

Abstract
Nitric oxide (NO) and its derivatives play important roles in the physiology and pathophysiology of the liver. Despite its diverse and complicated roles, certain patterns of the effect of NO on the pathogenesis and progression of liver diseases are observed. In general, NO derived from endothelial NO synthase (eNOS) in liver sinusoidal endothelial cells (LSECs) is protective against disease development, while inducible NOS (iNOS)-derived NO contributes to pathological processes. This review addresses the roles of NO in the development of various liver diseases with a focus on recently published articles. We present here two recent advances in understanding NO-mediated signaling, nitrated fatty acids and S-guanylation, and conclude with suggestions on future directions of NO-related studies on the liver.
Keywords: eNOS, iNOS, endothelial cells, portal hypertension, inflammation
Nitric oxide in liver physiology and pathophysiology
Nitric oxide (NO) is an important mediator of liver physiology and pathophysiology. NO is generated by three isoforms of nitric oxide synthases (NOSs): neuronal NOS (nNOS; NOS1), inducible NOS (iNOS; NOS2), and endothelial NOS (eNOS; NOS3) [1]. NOS catalyzes the oxidation of L-arginine to NO and citrulline [2]. In the cell, nNOS and iNOS are predominantly found in the cytosol, while eNOS binds to the membrane via palmitoylation and myristoylation [3].
In liver biology, eNOS and iNOS are major players, whereas the role of nNOS is little known. eNOS is mainly expressed in liver sinusoidal endothelial cells (LSECs) and endothelial cells of the hepatic artery, portal vein, central vein, and lymphatic vessels. eNOS is constitutively expressed and produces small amounts of NO in response to stimuli such as flow shear stress and vascular endothelial growth factor (VEGF) (Figure 1A). eNOS-derived NO maintains liver homeostasis and inhibits pathological conditions in the liver. In contrast, iNOS is induced in various liver cells, including LSECs, hepatocytes, Kupffer cells (liver resident macrophages), hepatic stellate cells (HSCs), smooth muscle cells, cholangiocytes, and other immune cells [4-6]. Under many pathological conditions, iNOS produces large amounts of NO, which is a major source of reactive nitrogen species (RNS) (Figure 1B). Particularly, peroxynitrite (ONOO-) can damage a wide range of cellular molecules including DNA, lipids, and proteins, and can also facilitate protein nitration, affecting structure and function of many target proteins [7] (Figure 1C).
Figure 1. The role of nitric oxide (NO) in the hepatic sinusoids in normal and pathologic liver conditions.
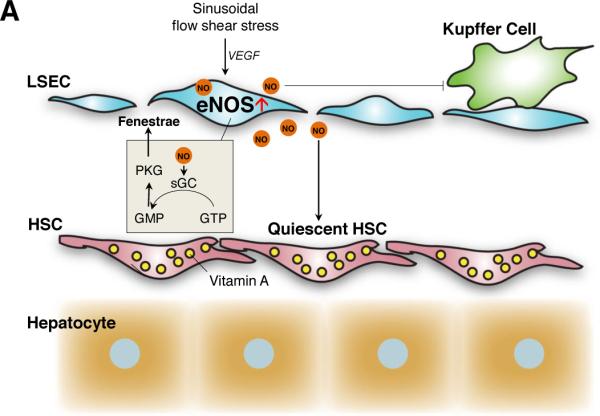
A. Homeostatic roles. Small amounts of NO constantly produced by eNOS in liver sinusoidal endothelial cells (LSECs) are essential for controlling intrahepatic sinusoidal vascular tone and blood flow. eNOS is activated by stimuli such as sinusoidal blood flow shear stress and vascular endothelial growth factor (VEGF) [116, 117]. NO keeps hepatic stellate cells (HSCs) [63, 64] and Kupffer cells quiescent [73]. NO maintains fenestrae of LSECs through activation of soluble guanylyl cyclase (sGC) that converts GTP to cGMP, which then stimulates protein kinase G (PKG) [112].
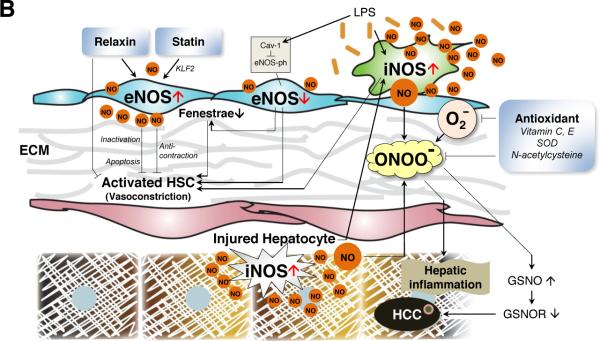
B. Pathological roles. Pro-inflammatory cytokines and bacterial endotoxin induce iNOS to produce large amounts of NO, facilitating liver injury. In pathological conditions, eNOS activity decreases, whereas iNOS is upregulated. Decreased NO production in LSECs causes endothelial cell capillarization and activation of HSCs, which is accompanied with extracellular matrix (ECM) deposition, HSC contraction and proliferation, and finally an increase in intrahepatic resistance and sinusoidal flow disturbance. Bacterial endotoxin lipopolysaccharide (LPS) secondary to gut bacterial translocation induces iNOS expression and NO production [130], which combines with superoxide to generate peroxynitrite. LPS also increases caveolin-1 and decreases eNOS phosphorylation (an active form of eNOS). These oxidative and nitrosative stresses also facilitate HSC activation and hepatocyte injury with decreased S-nitrosoglutathione reductase (GSNOR) and increased S-nitrosoglutathione (GSNO), which is related to the development of hepatocellular carcinoma (HCC) [41]. Several anti-oxidants such as superoxide dismutase (SOD) are important controllers of nitrooxidative stress. Recently, statin and relaxin were shown to restore LSEC function. Statin upregulated eNOS in LSECs via the transcription factor Krüppel-like factor 2 (KLF2) and reverted activated HSCs to the quiescent state [114]. Relaxin increased intraheptic NO levels and decreased portal pressure due to upregulation of relaxin receptor restricted to HSCs in fibrotic livers [128].
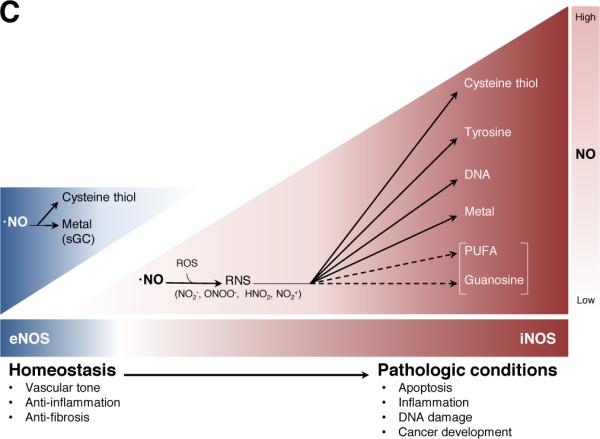
C. Mode of action of NO in homeostatic and pathological conditions in the liver. eNOS is constitutively expressed in endothelial cells and produces small amounts of NO, which play an important role in vascular homeostasis. In contrast, in pathological conditions, iNOS is upregulated and produces large amounts of NO, which cause cell apoptosis, inflammation, DNA damage, and cancer development. Nitration of polyunsaturated fatty acids (PUFA) and guanosines has not been studied in the liver, but may have protective roles in diseased liver. Abbreviations: reactive oxygen species (ROS), reactive nitrogen species (RNS).
This review summarizes the current understanding of NO biology including two new NO-mediated protein modifications, nitrated fatty acids (NO2-FA) and guanine nucleotides (8-nitroguanosine), and the roles of NO in liver diseases, concluding with suggestions on interesting areas of study to be explored in the context of NO and liver biology.
Mode of action of nitric oxide
The most recognized action of NO is through cyclic guanosine monophosphate (cGMP) as the second messenger. The cGMP-dependent action is initiated by binding of NO to a metal center of guanylyl cyclase (GC), which activates this enzyme, leading to increased production of cGMP. However, cGMP-independent actions of NO, such as protein tyrosine nitration and S-nitrosylation, have also been implicated in various disease conditions. For example, proteomic analysis of the liver has indicated the importance of S-nitrosylated proteins in the regulation of liver function [8, 9]. In pathological conditions, these protein modifications are generally enhanced by increased iNOS-derived NO in conjunction with increased free radicals. In addition to these conventional modes, this section also addresses novel mediators of NO-related action, nitrated fatty acids and nucleotides.
Metal ions
NO acts as a signaling molecule by binding to metal ions, proteins, lipids, and guanine nucleotides (Figure 2). Among metal ions, a primary target is ferrous heme iron [10], which forms highly stable NO complexes. A classic example is NO binding to ferrous heme iron of soluble guanylyl cyclase (sGC) to produce cGMP as the second messenger [11, 12]. Elevated cGMP directly modulates the activity of phosphodiesterases (PDEs), ion-gated channels, or cGMP-dependent protein kinases to regulate a wide range of physiological functions, including vasodilation, platelet aggregation, and neurotransmission [13].
Figure 2. Mode of action of nitric oxide (NO).

NO regulates a wide range of cellular activities by binding to metal ions, proteins, lipids, and guanine nucleotides. I) Metal ions. A primary target for NO is ferrous heme iron. The most known example is the binding of NO to ferrous heme iron of soluble guanylyl cyclase (sGC) to produce cGMP as the second messenger [11, 12]. II) Tyrosine nitration. Nitration of protein tyrosine residues results in irreversible and stable adducts and has been considered an indicator of nitrosative stress [35]. III) S-nitrosylation. S-nitrosylation occurs through the binding of oxidized NO to a specific cysteine thiol anion in proteins/peptides and is a reversible reaction [36]. IV) Nitration of unsaturated fatty acids (S-alkylation). NO or NO-derivatives can bind to unsaturated fatty acids, resulting in the formation of electrophilic compounds, nitrated fatty acids (NO2-FA). NO2-FA can readily react with nucleophilic amino acids, such as histidine and cysteine, in proteins and change protein function [148, 149]. V) Nitration of guanine nucleotides (S-guanylation). NO or NO-derivatives can also bind to guanine nucleotides and produce electrophilic NO derivatives (8-nitroguanosine), which conduct protein modification [65].
NO also binds to a heme/copper center of cytochrome c oxidase, also known as Complex IV, the terminal enzyme in the mitochondrial respiratory chain [14, 15] and irreversibly inhibits its activity. This inhibition facilitates the reduced state of electron carriers in the respiratory chain, increasing O2− generation as well as reducing adenosine triphosphate (ATP) production [14, 15] and thereby influencing energy metabolism.
Another important example is the binding of NO to the heme of NOS, which inhibits NOS enzyme activity [16]. The binding of NO to Fe(II) or Fe(III) heme attenuates oxygenase activity of NOS. This may serve as a negative feedback system of NOS activity. It is also possible that increased iNOS-derived NO observed in many types of liver diseases may inhibit eNOS activity through this mechanism, contributing to decreased eNOS-derived NO in LSECs and thus LSEC dysfunction.
Free radicals
NO reacts with reactive oxygen species (ROS) to form RNS such as •NO2, ONOO−, HNO2, and NO2+ (Figure 1C). Perhaps, the most studied RNS-forming reaction is the reaction between •NO and superoxide radical O2•-, which generates peroxynitrite (ONOO−) [17], a strong biological oxidant generally implicated in toxic effects on cells and tissues. Mitochondria have been considered the main source of reactive oxygen species (ROS), which mostly originate from the mitochondrial respiratory chain [18]. Excess mitochondrial ONOO- can impair oxidative phosphorylation by inhibiting the respiratory chain complexes (Complex I, Complex IV, and ATP synthase) and MnSOD activity [19]. In addition, ONOO- can oxidize DNA bases, tyrosine residues of proteins, and thiol groups [i.e., cysteine thiol and glutathione (GSH)], causing hepatocyte death [20, 21].
The depletion of the GSH pool and an increase in an oxidized form of GSH (GSSG) are closely related to action of ROS and RNS on cell and liver function [22]. GSH is a triple peptide (γ-L-glutamiyl-L-cysteinyl-glycin), synthesized in the cytosol and transported to cellular organelles, including the endoplasmic reticulum (ER), nucleus, and mitochondrion. With cysteine in its backbone, GSH plays an important role in the reduction of electrophiles and oxidants [23]. GSH becomes oxidized to GSSG when it reduces target molecules. Thus, the ratio of GSH to GSSG is a good indicator of oxidative stress and redox balance [24]. The depletion of the GSH pool is associated with the pathogenesis of liver diseases, such as drug-induced liver toxicity (i.e., acetaminophen overdose) [25], chronic alcohol intake [26], non-alcoholic fatty liver disease (NAFLD) [27] and biliary cirrhosis [28]. Chronic alcohol intake, NAFLD or biliary cirrhosis deplete the mitochondrial GSH pool by impairing GSH transport to the mitochondrial matrix, which is largely caused by alterations of lipid compositions (i.e., increased cholesterol contents) in mitochondrial membranes [22].
NOS generates superoxide anion (O2-) instead of NO [29, 30] in a condition known as “NOS uncoupling”. NOS uncoupling occurs when tetrahydrobiopterin (BH4), an essential cofactor, is depleted [31]. NOS can convert L-arginine to NO and L-citrulline only as a dimer because BH4 binding site is located at the dimer interface and monomeric NOS cannot bind BH4 or L-arginine [32]. Oxidative stress plays a major role in depleting the cellular BH4 pool, causing NOS uncoupling and further exacerbating oxidative stress [33]. Reduced bioavailability of NO as a result of NOS uncoupling has also been implicated in endothelial cell dysfunction [34].
Protein modification (tyrosine nitration and S-nitrosylation)
Nitration of protein tyrosine residues creates irreversible and stable adducts and has been considered an indicator of nitrosative stress (Figure 2). Tyrosine nitration of proteins often occurs in inflammatory conditions and mediates peroxynitrite-induced cell death signaling [35].
Unlike tyrosine nitration, S-nitrosylation is a reversible reaction (Figure 2), in which oxidized NO binds to a specific cysteine thiol anion in proteins and peptides [36]. Protein S-nitrosylation is enhanced by NOS activity, but decreased by S-nitrosoglutathione reductase (GSNOR), a ubiquitous and highly conserved denitrosylase, thus serving as a key regulator of protein S-nitrosylation [37]. eNOS facilitates S-nitrosylation in endothelial cells by specifically localizing at the Golgi apparatus and generating a high concentration of NO pool locally [38, 39].
A number of potential targets, such as CD147 and various mitochondrial proteins, for protein S-nitrosylation have been identified in the liver [8, 9]. It has been suggested that the dysregulation of protein S-nitrosylation, mostly mediated by iNOS, is associated with the pathogenesis of various liver diseases, including hepatocellular carcinoma (HCC) [40, 41], hepatic steatosis [9], and cholestasis [42, 43].
For example, lack of GSNOR resulted in S-nitrosylation, ubiquitination, and proteosomal degradation of O6-alkylguanine-DNA-alkyltransferase (AGT). AGT is a key DNA repair enzyme and was reported for protection against dialkylnitrosamine-induced HCC [44]. Both pharmacological inactivation and genetic deletion of iNOS prevented S-nitrosylation and inactivation of AGT in GSNOR knockout mice, suggesting a role of iNOS in S-nitrosylation and liver carcinogenesis in the absence of GSNOR gene [40]. Similarly, decreased expression and activity of GSNOR as well as increased S-nitrosylated proteins were observed in cholestatic livers isolated from mice with bile duct ligation surgery (an experimental model of cholestasis). Treatment with an iNOS inhibitor, S-methylisothiourea, ameliorated hepatocellular injury with restoration of GSNOR activity and decreased S-nitrosylated proteins, suggesting a contribution of increased S-nitrosylated proteins generated by increased iNOS-derived NO to cholestatic liver disease [43].
Another S-nitrosylated protein was also reported for its possible contribution to cholestasis. Taurocholic acid, a form of bile acids, is involved in the emulsification of fat. Its uptake by hepatocytes is mediated by a transporter NTCP/Ntcp, a Na+-dependent taurocholate cotransporting polypeptide. NO derived from iNOS S-nitrosylated Ntcp at Cys96 and inhibited taurocholic acid uptake in hepatocytes in vitro. This result implies that S-nitrosylation of Ntcp causes sepsis-associated cholestasis by inhibiting taurocholic acid uptake and thus decreasing bile flow [42].
NO signaling also appears to be an important regulator of the bile salt pool in hepatocytes [45]. Excess bile salts stimulated NO production in hepatocytes, leading to S-nitrosylation and nuclear translocation of GAPDH. Interestingly, S-nitrosylated GAPDH (SNO-GAPDH) transnitrosylated HDAC2 and SIRT1 in the nuclei of hepatocytes. S-nitrosylated HDAC2 formed a complex with SHP, which repressed gene expression of CYP7A1, an enzyme that converts cholesterol to bile acids. Thus, this mechanism could work as a negative feedback by decreasing bile acid production.
Nitrated fatty acids
Mitochondria keep polyunsaturated fatty acids (PUFAs) in their inner membranes [18]. NO-derived species (reactive nitrogen species, RNS), such as •NO2, ONOO−, HNO2, and NO2+, can oxidize or nitrate PUFAs and yield nitrated fatty acids (NO2-FA) (Figure 2). NO2-FA is an electrophile, which readily reacts with nucleophilic amino acids such as histidine and cysteine and changes protein function and catalytic activity [46, 47]. Importantly, NO2-FA appears to target specific proteins [48]. Although which RNS predominate the reaction is not known, NO2-FA production occurs in pathological conditions, such as cardiac ischemia [49], pulmonary hypertension [50], and diabetes [50, 51].
Unlike other electrophiles that exert cytotoxicity or pro-inflammatory effects by oxidizing lipids or lipoproteins in the vasculature [52], NO2-FA has been reported to exhibit anti-inflammatory properties through inhibition of leukocyte and platelet activation [53], vascular smooth muscle proliferation [54], and lipopolysaccharide (LPS)-stimulated macrophage cytokine secretion [55]. Among an increasing number of NO2-FA activated signaling pathways, reactions with transcription factors, such as Nrf2/Keap 1 (nuclear erythroid 2-related factor 2/Kelch ECH associating protein) and NFkB (nuclear factor kappa-light-chain-enhancer of activated B cells), are particularly important. Keap 1 binds to a transcription factor Nrf2 and prevents its nuclear translocation. Covalent binding of NO2-FA to cysteine residues of Keap1 allowed nuclear translocation of Nrf2, inducing expression of antioxidant-associated genes, such as glutathione S-transferases, heme oxygenase-1, thioredoxin, and components of the glutathione synthesis pathways [56]. Impaired Nrf2/Keap1 signaling has been observed in various liver abnormalities including hepatomegaly [57, 58], liver cirrhosis [59], and immune mediated hepatitis [56]. Thus, it is interesting to examine the involvement of NO2-FA.
NFkB induces major inflammatory cytokines involved in liver diseases. NO2-FA was shown to cause nitroalkylation of p65 subunit (an active subunit of NFkB) and inhibit its activity, thereby decreasing expression of inflammatory cytokines [55]. Supplementation of NO2-FA could therefore protect the liver from injury like the case of diabetic nephropathy in which administration of NO2-FA via osmotic pump showed moderate amelioration [60].
NO2-FA could also facilitate glucose homeostasis and lipid metabolism as a ligand of peroxisome proliferator-activated receptor-γ (PPAR-γ) [61]. NO2-FA bound to the ligand-binding domain Cys285 of PPAR-γ [62] and induced PPAR-γ downstream gene expression [61]. Since nitrated linoleic acids, a type of NO2-FA, increased glucose uptake and adipocyte differentiation through PPAR-γ activation [61], it is tempting to speculate that NO2-FA could be involved in the pathological process of non-alcoholic fatty liver disease (NAFLD). NO2-FA and nitrated guanine nucleotides, which are discussed below, are relatively new discoveries of NO's modifications. Their roles in liver biology are largely unknown and should be explored.
Nitrated guanine nucleotides (8-nitroguanosine)
Nitration of guanine nucleotides was originally found to occur in microbial infection in an iNOS-derived NO-dependent manner [63]. Among nitrated guanines, 8-nitroguanosine 3′,5′-cyclic monophosphate (8-nitro-cGMP) showed the highest biological activity [63, 64] and readily reacted with sulfhydryls of glutathione and proteins to form 8-thioalkoxy-cGMP adducts (Figure 2). Similar to NO2-FA, this NO-mediated protein modification, termed “S-guanylation” [65], occurs in two steps. Using cGMP as an example, the first step is the nitration of GTP (i.e., 8-nitro-GTP), which is then catalyzed by guanylyl cyclase (GC) to generate nitrated cGMP (i.e., 8-nitro-cGMP) [66]. The second is the modification of target proteins by nitrated cGMP (i.e., 8-nitro-cGMP). A number of target molecules for S-guanylation have been identified [67]. One remarkable finding was the role of 8-nitro-cGMP in autophagy in macrophages [68]. 8-nitro-cGMP bound to cysteine residues (i.e., S-guanylation) of invading bacteria such as group A streptococcus. Then, these S-guanylated bacteria were tagged with polyubiquitin, a molecular tag known to allow selective delivery to autophagosomes for degradation, resulting in the clearance of the bacteria.
Biological activity of 8-nitro-cGMP could be regulated by cysteine persulfide (CysSSH). CysSSH reacted with 8-nitro-cGMP to generate 8-SH-cGMP. An addition of CysSS-, called electrophile sulfhydration, inhibited the action of 8-nitro-cGMP [69, 70].
8-nitro-cGMP may represent the mechanism of adaptive response to oxidative stress [71]. Similar to NO2-FA, 8-nitro-cGMP helped to induce expression of antioxidant-associated genes by binding to Keap1 and thus allowing activation and nuclear translocation of Nrf2. The pathogenesis of liver diseases is largely related to increased oxidative stress. Thus, a role of S-guanylation is anticipated.
NO and liver diseases
RNS and ROS play a central role in the pathogenesis of a variety of liver diseases. iNOS-derived NO is closely related to this process. This section addresses how NO is involved in the development of various types of liver diseases (Figure 3).
Figure 3. The natural course of acute and chronic liver disease.
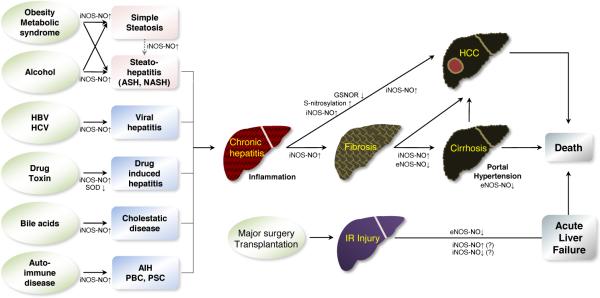
Chronic liver disease includes various heterogeneous disease conditions depending on etiology and time. Chronic exposures to different insults lead to various kinds of chronic hepatitis. If causal factors are not corrected and continue the harmful effects on the liver, hepatic inflammation progresses to hepatic fibrosis and further cirrhosis. In this stage, cirrhosis often accompanies portal hypertension, which is a main pathophysiologic cause of cirrhosis-related death and complications such as variceal hemorrhage and ascites formation. Hepatocellular carcinoma can develop at any stage of chronic liver disease even though advanced cirrhosis is the most risky stage. Ischemic-reperfusion injury after major liver surgery or transplantation can progress to sudden acute hepatic liver failure and death. The roles of NO derived from eNOS or iNOS vary, depending on disease etiologies and conditions. In general, NO produced by eNOS can be protective, but iNOS-derived NO or its derivatives such as peroxynitrite have been shown to promote inflammation.
Non-alcoholic fatty liver disease
NAFLD has emerged as the most common liver disorder in developed countries in recent years. In the US, it is estimated to affect approximately 30% of the general population [72]. NAFLD is a progressive disease that starts with fat accumulation in the liver (steatosis) and can advance to non-alcoholic steatohepatitis (NASH), fibrosis, cirrhosis, and eventually cancer over time (Figure 3). It is not fully understood what causes steatosis to progress to NASH and further to cirrhosis and HCC. NAFLD is strongly associated with obesity and type 2 diabetes. NO can be protective or promotive for NAFLD depending on its source as discussed below.
NO produced by eNOS can be protective through the inhibition of inflammatory activation of Kupffer cells [73] (Figure 1A) and enhancement of mitochondrial fatty acid β-oxidation [9]. Simvastatin [3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitor], which is known to increase eNOS activity and thus NO production, was also shown to mitigate NASH-related liver fibrosis by inhibiting HSC activation [74]. NO donors also showed preventive roles in hepatic steatosis by facilitating fatty acid β-oxidation [75] and by blocking cytochrome P450 2E1 enzyme (CYP2E1)-mediated oxidative stress [76].
Contrary to the protective effects of eNOS-derived NO or NO donors mentioned above, iNOS-derived NO or its derivatives such as peroxynitrite have been shown to promote NAFLD [77-79]. For example, Arg2−/− mice showed increased hepatic steatosis and injury in response to high-fat diet compared with their wildtype counterparts, accompanied with increased macrophage activation and iNOS expression. Given that arginase 2 competes with iNOS for its substrate (i.e., arginine), lack of arginase 2 favored iNOS induction, contributing to the increased steatosis and injury in Arg2−/− mice [78].
Leptin, a pro-inflammatory adipocytokine, was shown to contribute to steatohepatitic lesions through induction of iNOS and NADPH oxidase, which caused peroxynitrite-mediated oxidative stress and thereby activated Kupffer cells [79]. Insulin resistance, which is commonly found in metabolic syndrome and NAFLD, was also related to iNOS induction [80]. iNOS may cause lipid-induced hepatic insulin resistance by inhibiting key insulin signaling proteins such as Akt, insulin receptor-β (IRβ), insulin receptor substrate-1 (IRS-1), and 2 (IRS-2), through functional alterations by tyrosine (Tyr) nitration [81].
Overall, eNOS-derived NO or NO donors can be protective, while iNOS-derived NO is generally pathological (Figure 1B). This may be related to the microenvironments at the sites of iNOS induction where generated NO can facilitate the formation of ROS and RNS with other available radicals. While the pathogenesis of NASH remains to be elucidated, a “double-hit theory” has been proposed in which the “first hit” involves hepatic fat accumulation and the “second hit” could be any additional insult to the liver. iNOS-derived NO could be an important second hit.
Alcoholic liver disease
Alcoholic liver disease (ALD) resembles NAFLD but is caused by overconsumption of alcohol. Like NAFLD, ALD also starts with steatosis and can develop into advanced stages (Figure 3). The histological features of ALD are considerably similar to those of NAFLD [82]. Reported roles of NO in ALD are also comparable, showing the beneficial effect of eNOS-derived NO [83] and the harmful effect of iNOS-derived NO [84]. Chronic or acute ethanol overconsumption increases NO levels in the blood and liver [85, 86], with the latter in particular attributed to the increased activity of iNOS.
Caveolin-1, a cell membrane protein, was reported to suppress iNOS induction through inhibition of the epidermal growth factor receptor (EGFR)/signal transducer and activator of transcription 3 (STAT3) signaling cascade in the liver [87], and decrease iNOS-induced nitrosative stress, leading to the protection of the liver from alcohol-induced injury [88].
Argininosuccinate synthase (ASS), the rate-limiting enzyme in the urea cycle responsible for the conversion of L-citrulline to L-arginine (a NOS substrate), was also implicated in iNOS regulation in response to alcohol over-intake. In response to ethanol binge drinking, partial ASS ablation (ASS+/− mice) resulted in reduced liver injury by decreasing nitrosative stress through lower iNOS induction and less 3-nitrotyrosine (3-NT) protein residues but, in a chronic ethanol-feeding model, enhanced liver injury due to hyperammonemia, increased oxidative stress, and impairment of fatty acid β-oxidation despite no differences in nitrosative stress. The role of ASS may be distinctive depending on the disease process.
Alcohol-related increases of iNOS-derived NO could also cause hepatic injury by changing protein function through S-nitrosylation of important mitochondrial proteins, including aldehyde dehydrogenase 2 (ALDH2), ATP synthase, and 3-ketoacyl-CoA thiolase. These proteins play important roles in ethanol detoxification, ATP synthesis, and fatty acid β-oxidation, and their activities were inhibited in an experimental model of ALD [89].
Viral hepatitis
Infections with the hepatitis B and hepatitis C viruses (HBV/HCV) are common causes of hepatic inflammation, fibrosis, and HCC (Figure 3). iNOS-derived NO mediates important pathogenic events, such as DNA damage and gene mutation, in these infectious diseases.
Among the structural and functional components of HBV, hepatitis B virus X protein (HBx), a multi-functional viral regulator, has been implicated in hepatic metabolic disorder [90]. HBx-overexpressing transgenic (Tg) mice exhibited increased hepatic glucose production and hyperglycemia (high blood glucose) due to elevated expression of key gluconeogenic enzymes including PEPCK, G6Pase, PGC1α, and FOXO1. However, deletion of the iNOS gene in HBx Tg mice [HBx Tg/iNOS(−/−) mice] corrected these elevated gluconeogenic gene expressions and normalized hyperglycemia. Conversely, treatment with an NO donor, sodium nitroprusside, significantly increased the expression of these gluconeogenic genes in HBx-expressing HepG2 cells (human liver carcinoma cells).
Close connections between HCV infection and iNOS induction have also been described. iNOS induction has been reported in liver tissues from HCV patients [91, 92]. HCV, specifically the HCV core protein or nonstructural protein 3 (NS3), was shown to induce iNOS in human B lymphocytes or Hep G2 cells and cause DNA double-strand breaks that enhance DNA mutation. It was suggested that NO generated by iNOS is responsible for DNA damage and mutations of cellular genes, facilitating HCV pathogenesis and oncogenesis [93].
Current treatment regimens of chronic hepatitis C consist of pegylated interferon alpha (PEG-IFN-α), ribavirin, and direct-acting antivirals (i.e., viral protease/polymerase inhibitors) [94]. Potential mechanisms of ribavirin's anti-HCV activity are through its anti-angiogenic [95] and anti-inflammatory [96] properties. Ribavirin was shown to inhibit endothelial cell proliferation, endothelial cell tube formation, and vessel formation by decreasing eNOS-derived NO through a sequence of events (decreased intracellular guanosine-5′-triphosphate (GTP) and decreased cofactor tetrahydrobiopterin (BH4)) initiated by the inhibition of intracellular inosine-5′-monophosphate dehydrogenase. In cultured macrophages (RAW264.7), ribavirin also inhibited iNOS-derived NO, suggesting its anti-inflammatory activity [96]. However, use of ribavirin may need caution. Given an important role of eNOS-derived NO in liver homeostasis (Figure 1A), decreased eNOS-derived NO by ribavirin may cause LSEC dysfunction. In addition, decreased BH4 may induce eNOS uncoupling, which generates oxidative stress in the liver.
Interestingly, iNOS was also described as a main effector of IFN-γ-mediated anti-HCV activity in hepatocytes [97]. It was speculated that iNOS might be beneficial in a very early event of the viral replication cycle, but less protective in later persistent stages.
The role of NO in the other types of hepatitis virus infection such as hepatitis A virus (HAV) or E virus (HEV) is largely unknown. It was recently reported that the iNOS C150T polymorphism, the eNOS G894T polymorphism, and high levels of iNOS and eNOS were associated with the risk of HEV-related acute hepatitis and acute liver failure [98]. However, further studies are needed to understand the role of these polymorphisms in iNOS and eNOS in HEV pathogenesis.
Hepatocellular carcinoma
HCC development is also associated with elevated expression of iNOS and increased nitrosative stress [41] (Figure 3). GSNOR is a denitrosylase key in controlling protein S-nitrocylation. By preventing excessive protein S-nitrosylation, GSNOR appears to have a beneficial effect on health (Figure 1B). The human GSNOR gene (ADH5) is located approximately in the 4q23 region, where chromosomal deletion frequently occurs in HCC [99, 100]. Approximately 50% of liver samples from patients with HCC showed decreased GSNOR level and activity. In a diethylnitrosamine-induced HCC model, deletion of GSNOR gene substantially increased S-nitrosylation and degradation of O6-alkylguanine-DNA alkyltransferase (AGT), a key DNA repair protein, as well as phosphorylation of histone H2AX, a well-established marker of DNA double-strand breaks. These impairments were attenuated in GSNOR/iNOS double knockout mice [40, 101] as well as by administration of an iNOS inhibitor (1400W) [41]. Interestingly, gene-expression profiling of fixed tissues from patients with HCC correlated GSNOR and iNOS to good and poor survival, respectively [102].
GSNOR is also known as alcohol dehydrogenase class III and was originally recognized as glutathione-dependent formaldehyde dehydrogenase [103, 104]. Thus, its deletion may also cause accumulation of endogenous formaldehyde, which in turn may induce DNA injury, accelerating cancer progression. It is possible that dysregulated S-nitrosylation may not be the only mechanism through which GSNOR deficiency contributes to the development of HCC [41].
HCC frequently develops in patients with HBV and HCV. HBx has been implicated in hepatitis B-related HCC development through iNOS induction [105, 106]. iNOS induction by HBx was related to NFκ-B signaling [107]. Interestingly, iNOS also upregulated HBx expression and activated HBx-mediated c-jun n-terminal protein kinase (JNK) signaling, thereby facilitating HCC development.
Likewise, HCV core protein is known to facilitate oncogenic events by causing double-strand DNA breaks, which potentiate the mutation of proto-oncogenes and tumor suppressors. iNOS and RNS/ROS induced by HCV core protein were indicated to play a role [93, 108]. In transgenic mice expressing HCV core protein, treatment with butylated hydroxyanisole, an antioxidant scavenger of ROS and RNS, significantly decreased liver tumor size and number as well as mutation frequency of p53, a major tumor suppressor. Furthermore, HCV core-induced iNOS/NO was implicated in the impairment of DNA damage repair through the inhibition of DNA glycosylase activity [109]. This impairment of DNA glycosylase is likely mediated by tyrosine nitration or S-nitrosylation [110, 111].
Hepatic fibrosis
Under normal conditions, NO is constitutively synthesized by eNOS in LSECs, keeping HSCs quiescent in a paracrine manner [112, 113] (Figure 1A). In pathological conditions, however, LSECs become dysfunctional, accompanied by decreased eNOS-derived NO, which activates quiescent HSCs, causing extracellular matrix deposition and vasoconstriction and leading to fibrosis and further cirrhosis (Figure 1B).
It was reported that, in vitro, HSC quiescence required both LSECs (or eNOS) and VEGF [113] (Figure 1A). VEGF was thought to maintain LSEC integrity, partly by stimulating activation of eNOS and sGC. In rats with cirrhosis, an sGC activator, BAY 60-2770, restored LSEC structure, followed by increased quiescent HSCs and regressed fibrosis [112]. Statin (Simvastatin) was also reported to revert activated HSCs to quiescence by upregulating eNOS in LSECs via the transcription factor Krüppel-like factor 2 (KLF2) [114] (Figure 1B).
NO may also limit activated HSCs through apoptosis. Apoptosis of activated HSCs is one of the key events that facilitate regression of liver fibrosis. NO was reported to trigger HSC apoptosis via nitrosative stress-mediated mitochondrial membrane depolarization [115].
Portal hypertension
Portal hypertension, which is defined as an increase in pressure in the portal vein that carries blood to the liver, is a detrimental complication of liver disease. The most common cause of portal hypertension is liver cirrhosis due to increased intrahepatic vascular resistance (Figure 3). LSEC is a key player in the regulation of intrahepatic vascular resistance through production of eNOS-derived NO [116, 117] (Figure 1).
eNOS is regulated by complex post-translational modifications and protein-protein interactions in LSECs [117, 118]. Those regulators include phosphatidylinositol 3-kinase /Akt, caveolin-1, Hsp-90, G-protein-coupled receptor kinase 2 (GRK2), G-protein-coupled receptor kinase-interacting protein 1 (GIT1) [119, 120], dimethylarginine dimethylaminohydrolase-1 (DDAH-1) [121], and a cofactor BH4 [122]. In cirrhotic conditions, all these eNOS regulators, by suppression or activation, work toward the inhibition of eNOS activity and favor increased vasoconstriction by activated HSCs, leading to increased intrahepatic vascular resistance.
For example, in the livers of cirrhotic rats, the levels of guanosine triphosphate (GTP)-cyclohydrolase (BH4 rate-limiting enzyme) were significantly decreased, leading to decreased BH4 levels and eNOS uncoupling. eNOS uncoupling was associated with LSEC dysfunction, increased intrahepatic resistance and subsequent development of portal hypertension [123]. However, BH4 levels were significantly increased in the extrahepatic territory (i.e., mesenteric lymph nodes and mesenteric arteries) as a result of increased endotoxin levels via bacterial translocation, a pathological feature of liver cirrhosis [124, 125]. This resulted in increased NO production in the mesenteric arteries of cirrhotic rats. Although local BH4 production is an important determinant of NO bioavailability derived from eNOS in endothelial cells, the relationship between BH4 levels, iNOS uncoupling and NO production is largely unknown.
As mentioned above, in portal hypertension, regulation of NO is opposite inside and outside of the liver. Contrary to decreased NO availability in the intrahepatic circulation, NO production in splanchnic and systemic arteries increases and causes excessive vasodilation, resulting in increased blood flow to the portal vein and thereby worsening portal hypertension. Therefore, for a therapeutic purpose, liver-specific NO donors that do not cause the additional vasodilative effects on the splanchnic and systemic circulation are needed. Examples of NO donors can be found in Table 1 [117]. Some of them have demonstrated positive outcomes in experimental models. For example, NCX-1000, a hepatocyte-specific NO donor, ameliorated portal hypertension in cirrhotic rats [126], but failed in a clinical trial [127]. Although this discrepancy may come from differences between animals and humans, treatment of portal hypertension could benefit more from NO donor delivery to LSECs than to hepatocytes.
Table 1.
Nitric oxide delivery systems or pro-drugs for liver disease
| Drug (Pro-drug or delivery system) | Experimental model | Route | Effects | Reference |
|---|---|---|---|---|
| Murine model of autoimmune hepatitis, in vivo / HepG2 cell, in vitro | i.p. | Inhibition of apoptosis and inflammation via regulation of the caspase superfamily | Fiorucci. et al.[159] | |
| NCX-1000 ([2-(acetyloxy)benzoic acid 3-(nitrooxymethyl)phenyl ester]) | BDL cirrhotic rat model, in vivo | p.o. | Decrease of portal pressure and intrahepatic resistance without systemic effects along with increase of nitrite/nitrate and cGMP (cyclic guanosine monophosphate) concentrations in the liver | Fiorucci. et al.[126] |
| Clinical, patients with cirrhosis and portal hypertension | p.o. | Systemic hypotensive effects and lack of reduction in portal pressure | Berzigotti. et al.[127] | |
| O2-vinyl 1-(pyrrolidin-1-yl)diazen-1-ium- 1,2-diolate (V-PYRRO/NO) | Adult female CD-1 mice with acetaminophen (600 mg/kg, dissolved in saline, 25 mL/kg, i.p.) model / Osmotic pump of V-PYRRO/NO, i.v. | i.v. | Blocking of acetaminophen-induced hepatotoxicity by reduction of oxidative stress and apoptosis | Liu. et al.[160] |
| Male BALB/c mice with CCl4 (by gavage) hepatic fibrosis and portal hypertension model / V-PYRRO/NO, i.v. bolus | i.v. | Decrease of portal pressure and systemic mean arterial pressure | Edwards. et al.[161] | |
| C57BL/6J mice with high fat diet NAFLD model | i.p. | Anti-steatotic effects and improvement of postprandial glucose tolerance | Maslak. et al.[162] | |
| Pig ischemia/reperfusion liver model | i.v. | Decrease of hepatic resistance and improvement of hemodynamics. Improvement of plasma clearance of bile acids | Ricciardi. et al.[163] | |
| NO-releasing carbon-dot-based nanosystem (C-dot-triphenylphosphonium (TPP)- S-nitrosothiol) | Three cancer cell lines; HeLa (human cervical cancer cells), HepG2 (human liver carcinoma cells) and A549 (adenocarcinomic human alveolar basal epithelial cells), in vitro | - | Cytotoxic and apoptotic effects by mitochondria targeting, photo-enhanced NO release | Xu. et al.[164] |
| Gold and silica nanoparticles and NO donors conjugate *NO donors: S-nitroso-N-acetyl-DL-penicillamine (SNAP), glyco-SNAP, 3-morpholino-sydnonimine (SIN-1), S-nitrosoglutathione (GSNO) | Primary human HSCs and LX2 cell-line, in vitro | - | Decrease of HSC proliferation and vascular tube formation | Das. et al.[165] |
| Nitro-aspirin (NO-aspirin) (Cyclooxygenase-inhibiting nitric oxide donors (CINODs)) | Male Wistar albino rats with 2% cholesterol diet NAFLD model, in vivo | p.o. (gavage) | Decrease in expression of iNOS and cyclooxygenase-2 (COX-2). Blocking of NAFLD development | Ibrahim. et al.[166] |
| Furoxan-based NO releasing derivatives of farnesylthiosalicylic acid (FTS) | HCC SMMC-7721, Bel7402, LO2, HepG2 and Hep3B cells, in vitro | - | HCC cell apoptosis | Ling. et al.[167] |
| O2-(2.4-dinitroplienyl)-diazeniumdiolate and oleanolic acid (OA) activated by glutathione S-transferase π (GSTπ) | HepG2, in vitro / ICR mice inoculated with H22 (mice hepatoma cell line), in vivo | i.v. | anti-HCC via apoptosis and increase of intracellular ROS production | Fu. et al.[168] |
| Polymeric nanoparticles decorated with vitamin A conjugated with S-nitrosoglutathione (GSNO) | Primary rat HSCs and LX2 cell-line, in vitro / BDL rats, in vivo | i.v. | Inhibition of fibrogenic genes associated with HSC activation and attenuation of hemodynamic disorder | Duong. Et al.[129] |
| DETA NONOate [(Z)-1-[N-(2-aminoethyl)-N-(2-ammonioethyl) amino]diazen-1-ium-1,2-diolate] | Male C57BL/6J mice with MCD NASH model, CYP2E1 KO and iNOS KO mice with high fat diet NASH model, in vivo / BDCM-Induction of liver Injury | i.p. | Inhibition of CYP2E1-mediated oxidative stress | Seth. et al.[76] |
BDCM, bromodichloromethane; BDL, bile duct ligation; cGMP, cyclic guanosine monophosphate; CYP2E1, cytochrome P450 2E1; GSNO, S-nitrosoglutathione; HCC, hepatocellular carcinoma; HSC, hepatic stellate cell; MCD, methionine and choline–deficient; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis
Recently, relaxin, a peptide hormone, was reported to increase intraheptic NO levels and decrease portal pressure without changing systemic NO levels and pressure. This selective effect of relaxin was attributed to the upregulation of relaxin receptor restricted to HSCs in fibrotic livers [128].
In this line, a recent study explored if vitamin A-coupled polymeric nanoparticles encapsulated with an NO donor could be specifically delivered to HSCs and ameliorate liver fibrosis and portal hypertension [129]. Although this study showed decreased portal pressure without changing mean arterial pressure in cirrhotic rats treated with vitamin A-conjugated NO-nanoparticles, intake of vitamin A-coupled nanoparticles by each type of hepatic cells was not examined in vivo. Whether NO was delivered specifically to HSCs is not certain, so it is difficult to attribute the observed decrease in portal pressure in cirrhotic rats to the targeted delivery of NO to HSCs. To our best knowledge, there are no in vivo studies that have demonstrated cell-specific delivery of NO in the liver.
While decreased eNOS-derived NO in LSECs causes hepatic microvascular dysfunction and facilitates the development of fibrosis/cirrhosis and portal hypertension, iNOS-derived NO generally increases in this development. iNOS upregulation was shown to contribute to liver sinusoidal endothelial dysfunction in endotoxemia [130]. Unlike eNOS-derived NO, iNOS-derived NO does not appear to cause vasodilation. In addition, a paradoxical role of increased NO by iNOS in endothelial dysfunction, which is otherwise characterized by decreased NO by eNOS, may indicate that the effects of NO are highly dependent on its sources and surrounding microenvironments.
NO is also thought to regulate lymphatic flows by affecting the contractility of lymphatic smooth muscle cells. Increased levels of eNOS and NO were observed in endothelial cells isolated from mesenteric lymphatic vessels of cirrhotic rats, which was also attributed to decreased smooth muscle cell coverage of mesenteric lymphatic vessels in those rats [131]. Excessive NO-mediated relaxation of mesenteric lymphatic vessels may have implications in the formation of ascites (fluid accumulation in the peritoneal cavity), which is commonly associated with cirrhosis with portal hypertension [132].
Drug-induced liver injury (acetaminophen-induced liver injury)
Drug-induced liver injury causes acute hepatocellular injury with jaundice in patients, which showed a fatality rate of 10–50% depending on the drug involved (Figure 3). In severe cases, acute liver failure (ALF) with concomitant coagulopathy (blood clotting disorders) and encephalopathy (global brain dysfunction) occurs. In most Western countries, acetaminophen (paracetamol) overdose is the most frequent identified cause of ALF [133].
CYP2E1 enzyme plays a critical role in acetaminophen-induced liver injury. CYP2E1 metabolizes acetaminophen to N-acetyl-p-benzoquinone imine (NAPQI). This toxic compound is normally detoxified through conjugation with GSH. However, in the presence of large amounts of NAPQI, GSH is depleted and NAPQI remains unconjugated, which causes liver damage by binding to other proteins [25]. Oxidative stress resulting from depleted GSH is also thought to contribute to liver damage. NO produced from Kupffer cells, cytokines, ROS, and RNS are all involved in this process [134]. For example, the formation of nitrotyrosine protein adducts (3-NT), a hallmark of nitrosative stress, was observed in the centrilobular regions where acetaminophen-induced necrosis occurred. The formation of 3-NT was related to decreased levels/activities of nitrated proteins including superoxide dismutase 1(SOD1), an important O2- scavenger, through ubiquitin-mediated degradation. The formation of 3-NT and liver necrosis was dependent on the presence of CYP2E1 [135].
Acetaminophen was also reported to inhibit the activity of various mitochondrial enzymes through nitration, including aldehyde dehydrogenase, Mn-SOD (SOD2), glutathione peroxidase, ATP synthase, and 3-ketoacyl-CoA thiolase, which are involved in antioxidant defense, energy supply, or fatty acid metabolism [136].
Ischemia/reperfusion injury
Prolonged organ ischemia characterized by reduced tissue oxygenation induces the activation of anaerobic metabolic pathways and causes cell death (Figure 3). However, reperfusion after ischemia can also lead to cellular injury because the increase in oxygen delivery can generate ROS and RNS [137]. Hepatic ischemia/reperfusion (I/R) injury is a major problem in liver resection and transplantation.
Like in other liver diseases, NO has been known to have both protective and harmful effects on hepatic I/R injury. In general, eNOS-derived NO is beneficial [95-98], while iNOS-derived NO may be either protective or detrimental [5].
The protective effect of eNOS-derived NO is exerted at least in part through the sGC-cGMP-protein kinase G (PKG) pathway [138, 139]. Circulating adiponectin also showed protective property against hepatic I/R injury by reducing inflammatory response and apoptosis through activation of adenosine monophosphate-activated protein kinase (AMPK)/eNOS pathway [140].
The role of iNOS-derived NO in I/R injury is not straightforward because studies reported on its protective [141], harmful [142] or no [141, 143] effects. These studies differ in the length of ischemia or reperfusion, which may account for the observed different roles of iNOS in liver I/R injury.
iNOS-derived NO also contributed to hepatic I/R injury through S-nitrosylation or nitration of mitochondrial proteins. I/R-induced liver damage was associated with an increase in S-nitrosylated or oxidized mitochondrial proteins, including aldehyde dehydrogenase, 3-ketoacyl-CoA thiolases, and ATP synthase, and their activities were suppressed [144].
Many therapeutic and preventive trials have been conducted on hepatic I/R injury. NO inhalation to patients during the operative period of liver transplantation showed significant protection of hepatocytes from apoptotic death, quicker restoration of liver graft function, and reduced hospital length of stay [145]. Administration of sodium nitrite also reduced liver I/R injury with decreased hepatocyte apoptosis [146, 147].
Concluding remarks
NO has differential effects on the liver depending on its source. In general, eNOS-derived NO is protective against liver diseases, whereas iNOS-derived NO is deleterious because it mainly acts as a pro-inflammatory mediator. NO-mediated protein modifications such as tyrosine nitration and S-nitrosylation are significantly involved in liver biology. The new modifications, NO2-FA and S-guanylation, have increasingly been recognized and should be explored in the liver as well.
Due to its regulation of Nrf2/Keap1 and NFkB, which play important roles in the development of liver diseases [56, 59], NO2-FA [46] could be used as a potential therapeutic agent. For example, NO2-FA could block progression of NAFLD to NASH, in which inflammatory responses are involved. Given its vasoprotective role through induction of eNOS expression and activation. NO2-FA may also have an anti-fibrotic property. Because there is currently no effective anti-fibrotic drug, testing the potential of NO2-FA to ameliorate fibrosis/cirrhosis may have significant clinical implications. Lastly, given its high stability in the blood and urine [148, 149], a potential of NO2-FA as a biomarker of NAFLD, NASH, or fibrosis could be explored in combination with other non-invasive imaging systems.
Similarly, 8-nitro-cGMP exhibited an anti-inflammatory property through activation of Nrf2/Keap1 signaling to induce antioxidant enzymes [65, 66, 150]. Therefore, 8-nitro-cGMP could also be used as an anti-inflammatory agent for a wide range of liver diseases in which inflammation plays a central role.
The roles of NO in the liver have not been examined in relation to some important biological events such as autophagy and epigenetics. Since there are studies showing both its inductive [151] and inhibitory role [152], unraveling the effect of NO on autophagy is complicated. Autophagy has been implicated in a variety of liver diseases [153], including NAFLD [154], viral hepatitis [155], fibrosis [156], and HCC [157]. Given the importance of NO as a signaling molecule and its relation to ROS/RNS, examining the role of NO in the axis of autophagy and liver is interesting. Similarly, despite that NO is an epigenetic modulator in tumor cells [158], NO's epigenetic regulation has not been explored in HCC and other liver diseases. This is also an important area to be investigated.
As for NO-related therapy, NO's differential roles require liver/cell specificity. For reference, Table 1 lists NO donors that have been used in experimental models or clinical settings. Currently, there is no cell-specific NO donor delivery system. The development of such delivery systems will warrant effective treatments for liver diseases. At last, the role of nNOS in the liver is little known and should also be explored. There is much to do in NO-related studies in the liver.
Highlights.
eNOS-derived NO is protective against liver diseases, whereas iNOS-derived NO is deleterious.
In liver biology, eNOS and iNOS are major players, whereas the role of nNOS is little known.
NO regulates a wide range of cellular activities by binding to metal ions, proteins, lipids, and guanine nucleotides.
In liver biology, NO signaling through protein modifications by S-guanlyation or NO2-FA S-alkylation should be explored.
Acknowledgement
The authors would like to thank Dr. Teruo Utsumi for his critical comments on the manuscript.
Grant support: This work was supported by grant R01DK082600 from the National Institutes of Health (Iwakiri).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- 1.Knowles RG, Moncada S. Nitric oxide synthases in mammals. The Biochemical journal. 1994;298(Pt 2):249–258. doi: 10.1042/bj2980249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ignarro LJ. Physiology and pathophysiology of nitric oxide. Kidney international Supplement. 1996;55:S2–5. [PubMed] [Google Scholar]
- 3.Liu J, Hughes TE, Sessa WC. The first 35 amino acids and fatty acylation sites determine the molecular targeting of endothelial nitric oxide synthase into the Golgi region of cells: a green fluorescent protein study. The Journal of cell biology. 1997;137:1525–1535. doi: 10.1083/jcb.137.7.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carnovale CE, Ronco MT. Role of nitric oxide in liver regeneration. Annals of hepatology. 2012;11:636–647. [PubMed] [Google Scholar]
- 5.Abu-Amara M, Yang SY, Seifalian A, Davidson B, Fuller B. The nitric oxide pathway--evidence and mechanisms for protection against liver ischaemia reperfusion injury. Liver international : official journal of the International Association for the Study of the Liver. 2012;32:531–543. doi: 10.1111/j.1478-3231.2012.02755.x. [DOI] [PubMed] [Google Scholar]
- 6.Ishimura N, Bronk SF, Gores GJ. Inducible nitric oxide synthase up-regulates Notch-1 in mouse cholangiocytes: implications for carcinogenesis. Gastroenterology. 2005;128:1354–1368. doi: 10.1053/j.gastro.2005.01.055. [DOI] [PubMed] [Google Scholar]
- 7.Abdelmegeed MA, Song BJ. Functional roles of protein nitration in acute and chronic liver diseases. Oxidative medicine and cellular longevity. 2014;2014:149627. doi: 10.1155/2014/149627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sangwung P, Greco TM, Wang Y, Ischiropoulos H, Sessa WC, Iwakiri Y. Proteomic identification of S-nitrosylated Golgi proteins: new insights into endothelial cell regulation by eNOS-derived NO. PloS one. 2012;7:e31564. doi: 10.1371/journal.pone.0031564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doulias PT, Tenopoulou M, Greene JL, Raju K, Ischiropoulos H. Nitric oxide regulates mitochondrial fatty acid metabolism through reversible protein S-nitrosylation. Science signaling. 2013;6:rs1. doi: 10.1126/scisignal.2003252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henry Y, Lepoivre M, Drapier JC, Ducrocq C, Boucher JL, Guissani A. EPR characterization of molecular targets for NO in mammalian cells and organelles. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 1993;7:1124–1134. doi: 10.1096/fasebj.7.12.8397130. [DOI] [PubMed] [Google Scholar]
- 11.Arnold WP, Mittal CK, Katsuki S, Murad F. Nitric oxide activates guanylate cyclase and increases guanosine 3′:5′-cyclic monophosphate levels in various tissue preparations. Proceedings of the National Academy of Sciences of the United States of America. 1977;74:3203–3207. doi: 10.1073/pnas.74.8.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Underbakke ES, Iavarone AT, Chalmers MJ, Pascal BD, Novick S, Griffin PR, et al. Nitric oxide-induced conformational changes in soluble guanylate cyclase. Structure. 2014;22:602–611. doi: 10.1016/j.str.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Derbyshire ER, Marletta MA. Structure and regulation of soluble guanylate cyclase. Annual review of biochemistry. 2012;81:533–559. doi: 10.1146/annurev-biochem-050410-100030. [DOI] [PubMed] [Google Scholar]
- 14.Schweizer M, Richter C. Nitric oxide potently and reversibly deenergizes mitochondria at low oxygen tension. Biochemical and biophysical research communications. 1994;204:169–175. doi: 10.1006/bbrc.1994.2441. [DOI] [PubMed] [Google Scholar]
- 15.Giuffre A, Sarti P, D'Itri E, Buse G, Soulimane T, Brunori M. On the mechanism of inhibition of cytochrome c oxidase by nitric oxide. The Journal of biological chemistry. 1996;271:33404–33408. doi: 10.1074/jbc.271.52.33404. [DOI] [PubMed] [Google Scholar]
- 16.Hurshman AR, Marletta MA. Nitric oxide complexes of inducible nitric oxide synthase: spectral characterization and effect on catalytic activity. Biochemistry. 1995;34:5627–5634. doi: 10.1021/bi00016a038. [DOI] [PubMed] [Google Scholar]
- 17.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gutierrez J, Ballinger SW, Darley-Usmar VM, Landar A. Free radicals, mitochondria, and oxidized lipids: the emerging role in signal transduction in vascular cells. Circulation research. 2006;99:924–932. doi: 10.1161/01.RES.0000248212.86638.e9. [DOI] [PubMed] [Google Scholar]
- 19.Brown GC, Borutaite V. Inhibition of mitochondrial respiratory complex I by nitric oxide, peroxynitrite and S-nitrosothiols. Biochimica et biophysica acta. 2004;1658:44–49. doi: 10.1016/j.bbabio.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 20.Meguro M, Katsuramaki T, Kimura H, Isobe M, Nagayama M, Kukita K, et al. Apoptosis and necrosis after warm ischemia-reperfusion injury of the pig liver and their inhibition by ONO-1714. Transplantation. 2003;75:703–710. doi: 10.1097/01.TP.0000053400.42842.5C. [DOI] [PubMed] [Google Scholar]
- 21.Figueira TR, Barros MH, Camargo AA, Castilho RF, Ferreira JC, Kowaltowski AJ, et al. Mitochondria as a source of reactive oxygen and nitrogen species: from molecular mechanisms to human health. Antioxidants & redox signaling. 2013;18:2029–2074. doi: 10.1089/ars.2012.4729. [DOI] [PubMed] [Google Scholar]
- 22.Mari M, Morales A, Colell A, Garcia-Ruiz C, Fernandez-Checa JC. Mitochondrial glutathione, a key survival antioxidant. Antioxidants & redox signaling. 2009;11:2685–2700. doi: 10.1089/ars.2009.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hammond CL, Lee TK, Ballatori N. Novel roles for glutathione in gene expression, cell death, and membrane transport of organic solutes. Journal of hepatology. 2001;34:946–954. doi: 10.1016/s0168-8278(01)00037-x. [DOI] [PubMed] [Google Scholar]
- 24.Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Annual review of pharmacology and toxicology. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- 25.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 26.Fernandez-Checa JC, Garcia-Ruiz C, Ookhtens M, Kaplowitz N. Impaired uptake of glutathione by hepatic mitochondria from chronic ethanol-fed rats. Tracer kinetic studies in vitro and in vivo and susceptibility to oxidant stress. The Journal of clinical investigation. 1991;87:397–405. doi: 10.1172/JCI115010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mari M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A, et al. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell metabolism. 2006;4:185–198. doi: 10.1016/j.cmet.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 28.Krahenbuhl S, Talos C, Lauterburg BH, Reichen J. Reduced antioxidative capacity in liver mitochondria from bile duct ligated rats. Hepatology. 1995;22:607–612. doi: 10.1002/hep.1840220234. [DOI] [PubMed] [Google Scholar]
- 29.Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BS, Karoui H, et al. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:9220–9225. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xia Y, Tsai AL, Berka V, Zweier JL. Superoxide generation from endothelial nitric-oxide synthase. A Ca2+/calmodulin-dependent and tetrahydrobiopterin regulatory process. The Journal of biological chemistry. 1998;273:25804–25808. doi: 10.1074/jbc.273.40.25804. [DOI] [PubMed] [Google Scholar]
- 31.Marletta MA. Nitric oxide synthase structure and mechanism. The Journal of biological chemistry. 1993;268:12231–12234. [PubMed] [Google Scholar]
- 32.Crane BR, Arvai AS, Gachhui R, Wu C, Ghosh DK, Getzoff ED, et al. The structure of nitric oxide synthase oxygenase domain and inhibitor complexes. Science. 1997;278:425–431. doi: 10.1126/science.278.5337.425. [DOI] [PubMed] [Google Scholar]
- 33.Alp NJ, Channon KM. Regulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular disease. Arteriosclerosis, thrombosis, and vascular biology. 2004;24:413–420. doi: 10.1161/01.ATV.0000110785.96039.f6. [DOI] [PubMed] [Google Scholar]
- 34.Roe ND, Ren J. Nitric oxide synthase uncoupling: a therapeutic target in cardiovascular diseases. Vascular pharmacology. 2012;57:168–172. doi: 10.1016/j.vph.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 35.Franco MC, Estevez AG. Tyrosine nitration as mediator of cell death. Cellular and molecular life sciences : CMLS. 2014;71:3939–3950. doi: 10.1007/s00018-014-1662-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nature reviews Molecular cell biology. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 37.Liu L, Yan Y, Zeng M, Zhang J, Hanes MA, Ahearn G, et al. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell. 2004;116:617–628. doi: 10.1016/s0092-8674(04)00131-x. [DOI] [PubMed] [Google Scholar]
- 38.Iwakiri Y. S-nitrosylation of proteins: a new insight into endothelial cell function regulated by eNOS-derived NO. Nitric oxide : biology and chemistry / official journal of the Nitric Oxide Society. 2011;25:95–101. doi: 10.1016/j.niox.2011.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iwakiri Y, Satoh A, Chatterjee S, Toomre DK, Chalouni CM, Fulton D, et al. Nitric oxide synthase generates nitric oxide locally to regulate compartmentalized protein S-nitrosylation and protein trafficking. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:19777–19782. doi: 10.1073/pnas.0605907103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei W, Li B, Hanes MA, Kakar S, Chen X, Liu L. S-nitrosylation from GSNOR deficiency impairs DNA repair and promotes hepatocarcinogenesis. Science translational medicine. 2010;2:19ra13. doi: 10.1126/scitranslmed.3000328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang CH, Wei W, Hanes MA, Liu L. Hepatocarcinogenesis driven by GSNOR deficiency is prevented by iNOS inhibition. Cancer research. 2013;73:2897–2904. doi: 10.1158/0008-5472.CAN-12-3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramasamy U, Anwer MS, Schonhoff CM. Cysteine 96 of Ntcp is responsible for NO-mediated inhibition of taurocholate uptake. American journal of physiology Gastrointestinal and liver physiology. 2013;305:G513–519. doi: 10.1152/ajpgi.00089.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lopez-Sanchez LM, Corrales FJ, Barcos M, Espejo I, Munoz-Castaneda JR, Rodriguez-Ariza A. Inhibition of nitric oxide synthesis during induced cholestasis ameliorates hepatocellular injury by facilitating S-nitrosothiol homeostasis. Laboratory investigation; a journal of technical methods and pathology. 2010;90:116–127. doi: 10.1038/labinvest.2009.104. [DOI] [PubMed] [Google Scholar]
- 44.Iwakuma T, Sakumi K, Nakatsuru Y, Kawate H, Igarashi H, Shiraishi A, et al. High incidence of nitrosamine-induced tumorigenesis in mice lacking DNA repair methyltransferase. Carcinogenesis. 1997;18:1631–1635. doi: 10.1093/carcin/18.8.1631. [DOI] [PubMed] [Google Scholar]
- 45.Rodriguez-Ortigosa CM, Celay J, Olivas I, Juanarena N, Arcelus S, Uriarte I, et al. A GAPDH-mediated trans-nitrosylation pathway is required for feedback inhibition of bile salt synthesis in rat liver. Gastroenterology. 2014;147:1084–1093. doi: 10.1053/j.gastro.2014.07.030. [DOI] [PubMed] [Google Scholar]
- 46.Koenitzer JR, Freeman BA. Redox signaling in inflammation: interactions of endogenous electrophiles and mitochondria in cardiovascular disease. Annals of the New York Academy of Sciences. 2010;1203:45–52. doi: 10.1111/j.1749-6632.2010.05559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Trostchansky A, Bonilla L, Gonzalez-Perilli L, Rubbo H. Nitro-fatty acids: formation, redox signaling, and therapeutic potential. Antioxidants & redox signaling. 2013;19:1257–1265. doi: 10.1089/ars.2012.5023. [DOI] [PubMed] [Google Scholar]
- 48.Wong HL, Liebler DC. Mitochondrial protein targets of thiol-reactive electrophiles. Chemical research in toxicology. 2008;21:796–804. doi: 10.1021/tx700433m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rudolph V, Rudolph TK, Schopfer FJ, Bonacci G, Woodcock SR, Cole MP, et al. Endogenous generation and protective effects of nitro-fatty acids in a murine model of focal cardiac ischaemia and reperfusion. Cardiovascular research. 2010;85:155–166. doi: 10.1093/cvr/cvp275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kelley EE, Baust J, Bonacci G, Golin-Bisello F, Devlin JE, St Croix CM, et al. Fatty acid nitroalkenes ameliorate glucose intolerance and pulmonary hypertension in high-fat diet-induced obesity. Cardiovascular research. 2014;101:352–363. doi: 10.1093/cvr/cvt341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schopfer FJ, Cole MP, Groeger AL, Chen CS, Khoo NK, Woodcock SR, et al. Covalent peroxisome proliferator-activated receptor gamma adduction by nitro-fatty acids: selective ligand activity and anti-diabetic signaling actions. The Journal of biological chemistry. 2010;285:12321–12333. doi: 10.1074/jbc.M109.091512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Khoo NK, Rudolph V, Cole MP, Golin-Bisello F, Schopfer FJ, Woodcock SR, et al. Activation of vascular endothelial nitric oxide synthase and heme oxygenase-1 expression by electrophilic nitro-fatty acids. Free radical biology & medicine. 2010;48:230–239. doi: 10.1016/j.freeradbiomed.2009.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Coles B, Bloodsworth A, Clark SR, Lewis MJ, Cross AR, Freeman BA, et al. Nitrolinoleate inhibits superoxide generation, degranulation, and integrin expression by human neutrophils: novel antiinflammatory properties of nitric oxide-derived reactive species in vascular cells. Circulation research. 2002;91:375–381. doi: 10.1161/01.res.0000032114.68919.ef. [DOI] [PubMed] [Google Scholar]
- 54.Villacorta L, Zhang J, Garcia-Barrio MT, Chen XL, Freeman BA, Chen YE, et al. Nitro-linoleic acid inhibits vascular smooth muscle cell proliferation via the Keap1/Nrf2 signaling pathway. American journal of physiology Heart and circulatory physiology. 2007;293:H770–776. doi: 10.1152/ajpheart.00261.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cui T, Schopfer FJ, Zhang J, Chen K, Ichikawa T, Baker PR, et al. Nitrated fatty acids: Endogenous anti-inflammatory signaling mediators. The Journal of biological chemistry. 2006;281:35686–35698. doi: 10.1074/jbc.M603357200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Osburn WO, Yates MS, Dolan PD, Chen S, Liby KT, Sporn MB, et al. Genetic or pharmacologic amplification of nrf2 signaling inhibits acute inflammatory liver injury in mice. Toxicological sciences : an official journal of the Society of Toxicology. 2008;104:218–227. doi: 10.1093/toxsci/kfn079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nature cell biology. 2010;12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 58.Taguchi K, Hirano I, Itoh T, Tanaka M, Miyajima A, Suzuki A, et al. Nrf2 enhances cholangiocyte expansion in Pten-deficient livers. Molecular and cellular biology. 2014;34:900–913. doi: 10.1128/MCB.01384-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu T, Zhao F, Gao B, Tan C, Yagishita N, Nakajima T, et al. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes & development. 2014;28:708–722. doi: 10.1101/gad.238246.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu Y, Jia Z, Liu S, Downton M, Liu G, Du Y, et al. Combined losartan and nitro-oleic acid remarkably improves diabetic nephropathy in mice. American journal of physiology Renal physiology. 2013;305:F1555–1562. doi: 10.1152/ajprenal.00157.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schopfer FJ, Lin Y, Baker PR, Cui T, Garcia-Barrio M, Zhang J, et al. Nitrolinoleic acid: an endogenous peroxisome proliferator-activated receptor gamma ligand. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:2340–2345. doi: 10.1073/pnas.0408384102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rudolph V, Schopfer FJ, Khoo NK, Rudolph TK, Cole MP, Woodcock SR, et al. Nitro-fatty acid metabolome: saturation, desaturation, beta-oxidation, and protein adduction. The Journal of biological chemistry. 2009;284:1461–1473. doi: 10.1074/jbc.M802298200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Akaike T, Okamoto S, Sawa T, Yoshitake J, Tamura F, Ichimori K, et al. 8-nitroguanosine formation in viral pneumonia and its implication for pathogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:685–690. doi: 10.1073/pnas.0235623100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Terasaki Y, Akuta T, Terasaki M, Sawa T, Mori T, Okamoto T, et al. Guanine nitration in idiopathic pulmonary fibrosis and its implication for carcinogenesis. American journal of respiratory and critical care medicine. 2006;174:665–673. doi: 10.1164/rccm.200510-1580OC. [DOI] [PubMed] [Google Scholar]
- 65.Sawa T, Zaki MH, Okamoto T, Akuta T, Tokutomi Y, Kim-Mitsuyama S, et al. Protein S-guanylation by the biological signal 8-nitroguanosine 3′,5′-cyclic monophosphate. Nature chemical biology. 2007;3:727–735. doi: 10.1038/nchembio.2007.33. [DOI] [PubMed] [Google Scholar]
- 66.Fujii S, Sawa T, Ihara H, Tong KI, Ida T, Okamoto T, et al. The critical role of nitric oxide signaling, via protein S-guanylation and nitrated cyclic GMP, in the antioxidant adaptive response. The Journal of biological chemistry. 2010;285:23970–23984. doi: 10.1074/jbc.M110.145441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sawa T, Ihara H, Ida T, Fujii S, Nishida M, Akaike T. Formation, signaling functions, and metabolisms of nitrated cyclic nucleotide. Nitric oxide : biology and chemistry / official journal of the Nitric Oxide Society. 2013;34:10–18. doi: 10.1016/j.niox.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 68.Ito C, Saito Y, Nozawa T, Fujii S, Sawa T, Inoue H, et al. Endogenous nitrated nucleotide is a key mediator of autophagy and innate defense against bacteria. Molecular cell. 2013;52:794–804. doi: 10.1016/j.molcel.2013.10.024. [DOI] [PubMed] [Google Scholar]
- 69.Ida T, Sawa T, Ihara H, Tsuchiya Y, Watanabe Y, Kumagai Y, et al. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:7606–7611. doi: 10.1073/pnas.1321232111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nishida M, Sawa T, Kitajima N, Ono K, Inoue H, Ihara H, et al. Hydrogen sulfide anion regulates redox signaling via electrophile sulfhydration. Nature chemical biology. 2012;8:714–724. doi: 10.1038/nchembio.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fujii S, Akaike T. redox Signaling by 8-nitro-cyclic guanosine monophosphate: nitric oxide- and reactive oxygen species-derived electrophilic messenger. Antioxidants & redox signaling. 2013;19:1236–1246. doi: 10.1089/ars.2012.5067. [DOI] [PubMed] [Google Scholar]
- 72.Review T, LaBrecque DR, Abbas Z, Anania F, Ferenci P, Khan AG, et al. World Gastroenterology Organisation global guidelines: Nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Journal of clinical gastroenterology. 2014;48:467–473. doi: 10.1097/MCG.0000000000000116. [DOI] [PubMed] [Google Scholar]
- 73.Tateya S, Rizzo NO, Handa P, Cheng AM, Morgan-Stevenson V, Daum G, et al. Endothelial NO/cGMP/VASP signaling attenuates Kupffer cell activation and hepatic insulin resistance induced by high-fat feeding. Diabetes. 2011;60:2792–2801. doi: 10.2337/db11-0255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang W, Zhao C, Zhou J, Zhen Z, Wang Y, Shen C. Simvastatin ameliorates liver fibrosis via mediating nitric oxide synthase in rats with non-alcoholic steatohepatitis-related liver fibrosis. PloS one. 2013;8:e76538. doi: 10.1371/journal.pone.0076538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gu Q, Yang X, Lin L, Li S, Li Q, Zhong S, et al. Genetic ablation of solute carrier family 7a3a leads to hepatic steatosis in zebrafish during fasting. Hepatology. 2014;60:1929–1941. doi: 10.1002/hep.27356. [DOI] [PubMed] [Google Scholar]
- 76.Seth RK, Das S, Pourhoseini S, Dattaroy D, Igwe S, Ray JB, et al. M1 polarization bias and subsequent nonalcoholic steatohepatitis progression is attenuated by nitric oxide donor DETA NONOate via inhibition of CYP2E1-induced oxidative stress in obese mice. The Journal of pharmacology and experimental therapeutics. 2015;352:77–89. doi: 10.1124/jpet.114.218131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fujita K, Nozaki Y, Yoneda M, Wada K, Takahashi H, Kirikoshi H, et al. Nitric oxide plays a crucial role in the development/progression of nonalcoholic steatohepatitis in the choline-deficient, l-amino acid-defined diet-fed rat model. Alcoholism, clinical and experimental research. 2010;34(Suppl 1):S18–24. doi: 10.1111/j.1530-0277.2008.00756.x. [DOI] [PubMed] [Google Scholar]
- 78.Navarro LA, Wree A, Povero D, Berk MP, Eguchi A, Ghosh S, et al. Arginase 2 deficiency results in spontaneous steatohepatitis: A novel link between innate immune activation and hepatic de novo lipogenesis. Journal of hepatology. 2015;62:412–420. doi: 10.1016/j.jhep.2014.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chatterjee S, Ganini D, Tokar EJ, Kumar A, Das S, Corbett J, et al. Leptin is key to peroxynitrite- mediated oxidative stress and Kupffer cell activation in experimental non-alcoholic steatohepatitis. Journal of hepatology. 2013;58:778–784. doi: 10.1016/j.jhep.2012.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pasarin M, Abraldes JG, Rodriguez-Vilarrupla A, La Mura V, Garcia-Pagan JC, Bosch J. Insulin resistance and liver microcirculation in a rat model of early NAFLD. Journal of hepatology. 2011;55:1095–1102. doi: 10.1016/j.jhep.2011.01.053. [DOI] [PubMed] [Google Scholar]
- 81.Charbonneau A, Marette A. Inducible nitric oxide synthase induction underlies lipid-induced hepatic insulin resistance in mice: potential role of tyrosine nitration of insulin signaling proteins. Diabetes. 2010;59:861–871. doi: 10.2337/db09-1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Theise ND. Histopathology of Alcoholic Liver Disease. Clinical Liver Disease. 2013;2:64–67. doi: 10.1002/cld.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Deng XS, Deitrich RA. Ethanol metabolism and effects: nitric oxide and its interaction. Current clinical pharmacology. 2007;2:145–153. doi: 10.2174/157488407780598135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McKim SE, Gabele E, Isayama F, Lambert JC, Tucker LM, Wheeler MD, et al. Inducible nitric oxide synthase is required in alcohol-induced liver injury: studies with knockout mice. Gastroenterology. 2003;125:1834–1844. doi: 10.1053/j.gastro.2003.08.030. [DOI] [PubMed] [Google Scholar]
- 85.Maturu P, Reddy VD, Padmavathi P, Varadacharyulu N. Ethanol induced adaptive changes in blood for the pathological and toxicological effects of chronic ethanol consumption in humans. Experimental and toxicologic pathology : official journal of the Gesellschaft fur Toxikologische Pathologie. 2012;64:697–703. doi: 10.1016/j.etp.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 86.Baraona E, Zeballos GA, Shoichet L, Mak KM, Lieber CS. Ethanol consumption increases nitric oxide production in rats, and its peroxynitrite-mediated toxicity is attenuated by polyenylphosphatidylcholine. Alcoholism, clinical and experimental research. 2002;26:883–889. [PubMed] [Google Scholar]
- 87.Lo HW, Hsu SC, Ali-Seyed M, Gunduz M, Xia W, Wei Y, et al. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer cell. 2005;7:575–589. doi: 10.1016/j.ccr.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 88.Gao L, Zhou Y, Zhong W, Zhao X, Chen C, Chen X, et al. Caveolin-1 is essential for protecting against binge drinking-induced liver damage through inhibiting reactive nitrogen species. Hepatology. 2014;60:687–699. doi: 10.1002/hep.27162. [DOI] [PubMed] [Google Scholar]
- 89.Moon KH, Hood BL, Kim BJ, Hardwick JP, Conrads TP, Veenstra TD, et al. Inactivation of oxidized and S-nitrosylated mitochondrial proteins in alcoholic fatty liver of rats. Hepatology. 2006;44:1218–1230. doi: 10.1002/hep.21372. [DOI] [PubMed] [Google Scholar]
- 90.Shin HJ, Park YH, Kim SU, Moon HB, Park do S, Han YH, et al. Hepatitis B virus X protein regulates hepatic glucose homeostasis via activation of inducible nitric oxide synthase. The Journal of biological chemistry. 2011;286:29872–29881. doi: 10.1074/jbc.M111.259978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mihm S, Fayyazi A, Ramadori G. Hepatic expression of inducible nitric oxide synthase transcripts in chronic hepatitis C virus infection: relation to hepatic viral load and liver injury. Hepatology. 1997;26:451–458. doi: 10.1002/hep.510260228. [DOI] [PubMed] [Google Scholar]
- 92.Kandemir O, Polat A, Kaya A. Inducible nitric oxide synthase expression in chronic viral hepatitis and its relation with histological severity of disease. Journal of viral hepatitis. 2002;9:419–423. doi: 10.1046/j.1365-2893.2002.00382.x. [DOI] [PubMed] [Google Scholar]
- 93.Machida K, Cheng KT, Sung VM, Lee KJ, Levine AM, Lai MM. Hepatitis C virus infection activates the immunologic (type II) isoform of nitric oxide synthase and thereby enhances DNA damage and mutations of cellular genes. Journal of virology. 2004;78:8835–8843. doi: 10.1128/JVI.78.16.8835-8843.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dore GJ, Matthews GV, Rockstroh J. Future of hepatitis C therapy: development of direct-acting antivirals. Current opinion in HIV and AIDS. 2011;6:508–513. doi: 10.1097/COH.0b013e32834b87f8. [DOI] [PubMed] [Google Scholar]
- 95.Michaelis M, Michaelis R, Suhan T, Schmidt H, Mohamed A, Doerr HW, et al. Ribavirin inhibits angiogenesis by tetrahydrobiopterin depletion. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2007;21:81–87. doi: 10.1096/fj.06-6779com. [DOI] [PubMed] [Google Scholar]
- 96.Wohl BM, Smith AA, Kryger MB, Zelikin AN. Narrow therapeutic window of ribavirin as an inhibitor of nitric oxide synthesis is broadened by macromolecular prodrugs. Biomacromolecules. 2013;14:3916–3926. doi: 10.1021/bm401048s. [DOI] [PubMed] [Google Scholar]
- 97.Metz P, Dazert E, Ruggieri A, Mazur J, Kaderali L, Kaul A, et al. Identification of type I and type II interferon-induced effectors controlling hepatitis C virus replication. Hepatology. 2012;56:2082–2093. doi: 10.1002/hep.25908. [DOI] [PubMed] [Google Scholar]
- 98.Hazam RK, Deka M, Kar P. Role of nitric oxide synthase genes in hepatitis E virus infection. Journal of viral hepatitis. 2014;21:671–679. doi: 10.1111/jvh.12186. [DOI] [PubMed] [Google Scholar]
- 99.Raidl M, Pirker C, Schulte-Hermann R, Aubele M, Kandioler-Eckersberger D, Wrba F, et al. Multiple chromosomal abnormalities in human liver (pre)neoplasia. Journal of hepatology. 2004;40:660–668. doi: 10.1016/j.jhep.2003.12.020. [DOI] [PubMed] [Google Scholar]
- 100.Yeh SH, Chen PJ, Shau WY, Chen YW, Lee PH, Chen JT, et al. Chromosomal allelic imbalance evolving from liver cirrhosis to hepatocellular carcinoma. Gastroenterology. 2001;121:699–709. doi: 10.1053/gast.2001.27211. [DOI] [PubMed] [Google Scholar]
- 101.Wei W, Yang Z, Tang CH, Liu L. Targeted deletion of GSNOR in hepatocytes of mice causes nitrosative inactivation of O6-alkylguanine-DNA alkyltransferase and increased sensitivity to genotoxic diethylnitrosamine. Carcinogenesis. 2011;32:973–977. doi: 10.1093/carcin/bgr041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hoshida Y, Villanueva A, Kobayashi M, Peix J, Chiang DY, Camargo A, et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. The New England journal of medicine. 2008;359:1995–2004. doi: 10.1056/NEJMoa0804525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liu L, Hausladen A, Zeng M, Que L, Heitman J, Stamler JS. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature. 2001;410:490–494. doi: 10.1038/35068596. [DOI] [PubMed] [Google Scholar]
- 104.Jensen DE, Belka GK, Du Bois GC. S-Nitrosoglutathione is a substrate for rat alcohol dehydrogenase class III isoenzyme. The Biochemical journal. 1998;331(Pt 2):659–668. doi: 10.1042/bj3310659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yu DY, Moon HB, Son JK, Jeong S, Yu SL, Yoon H, et al. Incidence of hepatocellular carcinoma in transgenic mice expressing the hepatitis B virus X-protein. Journal of hepatology. 1999;31:123–132. doi: 10.1016/s0168-8278(99)80172-x. [DOI] [PubMed] [Google Scholar]
- 106.Park YH, Shin HJ, Kim SU, Kim JM, Kim JH, Bang DH, et al. iNOS promotes HBx-induced hepatocellular carcinoma via upregulation of JNK activation. Biochemical and biophysical research communications. 2013;435:244–249. doi: 10.1016/j.bbrc.2013.04.071. [DOI] [PubMed] [Google Scholar]
- 107.Bui-Nguyen TM, Pakala SB, Sirigiri DR, Martin E, Murad F, Kumar R. Stimulation of inducible nitric oxide by hepatitis B virus transactivator protein HBx requires MTA1 coregulator. The Journal of biological chemistry. 2010;285:6980–6986. doi: 10.1074/jbc.M109.065987. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 108.Machida K, Cheng KT, Lai CK, Jeng KS, Sung VM, Lai MM. Hepatitis C virus triggers mitochondrial permeability transition with production of reactive oxygen species, leading to DNA damage and STAT3 activation. Journal of virology. 2006;80:7199–7207. doi: 10.1128/JVI.00321-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Machida K, Tsukamoto H, Liu JC, Han YP, Govindarajan S, Lai MM, et al. c-Jun mediates hepatitis C virus hepatocarcinogenesis through signal transducer and activator of transcription 3 and nitric oxide-dependent impairment of oxidative DNA repair. Hepatology. 2010;52:480–492. doi: 10.1002/hep.23697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jaiswal M, LaRusso NF, Shapiro RA, Billiar TR, Gores GJ. Nitric oxide-mediated inhibition of DNA repair potentiates oxidative DNA damage in cholangiocytes. Gastroenterology. 2001;120:190–199. doi: 10.1053/gast.2001.20875. [DOI] [PubMed] [Google Scholar]
- 111.Tang CH, Wei W, Liu L. Regulation of DNA repair by S-nitrosylation. Biochimica et biophysica acta. 2012;1820:730–735. doi: 10.1016/j.bbagen.2011.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Xie G, Wang X, Wang L, Wang L, Atkinson RD, Kanel GC, et al. Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats. Gastroenterology. 2012;142:918–927. e916. doi: 10.1053/j.gastro.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Deleve LD, Wang X, Guo Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology. 2008;48:920–930. doi: 10.1002/hep.22351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Marrone G, Russo L, Rosado E, Hide D, Garcia-Cardena G, Garcia-Pagan JC, et al. The transcription factor KLF2 mediates hepatic endothelial protection and paracrine endothelial-stellate cell deactivation induced by statins. Journal of hepatology. 2013;58:98–103. doi: 10.1016/j.jhep.2012.08.026. [DOI] [PubMed] [Google Scholar]
- 115.Langer DA, Das A, Semela D, Kang-Decker N, Hendrickson H, Bronk SF, et al. Nitric oxide promotes caspase-independent hepatic stellate cell apoptosis through the generation of reactive oxygen species. Hepatology. 2008;47:1983–1993. doi: 10.1002/hep.22285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Iwakiri Y, Groszmann RJ. The hyperdynamic circulation of chronic liver diseases: from the patient to the molecule. Hepatology. 2006;43:S121–131. doi: 10.1002/hep.20993. [DOI] [PubMed] [Google Scholar]
- 117.Iwakiri Y. Endothelial dysfunction in the regulation of cirrhosis and portal hypertension. Liver international : official journal of the International Association for the Study of the Liver. 2012;32:199–213. doi: 10.1111/j.1478-3231.2011.02579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Iwakiri Y, Shah V, Rockey DC. Vascular Pathobiology in Chronic Liver Disease and Cirrhosis-Current Status and Future Directions. Journal of hepatology. 2014 doi: 10.1016/j.jhep.2014.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Liu S, Premont RT, Rockey DC. G-protein-coupled receptor kinase interactor-1 (GIT1) is a new endothelial nitric-oxide synthase (eNOS) interactor with functional effects on vascular homeostasis. The Journal of biological chemistry. 2012;287:12309–12320. doi: 10.1074/jbc.M111.320465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Liu S, Premont RT, Rockey DC. Endothelial Nitric-oxide Synthase (eNOS) Is Activated through G-protein-coupled Receptor Kinase-interacting Protein 1 (GIT1) Tyrosine Phosphorylation and Src Protein. The Journal of biological chemistry. 2014;289:18163–18174. doi: 10.1074/jbc.M113.521203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mookerjee RP, Mehta G, Balasubramaniyan V, Mohamed F, Davies N, Sharma V, et al. Hepatic Dimethylarginine-Dimethylaminohydrolase1 is Reduced in Cirrhosis and is a Target for Therapy in Portal Hypertension. Journal of hepatology. 2014 doi: 10.1016/j.jhep.2014.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Matei V, Rodriguez-Vilarrupla A, Deulofeu R, Garcia-Caldero H, Fernandez M, Bosch J, et al. Three-day tetrahydrobiopterin therapy increases in vivo hepatic NOS activity and reduces portal pressure in CCl4 cirrhotic rats. Journal of hepatology. 2008;49:192–197. doi: 10.1016/j.jhep.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 123.Matei V, Rodriguez-Vilarrupla A, Deulofeu R, Colomer D, Fernandez M, Bosch J, et al. The eNOS cofactor tetrahydrobiopterin improves endothelial dysfunction in livers of rats with CCl4 cirrhosis. Hepatology. 2006;44:44–52. doi: 10.1002/hep.21228. [DOI] [PubMed] [Google Scholar]
- 124.Wiest R, Cadelina G, Milstien S, McCuskey RS, Garcia-Tsao G, Groszmann RJ. Bacterial translocation up-regulates GTP-cyclohydrolase I in mesenteric vasculature of cirrhotic rats. Hepatology. 2003;38:1508–1515. doi: 10.1016/j.hep.2003.09.039. [DOI] [PubMed] [Google Scholar]
- 125.Wiest R, Das S, Cadelina G, Garcia-Tsao G, Milstien S, Groszmann RJ. Bacterial translocation in cirrhotic rats stimulates eNOS-derived NO production and impairs mesenteric vascular contractility. The Journal of clinical investigation. 1999;104:1223–1233. doi: 10.1172/JCI7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fiorucci S, Antonelli E, Brancaleone V, Sanpaolo L, Orlandi S, Distrutti E, et al. NCX-1000, a nitric oxide-releasing derivative of ursodeoxycholic acid, ameliorates portal hypertension and lowers norepinephrine-induced intrahepatic resistance in the isolated and perfused rat liver. Journal of hepatology. 2003;39:932–939. doi: 10.1016/s0168-8278(03)00393-3. [DOI] [PubMed] [Google Scholar]
- 127.Berzigotti A, Bellot P, De Gottardi A, Garcia-Pagan JC, Gagnon C, Spenard J, et al. NCX-1000, a nitric oxide-releasing derivative of UDCA, does not decrease portal pressure in patients with cirrhosis: results of a randomized, double-blind, dose-escalating study. The American journal of gastroenterology. 2010;105:1094–1101. doi: 10.1038/ajg.2009.661. [DOI] [PubMed] [Google Scholar]
- 128.Fallowfield JA, Hayden AL, Snowdon VK, Aucott RL, Stutchfield BM, Mole DJ, et al. Relaxin modulates human and rat hepatic myofibroblast function and ameliorates portal hypertension in vivo. Hepatology. 2014;59:1492–1504. doi: 10.1002/hep.26627. [DOI] [PubMed] [Google Scholar]
- 129.Duong HT, Dong Z, Su L, Boyer C, George J, Davis TP, et al. The Use of Nanoparticles to Deliver Nitric Oxide to Hepatic Stellate Cells for Treating Liver Fibrosis and Portal Hypertension. Small. 2015 doi: 10.1002/smll.201402870. [DOI] [PubMed] [Google Scholar]
- 130.La Mura V, Pasarin M, Rodriguez-Vilarrupla A, Garcia-Pagan JC, Bosch J, Abraldes JG. Liver sinusoidal endothelial dysfunction after LPS administration: A role for inducible-nitric oxide synthase. Journal of hepatology. 2014 doi: 10.1016/j.jhep.2014.07.014. [DOI] [PubMed] [Google Scholar]
- 131.Ribera J, Pauta M, Melgar-Lesmes P, Tugues S, Fernandez-Varo G, Held KF, et al. Increased nitric oxide production in lymphatic endothelial cells causes impairment of lymphatic drainage in cirrhotic rats. Gut. 2013;62:138–145. doi: 10.1136/gutjnl-2011-300703. [DOI] [PubMed] [Google Scholar]
- 132.Chung C, Iwakiri Y. The lymphatic vascular system in liver diseases: its role in ascites formation. Clinical and molecular hepatology. 2013;19:99–104. doi: 10.3350/cmh.2013.19.2.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bjornsson E. The natural history of drug-induced liver injury. Seminars in liver disease. 2009;29:357–363. doi: 10.1055/s-0029-1240004. [DOI] [PubMed] [Google Scholar]
- 134.Diesen DL, Kuo PC. Nitric oxide and redox regulation in the liver: part II. Redox biology in pathologic hepatocytes and implications for intervention. The Journal of surgical research. 2011;167:96–112. doi: 10.1016/j.jss.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Abdelmegeed MA, Moon KH, Chen C, Gonzalez FJ, Song BJ. Role of cytochrome P450 2E1 in protein nitration and ubiquitin-mediated degradation during acetaminophen toxicity. Biochemical pharmacology. 2010;79:57–66. doi: 10.1016/j.bcp.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Abdelmegeed MA, Jang S, Banerjee A, Hardwick JP, Song BJ. Robust protein nitration contributes to acetaminophen-induced mitochondrial dysfunction and acute liver injury. Free radical biology & medicine. 2013;60:211–222. doi: 10.1016/j.freeradbiomed.2013.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Datta G, Fuller BJ, Davidson BR. Molecular mechanisms of liver ischemia reperfusion injury: insights from transgenic knockout models. World journal of gastroenterology : WJG. 2013;19:1683–1698. doi: 10.3748/wjg.v19.i11.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Duranski MR, Elrod JW, Calvert JW, Bryan NS, Feelisch M, Lefer DJ. Genetic overexpression of eNOS attenuates hepatic ischemia-reperfusion injury. American journal of physiology Heart and circulatory physiology. 2006;291:H2980–2986. doi: 10.1152/ajpheart.01173.2005. [DOI] [PubMed] [Google Scholar]
- 139.Duranski MR, Greer JJ, Dejam A, Jaganmohan S, Hogg N, Langston W, et al. Cytoprotective effects of nitrite during in vivo ischemia-reperfusion of the heart and liver. The Journal of clinical investigation. 2005;115:1232–1240. doi: 10.1172/JCI22493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhang C, Liao Y, Li Q, Chen M, Zhao Q, Deng R, et al. Recombinant adiponectin ameliorates liver ischemia reperfusion injury via activating the AMPK/eNOS pathway. PloS one. 2013;8:e66382. doi: 10.1371/journal.pone.0066382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hines IN, Harada H, Bharwani S, Pavlick KP, Hoffman JM, Grisham MB. Enhanced post-ischemic liver injury in iNOS-deficient mice: a cautionary note. Biochemical and biophysical research communications. 2001;284:972–976. doi: 10.1006/bbrc.2001.5069. [DOI] [PubMed] [Google Scholar]
- 142.Hamada T, Duarte S, Tsuchihashi S, Busuttil RW, Coito AJ. Inducible nitric oxide synthase deficiency impairs matrix metalloproteinase-9 activity and disrupts leukocyte migration in hepatic ischemia/reperfusion injury. The American journal of pathology. 2009;174:2265–2277. doi: 10.2353/ajpath.2009.080872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kawachi S, Hines IN, Laroux FS, Hoffman J, Bharwani S, Gray L, et al. Nitric oxide synthase and postischemic liver injury. Biochemical and biophysical research communications. 2000;276:851–854. doi: 10.1006/bbrc.2000.3559. [DOI] [PubMed] [Google Scholar]
- 144.Moon KH, Hood BL, Mukhopadhyay P, Rajesh M, Abdelmegeed MA, Kwon YI, et al. Oxidative inactivation of key mitochondrial proteins leads to dysfunction and injury in hepatic ischemia reperfusion. Gastroenterology. 2008;135:1344–1357. doi: 10.1053/j.gastro.2008.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lang JD, Jr., Teng X, Chumley P, Crawford JH, Isbell TS, Chacko BK, et al. Inhaled NO accelerates restoration of liver function in adults following orthotopic liver transplantation. The Journal of clinical investigation. 2007;117:2583–2591. doi: 10.1172/JCI31892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Sinha SS, Shiva S, Gladwin MT. Myocardial protection by nitrite: evidence that this reperfusion therapeutic will not be lost in translation. Trends in cardiovascular medicine. 2008;18:163–172. doi: 10.1016/j.tcm.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 147.Li W, Meng Z, Liu Y, Patel RP, Lang JD. The hepatoprotective effect of sodium nitrite on cold ischemia-reperfusion injury. Journal of transplantation. 2012;2012:635179. doi: 10.1155/2012/635179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Baker PR, Schopfer FJ, Sweeney S, Freeman BA. Red cell membrane and plasma linoleic acid nitration products: synthesis, clinical identification, and quantitation. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:11577–11582. doi: 10.1073/pnas.0402587101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Baker PR, Lin Y, Schopfer FJ, Woodcock SR, Groeger AL, Batthyany C, et al. Fatty acid transduction of nitric oxide signaling: multiple nitrated unsaturated fatty acid derivatives exist in human blood and urine and serve as endogenous peroxisome proliferator-activated receptor ligands. The Journal of biological chemistry. 2005;280:42464–42475. doi: 10.1074/jbc.M504212200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sawa T, Ihara H, Akaike T. Antioxidant effect of a nitrated cyclic nucleotide functioning as an endogenous electrophile. Current topics in medicinal chemistry. 2011;11:1854–1860. doi: 10.2174/156802611796235080. [DOI] [PubMed] [Google Scholar]
- 151.Tripathi DN, Chowdhury R, Trudel LJ, Tee AR, Slack RS, Walker CL, et al. Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E2950–2957. doi: 10.1073/pnas.1307736110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sarkar S, Korolchuk VI, Renna M, Imarisio S, Fleming A, Williams A, et al. Complex inhibitory effects of nitric oxide on autophagy. Molecular cell. 2011;43:19–32. doi: 10.1016/j.molcel.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Czaja MJ, Ding WX, Donohue TM, Jr., Friedman SL, Kim JS, Komatsu M, et al. Functions of autophagy in normal and diseased liver. Autophagy. 2013;9:1131–1158. doi: 10.4161/auto.25063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Liu K, Zhao E, Ilyas G, Lalazar G, Lin Y, Haseeb M, et al. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy. 2015:0. doi: 10.1080/15548627.2015.1009787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hara Y, Yanatori I, Ikeda M, Kiyokage E, Nishina S, Tomiyama Y, et al. Hepatitis C virus core protein suppresses mitophagy by interacting with parkin in the context of mitochondrial depolarization. The American journal of pathology. 2014;184:3026–3039. doi: 10.1016/j.ajpath.2014.07.024. [DOI] [PubMed] [Google Scholar]
- 156.Hernandez-Gea V, Ghiassi-Nejad Z, Rozenfeld R, Gordon R, Fiel MI, Yue Z, et al. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142:938–946. doi: 10.1053/j.gastro.2011.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lan SH, Wu SY, Zuchini R, Lin XZ, Su IJ, Tsai TF, et al. Autophagy suppresses tumorigenesis of hepatitis B virus-associated hepatocellular carcinoma through degradation of microRNA-224. Hepatology. 2014;59:505–517. doi: 10.1002/hep.26659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Vasudevan D, Thomas DD. Insights into the diverse effects of nitric oxide on tumor biology. Vitamins and hormones. 2014;96:265–298. doi: 10.1016/B978-0-12-800254-4.00011-8. [DOI] [PubMed] [Google Scholar]
- 159.Fiorucci S, Mencarelli A, Palazzetti B, Del Soldato P, Morelli A, Ignarro LJ. An NO derivative of ursodeoxycholic acid protects against Fas-mediated liver injury by inhibiting caspase activity. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:2652–2657. doi: 10.1073/pnas.041603898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Liu J, Li C, Waalkes MP, Clark J, Myers P, Saavedra JE, et al. The nitric oxide donor, VPYRRO/NO, protects against acetaminophen-induced hepatotoxicity in mice. Hepatology. 2003;37:324–333. doi: 10.1053/jhep.2003.50063. [DOI] [PubMed] [Google Scholar]
- 161.Edwards C, Feng HQ, Reynolds C, Mao L, Rockey DC. Effect of the nitric oxide donor VPYRRO/NO on portal pressure and sinusoidal dynamics in normal and cirrhotic mice. American journal of physiology Gastrointestinal and liver physiology. 2008;294:G1311–1317. doi: 10.1152/ajpgi.00368.2007. [DOI] [PubMed] [Google Scholar]
- 162.Maslak E, Zabielski P, Kochan K, Kus K, Jasztal A, Sitek B, et al. The liver-selective NO donor, V-PYRRO/NO, protects against liver steatosis and improves postprandial glucose tolerance in mice fed high fat diet. Biochemical pharmacology. 2015;93:389–400. doi: 10.1016/j.bcp.2014.12.004. [DOI] [PubMed] [Google Scholar]
- 163.Ricciardi R, Foley DP, Quarfordt SH, Saavedra JE, Keefer LK, Wheeler SM, et al. VPYRRO/NO: an hepato-selective nitric oxide donor improves porcine liver hemodynamics and function after ischemia reperfusion. Transplantation. 2001;71:193–198. doi: 10.1097/00007890-200101270-00004. [DOI] [PubMed] [Google Scholar]
- 164.Xu J, Zeng F, Wu H, Hu C, Yu C, Wu S. Preparation of a mitochondria-targeted and NO-releasing nanoplatform and its enhanced pro-apoptotic effect on cancer cells. Small. 2014;10:3750–3760. doi: 10.1002/smll.201400437. [DOI] [PubMed] [Google Scholar]
- 165.Das A, Mukherjee P, Singla SK, Guturu P, Frost MC, Mukhopadhyay D, et al. Fabrication and characterization of an inorganic gold and silica nanoparticle mediated drug delivery system for nitric oxide. Nanotechnology. 2010;21:305102. doi: 10.1088/0957-4484/21/30/305102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ibrahim M, Farghaly E, Gomaa W, Kelleni M, Abdelrahman AM. Nitro-aspirin is a potential therapy for non alcoholic fatty liver disease. European journal of pharmacology. 2011;659:289–295. doi: 10.1016/j.ejphar.2011.03.016. [DOI] [PubMed] [Google Scholar]
- 167.Ling Y, Ye X, Zhang Z, Zhang Y, Lai Y, Ji H, et al. Novel nitric oxide-releasing derivatives of farnesylthiosalicylic acid: synthesis and evaluation of antihepatocellular carcinoma activity. Journal of medicinal chemistry. 2011;54:3251–3259. doi: 10.1021/jm1014814. [DOI] [PubMed] [Google Scholar]
- 168.Fu J, Liu L, Huang Z, Lai Y, Ji H, Peng S, et al. Hybrid molecule from O2-(2,4-dinitrophenyl)diazeniumdiolate and oleanolic acid: a glutathione S-transferase pi-activated nitric oxide prodrug with selective anti-human hepatocellular carcinoma activity and improved stability. Journal of medicinal chemistry. 2013;56:4641–4655. doi: 10.1021/jm400393u. [DOI] [PubMed] [Google Scholar]