

Abstract
Maternal obesity is associated with a high risk for gestational diabetes mellitus (GDM), which is a common complication of pregnancy. The influence of maternal obesity and GDM on the metabolic health of the offspring is poorly understood. We hypothesize that GDM associated with maternal obesity will cause obesity, insulin resistance and hepatic steatosis in the offspring. Female Sprague-Dawley rats were fed a high-fat (45%) and sucrose (HFS) diet to cause maternal obesity and GDM. Lean control pregnant rats received low-fat (LF; 10%) diets. To investigate the interaction between the prenatal environment and postnatal diets, rat offspring were assigned to LF or HFS diets for 12 weeks, and insulin sensitivity and hepatic steatosis were evaluated. Pregnant GDM dams exhibited excessive gestational weight gain, hyperinsulinaemia and hyperglycaemia. Offspring of GDM dams gained more weight than the offspring of lean dams due to excess adiposity. The offspring of GDM dams also developed hepatic steatosis and insulin resistance. The postnatal consumption of a LF diet did not protect offspring of GDM dams against these metabolic disorders. Analysis of the hepatic metabolome revealed increased diacylglycerol and reduced phosphatidylethanolamine in the offspring of GDM dams compared to offspring of lean dams. Consistent with altered lipid metabolism, the expression of CTP:phosphoethanolamine cytidylyltransferase, and peroxisomal proliferator activated receptor-α mRNA was reduced in the livers of GDM offspring. GDM exposure programs gene expression and hepatic metabolite levels and drives the development of hepatic steatosis and insulin resistance in young adult rat offspring.
Key points
Gestational diabetes mellitus is a common complication of pregnancy, but its effects on the offspring are poorly understood.
We developed a rat model of diet-induced gestational diabetes mellitus that recapitulates many of the clinical features of the disease, including excessive gestational weight gain, glucose intolerance, hyperinsulinaemia and mild hyperglycaemia.
Compared to the offspring of lean dams, exposure to gestational diabetes mellitus during the prenatal period resulted in obesity, hepatic steatosis and insulin resistance in young rat offspring that consumed a postnatal diet that was low in fat.
The combination of maternal gestational diabetes mellitus and the postnatal consumption of a high-fat diet by the offspring caused a more severe metabolic phenotype.
Metabolomic profiling of the liver tissues of the offspring of gestational diabetic dams revealed accumulation of lipotoxic lipids and reduced phosphatidylethanolamine levels compared to the offspring of lean dams.
The results establish that gestational diabetes mellitus is a driver of hepatic steatosis and insulin resistance in the offspring.
Introduction
Obesity and its associated co-morbidities are reaching epidemic proportions (Flegal et al. 2012). The proportion of young adults with obesity has increased dramatically in recent decades (Ogden et al. 2014) with several obesity-related pathologies, including hepatic steatosis, reaching record levels (Dabelea et al. 2000; Brumbaugh et al. 2013). While the consumption of calorie-dense foods and a sedentary lifestyle are driving forces behind the obesity epidemic, population health data and experimental animal model systems have established that alterations in the prenatal environment during crucial periods of development program biological systems and influence disease susceptibility in the offspring (Barker & Osmond, 1987; McMillen & Robinson, 2005). While this initial research focused on maternal under-nutrition, more research has begun to investigate maternal over-nutrition and obesity (reviewed in Pereira et al. 2015).
In the United States, up to 60% of women are overweight at the beginning of the gestational period and roughly 30% are obese (Hinkle et al. 2012). Being overweight or obese before or throughout pregnancy is a major risk factor for gestational diabetes mellitus (GDM) (Catalano et al. 1991, 1999). GDM is defined as a state of glucose intolerance and hyperglycaemia with first onset during pregnancy (Buchanan et al. 2012) and is the most common complication of pregnancy, affecting 5–10% of women, a figure that is expected to rise in parallel with the obesity epidemic (Reece et al. 2009; Moore, 2010). Overweight mothers diagnosed with GDM are more likely to have infants who are overweight at birth (Gillman et al. 2003; Franks et al. 2006; Shields, 2006; Moore, 2010) and are at higher risk for obesity and type 2 diabetes (T2D) both as adults (Dabelea et al. 2000; Clausen et al. 2008) and as children (Young et al. 2002; Franks et al. 2006; Clausen et al. 2009). Therefore, the trends in maternal obesity and GDM could be driving the development of metabolic disorders in the offspring.
For this study, our objective was to determine how exposure of the fetus to maternal GDM influences the metabolic health of rodent offspring. Unfortunately, studies using animal models of GDM with controlled prenatal and postnatal conditions are lacking. Several previous studies administered streptozotocin (STZ) to induce beta-cell failure in pregnant rodents; however, this approach results in severe hyperglycaemia, insulin deficiency and offspring with low birth weights (Jawerbaum & White, 2010), in contrast to the larger infants of the majority of obese and GDM mothers (Gillman et al. 2003; Franks et al. 2006; Shields, 2006; Moore, 2010). As maternal obesity appears to be the main factor driving the development of GDM in the majority of the population, a rodent model of GDM that is characterized by maternal obesity is necessary to more accurately investigate the development of metabolic disorders in the offspring. In this study, we utilized a rat model of high-fat and sucrose diet (HFS)-induced GDM to mimic several clinical features of GDM during human pregnancy, including mild hyperglycaemia, hyperinsulinaemia, hyperlipidaemia and glucose intolerance. Using this model, we demonstrate that GDM increased the susceptibility of young adult rat offspring to obesity, insulin resistance, dyslipidaemia and hepatic steatosis. Additionally, we report the results of the first metabolomic screen of hepatic metabolites in the offspring from GDM dams. Increased diacylglycerol and ceramide levels and reduced phosphatidylethanolamine (PE) were observed in the offspring of GDM dams, indicating that GDM alters hepatic lipid metabolism in the offspring.
Methods
Diet-induced GDM model
All procedures were approved by the Animal Welfare Committee of the University of Manitoba, which adheres to the principles for biomedical research involving animals developed by the Canadian Council on Animal Care and the Council for International Organizations of Medical Sciences. Rats were given ad libitum access to food and water and housed two per cage. The experimental design is presented in Fig.1. Female Sprague-Dawley rats were obtained at 3 weeks of age from the University of Manitoba colony and six were randomly assigned to a low-fat (LF) diet (10% fat, Research Diets D12450B; New Brunswick, NJ, USA) and six were randomly assigned to an HFS diet (45% fat, Research Diets D12451) for a period of 6 weeks and glucose and insulin tolerance tests were performed to confirm a glucose intolerant and pre-diabetic state. After 6 weeks of dietary intervention animals were mated with LF diet fed males and diets were continued in the same dams throughout pregnancy and the suckling period. Food intake and body weight of the dams were measured weekly and a glucose tolerance test was performed at embryonic day 15.5. Dams were allowed to deliver naturally and at birth litters were reduced to eight pups (i.e. four males and four females) to avoid competition for food. The offspring were weaned at 3 weeks of age and two male and two female offspring from each litter were randomly assigned to either the LF or the HFS postnatal diet. Therefore, four different experimental groups were created: offspring from LF-fed (‘Lean’) dams that were fed a post-weaning LF diet (n = 6 litters), offspring from the Lean dams receiving a post-weaning HFS diet (n = 6 litters), offspring from HFS-fed dams (‘GDM’) fed a post-weaning LF diet (n = 6 litters) and offspring from GDM dams fed a post-weaning HFS diet (n = 6 litters). Litters remained on specified diets for a total of 12 weeks after weaning, based on previous studies in rodents that demonstrated the induction of metabolic changes using the same diets (Dolinsky et al. 2011; Rueda-Clausen et al. 2011a). For all analyses, data from cagemates were averaged and the litter was used as the unit of analysis (i.e. n = 6 litters per group were generated). Food intake and body weight were measured weekly for all rodents. Rats were anaesthetized by intraperitoneal injection of a sodium pentobarbital overdose and blood was collected by cardiac puncture. Tissues were dissected, rinsed in PBS, weighed, and either fixed in 10% formalin or freeze clamped in liquid nitrogen and stored at −80°C for future analyses. For the analysis of insulin-responsive signalling pathways, offspring were administered human recombinant insulin that was injected intraperitoneally (1 mU kg–1) after a 4 h fast, and the tissues were collected 15 min following the injection.
Figure 1.
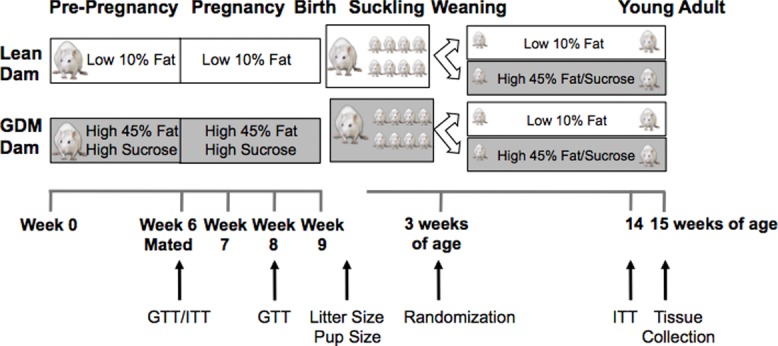
Experimental design
Three-week-old female Sprague-Dawley rats were randomly assigned to a low-fat or high-fat and high-sucrose diet for 6 weeks. Glucose and insulin tolerance were tested to confirm glucose intolerance and a pre-diabetic state. Female rats were mated with lean males and diets were continued throughout pregnancy and lactation. At the time of weaning offspring from each dam were randomly assigned to either a low-fat or high-fat and sucrose postnatal diet for a total of 12 weeks until they reached 15 weeks of age. Rats were subjected to experimental procedures at the indicated time points. GTT, glucose tolerance test; ITT, insulin tolerance test.
Analysis of mRNA and protein expression
RNA was isolated from tissues using a QIAshredder column and further purified using the RNeasy kit (Qiagen, Valencia, CA, USA). cDNA was synthesized using the Protoscript kit (New England Biolabs, Ipswich, MA, USA). The QuantiTect SYBR Green PCR kit (Qiagen) was used to monitor amplification of cDNA on an ABI-7500 real-time PCR detection machine (Applied Biosystems, Foster City, CA, USA). Expression of genes was assessed in duplicate using the 2-ΔΔCT method and data were normalized to eukaryotic initiation factor 2a (eIF2a) expression as the reference gene that was constant across all groups of offspring. All primer sequences were validated and are reported in Table1. For the analysis of protein expression, frozen tissue homogenates were prepared in ice-cold sucrose homogenation buffer (20 mM Tris-HCl (pH 7.4), 50 mM NaCl, 50 mM NaF, 5 mM sodium pyrophosphate, 0.25 M sucrose) containing protease and phosphatase inhibitor cocktails (Sigma, St Louis, MO, USA), 1 m dithiothreitol and 20 mM sodium orthovanadate, as previously described (Dolinsky et al. 2011; Rueda-Clausen et al. 2011a). In brief, tissues were homogenized on ice in a 20 s burst and the homogenate was centrifuged at 1000 g for 20 min at 4°C to remove nuclei and cellular debris. Protein concentrations were determined using a Bradford protein assay. In total, 15–20 μg of protein was subjected to 10% SDS-PAGE and transferred to nitrocellulose, and protein loading was controlled for by visualizing transferred proteins using Ponceau S red and normalizing immunoblots to tubulin expression. Immunoblots were visualized using the Perkin-Elmer enhanced chemiluminescence Western blotting detection system. All antibodies were purchased from Cell Signaling Technologies (Beverly, MA, USA).
Table 1.
Primer sequences used for quantitative real-time PCR
| Gene | Forward primer 5′–3′ | Reverse primer 5′–3′ |
|---|---|---|
| Acca | TCTATTGTGGCTCAAACTG | GCCTTTCTCATACAGATCCTG |
| Accb | AGAACTCGATGACCCTTCA | TCAGATTTTCCAAGACGC |
| G6pd | GGGCATCAATCTCCTCTG | GTCCAGGACCCACCAATACG |
| Pck1 | ATGCAGGAGGAGGGTGT | GTCTTGCTTTCGATCCT |
| Pgc1a | GACACGAGGAAAGGAAGACTAAA | GTCTTGGAGCTCCTGTGATATG |
| Ppara | GATGACCTGGAAAGTCCCTTATC | AGCTCCCTAAGTACTGGTAGTC |
| Srebp-1c | CCTCTCTGGAAGCCTT | ACTGGCTCCTCTTTGAT |
| Srebp-2 | AGGTCTAGGGATGGGTGAAA | GTGGGAAGGAACAGGACAATTA |
| Etnk1 | CTGCACCTGCTTCTCATACA | GAATCCCTTCCATGGTCGATAC |
| Etnk2 | CCGGGAGAATGAGATCAGAAAC | CCACACCCTGCATGTACTC |
| Pcyt2 | CCATCAAGTGGGTGGATGAA | CATTGCCATGAACGCAGAAG |
| eIF2a | CCTGAAGTGTGATCCTGTGTTT | CCAAATCCAGCCAGCACTAATA |
Determination of circulating factors
Blood samples were collected and placed on ice for a minimum of 10 min and serum was separated by centrifugation (10 min at 4000 r.p.m., 4°C) and stored at −80°C. Circulating concentrations of insulin, leptin and adiponectin were determined in duplicate using colourimetric ELISA assays (Millipore, St. Charles, MO, USA) according to the manufacturer’s instructions. Circulating concentrations of triacylglycerol (TG) and free fatty acids were determined in duplicate using a colorimetric assay (Wako Chemicals, Richmond, VA, USA).
Histology and morphometry
Histopathological preparations and haematoxylin/eosin (HE) staining were performed by the University of Manitoba Core Platform for Histology according to standard procedures. For the analysis of adipocyte size and number, the internal diameters of 10 consecutive adipocytes from 10 randomly selected fields on each HE-stained slide were measured under light microscopy using a digital micrometer under 40× magnification and averaged. Hepatic steatosis in HE-stained liver sections were scored as 0 if no steatosis (i.e. less than 5% of cells were steatotic) was detected, scored as 1 if 5–30% of cells were steatotic, scored as 2 if 30–50% steatosis was observed, and scored as 3 if greater than 50% of hepatocytes were steatotic. Steatosis scores are presented as the percentage of samples in each group that were scored in each category of steatosis. The location of steatosis relative to the distance from oxygenated blood was noted as zone 1 (encircles hepatic arteries), zone 3 (surrounds central veins) or zone 2 (the area between zone 1 and zone 3). Slides were evaluated and scored by a blinded and experienced pathologist.
Evaluation of glucose and insulin tolerance
Glucose tolerance tests (GTTs) and insulin tolerance tests (ITTs) were performed as described previously (Dolinsky et al. 2011; Rueda-Clausen et al. 2011a). A GTT was performed in female rats 6 weeks following the initiation of the LF or HFS diets to confirm glucose intolerance in the HFS diet group compared to the LF group prior to mating. Mid-gestational GTTs (embryonic day 15.5) were also performed in pregnant rats, following a 4 h fast to prevent prolonged hypoglycaemia in pregnancy that could influence the phenotype of the offspring. In 14-week-old offspring, ITTs were also performed. The homeostasis model assessment of insulin resistance (HOMA-IR) was calculated as the fasting insulin concentration × fasting glucose concentration.
Measurement of tissue metabolites
Liver tissue from rats fasted for 4 h prior to being killed were utilized for biochemical analyses. The determination of TG and glycogen content was measured by non-radioactive, colorimetric assays (Biovision, Milpitas, CA, USA) according to the manufacturer’s instructions. Phospholipid mass was determined in duplicate by phosphorus assay (Bartlett, 1959). The analysis of total liver tissue lipids was performed using a lipid soluble extraction as described previously (Folch et al. 1957), in duplicate and stored at −80°C until further analysis. Prior to analysis, liver lipid extracts were reconstituted with 100 μl of 80% acetonitrile prepared in deionized water. Metabolomic analysis was performed on a 1290 Infinity Agilent high-performance liquid chromatography (HPLC) system coupled to a 6538 UHD Accurate Quadrupole time-of-flight liquid chromatrography/mass spectrometry Q-TOF LC/MS from Agilent Technologies (Santa Clara, CA, USA) equipped with a dual electrospray ionization source. A 3 × 50 mm, 2.7μ Agilent Poroshell column (Agilent Technologies) was used to separate metabolites while the column temperature was maintained at 60°C. The mobile phases A and B were water and acetonitrile, with 0.1% formic acid. A sample size of 2 μl was injected by maintaining the HPLC flow rate at 0.7 ml min−1 with a gradient programme of: 0, 0.5, 16, 17 and 22 min with 30, 30, 100, 100 and 30% of solvent B, respectively. A post-run time of 2 min was buffered before injecting the next sample. The auto-sampler was maintained at a temperature of 15°C. The mass detection was operated using dual electrospray with reference ions of m/z 121.050873 and 922.009798 for positive mode, and m/z 119.03632 and 980.016375 for negative mode. The main parameters for MS were as follows: gas temperature, 300°C; drying N2 gas flow rate, 11 l min−1; nebulizer pressure, 50 p.s.i.; fragmentor voltage, 175 V; skimmer voltage; 50 V; and OCTRF Vpp voltage, 750 V. Targeted MS/MS mode was used to identify potential biomarkers. As part of the MassHunter Software, the collision energy was applied by setting an appropriate equation having a slope value of 5 and offset value of 2.5. A full range mass scan from 50 to 3000 m/z with an extended dynamic range of 2 GHz standardized at 3200 was applied. Data acquisition rate was maintained at 3 spectra s–1 at a time frame of 333.3 ms per spectrum with a transient/spectrum ratio of 1932.
Data processing and statistical analyses of metabolomics data
The workflow utilized for data processing comprised several algorithms used by Agilent Mass Hunter Qualitative (MHQ, B.05) and by Mass Profiler Professional (MPP, 12.6). The raw data files were first acquired and stored as ‘*.d’ files using an Agilent MassHunter Acquisition software (B.05) ready to be processed in MHQ. The Molecular Feature Extraction (MFE), considered a naïf extraction procedure, was the first algorithm applied to the total ion chromatogram (TIC) files. The MFE parameters were set to allow the extraction of detected features satisfying absolute abundances of > than 4000 counts. The MFE parameters were set to provide information regarding [M+H]+, isotopes and their corresponding Na+ adducts. The resulting extracted ions were treated as single features for which potential formulae were generated. The collected information summarizing retention time (RT), exact masses and ion abundances were converted into compound exchange format files (‘*.cef’) and were exported to MPP for further subsequent comparative and statistical analyses. Using alignment and normalization procedures, individual ‘*.cef’ files were binned and combined to generate new ‘*.cef’ files. These new files were reopened in MHQ for further data mining using a ‘Find by ion’ algorithm. This targeted feature algorithm helped with minimizing the false positive and negative features found by MFE. A second series of individual ‘*.cef’ files were created from original individual ‘*.d’ files and exported into MPP software for statistical and differential analysis. A frequency filtration was used to only accept features that were detected in at least one of the four conditions (treatments). This filtration step was employed to ensure elimination of the potential feature extraction artifacts. Other MPP filtering procedures such as number of detected ions (set to ‘2’) and charge states (set to ‘all charge states permitted’) were also applied. The RT compound alignment parameters were set to 0.15 min with a mass tolerance of 2.0 mDa. The data were normalized using a percentile shift algorithm set to 75 and were adjusted to the baseline values of the median of all samples.
Statistical analysis
Data are presented as mean ± SEM. Comparisons between two groups were evaluated using an unpaired t-test. Differences in measurements performed among four groups were analysed using two-way ANOVA and a Bonferroni post hoc test with both diet and GDM as sources of variation. P < 0.05 was considered statistically significant. For Q-TOF LC/MS analyses, a moderated t-test (P < 0.05) and volcano plots (> 2-fold changes and P < 0.05) were performed using MPP software (version 12.6).
Results
Maternal characteristics and glucose homeostasis
After 6 weeks, the female rats that consumed the HFS diet weighed 1.2-fold more (Fig.2A, P < 0.05) and were significantly more glucose intolerant (Fig.2B, C, P < 0.05) than the LF-fed females, although they had similar insulin tolerance (data not shown). During pregnancy, the GDM dams continued to consume the HFS diet and experienced a 24% increase in body weight whereas the Lean, LF-fed dams increased their body weight by 13% (Fig.2A). Thus, the HFS diet induced a remarkable 1.4-fold greater gestational weight gain in GDM dams compared to Lean dams (Fig.2D, P < 0.05). During pregnancy, differences in energy intake were not observed between the Lean and GDM dams (data not shown). Blood glucose levels were not different between Lean and GDM groups prior to mating (Fig.2E, P = 0.33), although the HFS-fed rats experienced significant mid-gestational hyperglycaemia (Fig.2E, P < 0.001). Prior to mating, the circulating insulin levels were not different between the groups, although hyperinsulinaemia was observed in the GDM group at mid-gestation, as reflected by the 2.3-fold increase in insulin levels compared with the Lean group (Fig.2F, P < 0.001). Furthermore, impaired glucose tolerance was observed during pregnancy in GDM dams compared to Lean dams (Fig.2G, H, P < 0.001). GDM also reduced mid-gestational levels of the insulin-sensitizing adipokine, adiponectin, by ∼40% (Fig.2I, P < 0.01).
Figure 2.
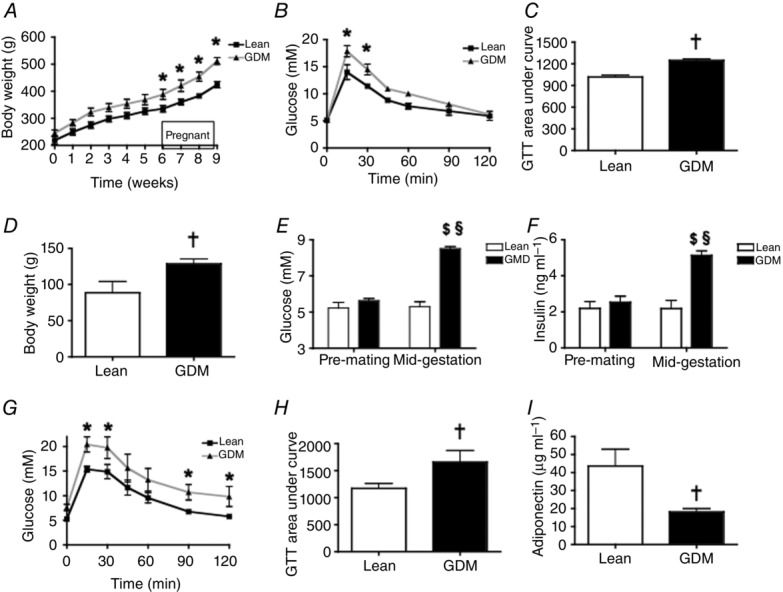
Effect of a maternal high-fat and sucrose diet (HFS) on maternal body composition and metabolic parameters during pregnancy
A, change in body weight over time. B, pre-mating glucose tolerance test (GTT); C, area under the curve of the GTT. D, gestational weight gain; E, pre-mating and mid-gestational (embryonic day (e)15.5) fasting blood glucose levels; F, pre-mating and mid-gestational (e15.5) fasting plasma insulin. G, mid-gestational (e15.5) GTT; H, area under the curve of the GTT; I, mid-gestational (e15.5) fasting plasma adiponectin levels. *P-values represent significant differences (<0.05) between LF diet (Lean dams) and HFS diet (GDM dams) as calculated by repeated measures ANOVA with a Bonferroni post hoc test. †P-values represent significant differences (<0.05) between Lean and GDM dams by Student’s t-test (n = 6). Significant difference $P < 0.05 pre-mating vs. mid-gestation; §P < 0.05 Lean vs. GDM using a two-way ANOVA followed by a Bonferroni post hoc test.
Characteristics of the offspring at birth
Maternal GDM did not influence the gestational length (data not shown), number of pups per litter or sex distribution in the litter (Table2). GDM did cause a 1.2-fold increase in birth weight, compared to pups from Lean mothers (Table2, P < 0.001). The body length of the newborn pups was also significantly increased (Table2, P < 0.001), indicating that HFS-induced GDM caused fetal macrosomia.
Table 2.
Characteristics of newborn rat litters
| Lean | GDM | |
|---|---|---|
| Litter size | 13 ± 1 | 13 ± 1 |
| Male offspring | 5 ± 1 | 7 ± 1 |
| Female offspring | 8 ± 2 | 6 ± 1 |
| Body weight (g) | 6.1 ± 0.2 | 7.2 ± 0.2* |
| Body length (mm) | 50.0 ± 0.7 | 53.9 ± 0.4* |
Data from all pups in a particular litter were averaged and the litter was used as the unit of analysis.
P-values represent significant differences (<0.05) between maternal environments: Lean vs.
GDM by Student’s t-test (n = 6 litters).
Postnatal body weight gain, energy intake and adiposity
By 7 weeks of age, consumption of the HFS diet increased the body weight in the male offspring of Lean and GDM mothers compared to the LF diet (Fig.3A, P < 0.001). Maternal GDM had a major influence on body weight in the male offspring since the GDM-LF group weighed significantly more than the Lean-LF group (Fig.3A, P < 0.001). In addition, there was an additive effect of GDM and the postnatal consumption of an HFS diet, given that male offspring of GDM dams that were fed an HFS diet weighed significantly more than the offspring of Lean dams fed a similar HFS diet (Fig.3A, P < 0.001). Interestingly, the weights of the male Lean-HFS group were not significantly different from the GDM-LF group (Fig.3A). While the HFS diet increased body weight in the female offspring (Fig.3B, P < 0.001), GDM did not influence the body weight of the female offspring over this range of ages (Fig.3B). In the male offspring of the Lean dams, energy intake per gram of body weight was not significantly different between offspring that consumed the HFS diet and the LF-fed controls (Table3). On the other hand, the energy intake per gram of body weight of the male offspring of GDM dams that consumed the LF diet was reduced compared to the male offspring from Lean dams that consumed the LF diet (Table3, P < 0.01). The energy intake of the male offspring of GDM dams that consumed a postnatal HFS diet was also significantly lower than that of male offspring of Lean dams that consumed a similar HFS diet postnatally (Table3, P < 0.05). However, significant differences in food consumption and energy intake were not observed between the female offspring of GDM dams and female offspring of Lean dams irrespective of the postnatal diet (Table3).
Figure 3.
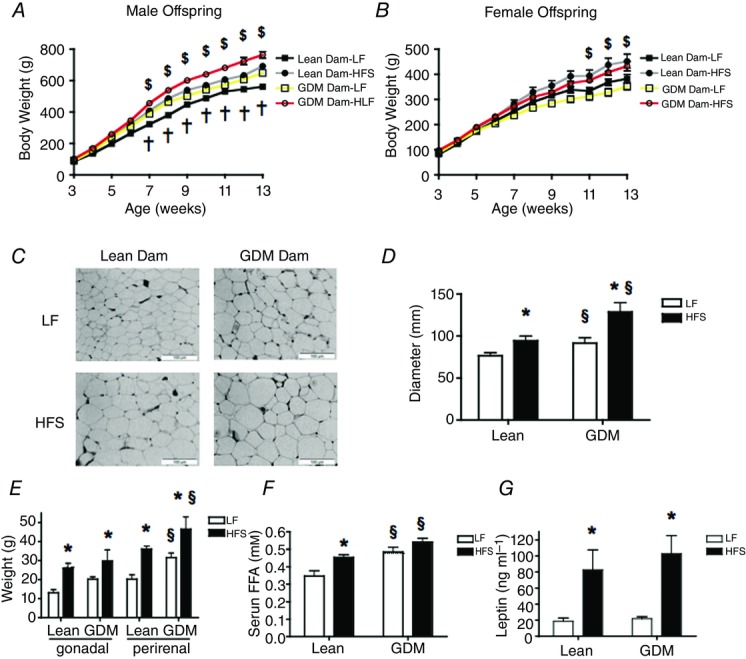
Effect of maternal GDM and postnatal diet on body composition of the offspring
A, change in body weight of male offspring over time. B, change in body weight of female offspring over time. C, representative images of perirenal fat tissue (magnification 40×) in male offspring. D, average adipocyte cell diameter in male offspring. E, individual fat pad weights in male offspring. F, serum free fatty acid (FFA) levels in male offspring. G, leptin concentrations in male offspring. LF, low-fat diet (10% fat 3.85 kcal g−1); HFS, high-fat and sucrose diet (45% fat, 4.73 kcal g−1). Significant difference *P < 0.05 LF vs. HFS; §P < 0.05 Lean vs. GDM using a two-way ANOVA followed by a Bonferroni post hoc test. $P-values represent significant differences (<0.05) between postnatal LF and HFS diet groups as calculated by repeated measures ANOVA with a Bonferroni post hoc test (n = 6 by group). †P-values represent significant differences (<0.05) between the offspring of Lean dams and the offspring of GDM dams as calculated by repeated measures ANOVA with a Bonferroni post hoc test (n = 6 by group).
Table 3.
Average daily energy intake by rat offspring
| Lean | GDM | |||
|---|---|---|---|---|
| LF diet | HFS diet | LF diet | HFS diet | |
| Male offspring | ||||
| Food consumption (g) | 23.38 ± 1.11 | 22.33 ± 1.03 | 18.94 ± 0.93§ | 18.32 ± 1.2§ |
| Energy intake (kcal day–1) | 157.5 ± 7.5 | 184.8 ± 8.5* | 127.6 ± 6.3 | 151.3 ± 10.1* |
| Energy intake (kcal day–1 (kg body weight)–1) | 249.5 ± 9.0 | 255 ± 11.3 | 194.1 ± 9.9§ | 200.3 ± 15.3§ |
| Female offspring | ||||
| Food consumption (g) | 20.48 ± 2.61 | 18.65 ± 2.85 | 15.74 ± 1.15 | 20.37 ± 0.7 |
| Energy intake (kcal day–1) | 138.0 ± 17.6 | 154.4 ± 23.6 | 106.0 ± 7.8 | 168.6 ± 4.7 |
| Energy intake (kcal day–1 (kg body weight)–1) | 333.1 ± 37.2 | 265.0 ± 18.8 | 215.5 ± 10.7 | 191.0 ± 15.2 |
P < 0.05 in Bonferroni post hoc test comparing postnatal LF vs.
HFS diet groups and
P < 0.05 in Bonferroni post hoc test comparing GDM and control offspring receiving the same diet (n = 6).
As expected, the consumption of a HFS diet by the male offspring of Lean dams increased perirenal adipocyte diameter; HFS-fed male offspring of GDM dams had larger adipocytes than the respective LF- and HFS-fed offspring of Lean mothers (Fig.3C, D, P < 0.05). Consumption of the HFS diet by the male offspring of Lean and GDM dams elevated the gonadal and perirenal fat pad mass compared to the respective LF-fed groups (Fig.3E, P < 0.01). The GDM-HFS group had similar gonadal fat pad mass to the Lean-HFS group, although the perirenal fat pad mass was increased in the GDM-HFS offspring compared to the Lean offspring (Fig.3E, P < 0.05). Interestingly, the effects of GDM on fat pad mass observed in the male offspring were absent in the female offspring (Table4). The male offspring of GDM dams also had elevated fasting serum levels of free fatty acids compared to the offspring of lean dams (Fig.3F, P < 0.05), although fasting serum TG levels were not different across the groups (data not shown). In addition, plasma levels of leptin were elevated in HFS-fed offspring of Lean and GDM dams compared to the respective LF-fed groups (Fig.3G, P < 0.001), although there were no differences in the plasma leptin concentrations between the offspring of Lean and GDM dams.
Table 4.
Metabolic characteristics of female rat offspring
| Lean | GDM | |||
|---|---|---|---|---|
| LF diet | HFS diet | LF diet | HFS diet | |
| Body weight gain (g) | 307.8 ± 17.2 | 270.8 ± 15.7 | 373.2 ± 26.9 | 352.3 ± 23.7 |
| Tibia length (mm) | 41.8 ± 1.0 | 42.2 ± 0.4 | 40.6 ± 0.7 | 42.3 ± 0.9 |
| Gonadal fat mass (g) | 12.73 ± 1.5 | 18.70 ± 1.6* | 11.14 ± 0.9 | 20.5 ± 4.1* |
| Perirenal fat mass (g) | 13.48 ± 2.3 | 25.02 ± 3.3* | 11.76 ± 0.8 | 19.03 ± 3.9* |
| Fasting glucose (mm) | 5.3 ± 0.1 | 5.7 ± 0.3 | 5.1 ± 0.3 | 5.3 ± 0.3 |
| Fasting insulin (ng ml–1) | 2.9 ± 0.3 | 3.2 ± 1.1 | 3.8 ± 0.8 | 5.5 ± 1.2 |
| HOMA-IR | 1.4 ± 0.2 | 1.7 ± 0.4 | 1.3 ± 0.3 | 1.4 ± 0.6 |
P < 0.05 in Bonferroni post hoc test comparing postnatal LF vs.
HFS diet groups (n = 6).
Effects of maternal GDM on glucose homeostasis in offspring
Eleven weeks of HFS feeding did not result in fasting hyperglycaemia in any of the experimental groups (Fig.4A). Although the offspring of Lean dams receiving an HFS diet tended to have a higher fasting plasma insulin concentration than offspring of Lean dams receiving the LF diet, this was not significant (Fig.4B). In contrast, offspring of GDM dams had much higher circulating insulin levels compared to the corresponding Lean offspring groups (Fig.4B, P < 0.01). The elevated HOMA-IR index of the offspring of GDM dams (Fig.4C, P < 0.01), suggested that GDM predisposes young rat offspring to develop whole body insulin resistance irrespective of the postnatal diet consumed. Consistent with this, the offspring of GDM dams exhibited impaired insulin tolerance (Fig.4D, E) compared to the offspring of Lean dams fed corresponding LF and HFS diets (Fig.4F, P < 0.05). To further investigate the molecular mechanisms underlying insulin resistance in the offspring of GDM dams, we measured expression of the insulin receptor and downstream kinases in the liver following acute administration of insulin to offspring immediately prior to the animals being killed. Expression of insulin receptor-β was markedly reduced in the offspring of GDM dams (Fig.4G, P < 0.05). While the postnatal consumption of a HFS diet did not alter the phosphorylation of phosphatidylinositol-3 kinase and Akt at activating sites in the offspring of Lean dams, phosphorylation at these activating sites was markedly reduced in the GDM offspring that consumed the HFS diet (Fig.4H, I, P < 0.05).
Figure 4.

Effect of maternal gestational diabetes mellitus (GDM) and postnatal diet on glucose homeostasis in male offspring
A, fasted blood glucose and B, fasted plasma insulin concentrations; C, HOMA index; D and E, insulin tolerance test (ITT) and F, area under the curve (AUC) of the ITT. G, levels of insulin receptor (IR)-β normalized against tubulin. H, levels of phosphorylated tyrosine-458 p85 subunit of phosphatidylinositol 3-kinase (P-PI3K) normalized against tubulin. I, levels of phosphorylated serine-473 Akt (P-Akt) were normalized against total Akt. Data in G–I were collected from rat offspring fasted for 4 h and administered insulin intraperitoneally (1 mU kg–1) prior to their death. Significant difference *P < 0.05 LF vs. HFS; §P < 0.05 Lean vs. GDM using a two-way ANOVA followed by a Bonferroni post hoc test. $P-values represent significant differences (<0.05) between postnatal LF and HFS diet groups as calculated by repeated-measures ANOVA with a Bonferroni post hoc test (n = 6 by group).
Effects of maternal GDM on liver morphology and metabolism in the offspring
Next, we examined the liver for signs of pathology using HE-stained sections. We detected histological evidence of steatosis in all of the sections from rat offspring that had consumed the HFS diet (Fig.5A, B). Steatosis appeared to be predominantly observed in zone 1 of the liver (i.e. hepatocytes around arteries) from HFS-fed offspring of Lean dams, whereas zone 3 steatosis (i.e. hepatocytes around central veins) was predominantly observed in the livers of HFS-fed offspring from GDM dams (Fig.5A). A larger proportion of the offspring from GDM dams had histologically detectable microvesicular and macrovesicular steatosis in the liver (Table5). However, liver weight and protein content were not significantly different between the groups (data not shown).
Figure 5.

Effect of maternal gestational diabetes mellitus (GDM) and postnatal diet on hepatic steatosis in the male offspring
A and B, 2× and 40× representative HE-stained liver sections. C and D, liver triacylglycerol (TG) and glycogen levels. E, heat map of metabolomic analysis (LC-MS) of hepatic metabolites extracted from the livers of young adult (15-week-old) offspring. In total, 286 metabolites with a significant change in abundance of 2-fold or greater (P < 0.05) were identified. F, heat map of diacylglycerol species from the metabolomic analysis of liver tissue. G and H, liver phosphatidylethanolamine (PE) and phosphatidylcholine (PC) phosphorus mass. P-values represent the significance *P < 0.05 comparing postnatal LF vs. HFS diet groups and §P < 0.05 comparing GDM and Lean offspring receiving the same diet as calculated by ANOVA with a Bonferroni post hoc test (n = 6 by group).
Table 5.
Hepatic steatosis scoring
| Lean | GDM | |||
|---|---|---|---|---|
| LF | HFS | LF | HFS | |
| Microvesicular Steatosis Score (%) | ||||
| 0 | 62.5 | 0 | 0 | 0 |
| 1 | 25 | 83 | 100 | 40 |
| 2 | 12.5 | 17 | 0 | 20 |
| 3 | 0 | 0 | 0 | 40 |
| Average Steatosis Score (1–3) | 0.5 ± 0.2 | 1.1 ± 0.1* | 1.0 ± 0.0§ | 2.0 ± 0.4*§ |
| Macrovesicular Steatosis Score | ||||
| 0 | 87.5 | 14.3 | 0 | 20 |
| 1 | 12.5 | 85.7 | 100 | 40 |
| 2 | 0 | 0 | 0 | 20 |
| 3 | 0 | 0 | 0 | 20 |
| Average Steatosis Score (1–3) | 0.1 ± 0.1 | 0.9 ± 0.1* | 1.0 ± 0.0 | 1.4 ± 0.5 |
P < 0.05 in chi-square test comparing postnatal LF vs.
HFS diet groups and
P < 0.05 in chi-square test comparing offspring from GDM and Lean dams receiving the same diet (n = 6).
Consistent with the development of hepatic steatosis, we observed that the offspring from GDM dams had higher TG levels compared to the offspring from Lean dams (Fig.5C, P < 0.05). The offspring of Lean and GDM dams had similar hepatic glycogen levels that were not influenced by the postnatal diets that were consumed (Fig.5D). To further investigate how in utero exposure to GDM influenced hepatic metabolism, we performed unbiased metabolic profiling of over 9000 metabolite entities with specific masses using LC-QTOF-MS to analyse hepatic lipid extracts. A total of 286 metabolite entities had a significant (P < 0.05) 2-fold change in abundance in the offspring of GDM dams compared to the offspring of Lean dams (Fig.5E). Among these entities, phospholipids were the most commonly observed metabolite, including 43 PE/lyso-PE species, 24 phosphatidylserine/lyso-phosphatidylserine species and 6 phosphatidylinositol species. The screen identified 11 species of diacylglycerol and 9 sphingolipids that were significantly elevated in the GDM offspring liver. In addition, 16 different fatty acids and 11 different steroids were increased by GDM in the liver of the offspring. Changes in the levels of all diacylglycerol species detected in the liver are represented by the heat map in Fig.5F, showing that the abundance of diacylglycerol tended to increase in the livers of GDM-LF offspring, although this appeared to be markedly increased in the GDM-HFS offspring. In general, alterations in levels of metabolites observed in the GDM offspring fed postnatal LF diets were further enhanced by the postnatal consumption of a HFS diet and we report representative data in Table6. Phosphorus assays revealed that the total PE content of the liver was reduced in the offspring of GDM dams (Fig.5G), while phosphatidylcholine levels were not significantly changed (Fig.5H).
Table 6.
Representative metabolic entities altered (< 2-fold) by GDM in the livers of 15-week-old male offspring (n = 12) identified by LC-QTOF-MS
| Fold change | |||||
|---|---|---|---|---|---|
| Entity | Formula | [M + H]+ | Lean-LF vs. Lean-HFS | Lean-LF vs. GDM-LF | Lean-LF vs. GDM-HFS |
| Ceramide | |||||
| Ceramide (d18:2/16:0) | C34H65NO3 | 536.5052 | +1.4 | +1.7 | +160.9 |
| Ceramide (d18:0/16:0) | C34H69NO3 | 540.5392 | +2.8 | +55.6 | +73.5 |
| Diacylglycerol (DG) | |||||
| DG (18:1/18:2/0:0) | C39H70O5 | 619.5312 | +2.8 | +35.7 | +35.7 |
| DG (18:1/18:3/0:0) | C39H68O5 | 617.5157 | +2.7 | +90 | +90 |
| DG (16:0/22:1/0:0) | C41H78O5 | 651.5922 | +4.1 | +57.7 | +57 |
| DG (18:0/18:1/0:0) | C39H74O5 | 623.0018 | +2.9 | +9.2 | +103.9 |
| DG (16:0/16:1/0:0) | C35H66O5 | 567.4983 | +3.2 | +1.5 | +613 |
| Phosphatidyl-ethanolamine (PE) | |||||
| PE (18:1/18:3) | C41H74NO8P | 739.5147 | +2.0 | −2.6 | −94.8 |
| PE (18:3/20:3) | C43H74NO8P | 764.5251 | −4.9 | −6.5 | −25.4 |
| PE (18:3/20:2) | C43H76NO8P | 766.5400 | +1.7 | −9.6 | −12.5 |
| PE (20:3/20:4) | C45H76NO8P | 790.5385 | +2.6 | −1.1 | −8.5 |
| PE (18:2/18:3) | C41H72NO8P | 738.5095 | −3.6 | −6.2 | −27.3 |
| PE (20:4/20:4) | C45H74NO8P | 788.5243 | −4.3 | −6.3 | −26.8 |
| Phosphatidylserine (PS) | |||||
| PS (18:0/20:2) | C44H82NO10P | 816.5749 | −1.6 | −1.6 | −5.6 |
| PS (18:0/20:3) | C44H80NO10P | 813.5514 | +3.0 | −4.0 | −4.6 |
| Phosphatidylinositol (PI) | |||||
| PI (18:1/20:3) | C47H83O13P | 887.5643 | −1.8 | −3.3 | −24.7 |
| PI (22:2/16:1) | C47H85O13P | 889.5781 | +1.1 | −45.1 | −58.3 |
Next, we investigated whether the altered metabolic phenotype of the offspring of GDM dams was a consequence of altered programming of gene expression in the liver and measured the mRNA expression of enzymes and transcription factors involved in hepatic lipid metabolism. While acetyl-coenzyme A carboxylase (ACC)-1 (Acca) was similar in all groups of offspring (Fig.6A), ACC-2 (Accb) expression was markedly increased in the livers of the offspring of GDM dams fed a postnatal HFS diet (Fig.6B, P < 0.05). Sterol response element binding protein (SREBP)-1c (Srebp1c) mRNA expression was significantly reduced in livers from the offspring of GDM dams fed a postnatal LF diet, whereas SREBP-1c expression was significantly increased in the livers from offspring of GDM dams fed a HFS diet (Fig.6C, P < 0.05). On the other hand, SREBP-2 gene expression was not significantly altered (Fig.6D). Notably, we observed that peroxisomal proliferator activated receptor (PPAR)-α (Ppara) expression was significantly reduced in the livers from the offspring of GDM dams (Fig.6E, P < 0.05), and a trend for reduced PPAR-γ coactivator-1α (Pgc1a) was also observed (Fig.6F). Despite reduced PE levels in the liver tissues of GDM offspring, the mRNA expression of ethanolamine kinase-1 and -2 (Etnk1 and Etnk2) were not different across all the groups of rat offspring (Fig.6G, H); however, expression of the rate limiting enzyme of PE synthesis, CTP:phosphoethanolamine cytidylyltransferase (Pcyt2), was reduced in livers from the offspring of GDM dams (Fig.6I), suggesting that silencing of Pcyt2 was probably responsible for reduced PE levels in the liver.
Figure 6.

Effect of maternal gestational diabetes mellitus (GDM) and postnatal diet on gene expression in the livers of the male offspring
A and B, acetyl-coenzyme A carboxylase-1 (Acca) and Acc-2 (Accb) mRNA expression. C and D, sterol response element binding protein (Srebp)-1c and Srebp-2 mRNA expression. E and F, peroxisomal proliferator activated protein receptor-α (Ppara) and PPAR-Coactivator-1α (Pgc1a) mRNA expression. G and H, ethanolamine kinase (Etnk)-1 and Etnk-2 mRNA expression. I, CTP:phosphoethanolamine cytidylyltransferase (Pcyt2) mRNA expression. Gene expression was evaluated by quantitative real-time PCR and normalized to the eukaryotic initiation factor 2a housekeeping gene. P-values represent the significance *P < 0.05 comparing postnatal LF vs. HFS diet groups and §P < 0.05 comparing GDM and Lean offspring receiving the same diet as calculated by ANOVA with a Bonferroni post hoc test (n = 6 by group).
Discussion
Environmental factors such as diet and lifestyle are important constituents of the developing obesity epidemic. In parallel with this trend, increasing numbers of women are consuming diets high in saturated fats and simple sugars that contribute to excess weight gain during their pregnancy (Kral, 2004). As a consequence, women with pre-existing obesity and glucose intolerance are at elevated risk for the development of GDM (Catalano et al. 1991, 1999). The majority of previous rodent studies of diabetes during pregnancy used STZ administered during pregnancy to induce insulin deficiency and a diabetic pregnancy (Jawerbaum & White, 2010); however, GDM is primarily characterized by the inability of the β-cell to adapt to the enhanced insulin resistance seen at mid-gestation rather than insulin deficiency. Therefore, blood glucose levels observed in rodent studies that utilize STZ typically exceeded 20 mM and diabetes of this severity is uncommon in pregnant women. In addition, STZ treatment results in low birth weight offspring (Jawerbaum & White, 2010), which is inconsistent with the weights of infants born to women with pre-existing obesity (Gillman et al. 2003; Franks et al. 2006; Shields, 2006; Moore, 2010). Low dose STZ models have also been used as a model for GDM, although a high variability in plasma glucose levels occurs with this approach (Caluwaerts et al. 2003). Several studies have examined how the consumption of a high calorie diet in the absence of hyperglycaemia during gestation affects metabolic health during pregnancy in rodents (reviewed by Pereira et al. 2015). In the present study, we initiated a HFS diet (45% kcal fat) in female rats 6 weeks prior to mating to induce weight gain and glucose intolerance before pregnancy. The HFS diet was continued throughout pregnancy and we observed excessive gestational weight gain, and mid-gestational hyperglycaemia that returned to normoglycaemic levels following birth of the litter. Using this approach, GDM was probably a result of the additive effects of pre-existing obesity and glucose intolerance, in combination with pregnancy, that is consistent with the etiology of GDM in many pregnant women (Catalano et al. 1999). In addition, we observed elevated gestational weight gain, mid-gestational hyperinsulinaemia and impaired glucose tolerance, as well as larger newborn pups. These findings suggest that the rodent model we report is more clinically relevant to human GDM because gestational hyperglycaemia occurs in association with maternal obesity. However, we recognize that differences in placentation, litter size and the level of maturity at birth are a limitation of using rodents as models of human disease during pregnancy (Rueda-Clausen et al. 2011b). Furthermore, it is unknown whether obesity influences the number of implantation sites. In this study, we did not observe differences in the number or sex of the offspring in the litters from GDM dams compared to offspring from Lean dams.
In our study, HFS diet-induced GDM consistently increased weight and length of newborn rat pups, probably as a response to maternal hyperinsulinaemia and hyperglycaemia as well as increased availability of calories from saturated fats and simple sugars through the maternal consumption of the HFS diet. This suggests that the association of maternal obesity with hyperglycaemia is an important contributor to the growth characteristics of newborn pups. We also observed that HFS-induced GDM did not alter the sex distribution and size of the litter, nor did we observe overt birth defects among the offspring. Our experimental design also investigated the influence of diet-induced GDM on the postnatal response to LF and HFS diets in young rat offspring. The male offspring of GDM dams fed post-weaning LF diets consistently weighed more than the LF-fed offspring of Lean dams, a body weight that was similar to the Lean offspring that consumed the HFS diet. Moreover, the male offspring of GDM dams gained a greater amount of weight more rapidly when they consumed a HFS diet than the offspring of Lean dams that also consumed the HFS diet. Interestingly, the body weight of the male offspring of GDM dams increased despite lower energy intake. In contrast, other reports have observed hyperphagia in offspring of mice that consumed HF diets during pregnancy (Samuelsson et al. 2008; Kjaergaard et al. 2014). While it is beyond the scope of the present study to investigate how GDM influences the regulation of feeding behaviour in the CNS, we may speculate that dysregulation of the fetal development of the satiety response or dysregulation of adipokine levels could be responsible. Similar to our findings, circulating leptin levels were unchanged or lower in offspring from dams that were fed an HF diet throughout pregnancy, despite increased adiposity (Chen et al. 2008; Morris & Chen, 2009; Benkalfat et al. 2011). These findings suggest that leptin levels could be programmed during fetal development and the inability to increase leptin in the circulation to a level that corresponds to the size of the adipocytes could be a factor that contributes to obesity in the male offspring of the GDM dams. Nonetheless, our data suggest that the increased body weight in the offspring of GDM dams was primarily a consequence of fetal programming in metabolic tissues. While postnatal caloric intake remains an important factor in the development of obesity, our results also highlight that the consumption of a LF diet did not prevent excessive postnatal weight gain following exposure to GDM in the gestational environment. In addition, our data suggest that GDM that occurs in association with maternal obesity is an important predictive factor for obesity in the offspring that exerts an additive influence on the postnatal response to an HFS diet. However, the larger increase in weight of offspring from GDM dams was significant only in the male offspring. This is in agreement with another recent study that showed that male offspring were more susceptible to elevated adiposity than female littermates early in life (Alfaradhi et al. 2014), but does suggest that sex hormones could modify the programming effects of GDM on adipose tissues. Future long-term studies will be performed to determine whether further ageing reveals a difference in the body weight of female offspring of GDM dams compared to the female offspring of Lean dams.
There is a clear link between the accumulation of lipids in tissues and the development of insulin resistance (McGarry, 2002; Sinha et al. 2002). The fetal liver and adipogenesis are key targets of altered in utero conditions (Symonds et al. 2009). Maternal obesity has been reported to increase hepatic steatosis and the TG content of the livers of adult chow-fed offspring (Buckley et al. 2005; Bouanane et al. 2009; Ronis et al. 2012). Previous studies also showed that maternal obesity did not cause insulin resistance in the chow-fed adult offspring, although these offspring were hyperinsulinaemic (Buckley et al. 2005; Caluwaerts et al. 2007; Samuelsson et al. 2008; Howie et al. 2009). Recent work using a non-obese model of maternal insulin resistance during pregnancy (insulin-receptor substrate-1 haploinsufficient mice) demonstrated that maternal insulin resistance alone caused hepatic steatosis, but did not confer obesity in the offspring (Isganaitis et al. 2014). In our study, even at the young adult stage (15 weeks old) the HFS-fed male offspring of Lean dams developed some hepatic steatosis, despite no alterations in liver TG levels or lipid biosynthetic genes, suggesting that reduced dietary fat catabolism was likely the driving factor. On the other hand, hepatic steatosis was observed in the LF-fed male offspring of GDM dams, suggesting that fetal programming of hepatic lipid metabolism contributed to this phenotype. Indeed, the data presented herein identified increased expression of genes involved in hepatic lipid synthesis (Accb, Srebp-1c) and reduced expression of a major regulator of lipid oxidation (Ppara) in the male offspring of GDM dams compared to the male offspring of Lean dams that could contribute to the overall increase in hepatic lipid levels. Moreover, consumption of an HFS diet by the male offspring of GDM dams contributed to a much more severe hepatic steatosis. These results suggest that the development of hepatic steatosis in the young adult offspring is multifactorial and involves interactions between diet, gestational hyperglycaemia and maternal insulin resistance.
Our study is the first to utilize QTOF-LC/MS technology to perform an unbiased metabolomic screen that compares changes in the levels of liver metabolites in the male offspring of Lean and GDM dams. Interestingly, we observed significant increases in the levels of several species of ceramide and diacylglycerol. Accumulation of these lipotoxic lipids occurs when fatty acid uptake exceeds the capacity of the tissue to utilize them for TG synthesis or oxidation, contributing to hepatic steatosis and insulin resistance (Weiss et al. 2003; Nagle et al. 2009). A particularly novel finding was the broad reduction in PE species and total PE levels in the liver. PE is an abundant inner membrane leaflet phospholipid that is synthesized via the cytidine diphosphate (CDP)–ethanolamine Kennedy pathway (Pavlovic & Bakovic, 2013). We also show that the expression of the rate-limiting enzyme for de novo PE synthesis (Pcyt2) expression is reduced in the livers of offspring from GDM dams compared to the offspring from Lean dams. Recent work has demonstrated that haplodeficient Pcyt2 mice have a significant decrease in PE synthesis and a marked increase in diacylglycerol and TG content, fatty liver and insulin resistance (Fullerton et al. 2009). Furthermore, increasing Pcyt2 levels in hepatocytes from Pcyt2+/− mice restored wild-type levels of PE, diacylglycerol and TG (Fullerton & Bakovic, 2010). These results suggest that Pcyt2 deficiency and reduced functioning of the CDP–ethanolamine Kennedy pathway could be a major factor contributing to the development of hepatic steatosis in the offspring of GDM dams. Future studies are required to establish how lipid metabolism is altered in the offspring and determine the extent that alterations in individual hepatic lipids contribute to hepatic and systemic insulin resistance.
The fat content of the liver is a significant determinant of insulin sensitivity independent of whole body and visceral fat mass in adolescents (Wicklow et al. 2012). In agreement with this finding, the ITT data demonstrated that the young adult male offspring of GDM dams had impaired insulin sensitivity, which was increased further by a postnatal HFS diet compared to the respective control offspring of the Lean dams. Therefore, our data show that the postnatal consumption of the HFS diet by the offspring of GDM dams contributes to obesity and insulin resistance in the male offspring. Future studies assessing tissue-specific insulin sensitivity and beta-cell function will allow us to identify whether alterations in insulin secretion, insulin signalling or altered insulin clearance contributes to hyperinsulinaemia in the offspring of GDM dams.
Although the pathophysiological mechanisms of GDM are probably multifactorial, our findings show that obese dams that develop GDM share many of the clinical characteristics of GDM (Reece et al. 2009; Buchanan et al. 2012), including poor metabolic health outcomes for the offspring (Gillman et al. 2003; Franks et al. 2006; Shields, 2006; Moore, 2010). Collectively, our findings highlight the effect of pre-pregnancy weight on the development of GDM and provide new mechanistic insights into how GDM influences the development of obesity, fatty liver and insulin resistance later in the life of the offspring. In the United States, ∼30% of reproductive age women are obese and > 40% of women gained excessive weight during their pregnancy and thus the prevalence of GDM has increased accordingly (Hinkle et al. 2012; Gaillard et al. 2013). Our findings highlight how this trend puts increasing numbers of children at risk of obesity and may contribute to the rising paediatric incidence of T2D (Young et al. 2002; Franks et al. 2006). While it is increasingly being recognized that lactation may also influence the phenotypes of the offspring, our study did not directly investigate whether the consumption of LF or HFS diets during the suckling period altered milk composition or the programming of metabolic disease in the offspring. Currently little is known about how GDM affects lactation and future research is necessary to investigate the influence of lactation on the offspring, using this model.
In summary, our findings emphasize the importance of treating GDM as a preventative measure against poor metabolic health outcomes in children. Furthermore, early treatment of altered lipid metabolism in the children of mothers diagnosed with GDM could prevent the development of obesity, hepatic steatosis and insulin resistance.
Glossary
- ACC
acetyl-CoA carboxylase
- CDP
cytidine diphosphate
- GDM
gestational diabetes mellitus
- GTT
glucose tolerance test
- HE
haematoxylin/eosin
- HFS
high-fat and sucrose diet
- ITT
insulin tolerance test
- LF
low-fat diet
- MFE
Molecular Feature Extraction
- PE
phosphatidylethanolamine
- PPAR
peroxisomal proliferator activated receptor
- SREBP
sterol response element binding protein
- STZ
streptozotocin
- T2D
type 2 diabetes
- TG
triacylglycerol
Additional information
Competing interests
None.
Author contributions
T.J.P., M.A.F., K.E.C., B.L.M., L.K.C. and V.W.D. developed the model and performed the experiments. J.K. evaluated liver slides for hepatic steatosis. M.A. performed LC/MS. T.J.P., M.A.F., K.E.C., B.L.M., L.K.C., G.M.H., C.A.D., J.K., M.A. and V.W.D. designed the experiments and wrote the manuscript.
Funding
This work was supported by research grants to V.W.D. from the Children’s Hospital Foundation of Manitoba and the Children’s Hospital Research Institute of Manitoba (CHF/CHRIM) and the Manitoba Medical Services Foundation (MMSF) Grant #812.10 and the Canadian Institutes of Health Research (CIHR) Grant #136885. T.J.P. was the recipient of a Manitoba Health Research Council (MHRC) studentship. L.K.C. is a recipient of a CIHR/HSFC IMPACT Fellowship. G.M.H. is a Canada Research Chair (Tier I) in Molecular Cardiolipin Metabolism. V.W.D. is the Amgen-Stewart Whitman Young Investigator and the Dr J. A. Moorhouse Fellow of the Diabetes Foundation of Manitoba.
References
- Alfaradhi MZ, Fernandez-Twinn DS, Martin-Gronert MS, Musial B, Fowden A. Ozanne SE. Oxidative stress and altered lipid homeostasis in the programming of offspring fatty liver by maternal obesity. Am J Physiol Regul Integr Comp Physiol. 2014;307:R26–R34. doi: 10.1152/ajpregu.00049.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ. Osmond C. Death rates from stroke in England and Wales predicted from past maternal mortality. Br Med J (Clin Res Ed) 1987;295:83–86. doi: 10.1136/bmj.295.6590.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett GR. Colorimetric assay methods for free and phosphorylated glyceric acids. J Biol Chem. 1959;234:469–471. [PubMed] [Google Scholar]
- Benkalfat NB, Merzouk H, Bouanane S, Merzouk SA, Bellenger J, Gresti J, Tessier C. Narce M. Altered adipose tissue metabolism in offspring of dietary obese rat dams. Clin Sci (Lond) 2011;121:19–28. doi: 10.1042/CS20100534. [DOI] [PubMed] [Google Scholar]
- Bouanane S, Benkalfat NB, Baba Ahmed FZ, Merzouk H, Mokhtari NS, Merzouk SA, Gresti J, Tessier C. Narce M. Time course of changes in serum oxidant/antioxidant status in overfed obese rats and their offspring. Clin Sci (Lond) 2009;116:669–680. doi: 10.1042/CS20080413. [DOI] [PubMed] [Google Scholar]
- Brumbaugh DE, Tearse P, Cree-Green M, Fenton LZ, Brown M, Scherzinger A, Reynolds R, Alston M, Hoffman C, Pan Z, Friedman JE. Barbour LA. Intrahepatic fat is increased in the neonatal offspring of obese women with gestational diabetes. J Pediatr. 2013;162:930–936. doi: 10.1016/j.jpeds.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan TA, Xiang AH. Page KA. Gestational diabetes mellitus: risks and management during and after pregnancy. Nat Rev Endocrinol. 2012;8:639–649. doi: 10.1038/nrendo.2012.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley AJ, Keseru B, Briody J, Thompson M, Ozanne SE. Thompson CH. Altered body composition and metabolism in the male offspring of high fat-fed rats. Metabolism. 2005;54:500–507. doi: 10.1016/j.metabol.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Caluwaerts S, Holemans K, van Bree R, Verhaeghe J. Van Assche FA. Is low-dose streptozotocin in rats an adequate model for gestational diabetes mellitus? J Soc Gynecol Investig. 2003;10:216–221. doi: 10.1016/s1071-5576(03)00044-3. [DOI] [PubMed] [Google Scholar]
- Caluwaerts S, Lambin S, van Bree R, Peeters H, Vergote I. Verhaeghe J. Diet-induced obesity in gravid rats engenders early hyperadiposity in the offspring. Metabolism. 2007;56:1431–1438. doi: 10.1016/j.metabol.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Catalano PM, Huston L, Amini SB. Kalhan SC. Longitudinal changes in glucose metabolism during pregnancy in obese women with normal glucose tolerance and gestational diabetes mellitus. Am J Obstet Gynecol. 1999;180:903–916. doi: 10.1016/s0002-9378(99)70662-9. [DOI] [PubMed] [Google Scholar]
- Catalano PM, Vargo KM, Bernstein IM. Amini SB. Incidence and risk factors associated with abnormal postpartum glucose tolerance in women with gestational diabetes. Am J Obstet Gynecol. 1991;165:914–919. doi: 10.1016/0002-9378(91)90438-w. [DOI] [PubMed] [Google Scholar]
- Chen H, Simar D, Lambert K, Mercier J. Morris MJ. Maternal and postnatal overnutrition differentially impact appetite regulators and fuel metabolism. Endocrinology. 2008;149:5348–5356. doi: 10.1210/en.2008-0582. [DOI] [PubMed] [Google Scholar]
- Clausen TD, Mathiesen ER, Hansen T, Pedersen O, Jensen DM, Lauenborg J. Damm P. High prevalence of type 2 diabetes and pre-diabetes in adult offspring of women with gestational diabetes mellitus or type 1 diabetes: the role of intrauterine hyperglycemia. Diabetes Care. 2008;31:340–346. doi: 10.2337/dc07-1596. [DOI] [PubMed] [Google Scholar]
- Clausen TD, Mathiesen ER, Hansen T, Pedersen O, Jensen DM, Lauenborg J, Schmidt L. Damm P. Overweight and the metabolic syndrome in adult offspring of women with diet-treated gestational diabetes mellitus or type 1 diabetes. J Clin Endocrinol Metab. 2009;94:2464–2470. doi: 10.1210/jc.2009-0305. [DOI] [PubMed] [Google Scholar]
- Dabelea D, Hanson RL, Lindsay RS, Pettitt DJ, Imperatore G, Gabir MM, Roumain J, Bennett PH. Knowler WC. Intrauterine exposure to diabetes conveys risks for type 2 diabetes and obesity: a study of discordant sibships. Diabetes. 2000;49:2208–2211. doi: 10.2337/diabetes.49.12.2208. [DOI] [PubMed] [Google Scholar]
- Dolinsky VW, Rueda-Clausen CF, Morton JS, Davidge ST. Dyck JR. Continued postnatal administration of resveratrol prevents diet-induced metabolic syndrome in rat offspring born growth restricted. Diabetes. 2011;60:2274–2284. doi: 10.2337/db11-0374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch J, Lees M. Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- Flegal KM, Carroll MD, Kit BK. Ogden CL. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999–2010. JAMA. 2012;307:491–497. doi: 10.1001/jama.2012.39. [DOI] [PubMed] [Google Scholar]
- Franks PW, Looker HC, Kobes S, Touger L, Tataranni PA, Hanson RL. Knowler WC. Gestational glucose tolerance and risk of type 2 diabetes in young Pima Indian offspring. Diabetes. 2006;55:460–465. doi: 10.2337/diabetes.55.02.06.db05-0823. [DOI] [PubMed] [Google Scholar]
- Fullerton MD. Bakovic M. Complementation of the metabolic defect in CTP:phosphoethanolamine cytidylyltransferase (Pcyt2)-deficient primary hepatocytes. Metabolism. 2010;59:1691–1700. doi: 10.1016/j.metabol.2010.03.022. [DOI] [PubMed] [Google Scholar]
- Fullerton MD, Hakimuddin F, Bonen A. Bakovic M. The development of a metabolic disease phenotype in CTP:phosphoethanolamine cytidylyltransferase-deficient mice. J Biol Chem. 2009;284:25704–25713. doi: 10.1074/jbc.M109.023846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard R, Durmus B, Hofman A, Mackenbach JP, Steegers EA. Jaddoe VW. Risk factors and outcomes of maternal obesity and excessive weight gain during pregnancy. Obesity (Silver Spring) 2013;21:1046–1055. doi: 10.1002/oby.20088. [DOI] [PubMed] [Google Scholar]
- Gillman MW, Rifas-Shiman S, Berkey CS, Field AE. Colditz GA. Maternal gestational diabetes, birth weight, and adolescent obesity. Pediatrics. 2003;111:e221–226. doi: 10.1542/peds.111.3.e221. [DOI] [PubMed] [Google Scholar]
- Hinkle SN, Sharma AJ, Kim SY, Park S, Dalenius K, Brindley PL. Grummer-Strawn LM. Prepregnancy obesity trends among low-income women, United States, 1999–2008. Matern Child Health J. 2012;16:1339–1348. doi: 10.1007/s10995-011-0898-2. [DOI] [PubMed] [Google Scholar]
- Howie GJ, Sloboda DM, Kamal T. Vickers MH. Maternal nutritional history predicts obesity in adult offspring independent of postnatal diet. J Physiol. 2009;587:905–915. doi: 10.1113/jphysiol.2008.163477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isganaitis E, Woo M, Ma H, Chen M, Kong W, Lytras A, Sales V, Decoste-Lopez J, Lee KJ, Leatherwood C, Lee D, Fitzpatrick C, Gall W, Watkins S. Patti ME. Developmental programming by maternal insulin resistance: hyperinsulinemia, glucose intolerance, and dysregulated lipid metabolism in male offspring of insulin-resistant mice. Diabetes. 2014;63:688–700. doi: 10.2337/db13-0558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jawerbaum A. White V. Animal models in diabetes and pregnancy. Endocr Rev. 2010;31:680–701. doi: 10.1210/er.2009-0038. [DOI] [PubMed] [Google Scholar]
- Kjaergaard M, Nilsson C, Rosendal A, Nielsen MO. Raun K. Maternal chocolate and sucrose soft drink intake induces hepatic steatosis in rat offspring associated with altered lipid gene expression profile. Acta Physiol (Oxf) 2014;210:142–153. doi: 10.1111/apha.12138. [DOI] [PubMed] [Google Scholar]
- Kral JG. Preventing and treating obesity in girls and young women to curb the epidemic. Obes Res. 2004;12:1539–1546. doi: 10.1038/oby.2004.193. [DOI] [PubMed] [Google Scholar]
- McGarry JD. Banting lecture 2001: dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes. 2002;51:7–18. doi: 10.2337/diabetes.51.1.7. [DOI] [PubMed] [Google Scholar]
- McMillen IC. Robinson JS. Developmental origins of the metabolic syndrome: prediction, plasticity and programming. Physiol Rev. 2005;85:571–633. doi: 10.1152/physrev.00053.2003. [DOI] [PubMed] [Google Scholar]
- Moore TR. Fetal exposure to gestational diabetes contributes to subsequent adult metabolic syndrome. Am J Obstet Gynecol. 2010;202:643–649. doi: 10.1016/j.ajog.2010.02.059. [DOI] [PubMed] [Google Scholar]
- Morris MJ. Chen H. Established maternal obesity in the rat reprograms hypothalamic appetite regulators and leptin signaling at birth. Int J Obes (Lond) 2009;33:115–122. doi: 10.1038/ijo.2008.213. [DOI] [PubMed] [Google Scholar]
- Nagle CA, Klett EL. Coleman RA. Hepatic triacylglycerol accumulation and insulin resistance. J Lipid Res. 2009;50(Suppl):S74–79. doi: 10.1194/jlr.R800053-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden CL, Carroll MD, Kit BK. Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011–2012. JAMA. 2014;311:806–814. doi: 10.1001/jama.2014.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlovic Z. Bakovic M. Regulation of phosphatidylethanolamine homeostasis – the critical role of CTP:phosphoethanolamine cytidylyltransferase (Pcyt2) Int J Mol Sci. 2013;14:2529–2550. doi: 10.3390/ijms14022529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira TJ, Moyce BL, Kereliuk SM. Dolinsky VW. Influence of maternal overnutrition and gestational diabetes on the programming of metabolic health outcomes in the offspring: experimental evidence. Biochem Cell Biol. 2015 doi: 10.1139/bcb-2014-0141. in press. [DOI] [PubMed] [Google Scholar]
- Reece EA, Leguizamon G. Wiznitzer A. Gestational diabetes: the need for a common ground. Lancet. 2009;373:1789–1797. doi: 10.1016/S0140-6736(09)60515-8. [DOI] [PubMed] [Google Scholar]
- Ronis MJ, Baumgardner JN, Marecki JC, Hennings L, Wu X, Shankar K, Cleves MA, Gomez-Acevedo H. Badger TM. Dietary fat source alters hepatic gene expression profile and determines the type of liver pathology in rats overfed via total enteral nutrition. Physiol Genomics. 2012;44:1073–1089. doi: 10.1152/physiolgenomics.00069.2012. [DOI] [PubMed] [Google Scholar]
- Rueda-Clausen CF, Dolinsky VW, Morton JS, Proctor SD, Dyck JR. Davidge ST. Hypoxia-induced intrauterine growth restriction increases the susceptibility of rats to high-fat diet-induced metabolic syndrome. Diabetes. 2011a;60:507–516. doi: 10.2337/db10-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueda-Clausen CF, Morton JS. Davidge ST. The early origins of cardiovascular health and disease: who, when, and how. Semin Reprod Med. 2011b;29:197–210. doi: 10.1055/s-0031-1275520. [DOI] [PubMed] [Google Scholar]
- Samuelsson AM, Matthews PA, Argenton M, Christie MR, McConnell JM, Jansen EH, Piersma AH, Ozanne SE, Twinn DF, Remacle C, Rowlerson A, Poston L. Taylor PD. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: a novel murine model of developmental programming. Hypertension. 2008;51:383–392. doi: 10.1161/HYPERTENSIONAHA.107.101477. [DOI] [PubMed] [Google Scholar]
- Shields M. Overweight and obesity among children and youth. Health Reports/Statistics Canada, Canadian Centre for Health Information. 2006;17:27–42. [PubMed] [Google Scholar]
- Sinha R, Dufour S, Petersen KF, LeBon V, Enoksson S, Ma YZ, Savoye M, Rothman DL, Shulman GI. Caprio S. Assessment of skeletal muscle triglyceride content by 1H nuclear magnetic resonance spectroscopy in lean and obese adolescents: relationships to insulin sensitivity, total body fat, and central adiposity. Diabetes. 2002;51:1022–1027. doi: 10.2337/diabetes.51.4.1022. [DOI] [PubMed] [Google Scholar]
- Symonds ME, Sebert SP. Budge H. The impact of diet during early life and its contribution to later disease: critical checkpoints in development and their long-term consequences for metabolic health. Proc Nutr Soc. 2009;68:416–421. doi: 10.1017/S0029665109990152. [DOI] [PubMed] [Google Scholar]
- Weiss R, Dufour S, Taksali SE, Tamborlane WV, Petersen KF, Bonadonna RC, Boselli L, Barbetta G, Allen K, Rife F, Savoye M, Dziura J, Sherwin R, Shulman GI. Caprio S. Prediabetes in obese youth: a syndrome of impaired glucose tolerance, severe insulin resistance, and altered myocellular and abdominal fat partitioning. Lancet. 2003;362:951–957. doi: 10.1016/S0140-6736(03)14364-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicklow BA, Wittmeier KD, MacIntosh AC, Sellers EA, Ryner L, Serrai H, Dean HJ. McGavock JM. Metabolic consequences of hepatic steatosis in overweight and obese adolescents. Diabetes Care. 2012;35:905–910. doi: 10.2337/dc11-1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young TK, Martens PJ, Taback SP, Sellers EA, Dean HJ, Cheang M. Flett B. Type 2 diabetes mellitus in children: prenatal and early infancy risk factors among native canadians. Arch Pediatr Adolesc Med. 2002;156:651–655. doi: 10.1001/archpedi.156.7.651. [DOI] [PubMed] [Google Scholar]