

Abstract
Background
Platelets have a pathophysiologic role in the ischemic microvascular environment of acute coronary syndromes (ACS). Compared to platelet activation in normal healthy conditions, less attention is given to mechanisms of platelet activation in diseased states. Platelet function and mechanisms of activation in ischemic and reactive oxygen species (ROS) rich environments may not be the same as in normal healthy conditions. Extracellular Regulated Protein Kinase 5 (ERK5) is a Mitogen Activated Protein Kinase (MAPK) family member activated in hypoxic, ROS rich environments, and in response to receptor signaling mechanisms. Prior studies suggest a protective effect of ERK5 in endothelial and myocardial cells following ischemia. We present evidence that platelets express ERK5 and platelet ERK5 has an adverse effect on platelet activation via selective receptor-dependent and receptor-independent ROS mediated mechanisms in ischemic myocardium.
Methods and Results
Using isolated human platelets and a mouse model of myocardial infarction (MI), we found that platelet ERK5 is activated post-MI and platelet specific ERK5−/− mice have less platelet activation, reduced MI size, and improved post-MI heart function. Furthermore, the expression of downstream ERK5 regulated proteins is reduced in ERK5−/− platelets post-MI.
Conclusions
ERK5 functions as a platelet activator in ischemic conditions and platelet ERK5 maintains the expression of some platelet proteins following MI, leading to infarct expansion. This demonstrates that platelet function in normal healthy conditions is different from platelet function in chronic ischemic and inflammatory conditions. Platelet ERK5 may be a target for acute therapeutic intervention in the thrombotic and inflammatory post-MI environment.
Keywords: Platelets, Infarction, Microcirculation, Echocardiography
Introduction
Platelet activation occurs at sites with vascular inflammation and in the presence of increased concentrations of reactive oxygen species (ROS) 1. Platelet agonists, inflammatory molecules and ROS are all greatly increased in the ischemic microenvironment following myocardial infarction (MI) or stroke. This results in sustained platelet activation and the secretion of inflammatory molecules and degradative enzymes such as matrix metalloproteinases (MMPs), leading to more leukocyte recruitment, extracellular matrix degradation, infarct expansion, and a decline in heart function beyond the primary infarct event2–5. Acute coronary syndromes (ACS) are a continuum with acute thrombosis at one end of the spectrum (ST elevated myocardial infarction/STEMI) and chronic, ongoing myocardial ischemia at the other (unstable angina). Platelet responses also represent a continuum in this process, beginning with plaque rupture and major vessel thrombosis, and continuing with microvascular occlusion and vascular inflammation in the peri-infarct tissue. This contributes to infarct expansion and a continued decline in heart function. The development of pharmacological agents to blunt platelet activation is largely based on platelet function in normal states, with less attention paid to ongoing platelet activation following an ischemic event.
Aspirin and clopidogrel are the major platelet inhibitors used to treat ACS, yet one prospective study showed only a 20% reduction in adverse vascular events and increased bleeding complications with the addition of clopidogrel to aspirin 6, 7. Variability in the patient response to platelet inhibitors is partly attributed to polymorphisms in drug metabolism 8, 9 and can result in sub-optimal treatment regimens for patients with ACS. Approximately 1/3 of patients suffering a STEMI, even with coronary stenting, develop a ‘no reflow’ phenomenon thought to arise from microvascular obstruction. Enhanced platelet activity or inadequate platelet inhibition at the time of MI is more common in patients with no reflow. A more complete understanding of mechanisms leading to dysregulated platelets in disease states may reveal additional therapeutic strategies 10.
ERK5 (also called BMK1) is a mitogen activated kinase (MAPK) family member activated in the myocardium and endothelial cells upon ROS exposure 11–14. Targets of ERK5 are largely nuclear 13, 15, but ERK5 also has non-transcriptional, cell signaling functions. We have identified ERK5 in platelets, and using human platelets and platelets isolated from platelet specific ERK5−/− mice, we have investigated whether ERK5 activation following exposure to ROS and specific platelet ligands is elevated in ischemic tissues and contributes to myocardial dysfunction and remodeling. Our data establishes a potential mechanistic link between platelet function in ischemic myocardial tissue and adverse myocardial remodeling following MI. We reveal that ERK5 is an ischemic sensor in platelets, contributing to ongoing post-MI platelet activation that accelerates ventricular remodeling and systolic dysfunction. Our data also demonstrates that ERK5 regulates platelet protein expression post-MI. Together this study indicates that platelet ERK5 has a central role in platelet activation in the ischemic post-MI environment.
Methods
Complete reagent list, buffers and methods are detailed in the supplementary data section.
Antibodies and reagents
A list if the antibodies and reagents used is indicated in the supplementary data section.
Platelet Isolation
Human platelets were obtained using laboratory protocols approved by the University of Rochester School of Medicine and Dentistry Institutional Review Board (IRB), with the isolation protocol as noted in the Supplemental Data Section.
Protein studies
Primary antibody was 1:1000 overnight at 4 °C in 3% BSA/TBS-T. Secondary antibody (GE Healthcare, Buckinghamshire, UK) was used in a 1:2000 titer in 5% milk/TBS-T for 1 hour at room temperature. Final autoradiographic films (Bioblot BXR, Laboratory Product Sales, Rochester NY) were quantified by densitometry using ImageJ software (NIH).
MMP activity assay
Platelets were isolated from 120 μL whole blood and ventricular apex isolates were homogenized in extraction buffer (supplemental). Final supernatants were protein normalized, lysates centrifuged for 15 mins at 4°C, and supernatants were placed in 50% vol/vol 2x non-denaturing sample buffer (supplemental). 50 μg of total cell lysate per lane was separated by SDS-PAGE, the gel was renatured by gently rocking in 2.5% Triton-X-100 for 30 mins at room temperature, then allowed to equilibrate at room temperature with gentle rocking in zymogram buffer (supplemental). The zymogram buffer was decanted, and the gel was rocked at room temperature for 4 hours in Simply Blue Safestain (Invitrogen). MMP activity was noted by clear bands in the final gel (a reverse image). Total MMP activity in each lane was quantified by densitometry using ImageJ software (NIH).
Mouse colony
ERK5flox/flox mice on a C57Bl6 (B6) background were mated with B6 PF4-Cre mice. Animal studies were performed in accordance with the University Committee on Animal Research at University of Rochester Medical Center.
Intravital microscopy (thrombosis model)
Briefly, WT or ERK5−/− mice were anesthetized, platelets were labeled in vivo with platelet specific fluorescent antibody (Emfret). Mesenteric arterioles (80–100 μm in diameter) were selected and damaged by the addition of Whatman’s paper soaked in 15% FeCl3 to the vessel surface for 45 sec. Thrombus formation was captured with a digital camera (Nikon).
Pulmonary Thromboembolism Model
Mice were briefly anesthetized and injected with anti GPIb beta antibody (Emftret) conjugated to Alexa 750 (Invitrogen). After 20 minutes the external jugular (EJ) vein was isolated and collagen (Chrono-log #385, 0.8mg/kg) and epinephrine (60ug/kg) were injected into the EJ vein to induce thromboembolism. Three minutes later the animal was perfused and the lungs were harvested. The harvested lungs were imaged on an Odyssey CLx Imager (Li-Cor Biotech.). The PE burden was expressed as mean fluorescence intensity (MFI).
Tail bleeding assay (hemostasis model)
Mice were anesthetized and 3 mm at the tail tip was amputated with a scalpel and a steady stream of blood was visualized in 37°C PBS until the time of hemostasis.
Myocardial Infarction Model
Chronic MI was induced by permanent ligation the left anterior descending (LAD) coronary artery. The anesthetized mouse was placed on a heating pad and endotracheal intubated for mechanical ventilation (inspiratory tidal volume of 250μl at 130 breaths/min). A left thoracotomy was performed in the fourth intercostal space. The mouse heart was exposed, and the LAD coronary artery was ligated 2 mm from its ostial origin with 9-0 silk suture. A sham operation involved the same procedure, but a suture was passed under the LAD coronary artery without ligation.
LV Infarct assessment
Hearts were harvested at the end of the study after perfusion and fixation in methanol/acetic acid fixative. Parasternal short axis section were cut before mounting and staining with Masson’s trichrome reagent. Slides were analyzed and photographed using an Olympus light microscope (Model BX41). The infarcted area (blue collagen staining for scar tissue) was expressed as percentage of LV surface area using ImageJ software (NIH). Echocardiography: Echocardiographic analysis using M-mode was performed using a Vevo2100 echocardiography machine (VisualSonics, Toronto, Canada) and a linear-array 40MHz transducer (MS-550D). LV systolic and diastolic measurements were captured in M-mode from the parasternal short axis. Heart function analysis completely described in supplemental information.
Statistical Analysis
Data were analyzed using SAS software (version 9.4, SAS Institute, Cary, NC, USA). For experiments involving repeated measurements for a given subject, Friedman’s chi-square test, a non-parametric test, was computed. Additionally, pairwise comparisons between the baseline and various concentration or time point values were made using the paired t-test. When independent samples were compared, the standard Student’s t-test was used as noted. A p-value of less than 0.05 was considered statistically significant.
Our in vivo animal studies report data from a relatively small population size. A limitation is that normality cannot be assumed in the sample distribution. For experiments involving repeated measurements for a given subject, Friedman’s chi-square test, a non-parametric test, was computed and reported to provide a more robust analysis.
Results
Expression and Function of ERK5 in Platelets
ERK5 is phosphorylated in the activated state (p-ERK5). To demonstrate that platelets express ERK5 that becomes activated, washed human platelets were isolated and left resting, or stimulated with multiple concentrations of the thrombin receptor agonist TRAP. P-ERK5 was determined by Western blot and quantified as ratio of total pERK5/ERK5. TRAP induced platelet ERK5 activation (Figure 1A). Platelet ERK5 was also strongly activated by the thromboxane receptor agonist U46619 (Figure 1B). However, the P2Y12 receptor agonist 2-methyl-ADP and the collagen receptor (GPVI) agonist convulxin only weakly activated ERK5 (Figure 1C–D, blots over-exposed for emphasis). The same concentrations of 2-methyl ADP and convulxin activated platelet ERK1 (Figure S1). These data indicate that ERK5 is selectively activated by specific platelet receptor agonists.
Figure 1.




ERK5 is expressed in human platelets and activated by thrombin and thromboxane. Washed human platelets were stimulated with A) 1–5 μM TRAP, B) 0.01–10 μM U46619, C) 1–10 μM 2-methyl adenosine diphosphate (ADP), or D) 5–5000 ng/mL convulxin, each for 10 mins. Western blotting was performed for p-ERK5 (blots over-exposed for emphasis in C and D, and total ERK5 as loading control). ERK5 activation reported as mean p-ERK5/ERK5 (± SEM, N=4–7, * P <0.05 vs 0, paired t-test). P=0.08 for A), P=0.06 for B), P=0.35 for C), P =0.85 for D) by Friedman’s test.
Global ERK5 deficient mice (ERK5−/−) are embryonic lethal 16. To determine the role of ERK5 in platelet function, platelet ERK5−/− mice were created by crossing PF4-Cre mice with ERK5 flox mice (Figure 2A). No change in ERK5 protein expression was noted in other major organs (data not shown). Washed platelets from WT and platelet ERK5−/− mice were isolated and stimulated with thrombin. ERK5−/− platelets had dramatically attenuated thrombin-mediated platelet activation as shown by decreased activated GPIIbIIIa (JON/A antibody binding) and decreased surface P-selectin expression compared to WT platelets (Figure 2B–C). ERK5−/− platelets also had attenuated thromboxane receptor-mediated platelet activation (Figure 2D). Similar to human platelet ERK5 activation, WT and ERK5−/− platelets had no significant change in activation in response to ADP or convulxin in (Figure 2E). These data confirm that platelet ERK5 is activated by select platelet agonists.
Figure 2.
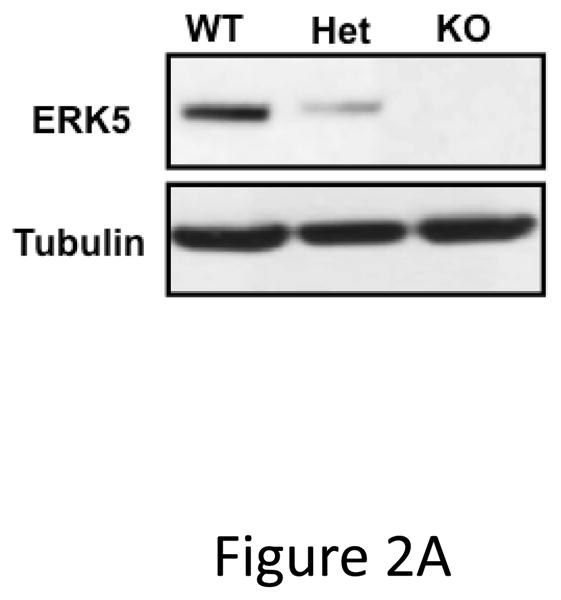




ERK5−/− platelets have decreased activation in response to PAR and thromboxane receptor stimulation. A) Washed platelets were isolated from WT, ERK5+/− (Het) or ERK5−/− mice and immunoblotted for ERK5. ERK5−/− platelets do not express ERK5 (representative blot). B–D) Washed platelets from WT and ERK5−/− mice were stimulated with B–C) thrombin, D) U46619 which is a thromboxane receptor agonist, E) ADP or convulxin. Platelet activation was determined by JON/A antibody staining or surface P-selectin expression via FACS (MFI ± SD, N=4, * P < 0.05. vs. WT at same agonist concentration and **P < 0.01. vs WT at same agonist concentration, t-test for equal variances).
ERK5 can be activated by redox stress in ischemic pathologic states 12, 17, 18. To determine whether platelet ERK5 is activated by redox stress, we incubated human platelets with hydrogen peroxide (H2O2) as a surrogate exogenous ischemia mediator. H2O2 activated human platelet ERK5 (Figure 3A). To determine whether platelet derived (endogenous) ROS generated in hypoxic conditions such as in ischemia activates platelet ERK5, human and mouse platelets were incubated under normoxic (21% O2) or reduced oxygen (5% O2) conditions and immunoblotted for p-ERK5. P-ERK5 is acutely increased by hypoxia in both human (Figure 3B) and mouse (Figure 3C) platelets. These data demonstrate that ERK5 is a mediator of platelet activation in response to thrombin and thromboxane, and that platelet ERK5 is activated directly by exogenous ROS and relative hypoxia.
Figure 3.



Platelet ERK5 is activated by ROS. A) Dose-dependent platelet ERK5 ROS activation. Washed human platelets were stimulated with H2O2 (0.5–1000 μM) for 5 mins and p-ERK5 determined at multiple time points by p-ERK5 immunoblotting normalized to total ERK5 (* P<0.05 vs 0, paired t-test; P =0.12 by Friedman’s test).
B) Washed human platelets were incubated under normoxic or reduced oxygen (5% O2) conditions for 2–10 mins (P <0.05 vs 0, paired t-test and P =0.02, Friedman’s test). C) Washed mouse platelets were incubated under normoxic or reduced oxygen (5% O2) conditions for 2–10 mins. (P <0.05 vs 0, paired t-test; P=0.02, Friedman’s test). ERK5 activation reported by Western blotting as mean p-ERK5/ERK5 (± SEM, N=4–6).
Platelet ERK5 In Vivo Function
To investigate whether ERK5 is a terminal platelet effecter or an intermediary, molecules controlling platelet activation that are known ERK5 targets in other tissues were examined. Platelets were isolated from WT and platelet ERK5−/− mice and RAC phosphorylation was determined by Western blot 19–21. Phosphorylation of RAC1 at serine 71, a signature of decreased RAC1 activity, is implicated in regulating cellular activity including ROS responses22–25. Basal RAC1 phosphorylation at inhibitory serine 71 was increased in ERK5−/− mouse platelets by almost five-fold compared to WT mouse platelets (Figure 4A), consistent with ERK5 maintaining platelets in an activation responsive state. This is supported by ERK5−/− platelets having reduced basal JON/A binding and P-selectin expression upon isolation and decreased plasma platelet factor 4 (PF4) and thromboxane B2 (TbxB2) in ERK5−/− mice under resting conditions (Figure S2). These data indicate that ERK5 plays a role in regulating downstream platelet activation pathways.
Figure 4.




ERK5 mediates in vivo platelet activation. A) RAC1 basal activation is reduced in ERK5−/− platelets. Washed WT and ERK5−/− platelet lysates were prepared and immunoblotted for total RAC or RAC1 phosphorylated on serine 71 (p-RAC S71 is inactive RAC1). Representative blots (each lane is an individual mouse platelet extract) run on separate protein gels (top), and densitometry (bottom). GAPDH was determined by re-probing as an additional loading control (mean ± SEM, N=4–7, *P < 0.05 vs WT, t-test for unequal variances). B) Platelet ERK5−/− mice have normal bleeding times. Mouse tail bleeding times were determined by time to visual cessation of bleeding (mean ± SEM, N=5–6). C) Platelet ERK5−/− mice have prolonged time to in vivo thrombus formation. Mesenteric ferric chloride injury model with time to vessel occlusion recorded (mean ± SEM, N=5, *p = 0.004 vs. WT, t-test for unequal variances). D) WT and platelet ERK5−/− mice have similar thrombus burden in a PE model. Mice were injected with Alexa750 labeled anti-GP1b antibody and then treated with collagen/epinephrine or vehicle control via the jugular vein to induce PE. Perfused lungs were isolated and imaged to quantify lung thrombus burden (MFI ± SEM, N=4, *p =0.02 vs. vehicle=PBS, t-test for unequal variances. Representative image from are from individual mice).
To determine whether platelet ERK5 deficiency affects hemostasis and thrombosis, we performed timed tail-bleeding studies in WT and platelet ERK5−/− mice and found no difference (Figure 4B). We also performed thrombosis studies using a mesenteric injury model that induces oxidative vessel injury 26. The time to ferric chloride-induced vessel occlusion was significantly prolonged in platelet ERK5−/− mice compared to WT mice (Figure 4C), but platelet ERK5−/− mice still exhibited vessel occlusion. To explore thrombus formation in a non-ROS dependent model, WT and ERK5−/− mice were injected with collagen/epinephrine via the jugular vein with to induce pulmonary embolism (PE)27. PE size was determined using an infrared fluorescent tagged platelet antibody. WT and platelet ERK5−/− mice had similar PE size (Figure 4D). Together these data indicate that platelet ERK5 mediates thrombus formation in response to some forms of vessel injury, but is not definitively necessary for physiologic thrombosis.
Post-MI Cardiac Function in Platelet ERK5−/− Mice
Because platelet ERK5 is activated by ROS, thrombin, and thromboxane, ERK5 activation in an ischemic environment was explored using a mouse myocardial infarction (MI) model in which these mediators have significant pathologic roles 28–30. As a chronic MI model the left anterior descending (LAD) coronary artery was permanently ligated. LAD ligation in WT C57Bl/6J (B6) mice lead to increased ERK5 activation in platelets isolated on day 3 post ligation as compared to sham mice (open chest, no LAD ligation) (Figure 5A). Similarly, ERK5 activation was noted post LAD ligation in platelets from mice on a mixed strain background (B6/129) (Figure S3). This demonstrates that persistent cardiac ischemia promotes platelet ERK5 activation.
Figure 5.




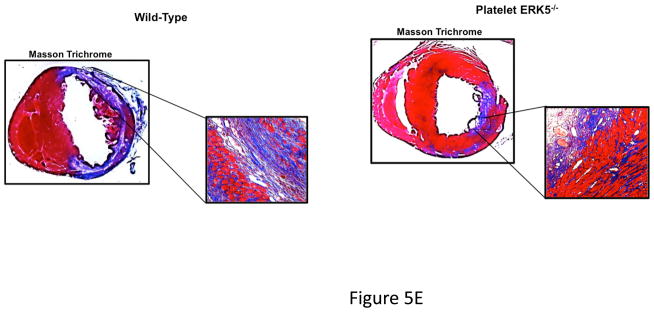

Platelet ERK5−/− mice have improved post-MI heart function. A) Washed mouse platelets isolated on day 3 following MI were assessed for activated ERK5 (p-ERK5) (mean p-ERK5/ERK5 ± SEM, N=5, *P = 0.03 vs. sham operated, t-test for unequal variances). B) ECG before and 30 seconds after LAD coronary artery ligation indicates transmural myocardial ischemia (ST-segment elevation). C) Representative M-mode echocardiograms for WT and ERK5−/− mice pre-LAD coronary ligation (day 0) and day 7 post-LAD coronary ligation. D) Echocardiography quantification of LVEF, FS, LVEDV, LVESV, LVIDd, LVIDs (mean ± SEM, N=4–6, *P < 0.05 vs. WT, t-test for unequal variances). E) Platelet ERK5−/− mice have smaller infarcts (reduced Masson Trichrome stain of LV, insert 40x magnification). F) Quantification of Masson Trichome positive staining (mean % of LV area, N=4 ± SEM,*P = 0.02 vs WT, t-test for equal variances).
Platelet inhibitors improve patient outcomes following transmural myocardial ischemia. LAD coronary ligation induces transmural ischemia as shown by clear ST-segment elevation on the electrocardiogram (ECG) in both WT and platelet ERK5−/− mice within 30 seconds of ligation (Figure 5B). Cardiac function was assessed following permanent coronary artery ligation in WT and platelet ERK5−/− mice. Baseline (pre-LAD ligation) echocardiograms of WT and platelet ERK5−/− mice showed no difference in left ventricular (LV) dimensions or function (Figure S4). The ischemic area at risk was also very similar between WT and platelet ERK5−/− mice following LAD coronary ligation (Figure S5). Platelet ERK5−/− mice had improved heart function starting on day 3 post LAD coronary artery ligation as evidenced by echocardiographic parameters of LV dimension and myocardial contractility. These include improved LV ejection fraction, improved fractional shortening, a less dilated LV cavity, thicker LV walls, and lower LV volume indices (LVEDV, LVESV, LVIDd, LVIDs) (Figure 5, and Figure S6). Hearts from WT mice were more dilated, congested, and remodeled compared to platelet ERK5−/− mouse hearts following LAD coronary ligation (Figure 5C–D, Movie 1 and Movie 2). Histologic data demonstrated that the infarction pattern spread far beyond the anterior wall of the left ventricle in WT mice and this pattern was reduced and patchy in platelet ERK5−/− mice (Figure 5E–F). These data indicate ERK5-dependent platelet activation promotes infarct expansion, while platelet ERK5 deficiency has a post-MI cardioprotective effect.
Platelet activation was assessed following coronary artery ligation in WT and platelet ERK5−/− mice by measuring concentrations of plasma PF4 and the stable thromboxane metabolite TbxB2. WT and ERK5−/− mice had very similar platelet counts pre- and post-MI (Figure S7), but both PF4 and TbxB2 plasma concentrations were significantly greater in the blood of WT mice compared to platelet ERK5−/− mice (Figure 6A). Day 3 post-MI heart tissue was immunostained using a platelet specific marker CD42c to assess relative platelet localization, particularly at the edges of the infarcted tissue. There was increased platelet immunostaining in WT hearts compared to platelet ERK5−/− mouse hearts (Figure 6B and Supplemental Figure S8). To further confirm that the phenotype in platelet ERK5−/− mice is not genetic background related due to PF4-Cre, LAD ligation was performed in WT and B6 PF4-Cre mice. Both LV function and infarct size were similar in these strains of mice (Figure S9).
Figure 6.
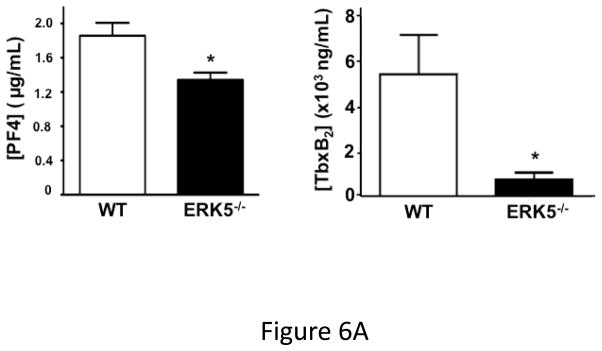




Post-MI platelet activation is reduced in platelet ERK5−/− mice. A) Plasma PF4 or TxB2 were measured by ELISA as markers of ongoing platelet activation 3 days following LAD coronary artery ligation (mean ± S.D, N=6. *P <0.05 vs. WT, t-test for unequal variances). B) Representative parasternal short axis sections of the heart on day 3 following LAD coronary ligation. CD42c (platelet) immunostaining is less in platelet ERK5−/− mouse hearts compared to WT hearts at the infarct border (additional images are shown in S11). C) Platelet MMP content by Western blotting on day 3 following LAD coronary ligation is reduced compared to sham-operated mice (mean MMP9/GAPDH ± SEM, N=4–5. *p = 0.02 vs. sham-operated, t-test for unequal variances). D) LV MMP9 and TIMP1 content is reduced in platelet ERK5−/− mice after MI (mean intensity ± SEM, N=5 each group, *P =0.02 vs. WT, t-test for unequal variances). LV extracts on day 3 following LAD coronary artery ligation were protein-normalized and run in duplicate on non-reducing gels with a gelatin matrix. Gelatinase activity is shown as light bands on the final zymogram. Total LV MMP activity (all isoforms) was calculated for each lane (mean intensity ± SEM, N=6–10, *P < 0.001 vs. WT, t-test for unequal variances). Washed platelet MMP9 content is increased in platelet ERK5−/− mice compared to WT mice on day 3 following LAD coronary ligation (mean MMP9/GAPDH ± SEM, N=5. *p < 0.05 vs. WT mouse platelets). E) Platelet MMP9 content and MMP activity are reduced in platelet ERK5−/− mice. Washed platelets were isolated on day 3 following LAD coronary artery ligation, homogenized, protein-normalized and immunoblotted for MMP9 content (mean intensity ± SEM, N=3–5, *P < 0.01 vs. WT, t-test for unequal variances). Washed platelets were isolated on day 3 following LAD coronary artery ligation and were also run on non-reducing gels with a gelatin matrix. Gelatinase activity is shown as light bands on the final zymogram. Total MMP activity (all isoforms) was calculated for each lane. (Protein expression normalized to GAPDH and gelatinase activity is demonstrated by light bands on the final zymogram (total platelet MMP activity, mean ± SEM, N=8–10, *P=0.04 vs. WT, t-test for unequal variances).
Platelet activation may promote infarct expansion by releasing pro-inflammatory molecules that recruit and activate white blood cells, or by platelet release of degradative enzymes such as MMPs. To assess post-MI leukocyte infiltrates WT and platelet ERK5−/− mouse hearts were immunostained for CD45 and CD45 positive cells quantified in the peri-infarct zone. Leukocyte infiltrates were unchanged in platelet ERK5−/− mice compared to WT mice (Figure S10). MMPs, including MMP9, are found in platelets and their secretion may contribute to tissue remodeling and loss 31, 32. Platelet MMP9 content was decreased in platelets from WT mice on day 3 post LAD artery ligation compared to sham mice, indicating that platelets release MMP9 post-MI (Figure 6C). In comparing platelet MMP9 expression and activity between WT and ERK5−/− mice post-MI, total MMP9 content is greater yet MMP activity less in ERK5−/− mouse platelets (Figure 6D) consistent with less platelet activation and secretion of MMPs. Basal MMP9 expression is also greater in platelet ERK5−/− mice (Figure S11), but basal MMP activity is not different between WT and ERK5−/− mice (Figure S12). Consistent with less platelet activation and less MMP release in platelet ERK5−/− mice, heart tissue isolated from the LV apex (myocardial region supplied by the LAD coronary artery) of WT mice had increased total MMP9 and TIMP1 (a MMP inhibitor increased by elevated local concentrations of MMP) and increased total MMP activity compared to platelet ERK5−/− mice (Figure 6E). These data indicate that reduced platelet MMP release into the post-MI coronary vasculature and subsequent reduced myocardial remodeling may contribute to the protective phenotype observed in platelet ERK5−/− mice.
To determine whether increased ERK5 activation results in increased platelet agonist sensitivity, washed platelets were isolated from WT and platelet ERK5−/− mice under sham operation or LAD coronary ligation on day 6 post-MI and platelets were then thrombin stimulated. WT mice had augmented post-MI thrombin-mediated activation compared to control WT mouse platelet activation. This effect was not observed in ERK5−/− platelets following MI (Figure 7A).
Figure 7.


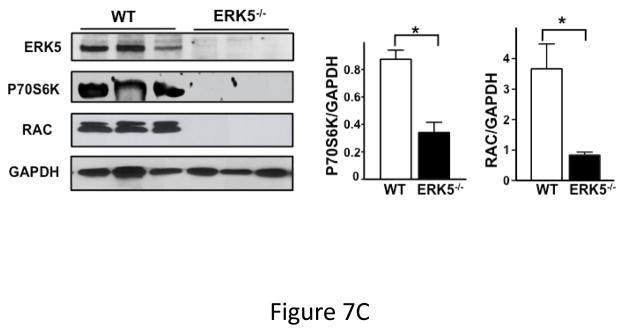

Dysregulated platelet activity following MI. A) Washed platelets from WT and platelet ERK5−/− mice on day 6 following LAD coronary ligation were examined for thrombin-induced activation. Platelet activation was determined by surface P-selectin expression via FACS (MFI ± SD, N=3–4. *P =0.03, Friedman’s test for WT + thrombin; P =0.04, Friedman’s test for WT/MI + thrombin; P =0.04, Friedman’s test for ERK5−/− + thrombin; P =0.03, Friedman’s test for ERK5−/−/MI + thrombin; ** P=0.04, t-test for unequal variance). B) Washed platelets on day 3 following LAD coronary ligation were examined for the expression of ERK5, P70S6K, RAC, TIMP1 and GAPDH by Western blotting. Protein gels were also stained by Coomassie Blue for total protein. Representative data (mean protein/GAPDH ± SEM, N=5. * P < 0.05 vs. platelets from sham-operated mice; # P =0.053 vs. platelets from sham-operated mice, t-test for unequal variances). C) Washed platelets from WT and platelet ERK5−/− mice on day 7 following LAD coronary ligation were examined for the expression of P70S6K and RAC by Western blotting (representative data are expressed as mean protein/GAPDH ± SEM, N=4–5. * P < 0.05 vs. WT, t-test for equal variances). D) Washed platelets at baseline and after 24hrs of LAD coronary ligation were examined for ubiquitinated proteins by Western blotting in WT and platelet ERK5−/− mice. Proteins ubiquitination was increased in ERK5−/− platelets, representative immunoblots.
While exploring potential downstream ERK5 signaling proteins we observed increased post-MI expression of several proteins also known to mediate platelet activation compared to platelets isolated from sham-operated mice. This included: ERK5, P70S6K and RAC (Figure 7B). The increased protein expression is specific to some proteins rather than a general affect as the expression of proteins such as TIMP1 and GAPDH did not change post-MI (Figure 7B). WT and ERK5−/− platelets had very similar basal protein expression including RAC (Figure 4A), P70S6K and TIMP1 (Figure S13). However, in post-MI conditions platelets taken from ERK5−/− mice had greatly reduced P70S6K and RAC expression compared to WT mice (Figure 7C). Because ERK5 is a regulator of ubiquitination in other cells 33, 34, and the ubiquitin-proteasome protein degradation pathway is active in platelets 35, 36 potentially accounting for decreased protein expression, we compared protein ubiquitination in platelets from WT and platelet ERK5−/− mice pre- or 24 hrs post-LAD ligation. ERK5−/− platelets had more protein ubiquitination both pre and post-MI (Figure 7D), a signature for proteasome targeting and degradation 37. This indicates that platelets may ‘sense and respond’ to a pathologic ischemic environment and that platelet ERK5 may be a key regulator of the expression of platelet proteins and platelet activity following MI.
Our data demonstrates that acute MI and subsequent chronic myocardial ischemia leads to ongoing platelet activation, infarct expansion, left ventricular remodeling, and severe contractile dysfunction, that is in part platelet ERK5 dependent. ERK5 promotes platelet activation in the dysregulated post-MI ischemic environment by initiating platelet activation pathways and regulating the expression of proteins important in platelet function. Inhibition of platelet ERK5 may therefore confer protection from ischemic ventricular remodeling by limiting platelet activation and targeting for degradation specific molecules important for platelet activation, thus limiting platelet MMP release (Figure 8).
Figure 8.

Proposed model. Following MI, platelet ERK5 is activated by both agonists and ROS. ERK5 activation leads to both platelet activation and increased expression of proteins important for platelet activation, in part via decreased protein ubiquitination.
Discussion
Our study indicates that in normal healthy conditions ERK5 mediates platelet activation in response to some agonists. Furthermore, in ischemic post-MI conditions platelet ERK5 is activated leading to increased platelet activation and ERK5 regulates the expression of some proteins important for platelet activation. Our data provides important evidence that platelet function and platelet protein expression is not static in diseased states. Current therapies may be severely limited by not accounting for a potential ‘moving therapeutic target’ that is apparent in platelets from the post-MI environment.
Platelet ERK5 activity contributes to ongoing myocardial dysfunction and tissue remodeling following MI. ERK5 is likely not the final effecter of increased platelet activation, but serves as an upstream platelet sensor of ROS in ischemic states and mediates downstream thrombin and thromboxane receptor signaling. ERK5 also has a central role in regulating the expression of platelet proteins post-MI in part though altered protein ubquitination. These findings strongly underscore the concept that mechanisms of platelet activation differ in pathologic states compared to normal healthy conditions. Targeting platelet ERK5 may be another strategy for anti-platelet therapy following acute MI.
ERK5 has been implicated in cell cycle progression and cellular transformation in many tissues 15, 38, 39. Since platelets are anucleate, the expression of ERK5 in platelets was initially somewhat surprising, though other members of the MAPK family regulate platelet maturation and activation 40. Various studies indicate that ERK5 can regulate cellular function via important cytosolic substrates including BAD, Cx43, PPARγ, and P90RSK 12, 41–43. It is reported that downstream P70S6K and RAC1 are integral signal transduction components required for platelet activation 19. Our study affirms this observation since ERK5 deficient platelets have attenuated activation coincident with reduced P70S6K and RAC1 expression only following MI. This adds evidence that ERK5 is not the final effecter for ischemia-mediated platelet dysregulation, but rather ERK5 may act as an upstream ischemic sensor or intermediary signaling molecule. How platelet ERK5 deficiency prevents infarct expansion and contractile dysfunction is especially important since this study is the first to ascribe a deleterious role for ERK5 following ischemia in the cardiovascular system. Endothelial ERK5 inhibition increases tissue damage following ROS exposure with a consequent protective effect on the heart and the vasculature 12, 34, 42, 43. The tissue specific difference in ERK5 functions underscores the need to better understand platelet activation in disease states. It also may make therapeutic targeting of ERK5 difficult because it must be targeted in a cell-specific manner. However, great advances are being made in drug delivery that may make specifically targeting platelets or downstream platelet ERK5 substrates possible.
ERK5 activation is agonist-specific. ERK5 deficiency alters platelet activation via PAR and the thromboxane receptor signaling, but not via collagen or P2Y12 receptors (Figures 1–2). A common downstream connection in PAR and thromboxane-mediated signaling in platelets is the Gαq subunit of G-Protein-Coupled Receptors (GPCRs) 44, 45. This is consistent with reports in other cell systems where ERK5 activation by surface agonists occurs in a Gαq-dependent manner 46, 47.
Microvascular occlusion and inflammatory cell infiltrates in the tissue adjacent to the initial infarcted tissue contributes significantly to infarct expansion and a decline in myocardial function 48. Because platelets are important immune modifiers we anticipated that WBC infiltration in myocardial sections following LAD ligation may differ in WT and platelet ERK5−/− hearts; however, we found no difference. MMP-mediated tissue damage and remodeling also contribute to infarct expansion. Platelets isolated from WT mice 3 days after LAD artery ligation had reduced MMP9 content and increased MMP activity compared to platelet ERK5−/− mice. An inverse pattern was noted in the LV apex. This suggests that platelet MMP release into the microvasculature of the myocardium leads to infarct expansion, at least in part in a platelet ERK5-dependent manner. Platelet MMP-mediated myocardial remodeling was proposed by Gresele et al. in a clinical study that showed a striking increase in platelet activity as well as MMP concentration in samples from the coronary sinus of patients during ACS compared to matched patients without ACS 49.
In our LAD ligation mouse model there is a marked decrease in P70S6K and RAC1 protein expression in ERK5−/− platelets following MI (Figure 7), while other proteins are unchanged. This suggests that ERK5 modulates either the synthesis or degradation of proteins important for sustained platelet activation in ischemic pathologies. Because ERK5 regulates the ubiquitin-proteasome pathway, and the platelet proteasome has been implicated in regulating platelet production and function, we focused on this mechanism and found that ERK5−/− platelets have increased protein ubiquitination and decreased expression of some proteins within 24 hrs of MI. This further emphasizes the need to consider mechanisms of platelet activation in pathologic states as fundamentally different compared to healthy conditions. Our studies focused on non-nuclear roles for ERK5 in circulating platelets, but ERK5 also has transcription regulatory functions that may influence gene expression patterns at the megakaryocyte level. Such ERK5 gene regulatory functions cannot be ruled out. Future studies are needed to fully consider megakaryocyte gene expression in healthy and post-MI conditions to determine whether ERK5 is also an ischemic sensor in the bone marrow, remote from the site of tissue injury.
Overall, these data indicate that ERK5 promotes ongoing platelet activation that augments myocardial damage following MI. Targeting ERK5 or its downstream mediators may represent a novel strategy for decreasing the thrombotic burden in patients with ischemic diseases such as MI and stroke.
Supplementary Material
Acknowledgments
We thank Dr. Jay Yang for introducing us to MMP biology.
Funding Sources: This study was supported by the following grants: NIH training grant 5T32HL066988-1 to SJC, Sable Heart Fund (Independent Order of Oddfellows) to SJC, American Heart Association (13EIA14250023) and National Institutes of Health R01HL124018 to CNM and National Institutes of Health R01HL108551 to JIA.
Footnotes
Disclosures: None.
References
- 1.Smyth SS, McEver RP, Weyrich AS, Morrell CN, Hoffman MR, Arepally GM, French PA, Dauerman HL, Becker RC, Platelet Colloquium P. Platelet functions beyond hemostasis. J Thromb Haemost. 2009;7:1759–1766. doi: 10.1111/j.1538-7836.2009.03586.x. [DOI] [PubMed] [Google Scholar]
- 2.Jia ZB, Tian H, Kang K, Miao HZ, Liu KY, Jiang SL, Wang LP. Expression of the tissue inhibitor of metalloproteinase-3 by transplanted vsmcs modifies heart structure and function after myocardial infarction. Transpl Immunol. 2014;30:149–158. doi: 10.1016/j.trim.2014.03.006. [DOI] [PubMed] [Google Scholar]
- 3.Kandalam V, Basu R, Abraham T, Wang X, Awad A, Wang W, Lopaschuk GD, Maeda N, Oudit GY, Kassiri Z. Early activation of matrix metalloproteinases underlies the exacerbated systolic and diastolic dysfunction in mice lacking timp3 following myocardial infarction. Am J Physiol Heart Circ Physiol. 2010;299:H1012–1023. doi: 10.1152/ajpheart.00246.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matsumura S, Iwanaga S, Mochizuki S, Okamoto H, Ogawa S, Okada Y. Targeted deletion or pharmacological inhibition of mmp-2 prevents cardiac rupture after myocardial infarction in mice. J Clin Invest. 2005;115:599–609. doi: 10.1172/JCI22304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heymans S, Luttun A, Nuyens D, Theilmeier G, Creemers E, Moons L, Dyspersin GD, Cleutjens JP, Shipley M, Angellilo A, Levi M, Nube O, Baker A, Keshet E, Lupu F, Herbert JM, Smits JF, Shapiro SD, Baes M, Borgers M, Collen D, Daemen MJ, Carmeliet P. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat Med. 1999;5:1135–1142. doi: 10.1038/13459. [DOI] [PubMed] [Google Scholar]
- 6.Wenger NK. 2011 accf/aha focused update of the guidelines for the management of patients with unstable angina/non-st-elevation myocardial infarction (updating the 2007 guideline): Highlights for the clinician. Clin Cardiol. 2012;35:3–8. doi: 10.1002/clc.20964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK Clopidogrel in Unstable Angina to Prevent Recurrent Events Trial I. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without st-segment elevation. N Engl J Med. 2001;345:494–502. doi: 10.1056/NEJMoa010746. [DOI] [PubMed] [Google Scholar]
- 8.Jia DM, Chen ZB, Zhang MJ, Yang WJ, Jin JL, Xia YQ, Zhang CL, Shao Y, Chen C, Xu Y. Cyp2c19 polymorphisms and antiplatelet effects of clopidogrel in acute ischemic stroke in china. Stroke. 2013;44:1717–1719. doi: 10.1161/STROKEAHA.113.000823. [DOI] [PubMed] [Google Scholar]
- 9.Lau WC, Gurbel PA, Watkins PB, Neer CJ, Hopp AS, Carville DG, Guyer KE, Tait AR, Bates ER. Contribution of hepatic cytochrome p450 3a4 metabolic activity to the phenomenon of clopidogrel resistance. Circulation. 2004;109:166–171. doi: 10.1161/01.CIR.0000112378.09325.F9. [DOI] [PubMed] [Google Scholar]
- 10.Aurigemma C, Scalone G, Tomai F, Altamura L, De Persio G, Stazi A, Lanza GA, Crea F. Persistent enhanced platelet activation in patients with acute myocardial infarction and coronary microvascular obstruction: Clinical implications. Thromb Haemost. 2014;111:122–130. doi: 10.1160/TH13-02-0166. [DOI] [PubMed] [Google Scholar]
- 11.Takeishi Y, Huang Q, Wang T, Glassman M, Yoshizumi M, Baines CP, Lee JD, Kawakatsu H, Che W, Lerner-Marmarosh N, Zhang C, Yan C, Ohta S, Walsh RA, Berk BC, Abe J. Src family kinase and adenosine differentially regulate multiple map kinases in ischemic myocardium: Modulation of map kinases activation by ischemic preconditioning. J Mol Cell Cardiol. 2001;33:1989–2005. doi: 10.1006/jmcc.2001.1463. [DOI] [PubMed] [Google Scholar]
- 12.Cameron SJ, Itoh S, Baines CP, Zhang C, Ohta S, Che W, Glassman M, Lee JD, Yan C, Yang J, Abe J. Activation of big map kinase 1 (bmk1/erk5) inhibits cardiac injury after myocardial ischemia and reperfusion. FEBS Lett. 2004;566:255–260. doi: 10.1016/j.febslet.2004.03.120. [DOI] [PubMed] [Google Scholar]
- 13.Pi X, Garin G, Xie L, Zheng Q, Wei H, Abe J, Yan C, Berk BC. Bmk1/erk5 is a novel regulator of angiogenesis by destabilizing hypoxia inducible factor 1alpha. Circ Res. 2005;96:1145–1151. doi: 10.1161/01.RES.0000168802.43528.e1. [DOI] [PubMed] [Google Scholar]
- 14.Hayashi M, Kim SW, Imanaka-Yoshida K, Yoshida T, Abel ED, Eliceiri B, Yang Y, Ulevitch RJ, Lee JD. Targeted deletion of bmk1/erk5 in adult mice perturbs vascular integrity and leads to endothelial failure. J Clin Invest. 2004;113:1138–1148. doi: 10.1172/JCI19890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kato Y, Tapping RI, Huang S, Watson MH, Ulevitch RJ, Lee JD. Bmk1/erk5 is required for cell proliferation induced by epidermal growth factor. Nature. 1998;395:713–716. doi: 10.1038/27234. [DOI] [PubMed] [Google Scholar]
- 16.Yan L, Carr J, Ashby PR, Murry-Tait V, Thompson C, Arthur JS. Knockout of erk5 causes multiple defects in placental and embryonic development. BMC Dev Biol. 2003;3:11. doi: 10.1186/1471-213X-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abe J, Kusuhara M, Ulevitch RJ, Berk BC, Lee JD. Big mitogen-activated protein kinase 1 (bmk1) is a redox-sensitive kinase. J Biol Chem. 1996;271:16586–16590. doi: 10.1074/jbc.271.28.16586. [DOI] [PubMed] [Google Scholar]
- 18.Suzaki Y, Yoshizumi M, Kagami S, Koyama AH, Taketani Y, Houchi H, Tsuchiya K, Takeda E, Tamaki T. Hydrogen peroxide stimulates c-src-mediated big mitogen-activated protein kinase 1 (bmk1) and the mef2c signaling pathway in pc12 cells: Potential role in cell survival following oxidative insults. J Biol Chem. 2002;277:9614–9621. doi: 10.1074/jbc.M111790200. [DOI] [PubMed] [Google Scholar]
- 19.Aslan JE, Tormoen GW, Loren CP, Pang J, McCarty OJ. S6k1 and mtor regulate rac1-driven platelet activation and aggregation. Blood. 2011;118:3129–3136. doi: 10.1182/blood-2011-02-331579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petzold T, Ruppert R, Pandey D, Barocke V, Meyer H, Lorenz M, Zhang L, Siess W, Massberg S, Moser M. Beta1 integrin-mediated signals are required for platelet granule secretion and hemostasis in mouse. Blood. 2013;122:2723–2731. doi: 10.1182/blood-2013-06-508721. [DOI] [PubMed] [Google Scholar]
- 21.Canault M, Ghalloussi D, Grosdidier C, Guinier M, Perret C, Chelghoum N, Germain M, Raslova H, Peiretti F, Morange PE, Saut N, Pillois X, Nurden AT, Cambien F, Pierres A, van den Berg TK, Kuijpers TW, Alessi MC, Tregouet DA. Human caldag-gefi gene (rasgrp2) mutation affects platelet function and causes severe bleeding. J Exp Med. 2014;211:1349–1362. doi: 10.1084/jem.20130477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feng H, Hu B, Liu KW, Li Y, Lu X, Cheng T, Yiin JJ, Lu S, Keezer S, Fenton T, Furnari FB, Hamilton RL, Vuori K, Sarkaria JN, Nagane M, Nishikawa R, Cavenee WK, Cheng SY. Activation of rac1 by src-dependent phosphorylation of dock180(y1811) mediates pdgfralpha-stimulated glioma tumorigenesis in mice and humans. J Clin Invest. 2011;121:4670–4684. doi: 10.1172/JCI58559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tong J, Li L, Ballermann B, Wang Z. Phosphorylation of rac1 t108 by extracellular signal-regulated kinase in response to epidermal growth factor: A novel mechanism to regulate rac1 function. Mol Cell Biol. 2013;33:4538–4551. doi: 10.1128/MCB.00822-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kwon T, Kwon DY, Chun J, Kim JH, Kang SS. Akt protein kinase inhibits rac1-gtp binding through phosphorylation at serine 71 of rac1. J Biol Chem. 2000;275:423–428. doi: 10.1074/jbc.275.1.423. [DOI] [PubMed] [Google Scholar]
- 25.Daugaard M, Nitsch R, Razaghi B, McDonald L, Jarrar A, Torrino S, Castillo-Lluva S, Rotblat B, Li L, Malliri A, Lemichez E, Mettouchi A, Berman JN, Penninger JM, Sorensen PH. Hace1 controls ros generation of vertebrate rac1-dependent nadph oxidase complexes. Nat Commun. 2013;4:2180. doi: 10.1038/ncomms3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barr JD, Chauhan AK, Schaeffer GV, Hansen JK, Motto DG. Red blood cells mediate the onset of thrombosis in the ferric chloride murine model. Blood. 2013;121:3733–3741. doi: 10.1182/blood-2012-11-468983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stolla M, Stefanini L, Andre P, Ouellette TD, Reilly MP, McKenzie SE, Bergmeier W. Caldag-gefi deficiency protects mice in a novel model of fcgamma riia-mediated thrombosis and thrombocytopenia. Blood. 2011;118:1113–1120. doi: 10.1182/blood-2011-03-342352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Figueras J, Monasterio Y, Lidon RM, Nieto E, Soler-Soler J. Thrombin formation and fibrinolytic activity in patients with acute myocardial infarction or unstable angina: In-hospital course and relationship with recurrent angina at rest. J Am Coll Cardiol. 2000;36:2036–2043. doi: 10.1016/s0735-1097(00)01023-8. [DOI] [PubMed] [Google Scholar]
- 29.Smid M, Dielis AW, Winkens M, Spronk HM, van Oerle R, Hamulyak K, Prins MH, Rosing J, Waltenberger JL, ten Cate H. Thrombin generation in patients with a first acute myocardial infarction. J Thromb Haemost. 2011;9:450–456. doi: 10.1111/j.1538-7836.2010.04162.x. [DOI] [PubMed] [Google Scholar]
- 30.Degousee N, Simpson J, Fazel S, Scholich K, Angoulvant D, Angioni C, Schmidt H, Korotkova M, Stefanski E, Wang XH, Lindsay TF, Ofek E, Pierre S, Butany J, Jakobsson PJ, Keating A, Li RK, Nahrendorf M, Geisslinger G, Backx PH, Rubin BB. Lack of microsomal prostaglandin e(2) synthase-1 in bone marrow-derived myeloid cells impairs left ventricular function and increases mortality after acute myocardial infarction. Circulation. 2012;125:2904–2913. doi: 10.1161/CIRCULATIONAHA.112.099754. [DOI] [PubMed] [Google Scholar]
- 31.Sawicki G, Salas E, Murat J, Miszta-Lane H, Radomski MW. Release of gelatinase a during platelet activation mediates aggregation. Nature. 1997;386:616–619. doi: 10.1038/386616a0. [DOI] [PubMed] [Google Scholar]
- 32.Trivedi V, Boire A, Tchernychev B, Kaneider NC, Leger AJ, O’Callaghan K, Covic L, Kuliopulos A. Platelet matrix metalloprotease-1 mediates thrombogenesis by activating par1 at a cryptic ligand site. Cell. 2009;137:332–343. doi: 10.1016/j.cell.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lim JH, Woo CH. Laminar flow activation of erk5 leads to cytoprotective effect via chip-mediated p53 ubiquitination in endothelial cells. Anat Cell Biol. 2011;44:265–273. doi: 10.5115/acb.2011.44.4.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Le NT, Takei Y, Shishido T, Woo CH, Chang E, Heo KS, Lee H, Lu Y, Morrell C, Oikawa M, McClain C, Wang X, Tournier C, Molina CA, Taunton J, Yan C, Fujiwara K, Patterson C, Yang J, Abe J. P90rsk targets the erk5-chip ubiquitin e3 ligase activity in diabetic hearts and promotes cardiac apoptosis and dysfunction. Circ Res. 2012;110:536–550. doi: 10.1161/CIRCRESAHA.111.254730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi DS, Smith MC, Campbell RA, Zimmerman PW, Franks ZB, Kraemer BF, Machlus KR, Ling J, Kamba P, Schwertz H, Rowley JW, Miles RR, Liu ZJ, Sola-Visner M, Italiano JE, Jr, Christensen H, Kahr WH, Li DY, Weyrich AS. Proteasome function is required for platelet production. J Clin Invest. 2014;124:3757–66. doi: 10.1172/JCI75247. Epub 2014 Jul 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gupta N, Li W, Willard B, Silverstein RL, McIntyre TM. Proteasome proteolysis supports stimulated platelet function and thrombosis. Arterioscler Thromb Vasc Biol. 2014;34:160–168. doi: 10.1161/ATVBAHA.113.302116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adler EM, Foley JF, Gough NR, Ray LB. 2007: Signaling breakthroughs of the year. Sci Signal. 2008;1:eg1. doi: 10.1126/stke.11eg1. [DOI] [PubMed] [Google Scholar]
- 38.Marinissen MJ, Servitja JM, Offermanns S, Simon MI, Gutkind JS. Thrombin protease-activated receptor-1 signals through gq- and g13-initiated mapk cascades regulating c-jun expression to induce cell transformation. J Biol Chem. 2003;278:46814–46825. doi: 10.1074/jbc.M305709200. [DOI] [PubMed] [Google Scholar]
- 39.Esparis-Ogando A, Diaz-Rodriguez E, Montero JC, Yuste L, Crespo P, Pandiella A. Erk5 participates in neuregulin signal transduction and is constitutively active in breast cancer cells overexpressing erbb2. Mol Cell Biol. 2002;22:270–285. doi: 10.1128/MCB.22.1.270-285.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Severin S, Ghevaert C, Mazharian A. The mitogen-activated protein kinase signaling pathways: Role in megakaryocyte differentiation. J Thromb Haemost. 2010;8:17–26. doi: 10.1111/j.1538-7836.2009.03658.x. [DOI] [PubMed] [Google Scholar]
- 41.Woo CH, Massett MP, Shishido T, Itoh S, Ding B, McClain C, Che W, Vulapalli SR, Yan C, Abe J. Erk5 activation inhibits inflammatory responses via peroxisome proliferator-activated receptor delta (ppardelta) stimulation. J Biol Chem. 2006;281:32164–32174. doi: 10.1074/jbc.M602369200. [DOI] [PubMed] [Google Scholar]
- 42.Pi X, Yan C, Berk BC. Big mitogen-activated protein kinase (bmk1)/erk5 protects endothelial cells from apoptosis. Circ Res. 2004;94:362–369. doi: 10.1161/01.RES.0000112406.27800.6F. [DOI] [PubMed] [Google Scholar]
- 43.Le NT, Heo KS, Takei Y, Lee H, Woo CH, Chang E, McClain C, Hurley C, Wang X, Li F, Xu H, Morrell C, Sullivan MA, Cohen MS, Serafimova IM, Taunton J, Fujiwara K, Abe J. A crucial role for p90rsk-mediated reduction of erk5 transcriptional activity in endothelial dysfunction and atherosclerosis. Circulation. 2013;127:486–499. doi: 10.1161/CIRCULATIONAHA.112.116988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rahman A, True AL, Anwar KN, Ye RD, Voyno-Yasenetskaya TA, Malik AB. Galpha(q) and gbetagamma regulate par-1 signaling of thrombin-induced nf-kappab activation and icam-1 transcription in endothelial cells. Circ Res. 2002;91:398–405. doi: 10.1161/01.res.0000033520.95242.a2. [DOI] [PubMed] [Google Scholar]
- 45.Nonne C, Lenain N, Hechler B, Mangin P, Cazenave JP, Gachet C, Lanza F. Importance of platelet phospholipase cgamma2 signaling in arterial thrombosis as a function of lesion severity. Arterioscler Thromb Vasc Biol. 2005;25:1293–1298. doi: 10.1161/01.ATV.0000163184.02484.69. [DOI] [PubMed] [Google Scholar]
- 46.Garcia-Hoz C, Sanchez-Fernandez G, Diaz-Meco MT, Moscat J, Mayor F, Ribas C. G alpha(q) acts as an adaptor protein in protein kinase c zeta (pkczeta)-mediated erk5 activation by g protein-coupled receptors (gpcr) J Biol Chem. 2010;285:13480–13489. doi: 10.1074/jbc.M109.098699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fukuhara S, Marinissen MJ, Chiariello M, Gutkind JS. Signaling from g protein-coupled receptors to erk5/big mapk 1 involves galpha q and galpha 12/13 families of heterotrimeric g proteins. Evidence for the existence of a novel ras and rho-independent pathway. J Biol Chem. 2000;275:21730–21736. doi: 10.1074/jbc.M002410200. [DOI] [PubMed] [Google Scholar]
- 48.Du XJ, Shan L, Gao XM, Kiriazis H, Liu Y, Lobo A, Head GA, Dart AM. Role of intramural platelet thrombus in the pathogenesis of wall rupture and intra-ventricular thrombosis following acute myocardial infarction. Thromb Haemost. 2011;105:356–364. doi: 10.1160/TH10-07-0449. [DOI] [PubMed] [Google Scholar]
- 49.Gresele P, Falcinelli E, Loffredo F, Cimmino G, Corazzi T, Forte L, Guglielmini G, Momi S, Golino P. Platelets release matrix metalloproteinase-2 in the coronary circulation of patients with acute coronary syndromes: Possible role in sustained platelet activation. Eur Heart J. 2011;32:316–325. doi: 10.1093/eurheartj/ehq390. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.