in vivo umbilical artery Doppler RI and PI measurements revealed a significant positive correlation in healthy pregnancy. FIHP at an inflow rate of 6 ml min−1 (Fig.1A) and 12 ml min−1 (data not shown) correlated with PI (r2 = 0.649, P < 0.01, and 0.515, P < 0.05 respectively). Correlations between FIHP and RI were even stronger with an r2 of 0.709 (P < 0.01) at a flow rate of 6 ml min−1 (Fig.1B) and 0.547 (P < 0.05) at 12 ml min−1 (data not shown). In the FGR group there was no overall correlation; in vivo velocimetry values were spread over a much wider range and the placentas with the highest Doppler indices did not always display the highest resistance values during perfusion at 6 ml min−1 (Fig.1C and D). The clinical characteristics of the patient study groups used in perfusion experiments are provided in Table1.
Figure 1.
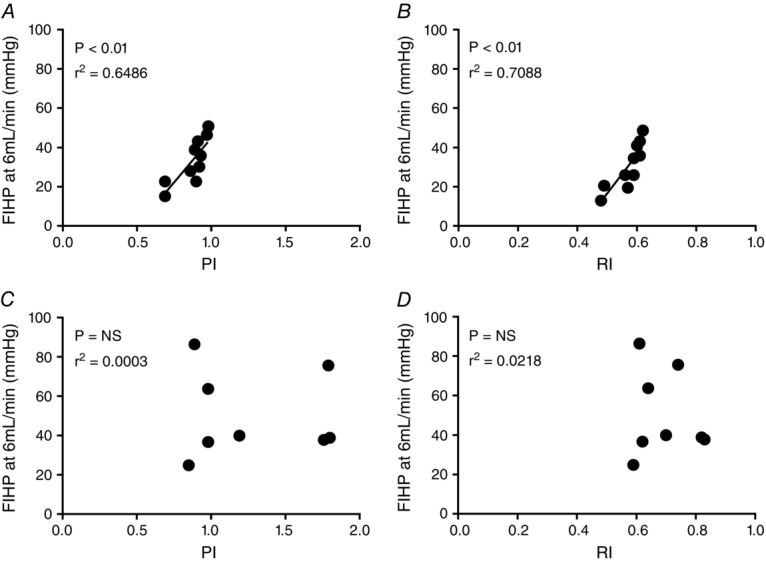
Correlations between in vivo pulsatility and resistance indices (PI and RI) in the umbilical artery and ex vivo perfused cotyledon resistance
Fetal-side inflow hydrostatic pressure (FIHP), which is proportional to vascular resistance in the fetoplacental circulation, was measured at a flow rate of 6 ml min−1 in normal (n = 10; A and B) and FGR placentas (n = 8, as RI and PI data cannot be obtained from two cases with absent or reversed end-diastolic flow; C and D) and correlated with clinical measures of both PI (A and C) and RI (B and D) in the umbilical artery obtained by Doppler velocimetry, using the Pearson coefficient correlation.
Ex vivo fetoplacental vascular resistance is higher in FGR placentas compared to normal placentas
As expected, vascular resistance increased with increasing flow rates in both the normal and the FGR perfused lobules (Fig.2A). However, in FGR placentas steady state FIHP was higher at each flow rate than in placentas from normal pregnancies, an effect particularly evident at higher inflow rates (Fig.2B; e.g. 72.3 ± 29.3 and 42.4 ± 19.0 mmHg, mean ± SD, respectively, at 12 ml min−1; 2-way ANOVA across 2–12 ml min−1 flow range, group effect: P < 0.05; Bonferroni post hoc test at inflow rates of 8, 10 and 12 ml min−1: P < 0.05, P < 0.01 and P < 0.01, respectively).
Figure 2.
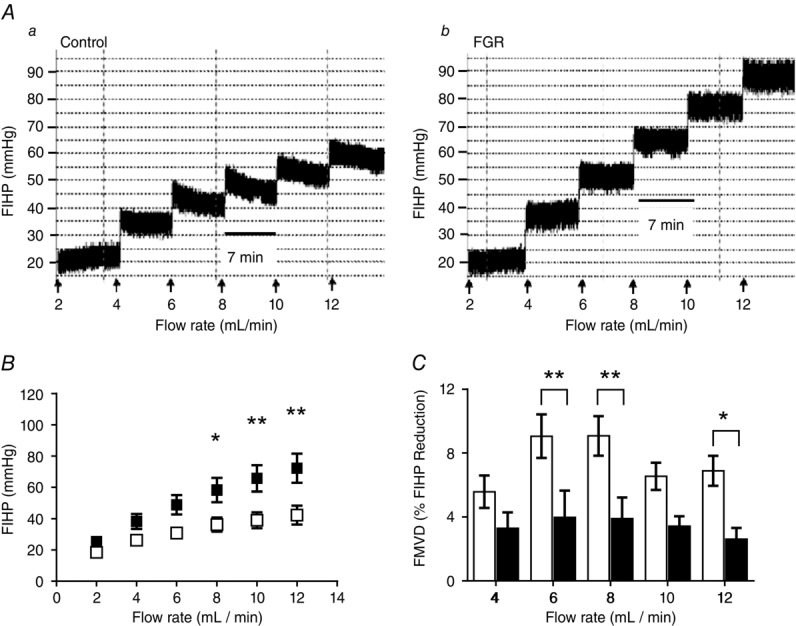
Vascular resistance in placentas from normal and fetal growth restricted (FGR) pregnancies
A, ex vivo dually perfused placental lobules were subjected to staged (7 min) incremental increases in fetal-side perfusate flow and fetal-side inflow hydrostatic pressure (FIHP) measured as an indication of vascular resistance. Traces are representative of FIHP in normal (Aa) and FGR (Ab) placentas. B, mean resistance values in placental lobules from normal (n = 10; open squares) and FGR (n = 10; filled squares) pregnancies were obtained for all flow rates during steady-state, 6–7 min following alterations in flow. Two-way ANOVA: P < 0.0001; Bonferonni test: *P < 0.05, **P < 0.01. C, flow-mediated vasodilatation (FMVD) was measured as the percentage decrease in FIHP, from peak FIHP to the new steady state following an elevation in flow, in normal (open bar) and fetal growth restricted (filled bar) placentas. Data show means ± SEM; 2-way ANOVA: P < 0.001; Bonferonni test: *P < 0.05, **P < 0.01.
FMVD in the placental vasculature is markedly reduced in FGR compared to normal pregnancy
Examining the data collected during the FIHP protocol noted above, we determined that placentas from the normal group demonstrated noticeable FMVD following each incremental increase in flow rate, with the largest responses occurring at 6 ml min−1 and 8 ml min−1 (9.06 ± 4.30% and 9.07 ± 3.92% reduction in FIHP, respectively; Fig.2C). In contrast, FMVD in placentas from FGR pregnancies was often absent or markedly reduced compared to controls (3.7 ± 5.30% at 6 ml min−1 and 3.95 ± 4.15% at 8 ml min−1, P < 0.01, mean ± SD; Fig.2C; 2-way ANOVA group effect across the full flow range: P < 0.0001; Bonferroni post hoc test at 6, 8 and 12 ml min−1: P < 0.01, P < 0.01 and P < 0.05, respectively) suggesting that placental vessels in FGR exhibit impaired FMVD.
In normal pregnancy, the extent of this FMVD response negatively correlated with the umbilical artery Doppler measurements when flow was elevated to 6 ml min−1 (Fig.3A and B) and 12 ml min−1 (data not shown). Both PI and RI significantly correlated with FMVD (r2 = 0.526, P < 0.05 and r2 = 0.619, P < 0.01, respectively) indicating that reduced FMVD is associated with increased vascular resistance in the placenta. In the FGR group, FMVD at an inflow rate of 6 ml min−1 was completely absent in four of the five placentas which had poor Dopplers, and one which had a normal Doppler (Fig.3C and D); results were similar at 12 ml min−1 (data not shown).
Figure 3.
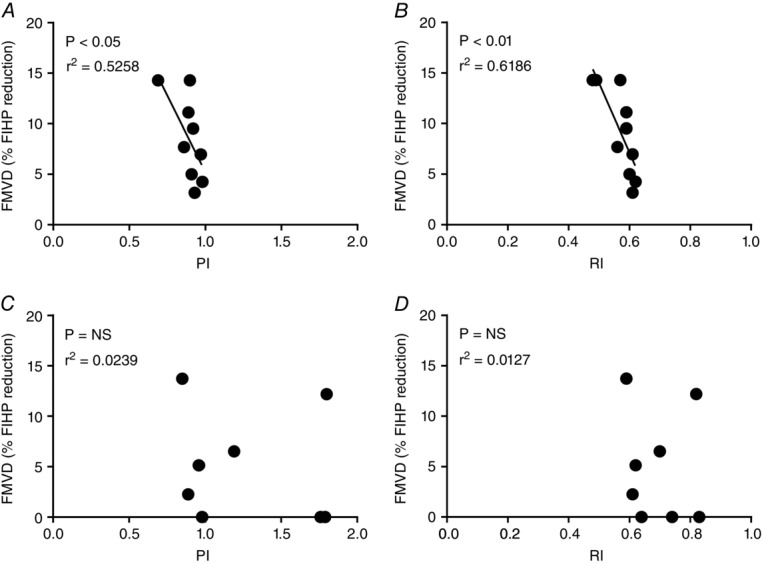
Correlations between in vivo pulsatility and resistance indices (PI and RI) and flow-mediated vasodilatation (FMVD)
FMVD, assessed as the percentage decrease in fetal-inflow hydrostatic pressure (FIHP) from peak FIHP to the new steady state following an elevation in flow, was measured in normal (n = 10; A and B) and FGR placentas (n = 8, as RI and PI data cannot be obtained from two cases with absent or reversed end-diastolic flow; C and D) during ex vivo dual perfusions at a flow rate of 6 ml min−1 and correlated with the umbilical artery Doppler pulsatility index (PI) (A and C) and resistance index (RI) (B and D) using the Pearson correlation coefficient. Two FGR cases had absent end-diastolic flow in the umbilical artery, preventing their inclusion.
NO has a role in regulating vascular resistance and flow-mediated vasodilatation in the placental vasculature
A second phase of perfusion in the same preparations in the presence of the NO synthase inhibitor l-NNA (100 μm) resulted in a small elevation in FIHP in healthy pregnancies, which was significant at higher flow rates (Fig.4A; 2-way ANOVA: P < 0.001). In contrast, inhibition of NO synthesis in FGR placentas resulted in a marked elevation in FIHP, which was significant at flow rates at and above 8 ml min−1 (Fig.4B; 2-way ANOVA: P < 0.0001), indicating a greater role for the NO signalling axis in lowering vascular resistance in FGR versus normal placentas.
Figure 4.
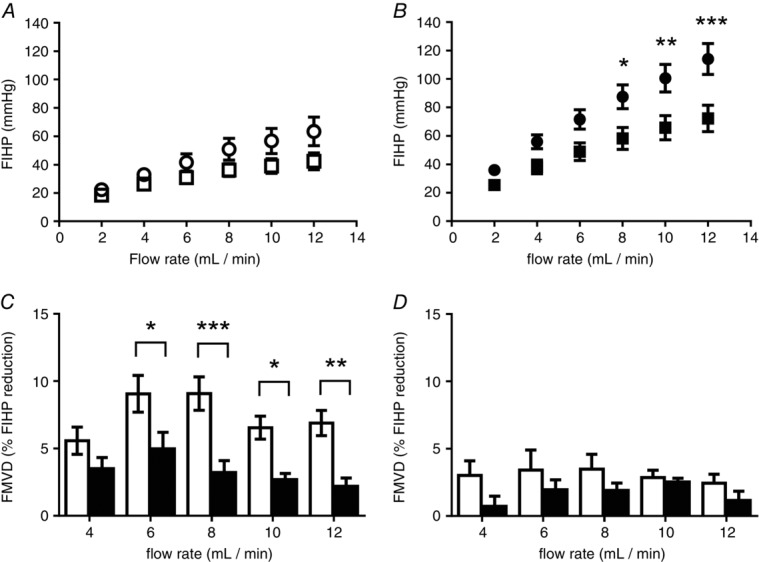
Contribution of nitric oxide (NO) to fetoplacental resistance and flow-mediated vasodilatation (FMVD)
Ex vivo dual perfusion of human placental lobules was performed following the inhibition of nitric oxide synthase. Vascular resistance was measured in normal placentas (n = 10; open symbols; A) or fetal growth restricted (FGR) placentas (n = 10; filled symbols; B) at increasing flow rates in the presence (circles) or absence (squares) of l-NNA (100 μm). FMVD, taken as the percentage decrease in fetal-inflow hydrostatic pressure (FIHP) from the initial peak to the new steady state following an elevation in flow, was measured in placentas from control (n = 10; C) and FGR pregnancies (n = 10; D) in the presence (filled bars) and absence (open bars) of l-NNA (100 μm). l-NNA had a significant effect on FIHP in normal and FGR perfused placental lobules (P < 0.001 and P < 0.0001, respectively). The flow-mediated vasodilatory effect was significantly reduced by l-NNA in normal and FGR perfused lobules (P < 0.0001 and P < 0.05, respectively). All data are presented as means ± SEM (ANOVA with Bonferroni post hoc test: *P < 0.05, **P < 0.01, ***P < 0.001).
In the normal placenta perfusions, FMVD was significantly reduced when NO release was inhibited with l-NNA (100 μm) indicating a pivotal role for NO in facilitating the FMVD vascular response to shear stress (Fig.4C). Since the FGR fetoplacental vasculature had displayed minimal flow-mediated vasodilatation under basal perfusion, l-NNA had little effect in reducing it further (Fig.4D).
Flow-mediated NO production by HPAECs is increased in FGR
To further investigate NO production by the fetoplacental endothelium in FGR, we turned our attention to fetoplacental endothelial cells isolated from chorionic plate arteries. The clinical characteristics of the patient study groups that HPAECs were isolated from are described in Table1. HPAECs from normal (Fig.5A) and FGR pregnancy (Fig.5B) demonstrated NO generation in a shear stress- and time-dependent manner. NO production was greater in the FGR group compared to normal cells (Fig.5C; both exposed to 48h of shear stress at 20 dyn cm–2). NO generation by FGR HPAECs was significantly inhibited by l-NNA (100 μm) (Fig.5D).
Figure 5.
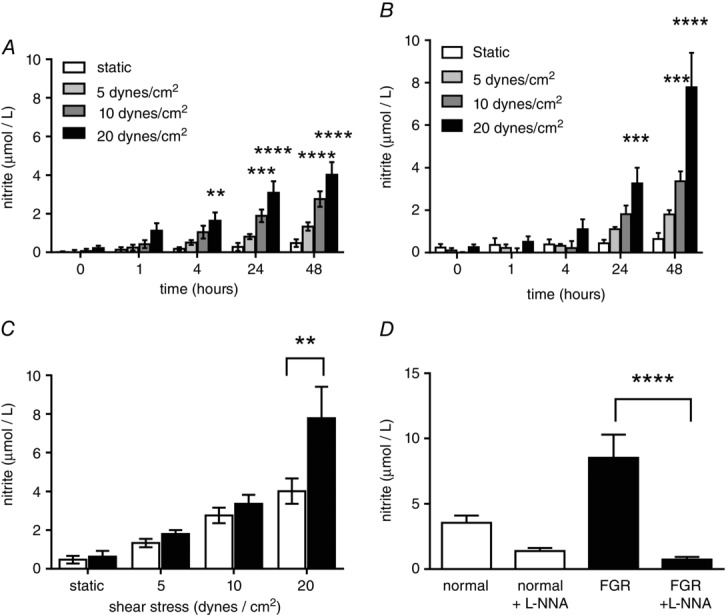
Shear stress-mediated NO production in HPAECs from control and growth restricted pregnancies
HPAECs isolated from control (A; n = 6) and FGR (B; n = 6) placentas were cultured under static or shear stress conditions for 48 h and nitrite concentration measured in flow conditioned media as an indication of nitric oxide production. There was a significant effect of time on nitrite formation in both groups; and at each time point, shear stress increased nitrite production compared to static culture (2-way ANOVA: P < 0.0001, both groups; Bonferonni multiple comparison test: **P < 0.01, ***P < 0.001, ****P < 0.0001, respectively). Nitrite, as a proportionate measure to NO, generated by FGR HPAECs (filled bars) was significantly greater than control HPAECs (open bars) following 48 h exposure to increasing shear stress conditions (C; 2-way ANOVA: P < 0.05; Bonferroni multiple comparison test: **P < 0.01). The production of nitrite, proportionate to NO release, by normal and FGR HPAECs in response to 48 h shear stress (at 20 dyn cm–2) was measured in the presence and absence of l-NNA (100 μm) (D); l-NNA caused a significant reduction only in the FGR group (Sidak’s multiple comparison test: ****P < 0.0001).
Expression and activation of eNOS in HPAECs are increased in FGR
Total eNOS expression was significantly higher in static cultured HPAECs isolated from FGR placentas (Fig.6A). There was also a trend (P = 0.065) towards increased levels of activated eNOS, assessed by Ser1177 phosphorylation in these cultures (data not shown), suggested by phosphokinase array data, which demonstrated a 10-fold increase in eNOS Ser1177 phosphorylation when comparing pooled lysates from control and FGR HPAECs (4 cell lines pooled for each group, data not shown). Inducible NO synthase (iNOS) was also observed at significantly higher levels in static cultured FGR HPAECs compared to normal cells (P < 0.05) (Fig.6B). Exposure of normal HPAECs to unidirectional laminar shear stress (20 dyn cm–2) did not elevate expression of eNOS (Fig.6C), but did significantly increase its activation (P < 0.05) (Fig.6D).
Figure 6.
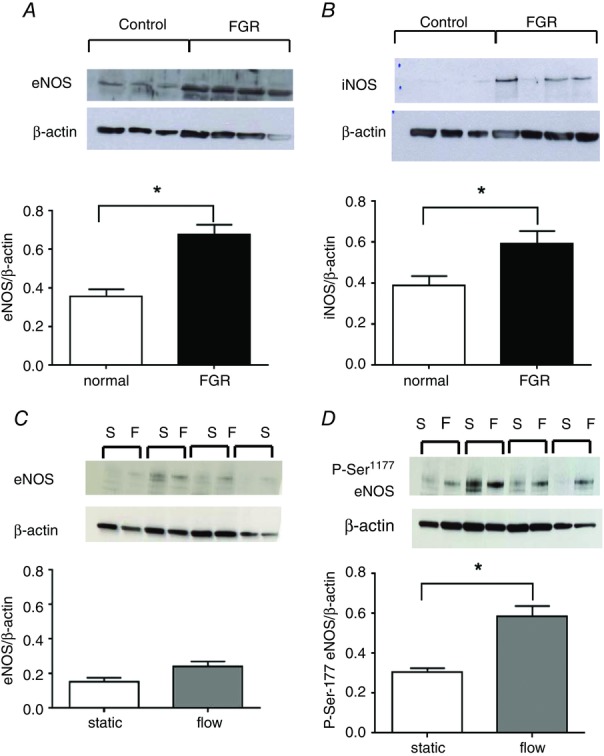
eNOS expression and activation in control and FGR HPAECs
Western blots were performed on lysates from normal and FGR HPAECs cultured under static conditions (passage 5–8) to detect eNOS (n = 3 and 4, respectively; A), activated and iNOS (n = 7, both groups; B). (Data represented as means ± SEM; *P < 0.05, unpaired t test.) eNOS (n = 7, both groups; C) and activated P-Ser1177-eNOS expression (n = 7, both groups; D) were compared in normal HPAECs following static and shear stress conditions (*P < 0.05, paired t test). Expression was normalised to β-actin expression and is shown as means ± SEM.
Discussion
In this study we provide data which support the concept that small resistance vessels and stem villi vessels of the fetoplacental circulation, as studied in ex vivo placental perfusion, are the loci that determine umbilicoplacental resistance, as measured via umbilical artery Doppler velocimetry in the clinic. In addition, we demonstrate that vascular resistance in the fetoplacental circulation measured ex vivo is significantly higher in FGR than in normal pregnancies. The key finding from the study, however, is the inability of FGR placentas to elicit vasodilatation in response to increases in flow, a response present in the normal fetoplacental circulation. We are not aware of any previous report demonstrating FMVD in the fetoplacental vasculature of the perfused organ, nor previous data demonstrating that it is severely perturbed in human FGR. However, NO is involved in flow-induced dilatation of isolated human small fetoplacental arteries (Learmont & Poston, 1996). Moreover in normal pregnancy, a direct negative correlation between FMVD and vascular resistance, as measured by umbilical artery Doppler velocimetry, was observed; placentas less able to adapt to increases in flow exhibited elevated resistance. Since flow- or shear stress-mediated vasodilatation represents a functional response predominantly orchestrated by the vascular endothelium, these results are consistent with our hypothesis that in FGR, the fetoplacental circulation exhibits endothelial dysfunction. This was supported by in vitro shear stress studies on HPAECs, which revealed altered NO signalling in cells from FGR pregnancies.
The remarkable correlation between umbilical artery resistance measurements determined in normal pregnancies in vivo and ex vivo perfusion pressure in the same placenta provides direct evidence that umbilical artery velocimetry values reflect vascular resistance in the fetoplacental circulation. Importantly, this correlation did not hold for the FGR group, and although generally the vascular resistance measured both in vivo and ex vivo was higher in this group, there was no clear correlation between the two measurements. Placental flow dysregulation in FGR is likely to be mainly caused by architectural vessel malformation and/or endothelial dysregulation. Alteration to placental capillary angiogenesis (Kingdom et al. 2000) and a general reduction in the luminal volume density of vasculature at the periphery of FGR placentas (Junaid et al. 2014) are likely to contribute to enhanced vascular resistance in some cases. This scenario suggests that the endothelia of the architecturally constrained vessels in FGR are not dysfunctional. However, the fact that resistance in vivo and ex vivo did not correlate well in the FGR placentas in this study, i.e. that some FGR cases showing high umbilical artery resistance found in the clinic did not manifest as a high ex vivo resistance during perfusion, does suggest that factors other than vessel malformation are involved and that an endothelial response is dysregulated. We suggest the lack of correlation is caused by the loss of ex vivo vasoreactivity in the FGR group, as the endocrine milieu with a pro-constriction bias becomes washed out during blood-free perfusion.
The significant increase in vascular resistance, which was observed in our FGR perfusion experiments, is in contrast to a previous study by Luria et al. (2012), which demonstrated no difference in the basal perfusion pressure in FGR and normal placentas during ex vivo perfusion. Discrepancy between the studies is likely to be due to the low fetal-side inflow rate used (6 ml min−1) by Luria et al. and FGR being defined as below the 10th centile of growth. In the current study, resistance was only significantly elevated at flow rates above 6 ml min−1, which are more akin to physiological flow rates when the ex vivo perfused tissue mass is standardised to in vivo values of blood flow to the whole placenta in the third trimester. This calculation was derived from an estimate that the normal placenta receives on average 21% of right ventricular output across the third trimester of pregnancy, equating to ca 180–250 ml min−1 for the whole placental mass (Kiserud et al. 2006). Hence, relating to the ex vivo perfusion of 35 g of term normal placental tissue, one might expect this mass to have experienced an average of 12 ml min−1 of in vivo blood flow after 32 weeks of gestation, although this value is likely to be reduced by approximately 25% in the FGR group (Kiserud et al. 2006). In a further difference to the Luria study, we defined FGR as the fifth centile and below to limit the inclusion of constitutively small fetuses, which are likely to display the normal ‘reactive’ phenotype.
In our cohort of 10 FGR placentas that was sufficiently intact for ex vivo organ perfusion, five had poor umbilical artery Doppler waveforms. However, as a whole, the FGR placentas (i.e. those with poor and normal umbilical artery waveforms) had higher vascular resistance ex vivo compared to the normal group. Umbilical artery Doppler indices indicate that fetoplacental vascular resistance falls with gestational age in normal pregnancy. As 60% of the perfused placentas from FGR pregnancies were below the minimum gestational age of the normal group, it is possible that gestational age contributed to the elevated vascular resistance observed in FGR. For the minimal gestational age case in the normal group (38 weeks + 4 days), the Doppler RI is estimated at 0.60, based on a linear regression plot of 1663 pregnancies between 24 and 42 weeks (Kurmanavicius et al. 1997). For the six FGR cases falling below this gestational age, a median gestation of 36 weeks + 1 day equates to an expected normal RI value of 0.62. Therefore, a higher resistance to flow in the FGR group compared to normal of just over 3% is expected as a consequence of the gestational age difference. However, the actual difference in basal perfusion pressure was 70% at a flow of 10 and 12 ml min−1 (Fig.2), indicating that the elevated resistance in FGR is caused predominantly by abnormal vascular function associated with this pregnancy complication.
Correlations between FMVD in vitro and resistance values measured in vivo demonstrate that placentas from normal pregnancy which exhibit low resistance to flow in the fetoplacental circulation exhibit the largest compensatory responses when flow is increased. FMVD is largely regulated by endothelial cells and is often used as a measure of endothelial function (Poredos & Jezovnik, 2013). These data therefore provide indirect evidence of endothelial dysfunction in FGR, resulting in perturbed FMVD and increased vascular resistance. Vasodilatation in response to changes in blood flow is not only significant as a measure of endothelial function, but also physiologically relevant; blood volume and subsequently flow are increased as gestation progresses (Reynolds et al. 2006). Furthermore, the spatial nature of fetoplacental blood flow may alter acutely during fetal- and maternal-side ‘blood flow matching’ – a dynamic occurrence, where versatility in the spatiotemporal fetoplacental blood flow is suggested to be a key element in the optimisation of placental oxygen transfer (Carter, 1989). In this regard, FMVD in fetoplacental vessels may be an important secondary event, following the initial agonist-evoked vasodilatory signalling mechanism that redefines the fetoplacental villous blood flow pattern during ‘blood flow matching’.
NO is arguably the most important vasodilator in the fetoplacental circulation, and plays a crucial role in facilitating the pregnancy-induced vascular adaptations involved in maintaining low resistance (Myatt, 1992; Myatt et al. 1992; Magness et al. 1997; Sladek et al. 1997). Evidence in support of this includes a significant increase in NO production by the fetoplacental vasculature during pregnancy and an elevation in the expression of secondary messenger cyclic guanosine monophosphate (cGMP) (Magness et al. 1997; Sladek et al. 1997). Moreover, prolonged inhibition of NO generation in animal models results in FGR (Baylis et al. 1992; Sladek et al. 1997), primarily through altered fetoplacental adaptations (Kulandavelu et al. 2012). Given the pivotal role of NO in regulating placental vascular tone and considering its well characterised role in FMVD in other vascular beds (Stoner et al. 2012), we investigated whether NO production was reduced in FGR, which could explain the lack of FMVD and increased vascular resistance. Interestingly our data suggest that the heightened resistance to flow in this pathology is not due to an inability of the endothelium to generate NO; indeed collectively, our organ perfusion and placental endothelial cell shear stress studies show an up-regulation of NOS/eNOS activity in this circulation; and remarkably in FGR HPAEC cultures, up-regulation of expression and activity was sustained through several passages and until the endothelial cells stopped dividing. These data suggest that the in vivo FGR environment can permanently change the endothelial cell phenotype. In this study, eNOS activation was assessed by phosphorylation of S1177; however, several other phosphorylation sites are associated with eNOS activity. Phosphorylation of the S633 amino acid residue results in enhanced NO production, whereas phosphorylation of T495 inhibits eNOS activity. While shear stress stimulates phosphorylation of both S1177 and S633 residues, it does not dephosphorylate T495 (Boo et al. 2002). Here we report a higher S1177 phosphorylation in the FGR HPAECs, complementing the finding of comparatively higher nitrite levels in the flow conditioned medium of this group. Ideally, other phosphorylation sites would be investigated had there been a sufficient quantity of recovered cell lysate.
Sustained iNOS up-regulation is normally associated with chronic conditions of vascular inflammation (Peng et al. 1998). The continued expression of iNOS in endothelial cells from FGR placentas suggests such inflammation is a feature of this condition, consistent with recent data on placentas associated with poor pregnancy outcome (Girard et al. 2014). Endothelial inflammation may also contribute to the general elevated resistance to flow in ex vivo perfused placental lobules from FGR pregnancy, through the loss of a smooth morphology at the luminal facing membrane, thus promoting turbulent flow (Leach & Firth, 1995). In some animal studies, nitric oxide bioavailability has been shown to be altered during labour, which could have a significant impact on our findings given the differences in mode of delivery between women having normal pregnancy and FGR. In human studies, however, nitric oxide synthase activity is not altered (Thomson et al. 1997), and eNOS and iNOS transcription in the placenta is unchanged (Cindrova-Davies et al. 2013). Moreover, there is some evidence to suggest that labour does not affect vascular reactivity of chorionic plate arteries (Abad et al. 2003; Mills et al. 2007). Collectively, these data indicate that the alterations in NO that we present are a direct result of FGR and not due to the difference in mode of labour between the two groups.
Our work is supportive of other evidence that shows up-regulation of eNOS activity in FGR (Rutherford et al. 1995; Lyall et al. 1996; Myatt et al. 1997) in addition to more recent studies that have demonstrated increased levels of nitrite in umbilical cord blood (Pisaneschi et al. 2012). However, the role of NO in FGR is controversial and there are numerous studies demonstrating reduced NO generation and reduced NO-dependent vasodilatation in FGR (Casanello & Sobrevia, 2002; Casanello et al. 2009; Krause et al. 2012, 2013). Studies by Krause and colleagues have shown reduced insulin-evoked NO vasodilatation in intra-uterine growth restricted human umbilical arteries (IUGR; 2nd–10th IBR) using wire myography (Krause et al. 2012). Enhanced l-arginase expression in human umbilical arterial endothelial cells isolated from IUGR placentas was also noted and the authors concluded that increased l-arginase expression reduced the availability of l-arginine to eNOS, which reduced NO production and compromised vasodilatation (Krause et al. 2012, 2013). There are various differences between the study of Krause and ours which may account for the contrasting results. Firstly, the loci and cell types studied: we focused on the fetoplacental vasculature, which determines vascular resistance, rather than the umbilical vessels. Secondly, our study also included physiological levels of eNOS substrate l-arginine in our buffers, which there is no mention of in the Krause studies. Thirdly, the definition of FGR in the studies also varies. It is important to note that despite the increased NO release in FGR placentas found here, these dilator levels failed to compensate for the high resistance found and the causative factor of increased vascular tone remains elusive.
Dysregulation of fetoplacental function through the imbalanced production of other vasoactive substances needs to be evaluated in future work. For example, angiotensin II (A-II), an acute vasoconstrictor of the human fetoplacental vasculature (Glance et al. 1986; Brownbill & Sibley, 2006), is reported to be elevated in the fetal circulation of human FGR pregnancies (Kingdom et al. 1993). Furthermore, A-II has the potential to up-regulate eNOS expression and activity in ovine fetoplacental arterial endothelial cells (Zheng et al. 2005). Hence, it would be interesting to determine any potential A-II effects on eNOS expression and assess possible changes in NO output under shear stress conditions in the human HPAEC cells.
In summary this study identified small blood vessels within the fetoplacental circulation as the primary loci which define vascular resistance in the umbilico-placental circulation in normal pregnancy. It is important to note that the ex vivo perfusion used in this study is largely exclusive of conduit arteries that may function differently to smaller microvessels. In FGR, these small blood vessels within the fetoplacental vasculature exhibit dysfunction resulting in reduced FMVD and increased vascular resistance. NO production is elevated in FGR, and goes some way to compensating for the increased resistance to flow, but cannot completely rescue levels back to within the normal range. It is unclear whether the NO compensation mechanism is at full capacity in FGR, or whether therapeutic interventions with the supplementation of l-arginine, the addition of the eNOS co-factor tetrahydrobiopterin, l-arginase inhibition, or sildenafil treatment to maintain cGMP levels in vascular smooth muscle would provide additional augmentation of this signalling pathway in humans. Further work is therefore required to determine whether the over-expression of NO can be therapeutically exploited or whether other signalling pathways should be considered as therapeutic targets to induce fetoplacental vasodilatation and reduce vascular resistance to rescue fetal growth. Elucidation of the constricting factor(s) which lead to elevated vascular resistance in FGR may also lead to the development of novel therapeutic strategies.
Translational perspective.
Understanding the mechanisms that lead to a fetus not meeting its predetermined genetic growth potential is a key step in identifying therapeutic targets and safe medicines for use in these pregnancies. There is considerable controversy in the literature regarding the up- or down-regulation of NO in pregnancies complicated by fetal growth restriction (FGR), since (i) some studies, such as nitrite measurement in fetal plasma conditioned by the whole fetal circulation, have used an assay prone to biochemical noise from Hb, and (ii) experimental designs have been used which are not reflective of resistance in the fetoplacental vascular bed, or are not inclusive of an endothelial response of placental origin, under shear stress conditions. To this end, our multi-model approach has overcome these caveats. We report an upregulation in NO synthesis, which represents a failed compensatory response to high fetoplacental vascular resistance in FGR. Furthermore, the ex vivo dual placenta model provides reliable resistance data for the fetoplacental microcirculation, tightly correlated to umbilical arterial in vivo resistance in the clinic, in normal pregnancy. Therefore our data support the use of the ex vivo systems, as described, as a useful testing platform for potential therapeutics to be used in the treatment of FGR. Our data also provide further support for targeting NO generating systems in the development of such therapies.
Acknowledgments
The authors are grateful to the staff of the delivery unit at St Mary’s Hospital, Manchester, for their assistance in obtaining placental tissue.
Glossary
- A-II
angiotensin II
- BMI
body mass index
- cGMP
cyclic guanosine monophosphate
- CS
Caesarean section
- DAPI
4’,6-diamidino-2-phenylindole
- eNOS
endothelial nitric oxide synthase
- EDHF
endothelial derived hyperpolarising factor
- FGF-2
fibroblast growth factor 2
- FGR
fetal growth restriction
- FIHP
fetal-side inflow hydrostatic pressure
- FMVD
flow-mediated vasodilatation
- HPAEC
human chorionic plate arterial endothelial cell
- IBR
individualised birth weight ratio
- iNOS
inducible nitric oxide synthase
- IUGR
intrauterine growth restriction
- l-NNA
Nω-nitro-l-arginine
- NO
nitric oxide
- PGI2
prostacyclin
- PI
pulsatility index
- RI
resistance index
- RIPA
radio-immunoprecipitation assay
- VEGF
vascular endothelial growth factor
Additional information
Competing interests
There authors have no competing interests.
Author contributions
P.B., C.P.S., S.L.G. and M.W. were involved in the conception and design of the study. All authors contributed to the collection, analysis and interpretation of data and drafting the article or revising it critically for important intellectual content.
Funding
This research was funded by the British Heart Foundation (grant number PG/11/11/28725) and facilitated by the Manchester Biomedical Research Centre and the Greater Manchester Comprehensive Local Research Network. The support of an Action Research Endowment Fund is also acknowledged.
References
- Abad A, Estan L, Morales-Olivas FJ. Serra V. Reactivity of isolated human chorionic vessels: analysis of some influencing variables. Can J Physiol Pharmacol. 2003;81:1147–1151. doi: 10.1139/y03-119. [DOI] [PubMed] [Google Scholar]
- Acharya G, Wilsgaard T, Berntsen GK, Maltau JM. Kiserud T. Doppler-derived umbilical artery absolute velocities and their relationship to fetoplacental volume blood flow: a longitudinal study. Ultrasound Obstet Gynecol. 2005;25:444–453. doi: 10.1002/uog.1880. [DOI] [PubMed] [Google Scholar]
- Alberry M. Soothill P. Management of fetal growth restriction. Arch Dis Child Fetal Neonatal Ed. 2007;92:F62–F67. doi: 10.1136/adc.2005.082297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainbridge SA, Farley AE, McLaughlin BE, Graham CH, Marks GS, Nakatsu K, Brien JF. Smith GN. Carbon monoxide decreases perfusion pressure in isolated human placenta. Placenta. 2002;23:563–569. doi: 10.1053/plac.2002.0845. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Martyn CN, Osmond C. Wield GA. Abnormal liver growth in utero and death from coronary heart disease. BMJ. 1995;310:703–704. doi: 10.1136/bmj.310.6981.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ, Osmond C, Golding J, Kuh D. Wadsworth ME. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ. 1989;298:564–567. doi: 10.1136/bmj.298.6673.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baschat AA. Pathophysiology of fetal growth restriction: implications for diagnosis and surveillance. Obstet Gynecol Surv. 2004;59:617–627. doi: 10.1097/01.ogx.0000133943.54530.76. [DOI] [PubMed] [Google Scholar]
- Baylis C, Mitruka B. Deng A. Chronic blockade of nitric oxide synthesis in the rat produces systemic hypertension and glomerular damage. J Clin Invest. 1992;90:278–281. doi: 10.1172/JCI115849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boo YC, Sorescu G, Boyd N, Shiojima I, Walsh K, Du J. Jo H. Shear stress stimulates phosphorylation of endothelial nitric-oxide synthase at Ser1179 by Akt-independent mechanisms: role of protein kinase A. Journal of Biological Chemistry. 2002;277:3388–3396. doi: 10.1074/jbc.M108789200. [DOI] [PubMed] [Google Scholar]
- Brownbill P. Sibley CP. Regulation of transplacental water transfer: the role of fetoplacental venous tone. Placenta. 2006;27:560–567. doi: 10.1016/j.placenta.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Carter AM. Factors affecting gas transfer across the placenta and the oxygen supply to the fetus. J Dev Physiol. 1989;12:305–322. [PubMed] [Google Scholar]
- Casanello P, Krause B, Torres E, Gallardo V, Gonzalez M, Prieto C, Escudero C, Farias M. Sobrevia L. Reduced l-arginine transport and nitric oxide synthesis in human umbilical vein endothelial cells from intrauterine growth restriction pregnancies is not further altered by hypoxia. Placenta. 2009;30:625–633. doi: 10.1016/j.placenta.2009.04.010. [DOI] [PubMed] [Google Scholar]
- Casanello P. Sobrevia L. Intrauterine growth retardation is associated with reduced activity and expression of the cationic amino acid transport systems y+/hCAT-1 and y+/hCAT-2B and lower activity of nitric oxide synthase in human umbilical vein endothelial cells. Circ Res. 2002;91:127–134. doi: 10.1161/01.res.0000027813.55750.e7. [DOI] [PubMed] [Google Scholar]
- Chaddha V, Viero S, Huppertz B. Kingdom J. Developmental biology of the placenta and the origins of placental insufficiency. Semin Fetal Neonatal Med. 2004;9:357–369. doi: 10.1016/j.siny.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Cindrova-Davies T, Herrera EA, Niu Y, Kingdom J, Giussani DA. Burton GJ. Reduced cystathionine gamma-lyase and increased miR-21 expression are associated with increased vascular resistance in growth-restricted pregnancies: hydrogen sulfide as a placental vasodilator. Am J Pathol. 2013;182:1448–1458. doi: 10.1016/j.ajpath.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demicheva E. Crispi F. Long-term follow-up of intrauterine growth restriction: Cardiovascular disorders. Fetal Diagn Ther. 2013;36:143–153. doi: 10.1159/000353633. [DOI] [PubMed] [Google Scholar]
- Figueroa-Diesel H, Hernandez-Andrade E, Acosta-Rojas R, Cabero L. Gratacos E. Doppler changes in the main fetal brain arteries at different stages of hemodynamic adaptation in severe intrauterine growth restriction. Ultrasound Obstet Gynecol. 2007;30:297–302. doi: 10.1002/uog.4084. [DOI] [PubMed] [Google Scholar]
- Gardosi J. New definition of small for gestational age based on fetal growth potential. Horm Res. 2006;65(3):15–18. doi: 10.1159/000091501. Suppl. [DOI] [PubMed] [Google Scholar]
- Ghosh GS. Gudmundsson S. Uterine and umbilical artery Doppler are comparable in predicting perinatal outcome of growth-restricted fetuses. BJOG. 2009;116:424–430. doi: 10.1111/j.1471-0528.2008.02057.x. [DOI] [PubMed] [Google Scholar]
- Girard S, Heazell AE, Derricott H, Allan SM, Sibley CP, Abrahams VM. Jones RL. Circulating cytokines and alarmins associated with placental inflammation in high-risk pregnancies. Am J Reprod Immunol. 2014;72:422–434. doi: 10.1111/aji.12274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glance DG, Elder MG. Myatt L. The actions of prostaglandins and their interactions with angiotensin II in the isolated perfused human placental cotyledon. Br J Obstet Gynaecol. 1986;93:488–494. [PubMed] [Google Scholar]
- Holwerda KM, Bos EM, Rajakumar A, Ris-Stalpers C, van Pampus MG, Timmer A, Erwich JJ, Faas MM, van Goor H. Lely AT. Hydrogen sulfide producing enzymes in pregnancy and preeclampsia. Placenta. 2012;33:518–521. doi: 10.1016/j.placenta.2012.02.014. [DOI] [PubMed] [Google Scholar]
- Junaid TO, Brownbill P, Chalmers N, Johnstone ED. Aplin JD. Fetoplacental vascular alterations associated with fetal growth restriction. Placenta. 2014;35:808–815. doi: 10.1016/j.placenta.2014.07.013. [DOI] [PubMed] [Google Scholar]
- Kingdom J, Huppertz B, Seaward G. Kaufmann P. Development of the placental villous tree and its consequences for fetal growth. Eur J Obstet Gynecol Reprod Biol. 2000;92:35–43. doi: 10.1016/s0301-2115(00)00423-1. [DOI] [PubMed] [Google Scholar]
- Kingdom JC, McQueen J, Connell JM. Whittle MJ. Fetal angiotensin II levels and vascular (type I) angiotensin receptors in pregnancies complicated by intrauterine growth retardation. Br J Obstet Gynaecol. 1993;100:476–482. doi: 10.1111/j.1471-0528.1993.tb15276.x. [DOI] [PubMed] [Google Scholar]
- Kiserud T, Kessler J, Ebbing C. Rasmussen S. Ductus venosus shunting in growth-restricted fetuses and the effect of umbilical circulatory compromise. Ultrasound Obstet Gynecol. 2006;28:143–149. doi: 10.1002/uog.2784. [DOI] [PubMed] [Google Scholar]
- Krause BJ, Carrasco-Wong I, Caniuguir A, Carvajal J, Farias M. Casanello P. Endothelial eNOS/arginase imbalance contributes to vascular dysfunction in IUGR umbilical and placental vessels. Placenta. 2013;34:20–28. doi: 10.1016/j.placenta.2012.09.015. [DOI] [PubMed] [Google Scholar]
- Krause BJ, Prieto CP, Munoz-Urrutia E, San Martin S, Sobrevia L. Casanello P. Role of arginase-2 and eNOS in the differential vascular reactivity and hypoxia-induced endothelial response in umbilical arteries and veins. Placenta. 2012;33:360–366. doi: 10.1016/j.placenta.2012.02.006. [DOI] [PubMed] [Google Scholar]
- Kulandavelu S, Whiteley KJ, Qu D, Mu J, Bainbridge SA. Adamson SL. Endothelial nitric oxide synthase deficiency reduces uterine blood flow, spiral artery elongation, and placental oxygenation in pregnant mice. Hypertension. 2012;60:231–238. doi: 10.1161/HYPERTENSIONAHA.111.187559. [DOI] [PubMed] [Google Scholar]
- Kurmanavicius J, Florio I, Wisser J, Hebisch G, Zimmermann R, Muller R, Huch R. Huch A. Reference resistance indices of the umbilical, fetal middle cerebral and uterine arteries at 24–42 weeks of gestation. Ultrasound Obstet Gynecol. 1997;10:112–120. doi: 10.1046/j.1469-0705.1997.10020112.x. [DOI] [PubMed] [Google Scholar]
- Lang I, Schweizer A, Hiden U, Ghaffari-Tabrizi N, Hagendorfer G, Bilban M, Pabst MA, Korgun ET, Dohr G. Desoye G. Human fetal placental endothelial cells have a mature arterial and a juvenile venous phenotype with adipogenic and osteogenic differentiation potential. Differentiation. 2008;76:1031–1043. doi: 10.1111/j.1432-0436.2008.00302.x. [DOI] [PubMed] [Google Scholar]
- Lawn JE, Cousens S, Zupan J. 4 million neonatal deaths: When? Where? Why? Lancet. 2005;365:891–900. doi: 10.1016/S0140-6736(05)71048-5. Lancet Neonatal Survival Steering Team. [DOI] [PubMed] [Google Scholar]
- Leach L. Firth JA. Advances in understanding permeability in fetal capillaries of the human placenta: a review of organization of the endothelial paracellular clefts and their junctional complexes. Reprod Fertil Dev. 1995;7:1451–1456. doi: 10.1071/rd9951451. [DOI] [PubMed] [Google Scholar]
- Learmont JG. Poston L. Nitric oxide is involved in flow-induced dilation of isolated human small fetoplacental arteries. Am J Obstet Gynecol. 1996;174:583–588. doi: 10.1016/s0002-9378(96)70432-5. [DOI] [PubMed] [Google Scholar]
- Li Y, Zheng J, Bird IM. Magness RR. Mechanisms of shear stress-induced endothelial nitric-oxide synthase phosphorylation and expression in ovine fetoplacental artery endothelial cells. Biol Reprod. 2004;70:785–796. doi: 10.1095/biolreprod.103.022293. [DOI] [PubMed] [Google Scholar]
- Luria O, Bar J, Barnea O, Golan A. Kovo M. Reactivity of blood vessels in response to prostaglandin E2 in placentas from pregnancies complicated by fetal growth restriction. Prenat Diagn. 2012;32:417–422. doi: 10.1002/pd.3827. [DOI] [PubMed] [Google Scholar]
- Lyall F, Barber A, Myatt L, Bulmer JN. Robson SC. Hemeoxygenase expression in human placenta and placental bed implies a role in regulation of trophoblast invasion and placental function. FASEB J. 2000;14:208–219. doi: 10.1096/fasebj.14.1.208. [DOI] [PubMed] [Google Scholar]
- Lyall F, Greer IA, Young A. Myatt L. Nitric oxide concentrations are increased in the feto-placental circulation in intrauterine growth restriction. Placenta. 1996;17:165–168. doi: 10.1016/s0143-4004(96)80009-9. [DOI] [PubMed] [Google Scholar]
- Magness RR, Shaw CE, Phernetton TM, Zheng J. Bird IM. Endothelial vasodilator production by uterine and systemic arteries. II. Pregnancy effects on NO synthase expression. Am J PhysiolHeart CircPhysiol. 1997;272:H1730–H1740. doi: 10.1152/ajpheart.1997.272.4.H1730. [DOI] [PubMed] [Google Scholar]
- Marzioni D, Tamagnone L, Capparuccia L, Marchini C, Amici A, Todros T, Bischof P, Neidhart S, Grenningloh G. Castellucci M. Restricted innervation of uterus and placenta during pregnancy: evidence for a role of the repelling signal Semaphorin 3A. Dev Dyn. 2004;231:839–848. doi: 10.1002/dvdy.20178. [DOI] [PubMed] [Google Scholar]
- Mills TA, Baker PN. Wareing M. The effect of mode of delivery on placental chorionic plate vascular reactivity. Hypertens Pregnancy. 2007;26:201–210. doi: 10.1080/10601330701274824. [DOI] [PubMed] [Google Scholar]
- Myatt L. Control of vascular resistance in the human placenta. Placenta. 1992;13:329–341. doi: 10.1016/0143-4004(92)90057-z. [DOI] [PubMed] [Google Scholar]
- Myatt L, Brewer AS, Langdon G. Brockman DE. Attenuation of the vasoconstrictor effects of thromboxane and endothelin by nitric oxide in the human fetal-placental circulation. Am J Obstet Gynecol. 1992;166:224–230. doi: 10.1016/0002-9378(92)91863-6. [DOI] [PubMed] [Google Scholar]
- Myatt L, Eis AL, Brockman DE, Greer IA. Lyall F. Endothelial nitric oxide synthase in placental villous tissue from normal, pre-eclamptic and intrauterine growth restricted pregnancies. Hum Reprod. 1997;12:167–172. doi: 10.1093/humrep/12.1.167. [DOI] [PubMed] [Google Scholar]
- Palinski W. Napoli C. Impaired fetal growth, cardiovascular disease, and the need to move on. Circulation. 2008;117:341–343. doi: 10.1161/CIRCULATIONAHA.107.750133. [DOI] [PubMed] [Google Scholar]
- Peng HB, Spiecker M. Liao JK. Inducible nitric oxide: an autoregulatory feedback inhibitor of vascular inflammation. J Immunol. 1998;161:1970–1976. [PubMed] [Google Scholar]
- Pisaneschi S, Strigini FA, Sanchez AM, Begliuomini S, Casarosa E, Ripoli A, Ghirri P, Boldrini A, Fink B, Genazzani AR, Coceani F. Simoncini T. Compensatory feto-placental upregulation of the nitric oxide system during fetal growth restriction. PLoS One. 2012;7:e45294. doi: 10.1371/journal.pone.0045294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poredos P. Jezovnik MK. Testing endothelial function and its clinical relevance. J Atheroscler Thromb. 2013;20:1–8. doi: 10.5551/jat.14340. [DOI] [PubMed] [Google Scholar]
- Poston L. The control of blood flow to the placenta. Exp Physiol. 1997;82:377–387. doi: 10.1113/expphysiol.1997.sp004033. [DOI] [PubMed] [Google Scholar]
- Reynolds LP, Caton JS, Redmer DA, Grazul-Bilska AT, Vonnahme KA, Borowicz PP, Luther JS, Wallace JM, Wu G. Spencer TE. Evidence for altered placental blood flow and vascularity in compromised pregnancies. J Physiol. 2006;572:51–58. doi: 10.1113/jphysiol.2005.104430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford RA, McCarthy A, Sullivan MH, Elder MG, Polak JM. Wharton J. Nitric oxide synthase in human placenta and umbilical cord from normal, intrauterine growth-retarded and pre-eclamptic pregnancies. Br J Pharmacol. 1995;116:3099–3109. doi: 10.1111/j.1476-5381.1995.tb15111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala C, Campise M, Ambroso G, Motta T, Zanchetti A. Morganti A. Atrial natriuretic peptide and hemodynamic changes during normal human pregnancy. Hypertension. 1995;25:631–636. doi: 10.1161/01.hyp.25.4.631. [DOI] [PubMed] [Google Scholar]
- Sibley CP, Turner MA, Cetin I, Ayuk P, Boyd CA, D’Souza SW, Glazier JD, Greenwood SL, Jansson T. Powell T. Placental phenotypes of intrauterine growth. Pediatr Res. 2005;58:827–832. doi: 10.1203/01.PDR.0000181381.82856.23. [DOI] [PubMed] [Google Scholar]
- Sladek SM, Magness RR. Conrad KP. Nitric oxide and pregnancy. Am J PhysiolRegulIntegrComp Physiol. 1997;272:R441–R463. doi: 10.1152/ajpregu.1997.272.2.R441. [DOI] [PubMed] [Google Scholar]
- Sprague B, Chesler NC. Magness RR. Shear stress regulation of nitric oxide production in uterine and placental artery endothelial cells: experimental studies and hemodynamic models of shear stresses on endothelial cells. International Journal of Developmental Biology. 2010;54:331–339. doi: 10.1387/ijdb.082832bs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoner L, Erickson ML, Young JM, Fryer S, Sabatier MJ, Faulkner J, Lambrick DM. McCully KK. There’s more to flow-mediated dilation than nitric oxide. J Atheroscler Thromb. 2012;19:589–600. doi: 10.5551/jat.11973. [DOI] [PubMed] [Google Scholar]
- Thomson AJ, Telfer JF, Kohnen G, Young A, Cameron IT, Greer IA. Norman JE. Vasoconstriction increases pulmonary nitric oxide synthesis and circulating cyclic GMP. Journal of Surgical Research. 1997;70:75–83. doi: 10.1006/jsre.1997.5095. [DOI] [PubMed] [Google Scholar]
- Thornburg KL, Shannon J, Thuillier P. Turker MS. In utero life and epigenetic predisposition for disease. Adv Genet. 2010;71:57–78. doi: 10.1016/B978-0-12-380864-6.00003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turan S, Miller J. Baschat AA. Integrated testing and management in fetal growth restriction. Semin Perinatol. 2008;32:194–200. doi: 10.1053/j.semperi.2008.02.008. [DOI] [PubMed] [Google Scholar]
- Vergani P, Roncaglia N, Ghidini A, Crippa I, Cameroni I, Orsenigo F. Pezzullo J. Can adverse neonatal outcome be predicted in late preterm or term fetal growth restriction? Ultrasound Obstet Gynecol. 2010;36:166–170. doi: 10.1002/uog.7583. [DOI] [PubMed] [Google Scholar]
- Wang K, Ahmad S, Cai M, Rennie J, Fujisawa T, Crispi F, Baily J, Miller MR, Cudmore M, Hadoke PW, Wang R, Gratacos E, Buhimschi IA, Buhimschi CS. Ahmed A. Dysregulation of hydrogen sulfide producing enzyme cystathionine gamma-lyase contributes to maternal hypertension and placental abnormalities in preeclampsia. Circulation. 2013;127:2514–2522. doi: 10.1161/CIRCULATIONAHA.113.001631. [DOI] [PubMed] [Google Scholar]
- Wieczorek KM, Brewer AS. Myatt L. Shear stress may stimulate release and action of nitric oxide in the human fetal-placental vasculature. Am J Obstet Gynecol. 1995;173:708–713. doi: 10.1016/0002-9378(95)90327-5. [DOI] [PubMed] [Google Scholar]
- Yanney M. Marlow N. Paediatric consequences of fetal growth restriction. Semin Fetal Neonatal Med. 2004;9:411–418. doi: 10.1016/j.siny.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Zheng J, Bird IM, Chen DB. Magness RR. Angiotensin II regulation of ovine fetoplacental artery endothelial functions: interactions with nitric oxide. J Physiol. 2005;565:59–69. doi: 10.1113/jphysiol.2004.082420. [DOI] [PMC free article] [PubMed] [Google Scholar]