

Abstract
The advent of next generation sequencing (NGS) technology has provided the means to directly analyze the genetic material in primary cells or tissues of any species in a high throughput manner for mutagenic effects of potential genotoxic agents. In principle, direct, genome-wide sequencing of human primary cells and/or tissue biopsies would open up opportunities to identify individuals possibly exposed to mutagenic agents, thereby replacing current risk assessment procedures based on surrogate markers and extrapolations from animal studies. NGS-based tests can also precisely characterize the mutation spectra induced by genotoxic agents, improving our knowledge of their mechanism of action. Thus far, NGS has not been widely employed in genetic toxicology due to the difficulties in measuring low-abundant somatic mutations. Here, we review different strategies to employ NGS for the detection of somatic mutations in a cost-effective manner and discuss the potential applicability of these methods in testing the mutagenicity of genotoxic agents.
Keywords: genetic toxicology, next generation sequencing, mutagenicity, mutation, genome rearrangement
1. Introduction
Evaluating the hazardous effects of chemicals, such as pharmaceutical, environmental, and industrial compounds, or other agents, such as ionizing radiation, on human health is among the most important problems facing humankind in the modern world. Human contact with these toxic agents is growing exponentially, and even low-level exposures to environmental toxins/pollutants pose serious long-term health risks.
The genome is considered the central governor of all cellular processes and any interference that affects genome integrity may lead to serious health consequences [1]. As such, DNA lesions caused by genotoxic agents may have two different outcomes, i.e., cell death, either actual (apoptosis) or functional (senescence), and acquisition of mutations, due to erroneous DNA replication or repair. The second outcome is arguably more important. The induced mutations, e.g., base-pair substitutions, small insertions and deletions (indels), genome rearrangements and chromosomal events, such as numerical chromosome changes, are generally considered to be a cause of many congenital diseases [2] and the multi-step process of malignant transformation [3]. Also the process of aging has been considered to be ultimately caused by the accumulation of mutations [4, 5]. Thus, an assessment of the somatic mutation frequency in cells after treatment with potentially genotoxic agents or in biopsied tissues of individuals potentially exposed to such agents is a critical step in hazard evaluation (Fig. 1).
Fig. 1.

NGS-based assays allow for the direct assessment of potential genotoxic agents for mutagenicity (left) and individual risk of exposure to possible mutagenic agents, such as radiation.
Historically, short-term tests (STTs) for genotoxic chemicals were established and validated decades ago. STTs include the Ames bacterial mutagenesis test [6], in vitro cytogenetics tests [7, 8], and the in vitro and in vivo micronucleus assays [9, 10]. More recently, transgenic animal models have been generated that enable testing for spontaneous or induced mutations in any target organ or tissue using reporter genes introduced into various loci of animal genomes[11–14]. However, these tests are indirect and do not provide information on the sequence integrity of the entire genome. Indeed, the field of genetic toxicology has always been based on surrogate markers and has never been able to assess human health risks based on systematic analysis of the entire genome in primary human cells or tissues. Now that the next-generation sequencing (NGS) era is well underway, new methods have been developed to directly analyze genetic material in a genome-wide manner with single nucleotide resolution. Moreover, there is no dependency on any particular gene or cell line and genetic material derived from any cell or tissue can be analyzed. This makes NGS-based mutagenicity assays particularly suitable for use in genetic toxicology. However, there are some serious obstacles that have thus far essentially constrained the application of NGS in genotoxicity testing.
Here, we discuss problems and pitfalls in the implementation of NGS in genetic toxicology. We will first explain why the application of NGS in measuring low-abundant somatic mutations is not straightforward, then describe how this obstacle can be overcome, albeit at high cost, by taking a single cell approach and, finally, review various NGS approaches for assessing mutations, both point mutations and genome structural variations, in small amounts of DNA at low cost.
2. Direct mutation assessment by next generation sequencing
Unlike conventional Sanger sequencing [15], next-generation sequencing is capable of processing hundreds of millions of DNA fragments in parallel, providing the previously unprecedented opportunity to decode the entire genome within days. Due to the relatively simple nature of genetic material, all possible mutations are, in principle, amenable to detection by direct sequencing. However, this is only true for mutations that are present in most or all cells in a given tissue or populations. Indeed, in genetic toxicology the mutations one wishes to detect are typically random, de novo mutations, turning the cell population under study into a mixture of genomes. In such genome mosaics each cell harbors hundreds if not thousands of unique, de novo mutations.
In principle, cellular heterogeneity in genome sequence integrity can be addressed by NGS in a straightforward way by sequencing at great depth. Sequence variants, even at very low abundance, should then be identifiable among the sequence reads at each locus. However, the reliable identification of mutations in this way is constrained by errors associated with each step of the NGS workflow (Fig. 2). Detection of different types of mutation, i.e., point mutations (base substitutions and small indels) and large structural variation (translocations, inversions, large insertions and deletions) is affected in different ways by these errors, which is why we will discuss each mutation type separately.
Fig. 2.
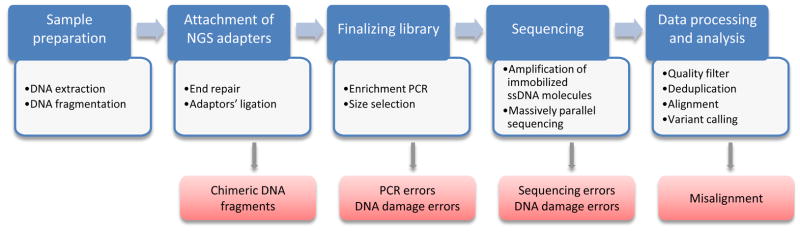
General workflow of NGS-based assays and putative errors associated with each step.
2.1. Assessment of point mutations and small indels
In principle, somatic point mutations and small indels that occur at low frequencies, i.e., down to 1 x 10−6 per locus, can be detected easily enough by sequencing the entire genome or part of it. However, straightforward detection of somatic mutations as variant reads after sequencing at great depth is essentially precluded by sequencing errors and artifacts introduced during library preparation (Fig. 2). For example, errors may result from base mis-incorporation during PCR amplification, which is often part of the library preparation protocol. PCR errors stem from less than absolute fidelity of polymerase. If they occur during the first round of amplification (the worst case scenario) they will be propagated and inherited by 50% of the daughter molecules of the starting template [16]. PCR errors may be exacerbated by the presence of damaged bases in the template molecule, which may readily lead to mis-incorporation of bases in the nascent strand, e.g., G->T mutations at 8-oxo-G lesions, which favor insertion of adenosine [17] or C->T at deaminated cytosines [18]. Sequencing errors, i.e. erroneous base calls, missed bases, or homopolymer-length errors, occur during sequencing. These types of errors are usually randomly distributed along the reads and will differ between the two strands (it's highly unlikely that the same error will occur when sequencing the opposing strands). The frequency of sequencing errors for contemporary platforms such as Illumina and Ion Torrent is estimated at 0.1–0.7 insertions/deletions and substitutions per 100 nucleotides of sequencing data [19]. Combined, these sources of error could result in an artifactual mutation frequency of up to 1% [20, 21] efficiently masking true mutations, which usually occur at a much lower frequency.
To address the issue of errors, all variant-calling algorithms utilize a consensus model.That is each analyzed region of the genome must be represented by several independent sequencing reads, i.e., independently sequenced fragments representing the same loci but originating from genomes of different cells. Randomly occurring errors are filtered out, while true mutations can be identified based on their presence in 50% of the reads (a hetereozygous mutation affects only one allele). This strategy works well if the same mutations are present in all cells, e.g., germline mutations or clonally amplified mutations in tumor tissue. However, ultra-low-abundant somatic mutations, often unique for each cell are discarded because like sequencing errors they are present in one read [22, 23] (Fig. 3). To address these issues several approaches have been developed with the core idea to identify and verify the variant by consensus analysis of dependent reads, i.e. independently sequenced copies/strands of one original DNA fragment.
Fig. 3.
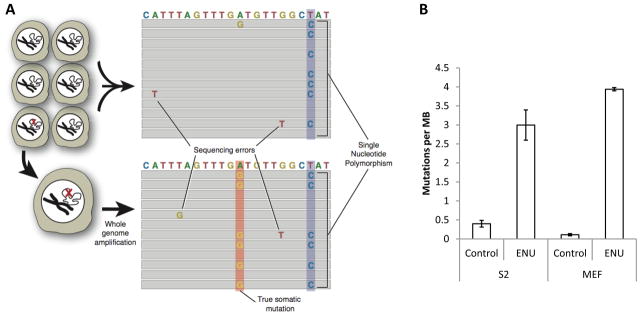
(A) Detecting point mutations and small indels in single cells. (B) ENU significantly elevates mutation frequency in both Drosophila S2 cells (n=3 for treated and untreated) cells and mouse embryonic fibroblasts (n=2 for treated and untreated). For details, see [22].
2.1.1. Single cell approach
The most logical approach to address the issue of low-abundant somatic mutations is the sequencing of single-cell genomes first developed in our laboratory [22]. The method is based on the sequencing of the genome of a single cell after whole genome amplification (WGA) using isothermal multiple displacement amplification protocol (MDA) [24–27]. This amplification provides multiple copies of the original single cell genome, which is sequenced and analyzed with standard analytical tools following the consensus model in NGS. True somatic mutations, which are always heterozygous, will now be present in about 50% of overlapping reads at any given locus (one mutant allele), whereas sequencing errors should only affect single reads scattered throughout the sequences (Fig. 3A). Single nucleotide polymorphisms between the sample and the reference genome can be identified by also sequencing the bulk, unamplified DNA and filtered out [22]. Amplification errors are not likely to be a serious problem here. Indeed, in the worst case scenario an amplification error is induced during the first round of amplification, in which case it will show up in only one of the 8 strands for an amplified diploid locus [28]. The single cell protocol was successfully used for detection of mutations induced by treatment with N-ethyl-N-nitrosourea (ENU), a powerful direct-acting mutagen, which causes mostly point mutations [29, 30] (Fig.3B).
The cost of whole genome sequencing in this single-cell approach is an issue for its routine application in genetic toxicology testing. While there are ways to reduce sequencing costs, for example, by using a reduced representation approach in which a randomly selected fraction of the genome is sequenced instead of the whole genome [31], sequencing costs are still high and for each determination multiple single cells plus the bulk, unamplified DNA must be sequenced. Therefore, we must consider alternative NGS approaches that greatly reduce this cost.
2.1.2. PCR copy consensus assay
The Safe-Sequencing System (Safe-SeqS) [32], introduced in 2011, was the first NGS-method for the detection of ultra-rare mutations in bulk DNA extracted from a population of cells. This approach utilizes a consensus analysis of independently sequenced, dependent fragments, i.e., a family of copies of one original template DNA molecule created during redundant PCR amplification of a sequencing library (Fig. 4). One of the key features of the Safe-SeqS assay is the assignment of a unique identifier (UID) to each DNA fragment. The UID is a molecular tag consisting of 12–14 random nucleotides uniquely marking each template DNA molecule [33]. Based on the presence of identical tags, the UID families containing copies of a particular starting molecule are identified and further analyzed. Consensus analysis of UID families with two or more members allows an effective means to filter out randomly scattered sequencing errors, whereas a true mutation is identified as a variant found in at least 95% of the UID family members. Theoretically, the Safe-SeqS approach allows elimination of PCR errors. Indeed, an error introduced at the first round of PCR during library amplification is inherited by 25% of all daughter molecules, whereas true mutations are expected in 100% of fragments (cut-off level –95%). However, possible errors introduced during the assignment of a UID, also performed by 2 cycles of PCR or DNA damage artifacts will be present in 100% of UID family members and, consequently, indistinguishable from true mutations.
Fig. 4.
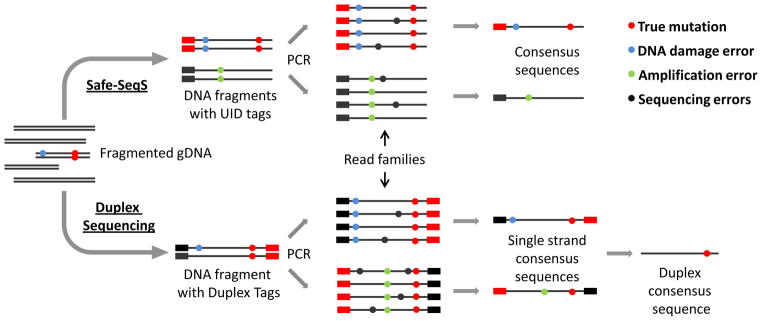
Schematic depiction of the Safe-Sequencing System and Duplex Sequencing assays.
The Duplex Sequencing approach [20] resolves the issue of sequencing errors by identifying and analyzing both strands of the DNA template (Fig. 4). Similar to the Safe-SeqS method, tagging of starting DNA fragments prior to PCR amplification is used. However, each of the two strands is tagged independently, amplified, and then sequenced. During data analysis, as in the Safe-SeqS approach, members of each PCR group, separate for each strand, are identified based on the presence of the same tag and used to create a single strand consensus sequence (SSCS) with all random sequencing errors eliminated. Only families containing three or more members and showing no less than 90% concordance for each position are used for the analysis. At the next step SSCSs representing two complementary strands are identified based on the presence of both molecular tags and used to form the so-called duplex consensus sequence (DCS), where only variants in agreement for both SSCSs are accepted as true mutations. Since the probability of the same error on both strands is negligible (estimated at 3.8 x 10−10) errors originating from damaged DNA bases, as well as first round PCR errors are effectively eliminated. The notion of comparative analysis of independently sequenced template strands allows to greatly reduce the frequency of errors from 2.0 x 10−4 errors/bp reported for Safe-SeqS down to 2.5 x 10−6 errors/bp when utilizing the Duplex Sequencing method. Of note, the latter value is in agreement with a previously reported 3.0 x 10−6 errors/bp determined by a well-established genetics methods [34].
Safe-SeqS and particularly Duplex Sequencing represent major advances in identifying random, low-abundant mutations in DNA from bulk cell populations or issues. However, they both suffer from a very low effective coverage due to the need for redundant PCR amplification. In practice, therefore, these two approaches are only applicable for analysis of small targets, such as mitochondrial DNA, plasmids, or individual genes.
2.1.3. Circle sequencing
A very elegant solution for the detection of somatic mutations that overcomes the limitations of previous methods was recently offered by Sawyer’s group [35]. The authors developed a new library preparation strategy – “circle sequencing” (Fig. 5). Genomic DNA, fragmented to the size of approximately 1/3 of the anticipated read length, is circularized by ligation of the fragment ends and isothermally amplified using a rolling circle amplification (RCA) approach [36]. During RCA, which is initiated from random hexamers, each circularized DNA fragment serves as a template that gives rise to a linear DNA stretch where the sequence of the template is copied multiple times in tandem. Sequencing libraries prepared from amplified DNA consists of fragments each containing ~3 copies of starting template. Next, these copies are used to compute a consensus sequence where sequencing and amplification errors are discarded and true mutations revealed, similar to approaches based on analysis of PCR duplicates. Errors originating from damaged bases are eliminated by the treatment of starting material with uracil-DNA glycosylase (UDG) and formamidopyrimidine-DNA glycosylase (Fpg), which recognize and remove uracil and oxidatively damaged bases [37, 38], respectively, preventing amplification of compromised templates.
Fig. 5.

Schematic depiction of the circle sequencing assay for mutation detection. Genomic DNA is ligated into circles and amplified by RCA. Sequenced DNA copies are collapsed into a consensus sequence to determine true point mutations/indels.
There are two major advantages in this approach. First, the starting DNA molecule is the only template for amplification and any possible errors are not amplified further. Since all copies are truly independent, there is no need for a large number of copies to form a reliable consensus sequence – assuming an error rate of 1%, or 1 per 100nt, the probability of getting the same error in three independent copies is less than 3 x 10−8. The other advantage is that since all the copies of the template are physically linked in one fragment, there is no need for molecular tagging to identify “read families”. That is, all the members belonging to the same group of supporting copies are automatically assembled in a single sequencing read. This makes it possible to avoid redundant amplification and sequencing, since the majority of reads contain all the information for consensus analysis, achieving a much higher efficiency in utilization of sequencing data – 20.2% of unique supported bases. This, as well as a low error rate (7.6 x 10−6), comparable to that reported for Duplex Sequencing, potentially make circle sequencing suitable for detecting point mutations and small indels in whole genomes.
2.2. Assessment of structural variants
Thus far, NGS methods were discussed to detect point mutations. However, genome structural variation as induced by clastogenic agents, i.e. agents capable of inducing DNA breakage, such as radiation and bleomycin, are often considered as more serious genotoxic risks [39–41]. Current methods to detect structural variants (SVs) by NGS are based on finding anomalous distributions of the paired ends or finding reads spanning breakpoints, i.e., the points of anomalous junction of genome fragments accompanying every SV [42–47] (Fig. 6). Thus, the identification of SVs is in a sense the identification of DNA breakpoints and wholly dependent on the efficiency and reliability of mapping sequencing data to the reference genome.
Fig. 6.

(A) Paired-end approach for detection of structural variants is based on discrepancies in mapping the ends of a sequenced fragment to the reference genome. When DNA fragments of a particular size are sequenced from both ends the paired reads should be positioned at a known distance from each other when aligned to a reference sequence. (B) Split-read approach is based on finding continuous sequencing reads with anomalous alignment of different parts.
For both approaches if the distance and orientation between the read pairs (or parts of the read) differs from that on the reference genome, then a rearrangement event, such as a deletion or insertion is implied. It is also possible that one of the two read pairs (part of the read) maps to another chromosome, indicating a chromosomal translocation. An event of 400 bp deletion is shown for both approaches. Generally the split-read approach relays on longer read length, but has enhanced resolution and, unlike the paired-end method, allows for the precise identification of DNA breakpoints.
There are many sophisticated analytical tools available for the detection of SVs [48], but all of them require that the breakpoint is detected in multiple overlapping reads at that locus. . Like a point mutation or small indel, an SV is an ultra-rare event, randomly occurring at the single-cell level. That is, each SV is unique and can be represented only by a single DNA fragment with no supporting reads, similar to point mutations. Similar to point mutations, ultra-low abundant SVs could be detected by sequencing at very high depth. However, identification of uniquely variant reads is essentially constrained by errors associated with the sequencing process, albeit of a very different nature. In principle, similar to point mutations, one could take a single-cell approach to address the genome mosaicism resulting from de novo SVs. However, this has the same disadvantage of the high cost associated with the need to sequence multiple cells. In addition, artifacts due to the necessary whole genome amplification are a much more serious problem with SVs than point mutations.
2.2.1. Erroneous SV calling
There are two main sources of errors during the identification of SVs: (i) the artificial creation of chimeric DNA molecules during ligation-based library preparation and (ii) mapping errors (Fig. 2). Both, Illumina and Ion Torrent, the most common sequencing platforms, require attachment of adapters, i.e., oligonucleotides of certain base composition, at the ends of DNA fragments to be sequenced. This step is usually performed by ligation of double-stranded adapters to the fragmented and end-repaired DNA. However, DNA fragments are capable of ligating to each other during this step, resulting in artificial chimeras. Since this is a random, relatively infrequent event, it does not create any problems for detecting germline SVs, but makes detection of rare somatic SVs virtually impossible. To address this problem, one approach uses two rounds of size selection before and after ligation of adapters [49]. Chimeric templates will be significantly longer and are removed during the second procedure of size selection. The other way to resolve the issue of chimeric templates is to completely avoid ligation but instead use a transposon-based method of library preparation [50]. Since this approach lacks ligase activity the resulting library is free from artificial chimeras.
The second source of SV miscalls, wrongful alignment, stems from the presence of repetitive elements and regions with low complexity within the mammalian genome that are not amenable to unique or unequivocal alignment. Indeed, the fraction of the human genome uniquely mappable is 79.6% for 30 nt sequence tags and 86.7% for 50 nt sequence tags [51]. Uncertainty in the placement of sequencing reads onto a reference genome often leads to aberrant mapping and, consequently, false positive SV calls. Since this is exclusively a computational problem it cannot be resolved by changes in sequencing protocols. This problem also cannot be resolved by consensus analysis of duplicates of the presumably aberrant read, as is done for point mutations and small indels, since errors are created at the analysis step and all the copies of the original fragment, representing a potential somatic SV, will be handled in the same manner by the alignment tool and will appear as aberrant too. While at the same time, independent DNA fragments overlapping the questionable DNA breakpoints simply do not exist. Thus, none of the available approaches are capable of reliably detecting somatic SVs.
2.2.2. Single read approach
As we discussed above, existing approaches to detect SVs rely on overlapping supporting reads and cannot be used for detecting ultra-rare somatic SVs, due to the absence of independent sequencing reads spanning the same somatic SV. As mentioned, the main source of artifacts, i.e., artificial creation of chimeric DNA molecules during ligation-based library preparation, can be circumvented by using a transposon-based method of library preparation. However the problem of misalignment remains unresolved. This inspired us to design a novel computational algorithm for the assessment of SVs based on low-coverage sequencing and finding a single read representing a true DNA breakpoint and validate this call computationally (Maslov et al., submitted).
3. Summary and future prospects
The advent of next generation sequencing technology now enables us, in principle, to directly analyze the mutational endpoints of environmental mutagens and carcinogens. This has several big advantages over existing genotoxicity assays. First, the nature of NGS allows to analyze genetic material for mutations independent of its source. Thus, wide application of NGS-based tests utilizing human or animal cell lines, cells or tissues from experimental animals and primary material from humans will greatly improve risk assessment procedures for genotoxic agents because they will no longer require the widespread safety assumptions associated with surrogate markers and mouse/human extrapolations.
Second, NGS provides direct information down to the base pair level, enabling the comprehensive characterization of all types of induced mutations and mutational landscapes on a genome-wide scale. The mutational signatures of tested genotoxic agents in their natural target cells or tissues will help to further elucidate their mechanism of action and potentially allows the identification of an unknown agent based on its mutational signature.
Third, the NGS-based tests can be easily automated. In fact, there are already many commercially available machines that perform automatic library preparation; data processing with established computational pipelines also do not require intervention. This makes it possible to utilize NGS-based assays in a high-throughput mode, which is critical for testing numerous compounds.
As we have shown in this review, the one single hindrance to the large-scale application of the new sequencing technology in the area of genetic toxicology, namely the high error rate associated with the sequencing procedure, which confounds the low-abundant mutations subject to genotoxicity studies, can now be addressed by various approaches. Indeed, even genome structural variation, generally considered to be the most difficult to detect when occurring as low-abundant events, can now be detected by NGS at low coverage. This is critically important in genotoxicity testing because there are few reliable ways of detecting clastogenic effects. Indeed, the detection of somatic structural variants for testing potential clastogens is not a trivial task. Assays are limited to cytogenetic damage, a small fraction of all possible SVs. Even the available transgenic reporter models are less suitable for detecting clastogenic effects. For instance, the currently commercially available lacI mouse model can only be used for detecting point mutations. The lacZ plasmid model, on the other hand, can be used for detecting SVs, but due to its small target the sensitivity of even this model for clastogenic agents is not great [52–54] which can be improved only by crossing it with DNA repair deficient mice [55].
The important metric to consider for the evaluation of practical applicability of any new test system is the cost. This is particularly important for genotoxicity assays that require the assessment of multiple potentially hazardous agents under different conditions and at different doses. Although the cost of NGS has greatly declined since it was introduced, it still remains high. This is why the efficiency of NGS-based assays to utilize all raw sequencing data is critical. We previously demonstrated that analysis of ~10% of the genome is sufficient for the reliable detection of point mutations introduced by a genotoxic intervention [22]. Given the current output of the Illumina HiSeq2500 platform, providing enough data for 10X human genome coverage per each flow cell lane, it is possible to perform genome-wide, point mutation analysis by multiplexing ~20 samples per one lane by using circle sequencing, assuming its demonstrated efficiency 20% and required 3X coverage at random loci (10X data output x 0.2 data utilization / 0.1X required coverage). This corresponds to approximately $100 per data point. As for detection of structural variants, taking the same requirement for coverage (10%) and expected sensitivity ~36%, it will be possible to combine ~25 samples per lane, if utilizing the single-read assay for SVs.
Hence, multiple approaches have now become available to apply NGS successfully in detecting low-abundant mutational events in genotoxicity testing with human and animal primary cells and tissues. Since the first next generation sequencing platform was introduced in 2005 [56] the concept of massively parallel sequencing has found application in many scientific fields as well as in clinical genetic testing. Thus far genetic toxicology has lagged behind. This gap is now on the verge of being fixed.
Acknowledgments
This work was supported by NIH grant P01 AG017242 (JV), Albert Einstein College of Medicine Human Genome Program Pilot project grant (AYM) and the Einstein-Nathan Shock Center of Excellence Pilot and feasibility grant 5P30AG038072–05 (AYM).
Footnotes
Conflict of Interest statement.
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Maslov AY, Vijg J. Genome instability, cancer and aging. Biochim Biophys Acta. 2009;1790(10):963–9. doi: 10.1016/j.bbagen.2009.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Veltman JA, Brunner HG. De novo mutations in human genetic disease. Nat Rev Genet. 2012;13(8):565–75. doi: 10.1038/nrg3241. [DOI] [PubMed] [Google Scholar]
- 3.Poon SL, et al. Mutation signatures of carcinogen exposure: genome-wide detection and new opportunities for cancer prevention. Genome Med. 2014;6(3):24. doi: 10.1186/gm541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361(15):1475–85. doi: 10.1056/NEJMra0804615. [DOI] [PubMed] [Google Scholar]
- 5.Vijg J, Suh Y. Genome instability and aging. Annu Rev Physiol. 2013;75:645–68. doi: 10.1146/annurev-physiol-030212-183715. [DOI] [PubMed] [Google Scholar]
- 6.McCann J, et al. Detection of carcinogens as mutagens in the Salmonella/microsome test: assay of 300 chemicals. Proc Natl Acad Sci U S A. 1975;72(12):5135–9. doi: 10.1073/pnas.72.12.5135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galloway SM, et al. Development of a standard protocol for in vitro cytogenetic testing with Chinese hamster ovary cells: comparison of results for 22 compounds in two laboratories. Environ Mutagen. 1985;7(1):1–51. doi: 10.1002/em.2860070102. [DOI] [PubMed] [Google Scholar]
- 8.Mitchell AD, Rudd CJ, Caspary WJ. Evaluation of the L5178Y mouse lymphoma cell mutagenesis assay: intralaboratory results for sixty-three coded chemicals tested at SRI International. Environ Mol Mutagen. 1988;12(Suppl 13):37–101. doi: 10.1002/em.2860120504. [DOI] [PubMed] [Google Scholar]
- 9.Schmid W. The micronucleus test. Mutat Res. 1975;31(1):9–15. doi: 10.1016/0165-1161(75)90058-8. [DOI] [PubMed] [Google Scholar]
- 10.Fenech M. The in vitro micronucleus technique. Mutat Res. 2000;455(1–2):81–95. doi: 10.1016/s0027-5107(00)00065-8. [DOI] [PubMed] [Google Scholar]
- 11.Gossen JA, et al. Efficient rescue of integrated shuttle vectors from transgenic mice: a model for studying mutations in vivo. Proc Natl Acad Sci U S A. 1989;86(20):7971–5. doi: 10.1073/pnas.86.20.7971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vijg J, van Steeg H. Transgenic assays for mutations and cancer: current status and future perspectives. Mutat Res. 1998;400(1–2):337–54. doi: 10.1016/s0027-5107(98)00030-x. [DOI] [PubMed] [Google Scholar]
- 13.Garcia AM, et al. A model system for analyzing somatic mutations in Drosophila melanogaster. Nat Methods. 2007;4(5):401–3. doi: 10.1038/NMETH1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dolle ME, et al. Evaluation of a plasmid-based transgenic mouse model for detecting in vivo mutations. Mutagenesis. 1996;11(1):111–8. doi: 10.1093/mutage/11.1.111. [DOI] [PubMed] [Google Scholar]
- 15.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977;74(12):5463–7. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanagawa T. Bias and artifacts in multitemplate polymerase chain reactions (PCR) J Biosci Bioeng. 2003;96(4):317–23. doi: 10.1016/S1389-1723(03)90130-7. [DOI] [PubMed] [Google Scholar]
- 17.Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991;349(6308):431–4. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- 18.Stiller M, et al. Patterns of nucleotide misincorporations during enzymatic amplification and direct large-scale sequencing of ancient DNA. Proc Natl Acad Sci U S A. 2006;103(37):13578–84. doi: 10.1073/pnas.0605327103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Junemann S, et al. Updating benchtop sequencing performance comparison. Nat Biotechnol. 2013;31(4):294–6. doi: 10.1038/nbt.2522. [DOI] [PubMed] [Google Scholar]
- 20.Schmitt MW, et al. Detection of ultra-rare mutations by next-generation sequencing. Proc Natl Acad Sci U S A. 2012;109(36):14508–13. doi: 10.1073/pnas.1208715109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quail MA, Swerdlow H, Turner DJ. Improved protocols for the illumina genome analyzer sequencing system. Curr Protoc Hum Genet. 2009;Chapter 18(Unit 18 ):2. doi: 10.1002/0471142905.hg1802s62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gundry M, et al. Direct, genome-wide assessment of DNA mutations in single cells. Nucleic Acids Res. 2012;40(5):2032–40. doi: 10.1093/nar/gkr949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gundry M, Vijg J. Direct mutation analysis by high-throughput sequencing: from germline to low-abundant, somatic variants. Mutat Res. 2012;729(1–2):1–15. doi: 10.1016/j.mrfmmm.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dean FB, et al. Rapid amplification of plasmid and phage DNA using Phi 29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res. 2001;11(6):1095–9. doi: 10.1101/gr.180501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dean FB, et al. Comprehensive human genome amplification using multiple displacement amplification. Proc Natl Acad Sci U S A. 2002;99(8):5261–6. doi: 10.1073/pnas.082089499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Panelli S, et al. Towards the analysis of the genomes of single cells: further characterisation of the multiple displacement amplification. Gene. 2006;372:1–7. doi: 10.1016/j.gene.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 27.Geigl JB, Speicher MR. Single-cell isolation from cell suspensions and whole genome amplification from single cells to provide templates for CGH analysis. Nat Protoc. 2007;2(12):3173–84. doi: 10.1038/nprot.2007.476. [DOI] [PubMed] [Google Scholar]
- 28.Dear PH. One by one: Single molecule tools for genomics. Brief Funct Genomic Proteomic. 2003;1(4):397–416. doi: 10.1093/bfgp/1.4.397. [DOI] [PubMed] [Google Scholar]
- 29.Russell WL, et al. Specific-locus test shows ethylnitrosourea to be the most potent mutagen in the mouse. Proc Natl Acad Sci U S A. 1979;76(11):5818–9. doi: 10.1073/pnas.76.11.5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hrabe de Angelis MH, et al. Genome-wide, large-scale production of mutant mice by ENU mutagenesis. Nat Genet. 2000;25(4):444–7. doi: 10.1038/78146. [DOI] [PubMed] [Google Scholar]
- 31.Altshuler D, et al. An SNP map of the human genome generated by reduced representation shotgun sequencing. Nature. 2000;407(6803):513–6. doi: 10.1038/35035083. [DOI] [PubMed] [Google Scholar]
- 32.Kinde I, et al. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci U S A. 2011;108(23):9530–5. doi: 10.1073/pnas.1105422108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCloskey ML, et al. Encoding PCR products with batch-stamps and barcodes. Biochem Genet. 2007;45(11–12):761–7. doi: 10.1007/s10528-007-9114-x. [DOI] [PubMed] [Google Scholar]
- 34.Thomas DC, et al. Fidelity of mammalian DNA replication and replicative DNA polymerases. Biochemistry. 1991;30(51):11751–9. doi: 10.1021/bi00115a003. [DOI] [PubMed] [Google Scholar]
- 35.Lou DI, et al. High-throughput DNA sequencing errors are reduced by orders of magnitude using circle sequencing. Proc Natl Acad Sci U S A. 2013;110(49):19872–7. doi: 10.1073/pnas.1319590110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fire A, Xu SQ. Rolling replication of short DNA circles. Proc Natl Acad Sci U S A. 1995;92(10):4641–5. doi: 10.1073/pnas.92.10.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lindahl T, et al. DNA N-glycosidases: properties of uracil-DNA glycosidase from Escherichia coli. J Biol Chem. 1977;252(10):3286–94. [PubMed] [Google Scholar]
- 38.Tchou J, et al. Substrate specificity of Fpg protein. Recognition and cleavage of oxidatively damaged DNA. J Biol Chem. 1994;269(21):15318–24. [PubMed] [Google Scholar]
- 39.Inaki K, Liu ET. Structural mutations in cancer: mechanistic and functional insights. Trends Genet. 2012;28(11):550–9. doi: 10.1016/j.tig.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 40.Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer. 2007;7(4):233–45. doi: 10.1038/nrc2091. [DOI] [PubMed] [Google Scholar]
- 41.Vijg J, Dolle ME. Large genome rearrangements as a primary cause of aging. Mech Ageing Dev. 2002;123(8):907–15. doi: 10.1016/s0047-6374(02)00028-3. [DOI] [PubMed] [Google Scholar]
- 42.Chen K, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009;6(9):677–81. doi: 10.1038/nmeth.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Korbel JO, et al. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318(5849):420–6. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang J, Wang J, Wu Y. An improved approach for accurate and efficient calling of structural variations with low-coverage sequence data. BMC Bioinformatics. 2012;13(Suppl 6):S6. doi: 10.1186/1471-2105-13-S6-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang ZD, et al. Identification of genomic indels and structural variations using split reads. BMC Genomics. 2011;12:375. doi: 10.1186/1471-2164-12-375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang Y, Wang Y, Brudno M. PRISM: pair-read informed split-read mapping for base-pair level detection of insertion, deletion and structural variants. Bioinformatics. 2012;28(20):2576–83. doi: 10.1093/bioinformatics/bts484. [DOI] [PubMed] [Google Scholar]
- 47.Rausch T, et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28(18):i333–i339. doi: 10.1093/bioinformatics/bts378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abel HJ, Duncavage EJ. Detection of structural DNA variation from next generation sequencing data: a review of informatic approaches. Cancer Genet. 2013;206(12):432–40. doi: 10.1016/j.cancergen.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quail MA, et al. A large genome center's improvements to the Illumina sequencing system. Nat Methods. 2008;5(12):1005–10. doi: 10.1038/nmeth.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adey A, et al. Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol. 2010;11(12):R119. doi: 10.1186/gb-2010-11-12-r119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rozowsky J, et al. PeakSeq enables systematic scoring of ChIP-seq experiments relative to controls. Nat Biotechnol. 2009;27(1):66–75. doi: 10.1038/nbt.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boerrigter ME, et al. Plasmid-based transgenic mouse model for studying in vivo mutations. Nature. 1995;377(6550):657–9. doi: 10.1038/377657a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mahabir AG, et al. Detecting genotoxic effects of potential clastogens: an in vivo study using the transgenic lacZ plasmid and the MutaMouse model. Mutat Res. 2008;652(2):151–7. doi: 10.1016/j.mrgentox.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 54.Mirsalis JC, Monforte JA, Winegar RA. Transgenic animal models for detection of in vivo mutations. Annu Rev Pharmacol Toxicol. 1995;35:145–64. doi: 10.1146/annurev.pa.35.040195.001045. [DOI] [PubMed] [Google Scholar]
- 55.Mahabir AG, et al. DNA-repair-deficient Rad54/Rad54B mice are more sensitive to clastogens than wild-type mice. Toxicol Lett. 2008;183(1–3):112–7. doi: 10.1016/j.toxlet.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 56.Margulies M, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437(7057):376–80. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]